

Herpetic keratitis (HK) is the leading cause of blindness by an infectious agent in the developed world. This disease can occur after reactivation of herpes simplex virus 1 in the trigeminal ganglia, leading to dissemination of virus to, and infection of, the cornea. A clinical paradox is evidenced by the bilateral presence of latent viral genomes in both trigeminal ganglia, while for any given patient the disease is unilateral with recurrences in a single eye. Our study links the kinetics of early infection to unilateral disease phenomenon and demonstrates protection against viral reactivation when kinetics are exploited. Our results have direct implications in the understanding of human disease pathogenesis and immunotherapeutic strategies for the treatment of HK and viral reactivations.
KEYWORDS: HSV-1, latency, reactivation, trigeminal ganglion, neurons, superinfection, keratitis
ABSTRACT
Ocular herpes simplex keratitis (HSK) is a consequence of viral reactivations from trigeminal ganglia (TG) and occurs almost exclusively in the same eye in humans. In our murine oro-ocular (OO) model, herpes simplex virus 1 (HSV-1) inoculation in one side of the lip propagates virus to infect the ipsilateral TG. Replication here allows infection of the brainstem and infection of the contralateral TG. Interestingly, HSK was observed in our OO model only from the eye ipsilateral to the site of lip infection. Thus, unilateral restriction of HSV-1 may be due to differential kinetics of virus arrival in the ipsilateral versus contralateral TG. We inoculated mice with HSV-1 reporter viruses and then superinfected them to monitor changes in acute- and latent-phase gene expression in TG after superinfection compared to the control (single inoculation). Delaying superinfection by 4 days after initial right lip inoculation elicited failed superinfecting-virus gene expression and eliminated clinical signs of disease. Initial inoculation with thymidine kinase-deficient HSV-1 (TKdel) completely abolished reactivation of wild-type (WT) superinfecting virus from TG during the latent stage. In light of these seemingly failed infections, viral genome was detected in both TG. Our data demonstrate that inoculation of HSV-1 in the lip propagates virus to both TG, but with delay in reaching the TG contralateral to the side of lip infection. This delay is responsible for restricting viral replication to the ipsilateral TG, which abrogates ocular disease and viral reactivations from the contralateral side. These observations may help to understand why HSK is observed unilaterally in humans, and they provide insight into vaccine strategies to protect against HSK.
IMPORTANCE Herpetic keratitis (HK) is the leading cause of blindness by an infectious agent in the developed world. This disease can occur after reactivation of herpes simplex virus 1 in the trigeminal ganglia, leading to dissemination of virus to, and infection of, the cornea. A clinical paradox is evidenced by the bilateral presence of latent viral genomes in both trigeminal ganglia, while for any given patient the disease is unilateral with recurrences in a single eye. Our study links the kinetics of early infection to unilateral disease phenomenon and demonstrates protection against viral reactivation when kinetics are exploited. Our results have direct implications in the understanding of human disease pathogenesis and immunotherapeutic strategies for the treatment of HK and viral reactivations.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) belongs to the Herpesviridae, which are widespread in nature and have the characteristic feature of establishing lifelong latency in the infected host (1). The unique natural host of HSV-1 is humans, in whom primary infection occurs almost always via the oral mucosae (2, 3). Subsequently, the nucleocapsid travels by retrograde axonal transport to neuronal cell bodies (4) located in the trigeminal ganglia (TG), where the virus establishes a latent infection. The main sites of HSV-1 latency are the TG, which drive sensitive innervation of the face (5, 6) and where the viral genome is retained for the entirety of the host’s life (7). Reactivations of these latent reservoirs can propagate infection of various ocular tissues, leading to ocular herpetic disease (OHD). The prevalence of OHD has been estimated at 150/100,000 inhabitants in the United States (8), and the annual incidence ranges between 12 and 30 per 100,000 inhabitants (8–10). Corneal recurrence of herpetic keratitis (HK) is the most common clinical presentation of OHD (11). While some HK can resolve without further complications, the cumulative risk of one or more recurrences is greater than 60% in persons over 20 years old (11). The risk of corneal opacities increases with the number of relapses. Hence, HK is a sight-threatening disease, and 10-year follow-up has shown that visual acuity is below 20/200 in more than 10% of affected eyes (10).
One of the most striking properties of HK is the unilateral nature of the recurrences in almost all patients (6, 11), despite similar latent HSV-1 DNA loads in both the left and right TG (5, 12, 13). This clinical-biological discordance suggests that the presence of viral genome in the TG is not sufficient for precipitating a reactivation event. Furthermore, different animal infection models and in vitro studies have confirmed that under conditions of sequential infection, immune (innate and adaptive) and HSV-1-mediated (superinfection exclusion) mechanisms influence the severity of acute-phase clinical signs and the establishment of latency by the superinfecting strains (14–18). However, these mechanisms are unable to control the spread of superinfecting HSV-1 to neuronal ganglia or to prevent reactivations completely. These studies support the idea that reactivations of HSV-1 to cause recurrent disease are heavily influenced by the sequence of infection: the primary virus infecting the host attenuates the infectivity of superinfections and their ability to reactivate from latency, while the primary virus retains its own ability to reactivate.
The profound unilateral reactivation phenomenon of HK, in light of bilateral latent HSV-1 infection, begs the question of how a primary infection can influence outcomes of viral superinfection of the neuronal network. To explore these concepts, and to investigate how the acute phase and virus latent state (linked to HK) were affected by infection kinetics, we took advantage of a well-defined mouse model of oral-ocular (OO) HSV-1 infection (19–22), which mimics several of the key aspects of OHD in humans. After precise primary inoculation into the oral tissue segregated to the left side of the upper lip, the ipsilateral TG (left) supports active virus replication from 4 days after primary infection and acute keratitis occurs in the ipsilateral eye from 6 days postinfection (DPI). During this time the virus also reaches the contralateral TG (right side), although there is no infection of the right eye. Once the acute phase of the infection has resolved, HSV-1 latency is detectable in both TG from 28 DPI. While the latent HSV-1 DNA loads are similar in the TG (22), as described for humans (5, 12, 13), neurons expressing high levels of latency associated transcript (LAT) are almost exclusively detected in the TG ipsilateral to the lip inoculation site (21, 22). We have observed that reactivation from latency occurred almost exclusively from one TG, the TG ipsilateral to the site of inoculation, mimicking the unilateral nature of HK in humans (6).
We hypothesized that unilateral viral reactivation in animals and the ocular disease patterns in humans could be related to the triggering of early events, induced during or just after the inoculation process, that eventually dictate the long-term establishment of latency and unilateral reactivation. We explored this idea using a dual-inoculation procedure in which HSV-1 reporter viruses were inoculated into the right upper lip of mice to initiate viral infection, while virulent wild-type virus was inoculated into the contralateral lip with delay. This study allowed us to determine how altered HSV-1 infection kinetics would influence superinfecting virus acute phase, viral dissemination, and latency establishment.
RESULTS
Unilateral lip inoculation of wild-type HSV-1 SC16 results in unilateral reactivation in mice.
HSV-1 inoculation in the oral-ocular (OO) model occurs in the upper lip and is localized to the center of one side, at the mucocutaneous border. After acute-phase replication in the lip, the virus disseminates to both left and right TG, as previously demonstrated by an abundance of viral DNA and bilateral transcription of ICP0 and LAT mRNA (21, 22). To demonstrate the unilateral HSV-1 reactivation phenomenon, mice (n =14) were inoculated in the left lip with wild-type SC16 virus. Most animals (12/14) survived the acute phase of infection and were sacrificed at 28 DPI. TG were harvested and HSV-1 reactivation from individual TG was induced by ex vivo culture. Infectious virus particles were generated from 50% of ipsilateral (left) TG cultures, while no contralateral (right) TG cultures generated infectious virus (Fig. 1A). A second group of mice (n = 5) infected via the same process were also harvested at 28 DPI, and HSV-1 genomes were detected by quantitative PCR (qPCR) targeting the gG coding sequence (Fig. 1B). HSV-1 DNA was detected in abundance in all TG, with no significant difference between HSV-1 DNA detected in left or right TG. These data demonstrate that after lip infection restricted to one side, HSV-1 is propagated to the both left and right TG. However, HSV-1 reactivations from latency occur unilaterally from the TG ipsilateral to HSV-1 inoculation.
FIG 1.

HSV-1 SC16 reactivation and viral DNA from TG harvested during latency. BALB/c mice were inoculated in the left upper lip with wild-type HSV-1 SC16. At 28 DPI (i.e., a time known to correspond with latent phase of TG infection in the OO model), mice were sacrificed. (A) TG from mice (n = 12) were harvested and incubated individually in culture medium (CM) for 14 days. Aliquots of medium were removed every 2 days and compensated with fresh CM. Infectious particle release (reactivation) from aliquots was measured by plaque assay. Bars represent the percentages of TG cultures from individual mice in which plaques developed at any given time. **, P < 0.01 (Kruskal-Wallis test). (B) DNA was extracted and qPCR performed targeting the gG coding sequence or mouse GAPDH gene, according to the method of Cavallero et al. (22). Results were determined as the average numbers of gG genome copies relative to GAPDH DNA, and bars represent the 95% confidence intervals. ns, not significant.
Delayed kinetics of infection block the transcriptional activity of superinfecting virus in the ipsilateral TG.
To further characterize the phenomenon of unilateral HSV-1 reactivation from the ipsilateral TG, we questioned whether the naturally delayed kinetics of viral arrival to the TG after lip infection (i.e., 4 DPI in TG ipsilateral and 6 DPI in TG contralateral to infection [19]) was involved in restricting virus reactivation to the ipsilateral TG. To explore this concept, we performed an initial virus inoculation in the right lip followed by superinfection in the left lip. The superinfecting left lip inoculations were performed in different groups of mice on different days, so that superinfecting virus would disseminate to the ipsilateral (left) TG at delayed time points in relation to the initial infection on day 0 (Fig. 2A). The right side infection aimed to produce day 4 right TG and day 6 left TG infection kinetics, while the delays in superinfection of the left lip would achieve infection of the left TG before, during, or after this 4- to 6-day window. To distinguish the initial infecting virus from superinfecting virus, we utilized reporter viruses all derived from the SC16 parent strain: gJ-GFP for the initial right-side inoculation and gJ-LacZ for superinfection in the left side. We previously observed that mice infected with gJ-GFP lack signs of clinical morbidity (no death or ocular signs) during the first 10 days after inoculation, despite the presence of replicating particles in the TG at day 4 after inoculation, albeit to a much lesser extent than with wild-type SC16 (unpublished observations).
FIG 2.

Quantification of HSV-1 superinfecting replication in the TG after initial infection and delayed superinfection. In each group, BALB/c mice (n = 3) were inoculated initially in the right upper lip with gJ-GFP (1 μl of PBS was delivered for mock infection) and superinfected in the left upper lip with gJ-LacZ. Superinfection was performed at different intervals per group after the primary infection on days 0 to 6, or mice received only gJ-LacZ in the left lip for group C. All mice were sacrificed during the acute phase of infection at 6 DPSI with gJ-LacZ. TG frontal cryosections (10 μm) were prepared and stained with X-Gal reactant and counterstained with neutral red. β-Galactosidase+ve neurons were enumerated. The bracket indicates groups corresponding to the period in which β-galactosidase+ve neurons were not detected (when superinfection occurred between 4 and 6 days after initial infection); data points indicate average numbers of cells ± SEMs.
A group of mice (n = 24) was inoculated with gJ-GFP in the right lip. Between 0 and 6 days later, subgroups of mice (n = 3) were superinfected with gJ-LacZ in the left lip. Left and right TG were harvested from these animals that were sacrificed 6 days postsuperinfection (DPSI), and fixed sections were analyzed for superinfecting virus gJ-LacZ reporter gene activity by enumeration of β-galactosidase-positive (β-galactosidase+ve) neurons in both TG (Fig. 2B). In the absence of an initial inoculation, or when superinfection was performed up to 3 days after initial inoculation, β-galactosidase+ve neurons were frequently observed in left (ipsilateral to the site of superinfection) TG (11/12). Under all conditions, and in the vast majority of mice (22/24), no β-galactosidase+ve neurons could be detected in the TG contralateral to the site of superinfection (right TG). Strikingly, no β-galactosidase+ve neurons could be detected in either the left or right TG from all mice that were superinfected at 4 DPI and beyond (9/9; highlighted by bracket in Fig. 2B). These data demonstrated that any delay between initial infection and contralateral superinfection in the lips leads to a reduction in the numbers of cells harboring viral replication in the TG ipsilateral to the site of superinfection during the acute phase. Furthermore, a 4-day delay or more abolished superinfecting viral gene expression in the TG ipsilateral to the site of superinfection.
To further explore this phenomenon, we reproduced this sequence of inoculation for all following experiments (i.e., an interval of 4 days between the initial infection in the right lip and superinfection in the left lip). To determine if the elimination of β-galactosidase activity of superinfecting gJ-LacZ virus was a consequence of restricted virus migration to TG, 2 additional groups of mice (n = 10) were initially inoculated in the right lip with either gJ-GFP or a mock inoculum. Superinfection with gJ-LacZ was performed at 4 DPI in the left lip in all mice. Enumeration of β-galactosidase+ve neurons at 6 DPSI identified gJ-LacZ virus transcriptional activity in mice receiving an initial mock inoculation with phosphate-buffered saline (PBS) (Fig. 3A). The vast majority of β-galactosidase+ve neurons were found in the TG ipsilateral (left) to the site of inoculation with gJ-LacZ (62 ± 33.5; range, 21 to 138) in initially mock-infected mice, compared to extremely low numbers of β-galactosidase+ve neurons in some (4/10) contralateral (right) TG (2 ± 3.9; range, 0 to 9). In contrast, both left and right TG from the 10 mice that received an initial gJ-GFP inoculation in the right lip before superinfection with gJ-LacZ in the left lip lacked β-galactosidase+ve neurons. This reduction in the number of cells harboring lytic viral replication, between initial mock- or gJ-GFP-infected groups, was significant (P < 0.01). Using an additional group of mice, which underwent the same inoculation procedure, qPCR for LacZ sequence was performed to detect superinfecting gJ-LacZ genomic DNA load from TGs harvested at 6 DPSI (n = 6 mice per group). Superinfecting virus DNA, relative to host glyceraldehyde-3-phosphate dehydrogenase (GAPDH), was abundant in both left and right TG whether mice received an initial right-lip gJ-GFP inoculation (left TG, 8.45E−03 ± 1.32E−02; right TG, 4.23E−03 ± 4.06E−03) or a mock inoculation (left TG, 1.73E−02 ± 1.34E−02; right TG, 9.21E−03 ± 1.68E−02) (Fig. 3B). Combined, these data demonstrated that superinfecting virus reporter gene activity was restricted to the TG ipsilateral to the site of lip infection (left). Thus, an initial infection in the opposite side (right) 4 days prior led to almost complete attenuation of superinfecting virus transcriptional activity in the TG without blocking the dissemination of virus to the TG.
FIG 3.
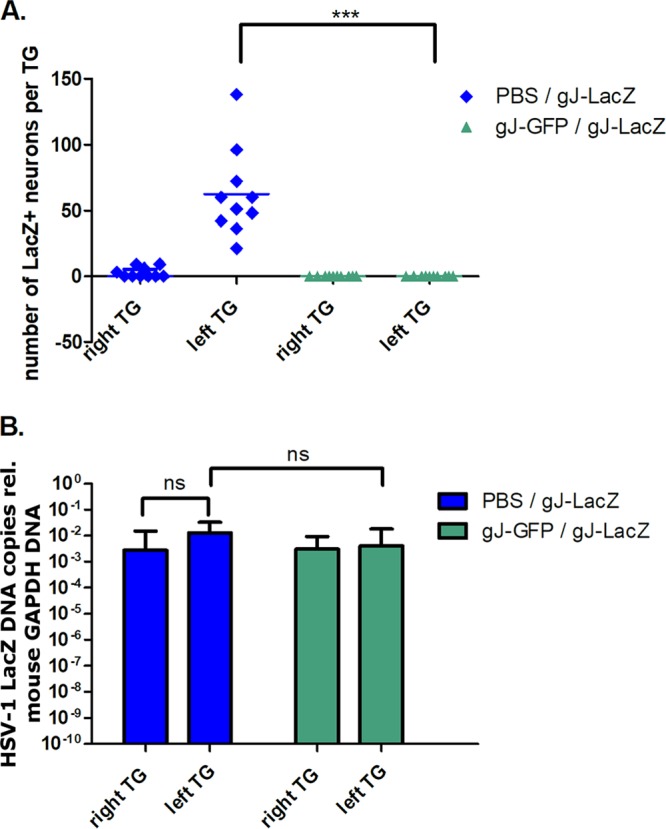
Transcriptional activity of superinfecting gJ-LacZ in TG of mice inoculated 4 days prior with gJ-GFP. Mice were initially infected in the right upper lip with gJ-GFP (or PBS for mock infection). Four days later, mice were superinfected with gJ-LacZ in the left upper lip and sacrificed 6 DPSI with gJ-LacZ. (A) TG frontal cryosections (10 μm) were prepared and stained with X-Gal reactant and counterstained with neutral red. β-Galactosidase+ve neurons were enumerated. Symbols represent the number of β-galactosidase+ve neurons from individual TG, and the horizontal bar represents the average. ***, P < 0.001 (Kruskal-Wallis test). (B) DNA was extracted from whole TG samples (n = 6 per group) and used as the template DNA for qPCR with primers targeting the lacZ gene or mouse GAPDH gene. The number of gJ-LacZ genome copies per TG was determined relative to GAPDH. Bars indicate the average numbers of gJ-LacZ genome copies relative to GAPDH DNA, and error bars represent the 95% confidence intervals.
HSV-1 infection in the lip leads to inhibition of mortality and disease and a major impact on the establishment of latency by a superinfecting virus.
In the following series of experiments, we used the LβA reporter virus for left-side superinfection to explore whether a primary infection with the attenuated gJ-GFP virus would translate into protection against LβA-associated diseases and mortality. While the LβA reporter virus has an insertion of the lacZ gene within the LAT locus, this reporter virus produces a virulent phenotype causing ocular disease and mortality in lip-infected mice comparable to that of the parental SC16 wild type (20).
Two groups of mice (n = 30 per group) were initially mock infected, or initially infected with gJ-GFP, in the right lip. All mice were then superinfected with LβA in the left lip 4 days later. During the acute phase, mice that received the initial mock inoculation exhibited signs of ocular diseases (67%), which included corneal clouding, corneal neovascularization, and/or iris atrophy with pupil mydriasis (Fig. 4A). Furthermore, 40% experienced death during the first 10 days, similar to previous observations of acute-phase LβA infection (Fig. 4B) (20). In contrast, when mice received the initial gJ-GFP inoculation before LβA, there were no signs of ocular morbidity, or mortality (Fig. 4A and B; endpoint chi-squared 2-tailed test, P < 0.001 for both panels A and B).
FIG 4.

Mortality, ocular disease, and LAT promoter activity during latency phase of LβA superinfection are extinguished in mice receiving an inoculation of gJ-GFP 4 days prior. BALB/c mice were infected in the right upper lip with gJ-GFP (or PBS for mock infection). At 4 DPI, mice were superinfected in the left upper lip with LβA. Ocular disease (corneal clouding, corneal neovascularization, and iris atrophy with pupil mydriasis) (A) and deaths (B) were recorded. At 28 DPSI with LβA, mice were sacrificed. Endpoint chi-squared 2-tailed test was performed for groups in panels A and B. *, P < 0.001. (C) TG frontal cryosections (10 μm) were prepared and stained with X-Gal reactant and counterstained with neutral red, and β-galactosidase+ve neurons were enumerated. Symbols represent the numbers of β-galactosidase+ve neurons from individual TG, and the horizontal bar represents the average. **, P < 0.01 (Kruskal-Wallis test). (D) DNA was extracted from whole TG samples (n = 6 per group) and used as the template DNA for qPCR with primers targeting the lacZ gene or GAPDH gene. The number of gJ-LacZ genome copies per TG was determined relative to GAPDH. Bars indicate the average numbers of gJ-LacZ genome copies relative to GAPDH DNA, and error bars represent the 95% confidence intervals.
At 28 DPSI, corresponding to the superinfecting virus latency phase, mice (n = 6) from each group were sacrificed and fixed TG sections were analyzed by microscopy for β-galactosidase+ve neurons, as LacZ expression is a surrogate marker of LAT promoter activity in the LβA strain (23). In initially mock-infected mice, significant numbers of β-galactosidase+ve neurons were counted in the left TG (199 ± 170.7; range, 48 to 498), the TG ipsilateral to the site of LβA superinfection (Fig. 4C). In stark contrast to the mock-infected mice, only a few β-galactosidase+ve neurons were found in the TG ipsilateral to LβA superinfection when mice were initially infected with gJ-GFP (3.5 ± 4.4; range, 0 to 9; mock versus gJ-GFP initial infection, P < 0.01). These reduced numbers of positive neurons were similar to those elicited in the TG contralateral to superinfection of initially mock-infected mice (mock right TG, 3.0 ± 3.8; range, 0 to 9) and also the TG contralateral to superinfection in initially gJ-GFP-infected mice (gJ-GFP right TG, 0). The load of superinfecting LβA DNA was quantified in left and right TG to rule out restricted viral dissemination as a cause for the reduction in numbers of β-galactosidase+ve neurons. LβA-superinfected animals (n = 6), which were initially mock or gJ-GFP infected, were sacrificed, and DNA was extracted from TGs. Quantification of LβA by qPCR, relative to host GAPDH, demonstrated no significant difference in superinfecting LβA genome loads between left and right TGs or between TGs of mice initially mock or gJ-GFP infected (gJ-GFP or mock: P > 0.05 for all comparisons) (Fig. 4D). These data demonstrated that while LβA virus disseminated to both TG during the acute phase, an initial infection 4 days prior in the lip opposite to superinfection prevented superinfecting LβA-mediated ocular disease and mortality without blocking viral dissemination and included the attenuation of LAT promoter activity during latent phase.
HSV-1 TKdel mutant infection in the lip attenuates acute and latent superinfecting virus activity and eliminates reactivation from latency.
Our original objective was to determine how altering kinetics of infection would influence superinfections, specifically. Thus, to avoid bias toward reactivation that might be elicited by initial reporter virus reactivations in the TG explant method (24), we sought a virus mutant that would be unable to reactivate from TG for use as the initial inoculating virus. Previous studies have shown that HSV-1 mutants deficient for thymidine kinase (TK) expression were unable to replicate in neurons (nonneurovirulent) but could still enter latency and persist in neuronal cells of immunocompetent animals (24). Ideally, these mutants could not reactivate from a latent state without rescue by nondefective virus (24–28). We utilized a TKdel mutant derived from the SC16 parent strain for initial infections in reactivation studies (26). Nonreactivatable TKdel allowed us to observe how reactivation from latency of the superinfecting virus alone would be influenced by the altered kinetics introduced by the earlier initial infection.
Mice were initially mock infected or infected with TKdel in the right lip. Four days postinfection, mice were superinfected with LβA (n = 17) or with SC16 wild-type virus (n = 20) in the left lip. During the acute phase of LβA or SC16 superinfection (and up to 15 DPSI), mice that received an initial TKdel inoculation lacked signs of ocular morbidity, mortality, and weight loss (LβA [Fig. 5A to C; endpoint chi-squared 2-tailed test, P < 0.001 for both panels A and B). In contrast, mice which were initially mock infected exhibited the expected rates of morbidity described earlier with LβA alone (20).
FIG 5.

Mortality, ocular disease, weight loss, and acute-phase LAT promoter activity of LβA superinfection are extinguished in mice when initially infected with TKdel 4 days prior. Mice initially infected in the right lip with TKdel (or PBS for mock infection) and again 4 DPI in the left lip with LβA were monitored for 15 days. Ocular disease signs (corneal clouding, corneal neovascularization, and iris atrophy with pupil mydriasis) (A) and deaths (B) were recorded. Endpoint chi-squared 2-tailed test was performed for groups in panels A and B. *, P < 0.05. (C) Weights of individual mice were recorded from before the first infection and daily for the duration of the experiment. Data represent the means of the lowest measure for each animal during the acute phase. At 6 DPSI with LβA mice were sacrificed. (D) TG frontal cryosections (10 μm) were prepared and stained with X-Gal reactant and counterstained with neutral red, and β-galactosidase+ve neurons were enumerated. Symbols represent the numbers of β-galactosidase+ve neurons from individual TG, and the horizontal bar represents the average. **, P < 0.01 (Kruskal-Wallis test).
Mice (n = 5) from both groups were randomly sampled at day 6 DPSI to observe LβA-associated LAT promoter activity during the acute phase (Fig. 5D). As expected, all of the left TG from mice that were initially mock infected, before superinfection in the left lip with LβA, contained β-galactosidase+ve neurons (272 ± 155.5; range, 99 to 483). Furthermore, TG contralateral to the LβA superinfection (4/5) contained extremely low numbers of β-galactosidase+ve neurons (6 ± 7.0; range, 0 to 18). In striking contrast, both the left and right TG from mice initially infected with TKdel before left lip superinfection with LβA were almost completely devoid of β-galactosidase+ve neurons (superinfected left TG, 14 ± 12.8; range, 3 to 21; superinfected right TG, 0). This reduction in β-galactosidase+ve neurons was statistically significant (P = 0.009). These data demonstrated that an initial infection with a virus unable to replicate in neurons could trigger protection against acute-phase disease, which corresponded with the attenuation of virulent superinfection viral gene expression.
To understand further the consequences of nonneurovirulent initial infection on the transcriptional activity of superinfecting virus during latency, LAT promoter activity was monitored during the latent phase (28 DPSI) of LβA virus infection (Fig. 6A). While β-galactosidase+ve neurons were frequently detected in all TG ipsilateral to LβA superinfection in initially mock-infected animals (132 ± 75.3; range, 72 to 219), there was a significant reduction in β-galactosidase+ve neurons of TG ipsilateral to LβA superinfection in initially TKdel-infected animals (5 ± 8.2; range, 0 to 21; P < 0.01 from 3/6 animals). β-Galactosidase+ve neurons in right TG, contralateral to the site of LβA superinfection, were detected only from mock-infected animals (1 ± 1.1; range, 1 to 3) and were not detected in right TG from superinfected mice that received the initial TKdel inoculation. Meanwhile, abundant loads of superinfecting LβA viral genome were detected from the left and right TG of mock-infected animals and in mice that received an initial TKdel inoculation (Fig. 6B). Additional groups of mice were either mock infected or infected with TKdel in the right lip and then left for 1 month before superinfection in the left lip with LβA. Mice were randomly sampled at day 28 DPSI (day 56) to observe LβA-associated LAT promoter activity during latency (Fig. 6C). Similar to the case with a 4-day delay, frequent β-galactosidase+ve neurons were detected in the TG ipsilateral to infection with LβA in mice that were initially mock infected, whereas in mice receiving an initial TKdel inoculation, β-galactosidase+ve neurons were almost undetectable from both TG. Meanwhile, LβA DNA was found in both left and right TG in groups of mice that were initially inoculated with TKdel or not (Fig. 6D). These data demonstrate that in the TG, LAT promoter activity of superinfecting virus was abrogated by initial infection with TKdel in the lip from 4 days before superinfection in the contralateral lip and were long-lived up to 28 DPSI.
FIG 6.

Reduced LAT promoter activity of LβA superinfection, with maintained viral DNA load, in mice initially infected with TKdel 4 or 28 days prior. Mice initially infected in the right lip with TKdel (or PBS for mock infection) and superinfected at 4 DPI or 28 DPI in the left lip with LβA. Mice were sacrificed 28 DPSI. TG frontal cryosections (10 μm) from mice, superinfected at 4 DPI (A) or at 28 DPSI (C), were prepared and stained with X-Gal reactant and counterstained with neutral red, and β-galactosidase+ve neurons were enumerated. Symbols represent the numbers of β-galactosidase+ve neurons from individual TG, and the horizontal bar represents the average. **, P < 0.01 (Kruskal-Wallis test). DNA was extracted from whole-TG samples (n = 5 per group) harvested from mice that were superinfected, at 4 DPI (B) or 28 DPSI (D), and used as the template DNA for qPCR with primers targeting the lacZ gene or murine GAPDH gene. The number of LβA genome copies per TG was determined relative to GAPDH. Bars indicate the average numbers of LβA genome copies relative to GAPDH DNA, and error bars represent the 95% confidence intervals.
Superinfecting virus reactivations from latency after the TKdel initial infection were quantified from explanted TG in 4 additional groups of mice sacrificed 28 DPSI. Groups were initially mock infected, infected with TKdel in the right lip or in the right footpad, and then superinfected with LβA or wild-type SC16 virus in the left lip 4 DPI. The supernatants from the TG cultures generated viral plaques only from cultures of TG ipsilateral to superinfection with LβA and SC16 virus (left) when initially mock infected (40% and 64% of TG, respectively [Fig. 7A and B, respectively]). Interestingly, no plaques were detected from any TG cultures from mice that were initially infected in the lip with TKdel and superinfected with LβA or the SC16 wild type. However, 53% of TG cultures from mice that were initially infected in the footpad with TKdel generated viral plaques. These data demonstrated that a fully virulent superinfecting HSV-1, which is normally capable of reactivating and was present in abundance bilaterally (at a viral load compatible with reactivation), was unable to reactivate if mice were initially infected in the lip, but not the footpad, with nonneurovirulent virus 4 days prior to the contralateral superinfection. We can infer that TKdel was not rescued by the superinfection 4 days later or during TG explant culture. These results indicate that an initial TKdel infection did not prevent superinfecting LβA from entering the TG to establish latency, as assessed by measurement of latent DNA loads. However, an initial TKdel infection in the lip led to control of transcriptional activity of the HSV-1 genomes and blocked reactivation. In contrast, an initial TKdel infection in the footpad did not abrogate reactivations from latency.
FIG 7.

Rates of HSV-1 reactivation from explanted TG from mice initially infected with TKdel and superinfected with HSV-1 SC16 wild-type or LβA reporter virus 4 days later. Groups of BALB/c mice were initially infected in the upper right lip with TKdel (or PBS for mock infection) or in the right footpad (TKdel FOOTPAD). At 4 DPI, mice were superinfected in the left lip with the LβA reporter virus (n = 10 mice) or the wild-type SC16 parent strain (TKdel lip initial n = 14 mice and TKdel footpad initial n = 13). All mice were sacrificed at 28 DPSI, corresponding to the latent phase of TG infection in the OO model. TG were harvested and incubated individually in CM during 14 days. Aliquots of medium were removed every 2 days and compensated with fresh CM. Infectious particles released (reactivation) from aliquots were quantified by plaque assay. Bars represent the percentages of cultured TGs that developed plaques at any given time. ***, P < 0.001 (Kruskal-Wallis test).
Attenuation of acute disease mediated by initial HSV-1 TKdel inoculation occurs systemically.
To further probe the ability of initial TKdel inoculation to dictate acute-phase and latent-phase superinfection outcomes, mice were initially mock infected or infected with TKdel in the right lip or right footpad. After 4 days, mice were challenged with virulent SC16 in the left lip and were monitored for signs of disease. Mice that were initially mock infected displayed rates of mortality (∼30%), ocular disease (∼60%), and weight loss as expected after a challenge with virulent SC16 in the lip (Fig. 8). In contrast, mice that received an initial inoculation of TKdel in the lip or footpad were completely protected from signs of disease. These data demonstrate that acute-phase disease, as a consequence of virulent superinfection in the lip, was attenuated systemically by an initial inoculation with TKdel 4 days prior to superinfection.
FIG 8.

Initial infection with TKdel attenuates mortality, ocular disease, and weight loss associated with virulent wild-type superinfection occurring 4 days later. Mice were initially mock infected with PBS or infected with TKdel in the right lip (n = 20 per group) or in the right footpad with TKdel (TKdelFOOT) (n = 12 per group). At 4 DPI mice were superinfected with SC16 wild-type HSV-1 in the left lip, and they were monitored for 15 days. Deaths (A) and ocular disease signs (corneal clouding, corneal neovascularization, and iris atrophy with pupil mydriasis) (B) were recorded. Endpoint chi-squared 2-tailed test was performed for groups in panels A and B. *, P < 0.001. Weights of individual mice were recorded from before the first infection and daily for the duration of the experiment. Data in panel C represent the means of the lowest measure for each animal during the acute phase.
DISCUSSION
In humans, almost all HK recurrences are unilateral (6, 11), while the viral load is not statistically different between left and right TG for a given patient (5, 12, 13). These data suggest that HSV-1 infection yields a different state of latency between left and right TG of the same infected individual. Using a mouse model of HSV-1 labial primary infection, in which infection is performed unilaterally at the orofacial cavity, we have previously demonstrated that both ipsilateral and contralateral TG are infected, with a 2-day delay between the former and the latter in any given animal (21, 22).
Using this model in the present study, we have shown that HSV-1 reactivation occurs solely from the TG ipsilateral to infection (Fig. 1A), while virus genomic DNA is present bilaterally (Fig. 1B). As symmetrical nerve networks innervating the left and right sides of the face are directly linked to a corresponding left or right TG (20), virus propagation from a primary lip infection, localized to one side of the face, is directly associated with axons of the TG aligned to that side of the face, thus explaining how this ipsilateral TG is infected before the contralateral TG. We therefore hypothesized that asymmetry of HSV-1 acute-phase activity and recurrences in humans are associated with differential kinetics of virus arrival in ipsilateral and contralateral TG during orofacial infection. Upon lip infection, the virus must disseminate through second- and third-order midbrain neurons of the trigeminal pathways to reach the contralateral TG, but with a delay in comparison to infection of the TG that is ipsilateral and directly connected to the infected lip site (as described in references 19 and 20). We confirmed this notion by utilizing a dual-inoculation process in which an initial HSV-1 infection in the lip aligned to one side was followed by a contralateral lip superinfection with different reporter viruses derived from the same HSV-1 strain. Delaying by 4 days or more the inoculation of superinfecting virus resulted in a complete inhibition of the acute phase of superinfection in the ipsilateral TG (Fig. 2 and 3A), with concomitant elimination of morbidity and mortality (Fig. 4A and B). Virus was still propagated to the ipsilateral and contralateral TG, as detected by viral genomic qPCR (Fig. 3B, 4D, and 6B and D). Interestingly, this 4-day window correlated with the time to develop adaptive immune surveillance, critical in the control of acute-phase HSV-1 infection in other mouse models (29–31). HSV-1-specific cytotoxic T cell function has been recorded in draining lymph nodes as early as 2 DPI, and proliferating HSV-1-specific T cells were found in the spleen 4 DPI (32); these cells provide long-lived protection by mounting rapid responses to a viral challenge. On the other hand, innate anti-HSV-1 mechanisms triggered by neurons, and driven by type I and type II interferons, have been reported to shut down viral transmission and repress acute-phase HSV-1 lytic activity (33, 34). The outcome of HSV-1 infection of the TG is further dictated by the neuronal subtypes that are infected, with NefH+ve neurons more efficient for harboring virus in latent state than NefH-negative (NefH−ve) neurons, which are more efficient in harboring lytic viral replication (35, 36). Even so, our observations from dual infection, in which altered kinetics of TG infection incurred attenuation of viral replication in the contralateral TG, may illustrate why acute-phase HSV-1 activity is restricted to the ipsilateral TG after orofacial infection. Indeed, a plausible explanation raised from our experiments is that while the TG ipsilateral to the site of orofacial inoculation becomes infected rapidly, the delay in travel time for virions to arrive in the contralateral TG enables the development of appropriate antiviral mechanisms in the host, which elicit protection against viral replication and associated disease of the contralateral side.
The use of a thymidine kinase-deficient mutant, TKdel, for the initial infection allowed us to explore further the implications of delayed superinfection of the contralateral TG on viral reactivations. The TKdel mutant HSV-1 can cause a fulminant infection in epithelial tissue during acute infection, is known for its capacity to establish latency and persist, but is unable to reactivate or replicate in neurons (24, 26, 37). Infectious particles can be found only at the site of lip infection during the acute phase and not from either TG (data available upon request). Hence, this mutant virus is unable to spread from one neuron to another in the TG and by extension cannot proceed to the TG contralateral to lip infection. Here we describe a major protective state elicited by TKdel infection. LAT promoter activity of the LβA superinfecting virus, ocular signs of disease, and mortality of the superinfected mice were eliminated by initial infection with the TKdel mutant virus (Fig. 5, 6, and 8). While efficient protection elicited by priming with attenuated virus is in line with previous experimental reports (37–42), of great interest was the complete absence of virulent wild-type superinfecting virus reactivations from ipsilateral and contralateral TG in initially TKdel-infected animals (Fig. 7). To establish protection against reactivation, footpad initial infection (not affecting the trigeminal neural network) was ineffective at blocking reactivations, while contralateral lip infection was effective. In contrast, initial inoculation with TKdel in the footpad was able to elicit attenuation of acute-phase disease (Fig. 8). These findings suggest that different immune- or virus-mediated mechanisms are required to protect against acute-phase lytic gene expression and associated disease versus blocking reactivations. Therefore, yet-unappreciated mechanisms exist for controlling viral reactivation that may involve properties unique to nonneurovirulent HSV-1 mutants. Further studies are required to determine whether the prophylaxis against reactivation elicited by TKdel is mediated by eliciting local adaptive cellular immunity (37) and/or by HSV-1-mediated cellular-level functions (14, 18). Defining these conditions may enable exploitation for the control of viral reactivation from the TG.
Our study was motivated by observations of unilateral HSV-1 infection and reactivation in the OO model and by clinical observations of recurrent unilateral human ocular disease. Altered lateral kinetics of infection, elicited by performing an initial inoculation in the lip contralateral to a delayed secondary infection, attenuated superinfection viral gene expression. A key interval of 4 days’ delay between infections was discovered, after which protective mechanisms had extended to include the lip and/or TG contralateral to the side of initial infection. These results are promising, as the natural origin of HSV-1-mediated ocular disease is from viral replication in the TG. However, superinfecting viral DNA was abundant in the opposing TGs, illustrating uncontained viral dissemination through the neural network. Initial infection with TKdel led to a profound absence of viral reactivations from latency in mice superinfected with virulent HSV-1. These results indicate that reactivation and, more particularly, unilateral recurrence are manifestations of latency establishment early during acute-phase infection in which the ipsilateral TG is directly linked to the orofacial site of inoculation and is infected before a critical state of immunity has occurred, in contrast to the contralateral TG, which is infected after this event. Further characterization of the mechanism restricting virus activity unilaterally will help to elucidate effective strategies for eliminating virus reactivation from the TG. These results are promising for understanding how the kinetics of HSV-1 infection influence the unilateral nature of virus activity and disease and for future studies aiming to develop preventive or therapeutic strategies. Future directions should include elucidation of local immune phenomena and inflammatory processes that are altered by the delayed orofacial infection kinetics, to discover how nonneurovirulent virus initial infection inhibits HSV-1 reactivation from the TG; these are the subjects of our ongoing studies.
MATERIALS AND METHODS
Virus strains and cells.
The wild-type SC16 strain of HSV-1 (43) was used as background for the following genetically modified viruses.
Reporter viruses SC16-BE8 (44) and SC16-C12 (45), termed gJ-LacZ and gJ-GFP here, respectively, contain an IE1CMV (immediate early promoter 1 of cytomegalovirus) promoter-driven lacZ or eGFP gene inserted in the US5 locus (encoding the gJ protein). β-Galactosidase activity or enhanced green fluorescent protein (eGFP) fluorescence was used as a virus-specific marker of active viral replication.
The recombinant LβA strain contains an encephalomyocarditis virus internal ribosomal entry site (IRES) inserted just before the lacZ gene of Escherichia coli (insertion into a 168-bp HpaI deletion [nucleotides 120301 to 120469]) within the major LAT locus (23). β-Galactosidase activity was used as a strain-specific marker for active viral replication and to assess LAT promoter activity.
The recombinant HSV-1 SC16 mutant TKDM21, termed TKdel here, harbors an 816-bp deletion within the thymidine kinase (TK) coding region (26). Viruses were propagated and titrated using the Vero cell line (ATCC) as previously described (19).
The expression of eGFP and lacZ cassettes from these strains faithfully reflects the transcription or expression of viral genes driven by the same promoters (20, 46). These strain-specific reporter genes were targeted for detection of specific viral genomes by qPCR, and strain-specific reporter gene expression was quantified as a surrogate marker for strain-specific viral activity.
Inoculation procedures.
Six-week-old inbred female BALB/c mice (Janvier Breeding, Le Genest-Saint-Isle, France), receiving unrestricted access to food and water, were used for the experiments. Procedures were performed under general anesthesia induced by intraperitoneal injection of 0.1 ml of a solution containing ketamine (25 mg/ml) and xylazine (20 mg/ml). Analgesic was added to drinking water during the first 10 days postsuperinfection (DPSI). Using a custom-made glass microneedle and Harvard microinjector, viral suspensions in saline solution of 1 × 106 PFU of virus in 1-μl volumes were injected below the epithelium of the right upper lip for initial infections. One microliter of saline was administered for mock inoculation. Superinfecting virus of identical virus load and volume was inoculated at defined later time points below the epithelium of the left upper lip, as previously described (19). Mice were observed daily for clinical signs of ocular infection from 0 to 28 DPSI, as previously described (20). Mice were randomly sacrificed at either 6 DPSI for acute infection or 28 DPSI for latent infection and reactivation studies. The experimental procedures involving animals were performed under general anesthesia. All procedures involving experimental animals conformed to ethical standards of the Association for Research in Vision and Ophthalmology (ARVO) statement for the use of animals in research and were approved by the local ethics committee (CEEA 59).
Quantification of HSV-1-infected cells and viral genome copy number in TG.
After sacrifice, TG were removed and processed according to the procedures for 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) assays on histological cryosections, or for qPCR on DNA extracts from whole tissues, as previously described (22). Primers used for quantification of reporter regions of HSV-1 strain genomic DNA gJ-LacZ and gJ-GFP are listed in Table 1, or as defined for gG by Cavallero et al. (22). qPCR conditions for viral DNA amplification involved activation for 15 min at 95°C, amplification for 15 s at 94°C and 1 min at 58°C (repeated 40 times), and a hold 5 min at 25°C; for GAPDH PCR, conditions involved activation for 15 min at 95°C, amplification for 15 s at 94°C, 1 min at 58°C, and 130 s at 72°C (repeated 30 times), and a melting curve for 1 min from 50°C to 90°C. Viral genome copy number was determined relative to murine GAPDH.
TABLE 1.
Sequences of primers and TaqMan probes used for quantitative PCR
| Target | Oligonucleotide | Sequence 5′–3′a |
|---|---|---|
| TK | Forward primer | GAAAGCTGTCCCCAATCCTC |
| Reverse primer | GGCTTGACCEGGCTATGTTG | |
| Taqman probe | FAM-TATTGGCAAGCAGCTCGTAA-TAMRA | |
| LacZ | Forward primer | TACTGTCGTCGTCCCCTCAAA |
| Reverse primer | TAACAACCCGTCGGATTCTCC | |
| Taqman probe | FAM-TATCCCATTACGGTCAATCCGCCG-TAMRA | |
| GFP | Forward primer | CAACAGCCACAACGTCTATATCATG |
| Reverse primer | ATGTTGTGGCGGATCTTGAAG | |
| Taqman probe | FAM-CAAGCAGAAGAACGGCATCAAGGTGA-TAMRA | |
| GAPDH | Forward primer | CAAGGTCATCCATGACAACTTTG |
| Reverse primer | GGCCATCCACAGTCTTCTGG |
Abbreviations: FAM, 6-carboxyfluorescein; TAMRA, 6-carboxytetramethylrhodamine.
Ex vivo explant reactivation assay.
In the OO model, a state of HSV-1 latency is achieved at 28 DPI (20). Latency is disrupted by culture of TG ex vivo, which results in the release of infectious virus into the culture supernatant, corresponding to ex vivo reactivation (47). Mice were sacrificed 28 DPSI, and both TG were removed and then cultured in culture medium (CM): Dulbeco’s modified eagle medium (DMEM) supplemented with 8% fetal calf serum and 1% streptomycin plus penicillin. An aliquot of medium (200 μl) was sampled from each culture well every 2 days for up to 14 days, and an equivalent 200 μl of medium was added to the TG culture to compensate for the loss. TG culture media from days 0, 4, 6, 8, 10, and 14 were plated on Vero cell monolayers to perform plaque assays using serial dilutions (10−1, 10−2, and 10−3). Reactivation was recorded for a single TG if plaques were observed for any of the time points measured. Generally, between 2 and 10,000 plaques were enumerated, and the peak yield of infectious virus usually occurred at day 8 of culture.
Plaque assay for identification and quantification of virus.
To identify the virus that reactivated following infection with gJ-GFP, and superinfection with gJ-LacZ or LβA, all GFP+ve and GFP−ve plaques were enumerated by fluorescence microscopy. β-Galactosidase+ve GFP−ve plaques were identified by staining cell monolayers with neutral red (0.02%) after fixation with paraformaldehyde 4% for 15 min at room temperature and counterstaining with X-Gal reactant [0.33 mg/ml X-Gal, 5 mM K4Fe(CN)6, 5 mM K3Fe(CN)6, 2 mM MgCl2, 0.01% sodium deoxycholate, and 0.1% Triton X-100] for 1 h at 37°C. Plaques that expressed β-galactosidase stained blue, whereas the GFP-expressing virus-, TKdel-, and SC16 wild-type-associated plaques remained transparent. When TKdel, SC16, or a mock inoculation was performed initially, the identification of SC16 or LβA strain reactivation plaques was performed after staining of the cell monolayer with crystal violet.
Infectious HSV-1 particle titration of homogenized TG.
As described previously (34), the TG harvested from sacrificed mice were homogenized in microtubes containing 250 μl of DMEM. Three cycles of freeze-thawing were performed with liquid nitrogen, and the samples were centrifuged and supernatants stored at –80°C until use. Serial dilutions were used to titrate the virus on Vero cells using plaque assays (as described above).
Statistical analyses.
The nonparametric Kruskal-Wallis test was used to analyze histological data and viral DNA loads (qPCR). Means and 95% confidence intervals (CI) were calculated with STATA 13.1 (StataCorp, USA). Endpoint chi-squared 2-tailed test was performed for survival curves.
ACKNOWLEDGMENTS
We are warmly thankful to Vincent Marechal and Flore Rozenberg for constructive discussions about results and to Rich Bains for careful proofreading of the manuscript.
This study was supported by research grant from Agence Nationale de la Recherche (ANR) (grant number ANR-18-CE15-0014-01 [EPIPRO project]), the Ministere de l’Education Nationale de la Recherche et de Technologie (MENRT), the Société Française d’Ophtalmologie (SFO), the Fondation pour les Aveugles de Guerre (FAG), and the Fondation de France (FAF).
REFERENCES
- 1.Roizman B, Knipe DM. 2001. Herpes simplex viruses and their replication, p 2399–2459. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 4th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Liesegang TJ. 1989. Epidemiology of ocular herpes simplex. Natural history in Rochester, Minn, 1950 through 1982. Arch Ophthalmol 107:1160–1165. doi: 10.1001/archopht.1989.01070020226030. [DOI] [PubMed] [Google Scholar]
- 3.Farooq AV, Shukla D. 2012. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol 57:448–462. doi: 10.1016/j.survophthal.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith GA, Gross SP, Enquist LW. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A 98:3466–3470. doi: 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richter ER, Dias JK, Gilbert JE, Atherton SS. 2009. Distribution of herpes simplex virus type 1 and varicella zoster virus in ganglia of the human head and neck. J Infect Dis 200:1901–1906. doi: 10.1086/648474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaye S, Choudhary A. 2006. Herpes simplex keratitis. Prog Retin Eye Res 25:355–380. doi: 10.1016/j.preteyeres.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Lewis ME, Leung WC, Jeffrey VM, Warren KG. 1984. Detection of multiple strains of latent herpes simplex virus type 1 within individual human hosts. J Virol 52:300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liesegang TJ, Melton LJ III, Daly PJ, Ilstrup DM. 1989. Epidemiology of ocular herpes simplex incidence in Rochester, Minn, 1950 through 1982. Arch Ophthalmol 107:1155–1159. doi: 10.1001/archopht.1989.01070020221029. [DOI] [PubMed] [Google Scholar]
- 9.Labetoulle M, Auquier P, Conrad H, Crochard A, Daniloski M, Bouee S, El Hasnaou IA, Colin J. 2005. Incidence of herpes simplex virus keratitis in France. Ophthalmology 112:888–895. doi: 10.1016/j.ophtha.2004.11.052. [DOI] [PubMed] [Google Scholar]
- 10.Young RC, Hodge DO, Liesegang TJ, Baratz KH. 2010. Incidence, recurrence, and outcomes of herpes simplex virus eye disease in Olmsted County, Minnesota, 1976–2007. Arch Ophthalmol 128:1178–1183. doi: 10.1001/archophthalmol.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Motani H, Sakurada K, Ikegaya H, Akutsu T, Hayakawa M, Sato Y, Yajima D, Sato K, Kobayashi K, Iwase H. 2006. Detection of herpes simplex virus type 1 DNA in bilateral human trigeminal ganglia and optic nerves by polymerase chain reaction. J Med Virol 78:1584–1587. doi: 10.1002/jmv.20742. [DOI] [PubMed] [Google Scholar]
- 13.Cohrs RJ, Randall J, Smith J, Gilden DH, Dabrowski C, van Der KH, Tal-Singer R. 2000. Analysis of individual human trigeminal ganglia for latent herpes simplex virus type 1 and varicella-zoster virus nucleic acids using real-time PCR. J Virol 74:11464–11471. doi: 10.1128/JVI.74.24.11464-11471.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Criddle A, Thornburg T, Kochetkova I, DePartee M, Taylor MP. 2016. gD-independent superinfection exclusion of alphaherpesviruses. J Virol 90:4049–4058. doi: 10.1128/JVI.00089-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meignier B, Norrild B, Roizman B. 1983. Colonization of murine ganglia by a superinfecting strain of herpes simplex virus. Infect Immun 41:702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Centifanto-Fitzgerald YM, Varnell ED, Kaufman HE. 1982. Initial herpes simplex virus type 1 infection prevents ganglionic superinfection by other strains. Infect Immun 35:1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price RW, Walz MA, Wohlenberg C, Notkins AL. 1975. Latent infection of sensory ganglia with herpes simplex virus: efficacy of immunization. Science 188:938–940. doi: 10.1126/science.166432. [DOI] [PubMed] [Google Scholar]
- 18.Mador N, Panet A, Steiner I. 2002. The latency-associated gene of herpes simplex virus type 1 (HSV-1) interferes with superinfection by HSV-1. J Neurovirol 8(Suppl 2):97–102. doi: 10.1080/13550280290167920. [DOI] [PubMed] [Google Scholar]
- 19.Labetoulle M, Kucera P, Ugolini G, Lafay F, Frau E, Offret H, Flamand A. 2000. Neuronal propagation of HSV1 from the oral mucosa to the eye. Invest Ophthalmol Vis Sci 41:2600–2606. [PubMed] [Google Scholar]
- 20.Labetoulle M, Maillet S, Efstathiou S, Dezelee S, Frau E, Lafay F. 2003. HSV1 latency sites after inoculation in the lip: assessment of their localization and connections to the eye. Invest Ophthalmol Vis Sci 44:217–225. doi: 10.1167/iovs.02-0464. [DOI] [PubMed] [Google Scholar]
- 21.Maillet S, Crepin S, Naas T, Roque-Afonso AM, Lafay F, Efstathiou S, Labetoulle M. 2006. Herpes simplex virus type 1 latently infected neurons differentially express the latency associated and ICP0 transcripts. J Virol 80:9310–9321. doi: 10.1128/JVI.02615-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cavallero S, Huot N, Francelle L, Lomonte P, Naas T, Labetoulle M. 2014. Biological features of herpes simplex virus type 1 latency in mice according to experimental conditions and type of neurones. Invest Ophthalmol Vis Sci 55:7761. doi: 10.1167/iovs.14-14673. [DOI] [PubMed] [Google Scholar]
- 23.Lachmann RH, Efstathiou S. 1997. Utilization of the herpes simplex virus type 1 latency-associated regulatory region to drive stable reporter gene expression in the nervous system. J Virol 71:3197–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. 1989. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci U S A 86:4736–4740. doi: 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilcox CL, Crnic LS, Pizer LI. 1992. Replication, latent infection, and reactivation in neuronal culture with a herpes simplex virus thymidine kinase-negative mutant. Virology 187:348–352. doi: 10.1016/0042-6822(92)90326-k. [DOI] [PubMed] [Google Scholar]
- 26.Efstathiou S, Kemp S, Darby G, Minson AC. 1989. The role of herpes simplex virus type 1 thymidine kinase in pathogenesis. J Gen Virol 70:869–879. doi: 10.1099/0022-1317-70-4-869. [DOI] [PubMed] [Google Scholar]
- 27.Thompson RL, Sawtell NM. 2000. Replication of herpes simplex virus type 1 within trigeminal ganglia is required for high frequency but not high viral genome copy number. J Virol 74:965–974. doi: 10.1128/JVI.74.2.965-974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen SH, Pearson A, Coen DM, Chen SH. 2004. Failure of thymidine kinase-negative herpes simplex virus to reactivate from latency following efficient establishment. J Virol 78:520–523. doi: 10.1128/jvi.78.1.520-523.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gurung HR, Carr MM, Carr DJ. 2017. Cornea lymphatics drive the CD8(+) T-cell response to herpes simplex virus-1. Immunol Cell Biol 95:87–98. doi: 10.1038/icb.2016.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vahed H, Agrawal A, Srivastava R, Prakash S, Coulon PA, Roy S, BenMohamed L. 2019. Unique type I interferon, expansion/survival cytokines and JAK/STAT gene signatures of multi-functional HSV-specific effector memory CD8(+) TEM cells are associated with asymptomatic ocular herpes in humans. J Virol 93:e01882-18. doi: 10.1128/JVI.01882-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greyer M, Whitney PG, Stock AT, Davey GM, Tebartz C, Bachem A, Mintern JD, Strugnell RA, Turner SJ, Gebhardt T, O’Keeffe M, Heath WR, Bedoui S. 2016. T cell help amplifies innate signals in CD8(+) DCs for optimal CD8(+) T cell priming. Cell Rep 14:586–597. doi: 10.1016/j.celrep.2015.12.058. [DOI] [PubMed] [Google Scholar]
- 32.Coles RM, Mueller SN, Heath WR, Carbone FR, Brooks AG. 2002. Progression of armed CTL from draining lymph node to spleen shortly after localized infection with herpes simplex virus 1. J Immunol 168:834–838. doi: 10.4049/jimmunol.168.2.834. [DOI] [PubMed] [Google Scholar]
- 33.Song R, Koyuncu OO, Greco TM, Diner BA, Cristea IM, Enquist LW. 2016. Two modes of the axonal interferon response limit alphaherpesvirus neuroinvasion. mBio 7:e02145-15. doi: 10.1128/mBio.02145-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maroui MA, Calle A, Cohen C, Streichenberger N, Texier P, Takissian J, Rousseau A, Poccardi N, Welsch J, Corpet A, Schaeffer L, Labetoulle M, Lomonte P. 2016. Latency entry of herpes simplex virus 1 is determined by the interaction of its genome with the nuclear environment. PLoS Pathog 12:e1005834. doi: 10.1371/journal.ppat.1005834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cabrera JR, Charron AJ, Leib DA. 2018. Neuronal subtype determines herpes simplex virus 1 latency-associated-transcript promoter activity during latency. J Virol 92:e00430-18. doi: 10.1128/JVI.00430-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP. 2011. A5-positive primary sensory neurons are nonpermissive for productive infection with herpes simplex virus 1 in vitro. J Virol 85:6669–6677. doi: 10.1128/JVI.00204-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato A, Suwanto A, Okabe M, Sato S, Nochi T, Imai T, Koyanagi N, Kunisawa J, Kawaguchi Y, Kiyono H. 2014. Vaginal memory T cells induced by intranasal vaccination are critical for protective T cell recruitment and prevention of genital HSV-2 disease. J Virol 88:13699–13708. doi: 10.1128/JVI.02279-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitchell BM, Stevens JG. 1996. Neuroinvasive properties of herpes simplex virus type 1 glycoprotein variants are controlled by the immune response. J Immunol 156:246–255. [PubMed] [Google Scholar]
- 39.Schrimpf JE, Tu EM, Wang H, Wong YM, Morrison LA. 2011. B7 costimulation molecules encoded by replication-defective, vhs-deficient HSV-1 improve vaccine-induced protection against corneal disease. PLoS One 6:e22772. doi: 10.1371/journal.pone.0022772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davido DJ, Tu EM, Wang H, Korom M, Gazquez Casals A, Reddy PJ, Mostafa HH, Combs B, Haenchen SD, Morrison LA. 2018. Attenuated herpes simplex virus 1 (HSV-1) expressing a mutant form of ICP6 stimulates a strong immune response that protects mice against HSV-1-induced corneal disease. J Virol 92:e01036-18. doi: 10.1128/JVI.01036-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan AA, Srivastava R, Chentoufi AA, Kritzer E, Chilukuri S, Garg S, Yu DC, Vahed H, Huang L, Syed SA, Furness JN, Tran TT, Anthony NB, McLaren CE, Sidney J, Sette A, Noelle RJ, BenMohamed L. 2017. Bolstering the number and function of HSV-1-specific CD8(+) effector memory T cells and tissue-resident memory T cells in latently infected trigeminal ganglia reduces recurrent ocular herpes infection and disease. J Immunol 199:186–203. doi: 10.4049/jimmunol.1700145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Royer DJ, Carr MM, Gurung HR, Halford WP, Carr D. 2017. The neonatal Fc receptor and complement fixation facilitate prophylactic vaccine-mediated humoral protection against viral infection in the ocular mucosa. J Immunol 199:1898–1911. doi: 10.4049/jimmunol.1700316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hill TJ, Field HJ, Blyth WA. 1975. Acute and recurrent infection with herpes simplex virus in the mouse: a model for studying latency and recurrent disease. J Gen Virol 28:341–353. doi: 10.1099/0022-1317-28-3-341. [DOI] [PubMed] [Google Scholar]
- 44.Proenca JT, Coleman HM, Connor V, Winton DJ, Efstathiou S. 2008. A historical analysis of herpes simplex virus promoter activation in vivo reveals distinct populations of latently infected neurones. J Gen Virol 89:2965–2974. doi: 10.1099/vir.0.2008/005066-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arthur JL, Scarpini CG, Connor V, Lachmann RH, Tolkovsky AM, Efstathiou S. 2001. Herpes simplex virus type 1 promoter activity during latency establishment, maintenance, and reactivation in primary dorsal root neurons in vitro. J Virol 75:3885–3895. doi: 10.1128/JVI.75.8.3885-3895.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Regge N, Van ON, Nauwynck HJ, Efstathiou S, Favoreel HW. 2010. Interferon alpha induces establishment of alphaherpesvirus latency in sensory neurons in vitro. PloS One 5:e13076. doi: 10.1371/journal.pone.0013076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sawtell NM, Thompson RL. 2004. Comparison of herpes simplex virus reactivation in ganglia in vivo and in explants demonstrates quantitative and qualitative differences. J Virol 78:7784–7794. doi: 10.1128/JVI.78.14.7784-7794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]