

HIV-1 has evolved multiple strategies to avoid humoral responses. One efficient mechanism is to keep its envelope glycoprotein (Env) in its “closed” conformation. Here, we report on a new family of small molecules that are able to “open up” Env, thus exposing vulnerable epitopes. This new family of molecules binds in the Phe43 cavity and contacts the highly conserved D368 residue. The structural and biological attributes of molecules of this family make them good candidates for drug development.
KEYWORDS: HIV-1, envelope glycoproteins, CD4 mimetics, ADCC, neutralization, Env, small-molecule inhibitors
ABSTRACT
The HIV-1 envelope glycoprotein (Env) trimer mediates virus entry into cells. The “closed” conformation of Env is resistant to nonneutralizing antibodies (nnAbs). These antibodies mostly recognize occluded epitopes that can be exposed upon binding of CD4 or small-molecule CD4 mimetics (CD4mc). Here, we describe a new family of small molecules that expose Env to nnAbs and sensitize infected cells to antibody-dependent cellular cytotoxicity (ADCC). These compounds have a limited capacity to inhibit virus infection directly but are able to sensitize viral particles to neutralization by otherwise nonneutralizing antibodies. Structural analysis shows that some analogs of this family of CD4mc engage the gp120 Phe43 cavity by contacting the highly conserved D368 residue, making them attractive scaffolds for drug development.
IMPORTANCE HIV-1 has evolved multiple strategies to avoid humoral responses. One efficient mechanism is to keep its envelope glycoprotein (Env) in its “closed” conformation. Here, we report on a new family of small molecules that are able to “open up” Env, thus exposing vulnerable epitopes. This new family of molecules binds in the Phe43 cavity and contacts the highly conserved D368 residue. The structural and biological attributes of molecules of this family make them good candidates for drug development.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) entry is mediated by the interaction of the HIV-1 envelope glycoprotein (Env) with the CD4 receptor and either the CCR5 or CXCR4 chemokine coreceptor on T cells. Env is exposed on the surface of viral particles and infected cells as three gp120 exterior glycoproteins noncovalently associated with three gp41 transmembrane glycoproteins [(gp120-gp41)3] (1–3). Binding of gp120 to the CD4 receptor leads to major conformational changes in gp120, resulting in the rearrangement of the V1, V2, and V3 loops and the formation of the coreceptor binding site (CoRBS) and the bridging sheet (4–11). CD4 interaction also leads to the exposure of a gp41 helical heptad repeat (heptad repeat 1 [HR1]) (12). The subsequent interaction of gp120 with the coreceptor triggers additional conformational changes in gp41, resulting in the formation of a six-helix bundle formed by HR1 and HR2 and the fusion of viral and cellular membranes (12–14).
The gp120 Env residues that contact CD4 are located in the vestibule of the Phe43 cavity, a highly conserved ∼150-Å3 pocket in gp120 located at the interface between the inner and outer domains (15). In addition to triggering the conformational changes required for viral entry, CD4 binding exposes vulnerable CD4-induced (CD4i) epitopes that are recognized by anti-Env antibodies (Abs) able to mediate antibody-dependent cellular cytotoxicity (ADCC) (16). Similar conformational changes can be triggered by small-molecule CD4-mimetic compounds (CD4mc) such as NBD-556 and NBD-557, which were discovered in a screen for inhibitors of the gp120-CD4 interaction (17). These small molecules (∼337 Da) and subsequently designed derivatives [JP-III-048 and (+)-BNM-III-170] engage gp120 via the Phe43 cavity and induce conformational changes in gp120 similar to those observed upon CD4 binding (18, 19). The binding of CD4mc thus sensitizes HIV-1 particles to neutralization by otherwise nonneutralizing CD4i Abs (20–22) and infected cells to ADCC (23–25). Recent in vivo studies have demonstrated that one of these CD4mc [(+)-BNM-III-170] synergizes with nonneutralizing antibodies (nnAbs) elicited by a monomeric gp120 to protect rhesus macaques for up to 6 months from multiple high-dose intrarectal challenges with a heterologous transmitted/founder simian-human immunodeficiency virus (SHIV) (20). These promising results not only highlight the potential of small-molecule CD4mc for drug development but also validate the prophylactic strategy of exposing vulnerable parts of Env to antibodies.
Here, we screened a library of small molecules for their capacity to expose vulnerable Env epitopes. We identified a new family of small molecules containing an N-substituted piperidine core linked to a halogenated aromatic ring via an amide bond. This new structural class of small molecules (∼450 Da) sensitizes viral particles and infected cells to CD4i Ab neutralization and to ADCC. Structural analyses of complexes formed between these compounds and the gp120 core revealed a binding mode within the gp120 Phe43 cavity similar to that of previously characterized CD4mc [(+)-BNM-III-170] but also unveiled new properties, including a close proximity to the highly conserved D368 residue involved in CD4 binding.
RESULTS
High-throughput screening of small molecules for their ability to expose the coreceptor binding site.
To identify new molecules that are able to expose vulnerable Env epitopes, we adapted a cell-based enzyme-linked immunosorbent assay (ELISA) (CBE), which is capable of measuring conformational changes of membrane-bound trimeric Env (26, 27), into a high-throughput screening (HTS) platform (Fig. 1A). Briefly, we expressed the cytoplasmic-tail-deleted HIV-1JR-FL tier 2 Env on the surface of human osteosarcoma (HOS) cells in a 384-well-plate format. The cytoplasmic tail of Env was deleted to enhance Env expression at the cell surface and therefore enhance the sensitivity of the CBE (26, 28). We used soluble CD4 (sCD4) as a positive control to induce conformational changes and evaluated the exposure of the CoRBS with the CD4i 17b antibody (29, 30). Using this system, we screened a library comprising ∼108,000 small molecules for their ability to expose the CoRBS. The addition of sCD4 enhanced 17b binding by ∼8-fold compared to the vehicle alone. The assay exhibited a Z factor of ∼0.55. After the first round of screening, we selected 2,500 molecules, which were retested by the CBE along with sCD4 as a positive control (Fig. 1B). All molecules that led to enhanced 17b binding of 25% over that induced by sCD4 were retested, and only one molecule was deemed a true positive, UM0059920, which proved to be a racemic mixture (Fig. 1C). Synthesis of the individual enantiomers and testing by a CBE revealed the active enantiomer to be (S)-MCG-II-153 (Fig. 1D), the structure of which shared some characteristics with the NBD-556 family of CD4mc (Fig. 2A). (S)-MCG-II-153 shared the halogenated aromatic ring attached via an amide (or oxalamide) linker to a piperidine (later indane) core. However, NBD-556 lacked the methyl sulfonamide substituent on the piperidine nitrogen. The straightforward access to (S)-MCG-II-153 via two-step synthesis made this lead structure a strategic candidate for building a library of related compounds by employing high-throughput parallel synthesis and purification. We first added a fluorine atom ortho to the chlorine atom on the aromatic ring and compared its ability to expose the CoRBS to those of early (NBD-556) and late [(+)-BNM-III-170] generations of CD4mc. As expected from previously reported CD4mc structure-activity relationships (18, 19), the addition of the fluorine enhanced the capacity of (S)-MCG-III-027-D05 to expose the CoRBS, compared with (S)-MCG-II-153 (Fig. 2B). Importantly, (S)-MCG-III-027-D05 exposed the CoRBS at levels similar to those reached by NBD-556 (Fig. 2B).
FIG 1.

High-throughput screening of small molecules for their ability to expose the coreceptor binding site. (A) A cell-based ELISA (CBE) was adapted to screen a library comprising ∼108,000 small molecules. In the assay, HOS cells expressing HIV-1JR-FL EnvΔCT were plated in a 384-well-plate format. Small molecules or, as a positive control, sCD4 were added to expose the HIV-1JR-FL EnvΔCT epitope that can be recognized by the CD4i antibody 17b. 17b binding was detected by a horseradish peroxidase (HRP)-conjugated second antibody, and HRP enzyme activity was measured by Western Lightning oxidizing and luminal reagents. (B) 17b binding in the presence of sCD4 was set as the control, small molecules that enhanced 17b binding above 25% of the one induced by sCD4 were retested in quadruplicate, and only one molecule (UM0059920) was deemed a true positive. (C) UM0059920 is a racemic mixture. (D) Addition of (S)-MCG-II-153 but not (R)-MCG-II-156 enhances 17b binding to levels similar to those of UM0059920 in the CBE. Data shown are mean relative light unit (RLU) values ± standard deviations (SD) from three independent experiments performed in quadruplicate, with the signal obtained from wells transfected with an empty pcDNA3.1 plasmid (no Env) subtracted, normalized to Env levels as determined by 2G12 binding. Statistical significance was evaluated by using an unpaired t test (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant).
FIG 2.

Chemical structures of different CD4mc and their capacity to expose the CoRBS. (A) (S)-MCG-II-153 shares some characteristics with CD4mc NBD-556 (17). Based on analogy to (+)-BNM-III-170, a fluorine group was added to (S)-MCG-II-153 to make (S)-MCG-III-027-D05. (B) The CBE was used to evaluate 17b binding to HIV-1JR-FL EnvΔCT expressed at the surface of HOS cells in the presence of the different compounds (50 μM), sCD4 (10 μg/ml), or the compounds’ vehicle (DMSO). Data shown are the mean RLU values ± SD from at least three independent experiments performed in quadruplicates, with the signal obtained from wells transfected with an empty pcDNA3.1 plasmid (no Env) subtracted, normalized to Env levels as determined by 2G12 binding. Statistical significance was evaluated using an unpaired t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, not significant).
A new family of small-molecule CD4-mimetic compounds.
We employed a parallel synthesis-and-purification strategy to introduce diversity to the core scaffold of (S)-MCG-II-153 via late-stage diversification of common intermediates to generate 118 analogs. Parallel synthetic library development involved incorporating modifications to the following regions: (i) a substituted aromatic ring, (ii) an amide linker, (iii) a piperidine core, and (iv) a sulfonamide substituent. All analogs were first screened by CBE for their ability to expose the CoRBS epitopes (Fig. 3A). Interestingly, we found that all analogs sharing a replacement of the sulfonamide substituent presented higher activity [(S)-MCG-III-188-A01, (S)-MCG-III-188-A02, (S)-MCG-IV-031-A05, and (S)-MCG-IV-210] (Fig. 3A and B). To gain a better understanding of their mode of action, we cocrystallized the higher-activity analogs in complex with a gp120 core (LM/HT gp120CRF01_AE coree) stabilized in the gp120 CD4-bound conformation (31).
FIG 3.

Optimization of MCG analogs for their capacity to expose the CoRBS. Based on the structure of (S)-MCG-III-027-D05, 118 analogs were synthesized and purified. (A) Binding of the CD4i antibody 17b to HIV-1JR-FL EnvΔCT in the presence of analogs (50 μM) was evaluated by CBE. Analogs with higher activity (shown in red) were selected for further analysis. The mean RLU values from at least three independent experiments performed in quadruplicate are shown, with the signal obtained from wells transfected with an empty pcDNA3.1 plasmid (no Env) subtracted, normalized to Env levels as determined by 2G12 binding. Analogs and the positive control (+)-BNM-III-170 are plotted in decreasing order. (B) Structures of the selected analogs.
Complex structures of gp120CRF01_AE coree with (S)-MCG-III-027-D05, (S)-MCG-III-188-A01, (S)-MCG-III-188-A02, (S)-MCG-IV-031-A05, and (S)-MCG-IV-210 were solved in the same P212121 orthorhombic space group (see Table S1 in the supplemental material) at 3.25-Å, 2.2-Å, 1.84-Å, 2.24-Å, and 2.65-Å resolutions, respectively. As shown in Fig. 4A, the tested MCG analogs bind within the CD4 binding site, with the aromatic ring anchoring deeply in the Phe43 cavity; the entrance to this cavity is occupied by Phe43 of CD4 in the gp120-CD4 receptor complex (15). Interestingly, the chloride group of the 4-chloro-3-fluoro-substituted aromatic ring packs against the Cβ atom of Thr375, contributing significantly to binding through hydrophobic forces (Fig. 4A). The compounds largely overlap when bound to the CD4 binding cavity (Fig. 4C). Also, they involve almost the same set of gp120 residues for binding, which include Val255, Ser265, Thr257, Asp368, Glu370, Ile371, Thr375, Phe376, Phe382, Tyr384, Ile424, Asn425, Gly429, Gly473, Asp474, and Met475. In addition, analogs sharing a replacement of the sulfonamide substituent, e.g., (S)-MCG-III-188-A01, (S)-MCG-III-188-A02, (S)-MCG-IV-031-A05, and (S)-MCG-IV-210, reach deep into the Phe43 cavity and are within van der Waals contact distance to Trp112 and the Gly472-Gly473 stretch immediately preceding the α5 helix of gp120 (Fig. 4A). The contribution of residues lining the Phe43 cavity to the binding of each compound is shown in Fig. 4D. Interestingly, all but (S)-MCG-III-188-A01 contact Asp474, with the highest buried surface area for this residue observed for the most potent compound in this group, (S)-MCG-IV-210 (Fig. 4D). Overall, (S)-MCG-IV-210 stands out among the compounds tested. (S)-MCG-IV-210 establishes the most contacts with gp120, with (i) the highest total area buried at the interface, 687 Å2 [compared to 641 Å2 for (S)-MCG-III-188-A01, 614 Å2 for (S)-MCG-III-027-D05, 604 Å2 for (S)-MCG-IV-031-A05, and 592 Å2 for (S)-MCG-III-188-A02], (ii) the addition of a hydrogen bond with the main-chain oxygen of Gly472, (iii) the highest buried surface area of Gly473 (Fig. 4C), and (iv) the farthest reach toward Thr283 (Fig. 4A and C). Overall, the binding mode of (S)-MCG-IV-210 is reminiscent of the binding mode of BNM-III-170 (19), with improved contacts in (S)-MCG-IV-210 to the highly conserved gp120 residue Asp368 (Fig. 4B).
FIG 4.

Crystal structures of MCG analogs in complex with gp120CRF01_AE coree. (A) Blow-up views into the gp120 CD4 binding pocket. Complexes are shown as ribbon-ball-stick representations, with the side chains of gp120 residues contributing to the compounds’ binding shown as sticks. The H bonds are shown as pink dashes. (B) Details of the compounds’ interactions with gp120 Asp368 and Glu370. The closest distances between each compound and the side chain atoms of Asp368 and Glu370 are shown as dashes. (C) Superposition of MCG compounds onto BNM-III-170 (PDB accession number 5F4P). (D) Analysis of the compounds’ binding interface. The relative contribution of the indicated gp120 residue to each compound’s binding is shown as the buried surface area, as calculated by PISA. The buried surface area represents the solvent-accessible surface area of the corresponding residue that is buried upon interface formation.
Figure 4B shows the network of interactions of these CD4mc with the side chain atoms of Asp368 and Glu370, two highly conserved CD4 binding-site residues (15). In all cases, there are extensive contacts mediated by C atoms of the 4-chloro-3-fluoro-substituted aromatic ring and the piperidine ring to the Cβ atom or carboxyl oxygen of Asp368 and Glu370 (as measured by distances below 5 Å). To test the contribution of Asp368 and Glu370 to CD4mc binding, we altered these residues alone and in combination and then evaluated the ability of the CD4mc to expose the CoRBS of these mutant Envs. Of note, none of the altered gp120 residues are part of the 17b epitope (30). In addition, we altered residue 375, which lines the Phe43 cavity. As expected, and as shown in Fig. 5, filling the Phe43 cavity with a tryptophan residue (S375W) almost completely abrogated the capacity of all CD4mc tested to expose the 17b epitope, as measured by CBE. Interestingly, we found that replacement of aspartic acid (D368) with alanine (A368) in HIV-1JR-FL EnvΔCT enhanced the ability of the MCG analogs to expose the CoRBS compared to the wild type (WT); this result was in contrast to that observed for a late-generation CD4mc [(+)-BNM-III-170], which exhibited no impact of D368A relative to the unmodified EnvΔCT. This indicates that a hydrophobic interaction mediated by a Cβ atom of the residue at position 368 is sufficient to stabilize CD4mc in the Phe43 cavity, and the interactions mediated by the carboxyl group of Asp368 may have a potentially disrupting effect. Additionally, replacement of the D368 residue with an arginine residue (D368R) reduced the ability of all compounds to induce exposure of the CoRBS for all compounds to the same extent. Moreover, replacement of E370 with an arginine (E370R) or addition of D368R to E370R abrogated the activity of all CD4mc tested.
FIG 5.
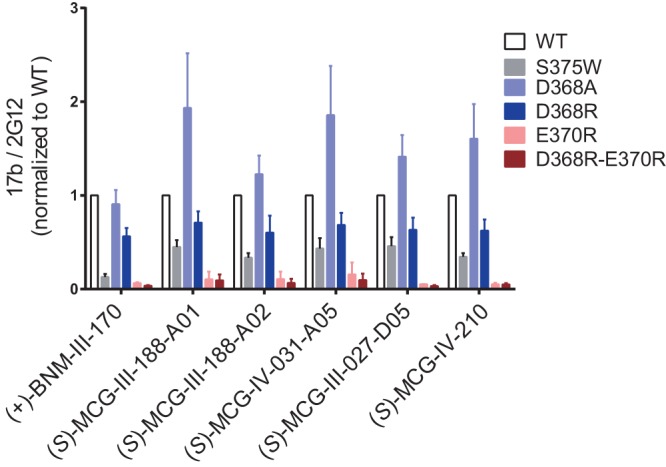
MCG analogs contact highly conserved residues D368 and E370. A CBE was used to determine CoRBS Ab 17b binding to WT HIV-1JR-FL EnvΔCT or mutants with different alterations, S375W, D368A, D368R, E370R, or D368R-E370R, expressed at the surface of HOS cells in the presence of CD4mc (50 μM) or the compounds’ vehicle (DMSO). Shown are the mean RLU values ± SD from at least three independent experiments performed in quadruplicates, with the signal obtained from wells transfected with an empty pcDNA3.1 plasmid (no Env) subtracted, normalized to Env levels as determined by bNAb 2G12 binding.
(S)-MCG-IV-210 stabilizes state 2A.
HIV-1 Env is a flexible molecule known to sample three different conformational states (states 1 to 3) (32). By exploring the Env conformational landscape in association with the epitopes recognized by different classes of CD4i Abs, we recently found that Env is able to sample a fourth conformation, state 2A, in the presence of the CD4mc (+)-BNM-III-170 and sera from HIV-1-infected individuals (33). The hallmark of state 2A is the exposure of otherwise occluded cluster A gp120 epitopes (33), which are exposed upon a sequential opening of the trimer (34). This opening requires initial triggering by CD4mc, followed by the interaction of CoRBS Abs, which then exposes the cluster A region (34). To evaluate whether (S)-MCG-IV-210 was able to stabilize state 2A, we infected primary CD4+ T cells with the primary CH58 transmitted/founder (CH58 TF) virus and evaluated the exposure of the cluster A region with an Alexa Fluor 647-conjugated A32 (A32-AF647) antibody in the presence or absence of CD4mc and the CoRBS 17b Ab, as reported previously (34). As a positive control, we used the CD4mc (+)-BNM-III-170, which was previously shown to stabilize state 2A and expose the A32 epitope in the presence of 17b (33). As expected, A32-AF647 failed to bind infected cells in the absence of CD4mc. The addition of 17b significantly enhanced the recognition of infected cells by A32-AF647 in the presence of (+)-BNM-III-170 or (S)-MCG-IV-210 (Fig. 6A to C). Since anti-cluster A and CoRBS Abs are present in a majority of HIV-1-infected individuals (35, 36), we evaluated whether HIV-positive (HIV+) plasma facilitated the exposure of the A32 epitope in the presence of (+)-BNM-III-170 and (S)-MCG-IV-210. Although (+)-BNM-III-170 exhibited higher activity, both CD4mc significantly enhanced the binding of infected cells by A32-AF647 in the presence of HIV+ plasma (Fig. 6D); thus, both CD4mc apparently stabilize state 2A at the surface of HIV-1-infected cells.
FIG 6.

(S)-MCG-IV-210 stabilizes state 2A at the surface of infected cells and virions. (A to D) For cell surface staining with A32-AF647, primary CD4 T cells isolated from PBMCs were infected with HIV-1CH58TF for 48 h. Cells were then incubated with A32-AF647 together with 5 μg/ml 17b (A to C) or 1:1,000-diluted HIV+ plasma from infected individuals (D) in the presence of DMSO, 50 μM (+)-BNM-III-170, or 50 μM (S)-MCG-IV-210 at 37°C. The mean fluorescence intensity (MFI) of A32-AF647 was measured by flow cytometry. (E) For the VCA, virus produced from HEK293T cells cotransfected with plasmids pNL4.3 Luc Env−, HIV-1CH58TF, and VSV-G was incubated with or without 5 μg/ml 17b in the presence of DMSO, 50 μM (+)-BNM-III-170, or 50 μM (S)-MCG-IV-210 at 37°C for 1 h. Virus was then applied to ELISA plates coated with Ab 2G12, 17b, A32, or C11 overnight at 4°C. Free virions were washed away, and HEK293T cells were added to the well. After 48 h, cells were lysed, and luciferase activity was measured. To compare the binding capacities of different Abs, the relative ratio of the luciferase activity to the luciferase activity of 2G12 was calculated. Data shown are the means ± SD from at least three independent experiments. Statistical significance was evaluated using a Mann-Whitney unpaired t test (C) or a Wilcoxon paired t test (D) (*, P < 0.05).
To verify that (S)-MCG-IV-210 also stabilizes state 2A at the surface of viral particles, we adapted a virus capture assay (VCA) to evaluate the Env conformation on virions (37, 38, 59). This VCA relies on the capture of HIV-1 virions by anti-Env Abs immobilized on ELISA plates. The viral particles used in this assay were generated by cotransfecting the pNL4.3 Luc Env− construct (38–41) with a plasmid encoding the Env of the transmitted/founder virus CH58 (42–45) and a plasmid encoding the G glycoprotein from vesicular stomatitis virus (VSV-G), resulting in a virus capable of a single round of infection. Virus-containing supernatants were added to the plate, and unbound virions were washed away. Retention of virions on the plate surface by anti-Env Abs was evaluated by the addition of HEK293T cells that do not express CD4. Infection was mediated by VSV-G and measured by luciferase activity 2 days after incubation with HEK293T cells. As recently reported and in agreement with the occluded nature of CD4i epitopes, CoRBS 17b and anti-cluster A A32 and C11 Abs failed to capture viral particles (59). The addition of both CD4mc exposed the CoRBS region, as shown by a dramatic increase in 17b-mediated capture of virions (Fig. 6E). In agreement with a sequential opening of the trimer required to expose the cluster A region, the addition of CD4mc in combination with 17b was required to permit virion capture by the A32 and C11 cluster A antibodies. Altogether, these results indicate that (S)-MCG-IV-210 is able to stabilize state 2A at the surface of infected cells and viral particles.
(S)-MCG-IV-210 sensitizes HIV-1-infected cells to ADCC and prevents killing of uninfected bystander CD4+ T cells.
We next evaluated the ADCC susceptibility of primary CD4+ T cells infected with CH58 TF mediated by HIV+ plasma with or without MCG analogs, using a fluorescence-activated cell sorter (FACS)-based assay as previously reported (46, 47). As shown in Fig. 7A and B, all tested MCG CD4mc [(S)-MCG-III-027-D05, (S)-MCG-III-188-A01, (S)-MCG-III-188-A02, (S)-MCG-IV-031-A05, and (S)-MCG-IV-210] enhanced the recognition of infected cells by the anti-CoRBS 17b antibody and by plasma from 15 HIV-1-infected individuals. Importantly, enhanced recognition of infected cells was translated into enhanced ADCC responses, supporting the positive correlation expected for these two activities (Fig. 7C and D).
FIG 7.

MCG analogs sensitize HIV-1-infected cells to ADCC and viral particles to neutralization by nnAbs. (A and B) Primary CD4 T cells isolated from PBMCs were infected with HIV-1CH58TF for 48 h. For cell surface staining, 5 μg/ml 17b (A) or 1:1,000-diluted HIV+ plasma (n = 15) (B) was used in the presence of the different MCG analogs (50 μM), (+)-BNM-III-170, or an equivalent volume of the vehicle (DMSO). An Alexa Fluor 647-conjugated anti-human IgG secondary Ab was then used for fluorescence labeling. (C) For ADCC, infected cells were used as target cells in a FACS-based ADCC assay that measures the killing of infected (p24+) cells (48). The assay determines susceptibility to ADCC mediated by a 1/1,000 dilution of plasma from 15 HIV-1-infected individuals in the presence of the different MCG analogs (50 μM), (+)-BNM-III-170 (50 μM), or an equivalent volume of the vehicle (DMSO). (D) The correlation between cell surface staining with HIV+ plasma and ADCC was calculated using the Spearman rank correlation. (E) To evaluate the direct virus-neutralizing ability of the analogs, HIV-1CH58TF was incubated with the indicated amounts of different compounds or DMSO for 1 h at 37°C and then added to TZM-bl cells. After incubation for 48 h at 37°C, luciferase activity was measured. Relative infectivity was calculated as the percentage of the value seen in the absence of the compound. (F) To measure the ability of (S)-MCG-IV-210 to sensitize viral particles to neutralization by otherwise nonneutralizing Ab 17b, HIV-1CH58TF was incubated with the indicated amounts of 17b in the presence of 0.5 μM (+)-BNM-III-170, 20 μM (S)-MCG-IV-210, or DMSO for 1 h at 37°C and then added to TZM-bl cells. After incubation for 48 h at 37°C, luciferase activity was measured. Relative infectivity was calculated as the percentage of the value seen in the absence of the compound. Data shown are the means ± SD from at least three independent experiments. Statistical significance was evaluated using a Mann-Whitney unpaired t test (A) or a Wilcoxon paired t test (B and C) (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
It was recently shown that gp120 shed from productively infected cells results in the coating of uninfected bystander CD4+ T cells, sampling of the open CD4-bound conformation, and exposure of ADCC-mediating epitopes, which are otherwise occluded in the native trimer (48). It has been proposed that this might represent a viral mechanism to divert cytotoxic immune responses to uninfected cells. By occupying the Phe43 cavity, CD4mc can block the coating of uninfected CD4+ T cells by shed gp120 and consequently protect these cells from ADCC responses (48). We therefore evaluated the capacity of (S)-MCG-IV-210 to protect uninfected bystander CD4+ T cells from ADCC responses. Briefly, primary CD4+ T cells were infected with the NL4-3.ADA.GFP WT virus. The cell proliferation dye eFluor-450 was used to stain uninfected autologous CD4+ T cells, which were then added to infected cells (eFluor-450-negative [eFluor-450−] cells) and cocultured for 72 h. The detection of autologous uninfected bystander CD4+ T cells, designated eFluor-450+ green fluorescent protein-negative (GFP−) cells, by HIV+ plasma was then evaluated by FACS analysis (Fig. 8A). When (+)-BNM-III-170 or (S)-MCG-IV-210 was added at the time of coculture between infected and eFluor-450-stained bystander cells, recognition of bystander cells by HIV+ plasma was significantly decreased, thereby protecting uninfected bystander CD4+ T cells from ADCC-mediated killing (Fig. 8B). This observation suggests that, besides sensitizing HIV-1-infected cells to ADCC, (S)-MCG-IV-210 analogs might have therapeutic utility by preventing the death of uninfected CD4+ T cells.
FIG 8.

(S)-MCG-IV-210 blocks uninfected bystander ADCC killing. Uninfected primary CD4+ T cells were stained with the cellular dye eFluor-450 and cocultured with unstained NL4-3.ADA.GFP WT virus-infected autologous cells for 72 h. (A) The ability of HIV+ plasma to recognize uninfected bystander cells in the presence of 50 μM (+)-BNM-III-170 or (S)-MCG-IV-210 was evaluated by FACS analysis. (B) These uninfected eFluor-450+ cells were also used as target cells for ADCC with autologous PBMCs and 5 HIV+ plasma samples in the presence of 50 μM (+)-BNM-III-170 or (S)-MCG-IV-210. Statistical significance was evaluated using a paired t test (*, P < 0.05; ****, P < 0.0001; ns, not significant).
(S)-MCG-IV-210 both neutralizes viral particles and sensitizes viral particles to neutralization by nnAbs.
Since (S)-MCG-IV-210 exhibited the highest activity among the different MCG analogs, we evaluated the capacity of (S)-MCG-IV-210 to neutralize HIV-1 particles bearing the primary HIV-1CH58TF Env in a standard TZM-bl assay. As a positive control, we used (+)-BNM-III-170, which has been reported to inhibit HIV-1 infection (19) at low-micromolar concentrations. As expected, (+)-BNM-III-170 neutralized HIV-1CH58TF with a 50% inhibitory concentration (IC50) of 0.11 μM. While all tested MCG analogs also neutralized HIV-1CH58TF, they did so with only modest potency (Fig. 7E). For example, MCG-IV-210 inhibited HIV-1CH58TF infection with an IC50 of 6.97 μM. Additional structural modifications will be required to improve the direct antiviral potency of the MCG compounds. Based on the cocrystal of (+)-BNM-III-170, which makes unique contacts with V430, G431, and L455 (Fig. 4D), reaching toward these residues might increase the potency of MCG analogs.
Another well-documented antiviral property of CD4mc is their capacity to sensitize viral particles to neutralization by otherwise nonneutralizing antibodies (20, 21, 49). We therefore evaluated if (S)-MCG-IV-210 shared this property. As expected, the anti-CoRBS 17b Ab did not neutralize infectious viral particles bearing the primary HIV-1CH58 Env but potently neutralized these same viral particles when they were sensitized with subinhibitory concentrations of (S)-MCG-IV-210 (Fig. 7F).
DISCUSSION
HIV-1 has evolved multiple strategies to avoid the humoral response elicited against Env, including extensive glycosylation, sequence-variable loops, and conformational masking of vulnerable epitopes (3, 50, 51). Despite the fact that the vast majority of antibodies elicited during natural infection have a limited HIV-1-neutralizing capacity and are thought to play a minimal role in long-term control of viral replication, these antibodies can exert selection pressure on HIV-1 through their weakly neutralizing activities and thus drive viral evolution (52). In addition to their nonneutralizing or weakly neutralizing activities, these antibodies possess Fc-mediated effector functions with the potential to eliminate HIV-1-infected cells through several immune mechanisms, including ADCC, provided that Env is exposed in the “open” CD4-bound conformation (35). Accordingly, small-molecule CD4mc, with the capacity to “push” Env to sample the CD4-bound conformation when combined with HIV+ sera, enhance the susceptibility of HIV-1-infected cells to ADCC (20, 23–25, 33), thus providing a new strategy to eliminate HIV-1-infected cells.
Through the use of an HTS CBE, we identified a new family of small-molecule CD4mc able to “open up” Env. In combination with CoRBS Abs or HIV+ plasma, these new small-molecule CD4mc stabilized the antibody-vulnerable state 2A conformation and sensitized HIV-1-infected cells to ADCC. Although these CD4mc have low direct neutralization capacities, they sensitize viral particles to neutralization by otherwise nonneutralizing antibodies. This family of CD4mc thus has some unique features that warrant additional efforts to improve their potency. The 3-substituted piperidine core is readily accessible from commercial (S)-3-piperidinecarboxylic acid via amide coupling with 4-chloro-3-fluoroaniline. The piperidine nitrogen can then be reacted with a variety of electrophiles to access N-substituted piperidine analogs. For example, the addition of an alkyl urea with a terminal amino group [(S)-MCG-IV-210] further improved the capacity of the compound to “open” Env. Accordingly, (S)-MCG-IV-210 was able to stabilize the vulnerable state 2A conformation at the surface of HIV-1-infected cells and viral particles. Thus, the small-molecule MCG compounds could serve as a new scaffold to develop more potent CD4mc that are able to sensitize HIV-1-infected cells to ADCC and viral particles to commonly elicited nonneutralizing antibodies.
Cocrystal structures of these small-molecule CD4mc with a modified gp120 core confirm that these compounds anchor deeply within the HIV-1 gp120 Phe43 cavity while also establishing contacts with the residues at the cavity rim. Most importantly, the mode of binding of this class of CD4mc allows contacts to the side chain atoms of Asp368 and Glu370. These two residues are highly conserved in circulating HIV-1 strains; therefore, targeting these sites by analogs capable of establishing side chain-specific contacts to these residues might ensure the breadth of this class of CD4mc.
In summary, (S)-MCG-IV-210 is a representative of a valuable new family of small-molecule CD4mc. The attractive structural and biological attributes of (S)-MCG-IV-210 make it a good candidate for further development. Structural and functional studies show that the 4-chloro-3-fluoro substitution on the aromatic ring, the amide linker, and the piperidine nitrogen substituent are important for the antiviral activities. Efforts to optimize each of these regions are ongoing.
MATERIALS AND METHODS
Ethics statement.
Written informed consent was obtained from all study participants (the Montreal Primary HIV Infection Cohort [53, 54] and the Canadian Cohort of HIV Infected Slow Progressors [55–57]), and research adhered to the ethical guidelines of the CRCHUM and was reviewed and approved by the CRCHUM institutional review board (ethics committee, approval number CE 16.164-CA). Research adhered to the standards indicated by the Declaration of Helsinki. All participants were adult and provided informed written consent prior to enrollment in accordance with institutional review board approval.
Cell lines and isolation of primary cells.
HEK293T human embryonic kidney cells and HOS cells (both obtained from the ATCC) were grown as previously described (16, 25, 41). Primary human peripheral blood mononuclear cells (PBMCs) and CD4+ T cells were isolated, activated, and cultured as previously described (16, 25). Briefly, PBMCs were obtained by leukapheresis. CD4+ T lymphocytes were purified from resting PBMCs by negative selection using immunomagnetic beads according to the manufacturer’s instructions (StemCell Technologies, Vancouver, BC, Canada). CD4+ T lymphocytes were activated with phytohemagglutinin-L (10 μg/ml) for 48 h and then maintained in RPMI 1640 complete medium supplemented with recombinant interleukin-2 (rIL-2) (100 U/ml).
Cell-based ELISA.
Detection of trimeric HIV-1JR-FL EnvΔCT at the surface of HOS cells was performed by a cell-based ELISA, as previously described (26, 39, 58). Briefly, HOS cells were seeded in T-75 flasks (3 × 106 cells per flask) and transfected the next day with 22.5 μg of Env-expressing plasmids using the standard polyethylenimine (PEI; Polysciences Inc., PA, USA) transfection method. Twenty-four hours after transfection, cells were plated in 384-wells plates (2 × 104 cells per well). One day later, cells were incubated in blocking buffer (washing buffer [25 mM Tris {pH 7.5}, 1.8 mM CaCl2, 1.0 mM MgCl2 {pH 7.5}, and 140 mM NaCl] supplemented with 10 mg/ml nonfat dry milk and 5 mM Tris [pH 8.0]) for 30 min and then coincubated for 1 h at room temperature with either the anti-CoRBS 17b Ab or the broadly neutralizing Ab (bNAb) 2G12 (1 μg/ml) and with the compounds (50 μM), soluble CD4 (sCD4) (10 μg/ml), or the compounds’ vehicle (dimethyl sulfoxide [DMSO]) diluted in blocking buffer. A horseradish peroxidase (HRP)-conjugated antibody specific for the Fc region of human IgG (Pierce) was then incubated with the samples for 45 min at room temperature. Under all conditions, cells were washed 5 times with blocking buffer and 5 times with washing buffer. HRP enzyme activity was determined after the addition of 20 μl per well of a 1:1 mix of Western Lightning oxidizing and luminol reagents (Perkin Elmer Life Sciences). Light emission was measured with an LB 941 TriStar luminometer (Berthold Technologies).
Identification of new chemical probes using a high-throughput cell-based ELISA.
A cell-based ELISA (CBE), capable of measuring conformational changes of membrane-bound trimeric Env (26, 27), was adapted to 384-well plates. Briefly, HOS cells were seeded in culture flasks (8 × 106 cells per flask [125-cm2 area]) on day 1. The culture medium Dulbecco’s modified Eagle medium (DMEM; Invitrogen) supplemented with 5% fetal bovine serum and 1% of a penicillin-streptomycin mix was used. Cells were transfected using the standard PEI transfection method (Polysciences Inc.) in the culture flask on the next day with the cytoplasmic-tail-deleted HIV-1JR-FL tier 2 Env. The cytoplasmic tail of Env was deleted to enhance Env expression on the cell surface, therefore enhancing the sensitivity of the CBE (26, 28). Twenty-four hours after transfection, cells were plated in 384-well plates (2 × 104 cells per well). Cells were given 20 h to attach to the plate surface. The medium was then removed by aspiration and replaced by a solution containing the compounds to be screened diluted in blocking buffer (10 mg/ml nonfat dry milk, 1.8 mM CaCl2, 1 mM MgCl2, 25 mM Tris [pH 7.5], and 140 mM NaCl) at a concentration of 10 μM. Cells were incubated for 1 h at room temperature in the presence of the compounds. We used sCD4 as our positive control to induce conformational changes and evaluated the exposure of the CoRBS with the CD4i 17b antibody (29, 30). Cells were then washed three times with blocking buffer before a solution containing the coreceptor binding-site antibody 17b diluted in blocking buffer at a concentration of 1 μg/ml was added. Cells were incubated in the presence of the antibody at room temperature for 1 h. Cells were washed three times with blocking buffer. An HRP-conjugated antibody specific for the Fc region of human IgG (Pierce) diluted in blocking buffer at a concentration of 0.33 μg/ml was then added. Cells were incubated with the antibody for 45 min at room temperature. Env antibody staining was assessed by measuring HRP enzyme activity with the addition of 20 μl per well of a 1:1 mix of Western Lightning oxidizing and luminol reagents (Perkin Elmer Life Sciences). Light emission was measured with a luminometer.
Compounds.
Small molecules were purchased from Chembridge (DiverSet, 60,000 compounds), Maybridge (32,000 compounds), and SPECS (16,000 compounds). A few compounds were synthesized using the synthetic procedures detailed in the chemical synthesis section in the supplemental material. All compounds were >95% pure.
Site-directed mutagenesis.
Mutations were introduced into the plasmid expressing the dCT HIV-1JR-FL Envs using the QuikChange II site-directed mutagenesis protocol or the QuikChange multisite-directed mutagenesis kit (Stratagene). The presence of the desired mutations was confirmed by DNA sequencing. All residues are numbered based on alignment with the HXBc2 prototypic sequence, according to convention.
Viral production and infections.
HIV-1 was produced and titrated as previously described (35). Viruses were then used to infect activated primary CD4 T cells from healthy HIV-1-negative donors by spin infection at 800 × g for 1 h in 96-well plates at room temperature.
Virus capture assay.
A VCA was performed as recently described (59). Briefly, viral particles were produced by transfecting 2 × 106 HEK293T cells with pNL4.3 Luc Env− (3.5 μg), HIV-1CH58TF (3.5 μg), and VSV-G (1 μg) using a standard calcium phosphate protocol. Forty-eight hours later, supernatants containing virions were collected, and cell debris was removed by centrifugation (1,500 rpm for 10 min). Supernatants were aliquoted and incubated with or without 5 μg/ml 17b in the presence of DMSO or 50 μM (+)-BNM-III-170 or (S)-MCG-IV-210 at 37°C for 1 h. To immobilize antibodies on ELISA plates, white MaxiSorp ELISA plates (Thermo Fisher Scientific) were incubated with 5 μg/ml of Ab 2G12, 17b, A32, or C11 in phosphate-buffered saline (PBS) overnight at 4°C. Unbound antibodies were removed by washing the plates twice with PBS. Plates were subsequently blocked with 3% bovine serum albumin (BSA) in PBS for 1 h at room temperature. After two washes with PBS, 200 μl of the virus-containing supernatant (with or without CD4mc and with or without 17b) was added to the wells. After a 4- to 6-h incubation, virions were removed, and the wells were washed with PBS 3 times. Viral capture by any given antibody was visualized by adding HEK293T cells (10 × 104) in full DMEM per well. At 48 h postinfection, cells were lysed by the addition of 30 μl of passive lysis buffer (Promega) and three freeze-thaw cycles. An LB 941 TriStar luminometer (Berthold Technologies) was used to measure the luciferase activity of each well after the addition of 100 μl of luciferin buffer (15 mM MgSO4, 15 mM KPO4 [pH 7.8], 1 mM ATP, and 1 mM dithiothreitol) and 50 μl of 1 mM d-luciferin potassium salt (Prolume).
All experiments using VSV-G-pseudotyped HIV-1 were done in a biosafety level 3 laboratory according to manipulation protocols accepted by the CRCHUM biosafety committee, which respect the requirements of the Public Health Agency of Canada.
Viral neutralization.
The viral infection assay was done as previously described (40). TZM-bl target cells were seeded at a density of 5 × 103 cells/well in 96-well luminometer-compatible tissue culture plates (Perkin Elmer) 24 h before infection. HIV-1CH58TF (10,000 reverse transcriptase units) in a final volume of 100 μl was incubated with the indicated amounts of different antibodies, compounds, or the mixture of Abs and compounds for 1 h at 37°C and then added to the target cells, followed by incubation for 48 h at 37°C; the medium was then removed from each well; and the cells were lysed by the addition of 30 μl of passive lysis buffer (Promega) and three freeze-thaw cycles. An LB 941 TriStar luminometer (Berthold Technologies) was used to measure the luciferase activity in each well after the addition of 100 μl of luciferin buffer (15 mM MgSO4, 15 mM KPO4 [pH 7.8], 1 mM ATP, and 1 mM dithiothreitol) and 50 μl of 1 mM d-luciferin potassium salt (Prolume).
Antibodies and plasma.
The following Abs were used alone or in combination with different compounds for cell surface staining: 2G12 and 17b. The anti-cluster A A32 and C11 antibodies were previously described (60, 61). Randomly selected plasma samples from different HIV-infected donors were collected, heat inactivated, and conserved as previously described (16, 25). Secondary goat anti-human antibodies coupled to Alexa Fluor 647 (Invitrogen) were used as secondary Abs.
Flow cytometry analysis of cell surface staining.
Cell surface staining was performed as previously described (23). Primary CD4 T cells were isolated from 3 different healthy donors and infected with HIV-1CH58TF. Binding of HIV-1-infected cells by plasma (1:1,000 dilution) or antibodies (5 μg/ml) in the presence or absence of 50 μM compounds was performed 48 h after infection. Cells were then incubated at 37°C for 1 h, followed by the addition of anti-human Alexa Fluor 647 (Invitrogen) secondary Abs for 20 min. Primary CD4 T cells infected with HIV-1CH58TF were then stained intracellularly for HIV-1 p24, using the Cytofix/Cytoperm fixation/permeabilization kit (BD Biosciences, Mississauga, ON, Canada) and a fluorescent anti-p24 monoclonal Ab (mAb) (phycoerythrin [PE]-conjugated anti-p24, clone KC57; Beckman Coulter/Immunotech). Binding of HIV-1-infected cells with Alexa Fluor 647-conjugated anti-cluster A Ab A32 (A32-AF647) was performed alone, in combination with 17b (5 μg/ml), or in combination with HIV+ plasma (1:1,000 dilution). The percentage of infected or transfected cells (p24+ cells or GFP+ cells, respectively) was determined by gating the live-cell population on the basis of AquaVivid viability dye staining. Samples were analyzed on an LSRII cytometer (BD Biosciences), and data analysis was performed using FlowJo vX.0.7 (TreeStar, Ashland, OR, USA).
ADCC FACS-based assay.
Measurement of ADCC using the FACS-based assay was performed at 48 h postinfection as previously described (16, 25, 47, 62). Briefly, HIV-1CH58TF-infected primary CD4+ T cells were stained with viability (AquaVivid; Thermo Fisher Scientific) and cellular (cell proliferation dye eFluor-670; eBioscience) markers and used as target cells. Autologous effector PBMCs, stained with another cellular marker (cell proliferation dye eFluor-450; eBioscience), were added at an effector-to-target cell ratio of 10:1 in 96-well V-bottom plates (Corning, Corning, NY). Briefly, mixed target and effector cells were incubated with HIV+ plasma (1:1,000) in the presence of 50 μM compounds or an equivalent volume of the vehicle (DMSO). The plates were subsequently centrifuged for 1 min at 300 × g and incubated at 37°C with 5% CO2 for 4 to 6 h before being fixed in a 2% PBS–formaldehyde solution. Samples were analyzed on an LSRII cytometer (BD Biosciences). Data analysis was performed using FlowJo vX.0.7 (TreeStar). The percentage of ADCC was calculated with the formula (% of p24+ cells in targets plus effectors) − (% of p24+ cells in targets plus effectors plus plasma)/(% of p24+ cells in targets) by gating on infected live target cells.
Uninfected bystander FACS-based analysis.
Activated primary CD4+ T cells were stained with the eFluor-450 cell marker (eBioscience) for 15 min at room temperature and washed twice with complete RPMI 1640 medium. eFluor-450+ cells were then cocultured with autologous cells infected for 72 h with the NL4-3.ADA.GFP WT virus, at a ratio of 1 uninfected cell to 4 infected cells (2 × 105 eFluor-450+ cells to 8 × 105 infected cells) in the presence or absence of 50 μM the CD4-mimetic compound (+)-BNM-III-170 or (S)-MCG-IV-210. After 2 days of coculture, cells were stained with 5 different HIV+ plasma samples (1:1,000 dilution), followed by the appropriate secondary Abs. The uninfected bystander cells were designated eFluor-450+ GFP− cells, as previously described (48). Cells were also used as target cells, while autologous PBMCs were used as effector cells to perform the ADCC measurement. In this specific context, target cells were stained with a viability dye (AquaVivid; Thermo Fisher Scientific), while autologous effector PBMCs were stained with the eFluor-670 cell marker (eBioscience). PBMCs were added to target cells at an effector-to-target cell ratio of 10:1 in 96-well V-bottom plates (Corning, Corning, NY). HIV+ plasma (1:1,000 final dilution) was added to appropriate wells, and cells were incubated for 15 min at room temperature. The plates were subsequently centrifuged for 1 min at 300 × g and incubated at 37°C with 5% CO2 for 5 to 6 h before being fixed with a PBS-formaldehyde solution (final concentration of 2% formaldehyde) containing a constant number of flow cytometry particles (5 × 104 particles/ml) (AccuCount blank particles, 5.3 μm; Spherotech, Lake Forest, IL, USA). As previously reported (48), these flow cytometry particles were used to calculate the relative cell count of viable target cells. The percentage of ADCC responses directed against the uninfected bystander cell population (eFluor-450+ eFluor670− GFP− viable cells) was calculated with the following formula: (relative cell counts in the targets + PBMC condition) − (relative cell counts in the targets + PBMC + plasma condition)/(relative cell counts in the target condition).
Statistical analysis.
Statistics were analyzed using GraphPad Prism version 7.0a (GraphPad, San Diego, CA, USA). Every data set was tested for statistical normality, and this information was used to apply the appropriate (parametric or nonparametric) statistical test. P values of <0.05 were considered significant; significance values are indicated in the figure legends.
CRF01_AE coree expression and purification.
Plasmids encoding the layer mutant gp120 extended core (coree) protein, LM/HT gp120CRF01_AE coree, were transfected into GnT1− cells using X-tremeGENE transfection reagent (Sigma-Aldrich) according to the manufacturer’s instructions. Following 7 days of culture growth at 37°C with 8% CO2, the cell supernatant was filtered and passed over a 17b affinity column to isolate expressed gp120. The gp120 glycoprotein was eluted with 0.1 M glycine (pH 3.0) into tubes containing 1 M Tris-HCl (pH 8.5) to immediately raise the pH. The protein was then deglycosylated with 10 U/μg of Endo Hf (NE Biolabs) overnight at 37°C. Endo Hf was removed by passage over an amylose resin column, followed by gel filtration chromatography on a Superdex 200 16/60 column (GE Healthcare, Piscataway, NJ) equilibrated with 5 mM Tris-HCl (pH 7.2) and 150 mM sodium chloride. The protein was concentrated to approximately 5 mg/ml for use in crystallization trials.
Crystallization of gp120 core complex with CD4mc.
Deglycosylated LM/HT gp120CRF01_AE coree (5 mg/ml) was crystalized by the hanging-drop method in a solution containing 5 to 10% polyethylene glycol 1500 (PEG 1500), 6% PEG 400, and 0.1 M HEPES (pH 7.5). Crystals were allowed to grow fully prior to soaking with CD4mc. All molecules were solubilized with DMSO at a concentration of 10 mM and diluted with crystallization buffer to 100 nM prior to use in crystal soaks. Briefly, 0.4 μl of 100 nM mimetic was added to the 0.4-μl hanging drop containing the gp120 crystals prior to incubation for 4 h. Crystals were then flash frozen in liquid nitrogen following a brief soak in crystallization buffer containing 15% 2-methyl-2,4-pentanediol (MPD) for cryoprotection and 50 nM the CD4mc.
Data collection, structure solution, and refinement.
Data were collected on Stanford Synchrotron Radiation Light Source (SSRL) beamline 12-2 on a Dectris Pilatus 6M detector. Data were integrated and processed with HKL2000 (63) or MOSFLM and SCALA from the CCP4 suite (64). Crystals were orthorhombic, belonging to space group P212121 with cell dimensions of an a of 66.3 to 67.1 Å, a b of 66.8 to 67.2 Å, and a c of 85.9 to 86.9 Å and diffracted to 3.25 Å [(S)-MCG-III-027-D05], 2.2 Å [(S)-MCG-III-188-A01], 1.84 Å [(S)-MCG-III-188-A02], 2.24 Å [(S)-MCG-IV-031-A05], and 2.65 Å [(S)-MCG-IV-210]. Structures were solved by molecular replacement with the program PHASER from the CCP4 suite using the structure under PDB accession number 3TGT as a starting model. Model building was done using the program COOT (65). Refinement was done using the program REFMAC from the CCP4 suite or PHENIX (66).
Structure validation and analysis.
The quality of the final refined models was monitored using the program MolProbity (67). Structural alignments were performed using the Dali server and the program LSQKAB from the CCP4 suite (64). The PISA Web server was used to determine contact surfaces and residues. All illustrations were prepared with the PyMOL molecular graphic suite (DeLano Scientific, San Carlos, CA, USA). The Ramachandran plot was obtained by using the validation program MolProbity and shows that 87 to 96.7% of the total amino acids are in the most favored region, 3 to 10.3% are in the generously allowed region, and 0 to 2.7% are in the disallowed region, depending on the structure, with better values corresponding to the higher-resolution structures. Complete data collection and refinement statistics can be found in Table S1 in the supplemental material.
Chemical synthesis: general considerations.
All reactions were conducted in oven-dried glassware under an inert atmosphere of nitrogen, unless otherwise stated. All solvents were of reagent or high-performance liquid chromatography (HPLC) grade. Anhydrous CH2Cl2, toluene, ether, and tetrahydrofuran (THF) were obtained from the Pure Solve PS-400 system under an argon atmosphere. All reagents were purchased from commercial sources and used as received. Reaction mixtures were magnetically stirred under a nitrogen atmosphere, unless otherwise noted, and reactions were monitored by either thin-layer chromatography (TLC) with 250-μm SiliaPlate precoated TLC plates or analytical ultraperformance liquid chromatography (UPLC). Yields refer to chromatographically or spectroscopically pure compounds. Optical rotations were measured on a Jasco P-2000 polarimeter. Proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III 500-MHz spectrometer. Chemical shifts (δ) are reported in parts per million relative to chloroform (δ 7.26), dimethyl sulfoxide (δ 2.50), acetone (δ 2.05), or methanol (δ 3.31) for 1H NMR and relative to chloroform (δ 77.0), dimethyl sulfoxide (δ 39.4), acetone (δ 29.8), or methanol (δ 49.0) for 13C NMR. High-resolution mass spectra (HRMS) were recorded at the University of Pennsylvania Mass Spectroscopy Service Center on either a Waters LCT Premier XE liquid chromatography-mass spectrometry (LC-MS) system or a Waters GC-TOF Premier system. Preparative-scale UPLC was performed with a Waters AutoPurification system equipped with a Sunfire C18 OBD column (10-μm packing material, 30- by 150-mm column dimensions), a 2767 sample manager, a 2545 binary gradient module, a system fluidics organizer, a 2489 UV-visible (UV-Vis) dual-wavelength (210- and 254-nm) detector, and MassLynx software with the FractionLynx application manager. Solvent systems were comprised of H2O and acetonitrile containing 0.1% trifluoroacetic acid. Supercritical fluid chromatography (SFC) analyses were performed with a Jasco system equipped with a PU-280-CO2 plus CO2 delivery system, a CO-2060 plus intelligent column thermostat/selector, an HC-2068-01 heater controller, a BP-2080 plus automatic back pressure regulator, an MD-2018 plus photodiode array detector (200 to 648 nm), and PU 2080 plus intelligent HPLC pumps. The purity of new compounds was judged by NMR and LC-MS (>95%). Chemical synthesis is detailed in the supplemental material.
Data availability.
Structures were deposited in the Protein Data Bank (PDB) with accession numbers 6ONV [(S)-MCG-III-027-D05], 6ONE [(S)-MCG-III-188-A01], 6ONF [(S)-MCG-III-188-A02], 6ONH [(S)-MCG-IV-031-A05], and 6P9N [(S)-MCG-IV-210].
Supplementary Material
ACKNOWLEDGMENTS
We thank Juliana Lanza from the CRCHUM for helpful discussions, Jean Duchaine and Dominic Salois from the IRIC High-Throughput Screening facility for performing the HTS screening, and Mathieu Coutu for technical assistance. We thank Patrick Blank and David Christianson for helpful discussions. We also thank the CRCHUM Flow Cytometry and NC3 Platform, the FRQS AIDS and Infectious Diseases Network, and Mario Legault for cohort coordination.
Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under contract number DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS). This research used resources of beamline 17-ID-1 of the National Synchrotron Light Source II, a U.S. DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under contract number DE-SC0012704. The Life Science Biomedical Technology Research resource is primarily supported by the National Institutes of Health, National Institute of General Medical Sciences, through a Biomedical Technology Research Resource P41 grant (P41GM111244) and by the DOE Office of Biological and Environmental Research (KP1605010). This work was supported by an internal CRCHUM grant, by a UNIVALOR grant, and by CIHR foundation grant number 352417 to A.F. Support for this work was also provided by NIH R01 grants to A.F. and M.P. (AI129769), to J.S. (AI124982), and to M.P. (AI116274). This study was also supported by grant P01-GM56550/AI150741 to C.A., J.S., A.B.S., and A.F. A.F. is the recipient of a Canada Research Chair on Retroviral Entry (number RCHS0235 950-232424). S.D. is the recipient of an FRSQ postdoctoral fellowship award. J.P. is the recipient of a CIHR doctoral fellowship. J.R. is the recipient of Mathilde Krim fellowships in basic biomedical research from the amfAR. The funders had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
The views expressed in this presentation are those of the authors and do not reflect the official policy or position of the Uniformed Services University, the U.S. Army, the Department of Defense, or the U.S. Government.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01325-19.
REFERENCES
- 1.Allan JS, Coligan JE, Barin F, McLane MF, Sodroski JG, Rosen CA, Haseltine WA, Lee TH, Essex M. 1985. Major glycoprotein antigens that induce antibodies in AIDS patients are encoded by HTLV-III. Science 228:1091–1094. doi: 10.1126/science.2986290. [DOI] [PubMed] [Google Scholar]
- 2.Robey WG, Safai B, Oroszlan S, Arthur LO, Gonda MA, Gallo RC, Fischinger PJ. 1985. Characterization of envelope and core structural gene products of HTLV-III with sera from AIDS patients. Science 228:593–595. doi: 10.1126/science.2984774. [DOI] [PubMed] [Google Scholar]
- 3.Wyatt R, Sodroski J. 1998. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 4.Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, Berger EA. 1996. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955–1958. doi: 10.1126/science.272.5270.1955. [DOI] [PubMed] [Google Scholar]
- 5.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 6.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 7.Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, Parmentier M, Collman RG, Doms RW. 1996. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 8.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 9.Feng Y, Broder CC, Kennedy PE, Berger EA. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 10.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. 1996. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 11.Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. 1996. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 384:184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- 12.Furuta RA, Wild CT, Weng Y, Weiss CD. 1998. Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Biol 5:276–279. doi: 10.1038/nsb0498-276. [DOI] [PubMed] [Google Scholar]
- 13.He Y, Vassell R, Zaitseva M, Nguyen N, Yang Z, Weng Y, Weiss CD. 2003. Peptides trap the human immunodeficiency virus type 1 envelope glycoprotein fusion intermediate at two sites. J Virol 77:1666–1671. doi: 10.1128/JVI.77.3.1666-1671.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koshiba T, Chan DC. 2003. The prefusogenic intermediate of HIV-1 gp41 contains exposed C-peptide regions. J Biol Chem 278:7573–7579. doi: 10.1074/jbc.M211154200. [DOI] [PubMed] [Google Scholar]
- 15.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. 1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veillette M, Desormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, Haynes BF, Robinson JE, Kaufmann DE, Bonsignori M, Sodroski J, Finzi A. 2014. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol 88:2633–2644. doi: 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. 2005. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology 339:213–225. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Courter JR, Madani N, Sodroski J, Schon A, Freire E, Kwong PD, Hendrickson WA, Chaiken IM, LaLonde JM, Smith AB III. 2014. Structure-based design, synthesis and validation of CD4-mimetic small molecule inhibitors of HIV-1 entry: conversion of a viral entry agonist to an antagonist. Acc Chem Res 47:1228–1237. doi: 10.1021/ar4002735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Melillo B, Liang S, Park J, Schon A, Courter JR, LaLonde JM, Wendler DJ, Princiotto AM, Seaman MS, Freire E, Sodroski J, Madani N, Hendrickson WA, Smith AB III. 2016. Small-molecule CD4-mimics: structure-based optimization of HIV-1 entry inhibition. ACS Med Chem Lett 7:330–334. doi: 10.1021/acsmedchemlett.5b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Madani N, Princiotto AM, Mach L, Ding S, Prevost J, Richard J, Hora B, Sutherland L, Zhao CA, Conn BP, Bradley T, Moody MA, Melillo B, Finzi A, Haynes BF, Smith AB III, Santra S, Sodroski J. 2018. A CD4-mimetic compound enhances vaccine efficacy against stringent immunodeficiency virus challenge. Nat Commun 9:2363. doi: 10.1038/s41467-018-04758-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madani N, Princiotto AM, Schon A, LaLonde J, Feng Y, Freire E, Park J, Courter JR, Jones DM, Robinson J, Liao HX, Moody MA, Permar S, Haynes B, Smith AB III, Wyatt R, Sodroski J. 2014. CD4-mimetic small molecules sensitize human immunodeficiency virus to vaccine-elicited antibodies. J Virol 88:6542–6555. doi: 10.1128/JVI.00540-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Madani N, Princiotto AM, Zhao C, Jahanbakhshsefidi F, Mertens M, Herschhorn A, Melillo B, Smith AB III, Sodroski J. 2017. Activation and inactivation of primary human immunodeficiency virus envelope glycoprotein trimers by CD4-mimetic compounds. J Virol 91:e01880-16. doi: 10.1128/JVI.01880-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding S, Verly MM, Princiotto A, Melillo B, Moody T, Bradley T, Easterhoff D, Roger M, Hahn BH, Madani N, Smith AB III, Haynes BF, Sodroski JMD, Finzi A. 1 May 2017. Small molecule CD4-mimetics sensitize HIV-1-infected cells to ADCC by antibodies elicited by multiple envelope glycoprotein immunogens in non-human primates. AIDS Res Hum Retroviruses doi: 10.1089/AID.2016.0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richard J, Prevost J, von Bredow B, Ding S, Brassard N, Medjahed H, Coutu M, Melillo B, Bibollet-Ruche F, Hahn BH, Kaufmann DE, Smith AB III, Sodroski J, Sauter D, Kirchhoff F, Gee K, Neil SJ, Evans DT, Finzi A. 2017. BST-2 expression modulates small CD4-mimetic sensitization of HIV-1-infected cells to antibody-dependent cellular cytotoxicity. J Virol 91:e00219-17. doi: 10.1128/JVI.00219-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Richard J, Veillette M, Brassard N, Iyer SS, Roger M, Martin L, Pazgier M, Schon A, Freire E, Routy JP, Smith AB III, Park J, Jones DM, Courter JR, Melillo BN, Kaufmann DE, Hahn BH, Permar SR, Haynes BF, Madani N, Sodroski JG, Finzi A. 2015. CD4 mimetics sensitize HIV-1-infected cells to ADCC. Proc Natl Acad Sci U S A 112:E2687–E2694. doi: 10.1073/pnas.1506755112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veillette M, Coutu M, Richard J, Batraville LA, Desormeaux A, Roger M, Finzi A. 2014. Conformational evaluation of HIV-1 trimeric envelope glycoproteins using a cell-based ELISA assay. J Vis Exp 2014:51995. doi: 10.3791/51995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haim H, Strack B, Kassa A, Madani N, Wang L, Courter JR, Princiotto A, McGee K, Pacheco B, Seaman MS, Smith AB III, Sodroski J. 2011. Contribution of intrinsic reactivity of the HIV-1 envelope glycoproteins to CD4-independent infection and global inhibitor sensitivity. PLoS Pathog 7:e1002101. doi: 10.1371/journal.ppat.1002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haim H, Si Z, Madani N, Wang L, Courter JR, Princiotto A, Kassa A, DeGrace M, McGee-Estrada K, Mefford M, Gabuzda D, Smith AB III, Sodroski J. 2009. Soluble CD4 and CD4-mimetic compounds inhibit HIV-1 infection by induction of a short-lived activated state. PLoS Pathog 5:e1000360. doi: 10.1371/journal.ppat.1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wyatt R, Moore J, Accola M, Desjardin E, Robinson J, Sodroski J. 1995. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor binding. J Virol 69:5723–5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rizzuto CD, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong PD, Hendrickson WA, Sodroski J. 1998. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280:1949–1953. doi: 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- 31.Zoubchenok D, Veillette M, Prevost J, Sanders-Buell E, Wagh K, Korber B, Chenine AL, Finzi A. 2017. Histidine 375 modulates CD4 binding in HIV-1 CRF01_AE envelope glycoproteins. J Virol 91:e02151-16. doi: 10.1128/JVI.02151-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB III, Kwong PD, Blanchard SC, Mothes W. 2014. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 346:759–763. doi: 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alsahafi N, Bakouche N, Kazemi M, Richard J, Ding S, Bhattacharyya S, Das D, Anand SP, Prevost J, Tolbert WD, Lu H, Medjahed H, Gendron-Lepage G, Ortega Delgado GG, Kirk S, Melillo B, Mothes W, Sodroski J, Smith AB III, Kaufmann DE, Wu X, Pazgier M, Rouiller I, Finzi A, Munro JB. 2019. An asymmetric opening of HIV-1 envelope mediates antibody-dependent cellular cytotoxicity. Cell Host Microbe 25:578–587.e5. doi: 10.1016/j.chom.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richard J, Pacheco B, Gohain N, Veillette M, Ding S, Alsahafi N, Tolbert WD, Prevost J, Chapleau JP, Coutu M, Jia M, Brassard N, Park J, Courter JR, Melillo B, Martin L, Tremblay C, Hahn BH, Kaufmann DE, Wu X, Smith AB III, Sodroski J, Pazgier M, Finzi A. 2016. Co-receptor binding site antibodies enable CD4-mimetics to expose conserved anti-cluster A ADCC epitopes on HIV-1 envelope glycoproteins. EBioMedicine 12:208–218. doi: 10.1016/j.ebiom.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veillette M, Coutu M, Richard J, Batraville LA, Dagher O, Bernard N, Tremblay C, Kaufmann DE, Roger M, Finzi A. 2015. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J Virol 89:545–551. doi: 10.1128/JVI.02868-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Decker JM, Bibollet-Ruche F, Wei X, Wang S, Levy DN, Wang W, Delaporte E, Peeters M, Derdeyn CA, Allen S, Hunter E, Saag MS, Hoxie JA, Hahn BH, Kwong PD, Robinson JE, Shaw GM. 2005. Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J Exp Med 201:1407–1419. doi: 10.1084/jem.20042510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore PL, Crooks ET, Porter L, Zhu P, Cayanan CS, Grise H, Corcoran P, Zwick MB, Franti M, Morris L, Roux KH, Burton DR, Binley JM. 2006. Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J Virol 80:2515–2528. doi: 10.1128/JVI.80.5.2515-2528.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kassa A, Finzi A, Pancera M, Courter JR, Smith AB III, Sodroski J. 2009. Identification of a human immunodeficiency virus type 1 envelope glycoprotein variant resistant to cold inactivation. J Virol 83:4476–4488. doi: 10.1128/JVI.02110-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Desormeaux A, Coutu M, Medjahed H, Pacheco B, Herschhorn A, Gu C, Xiang SH, Mao Y, Sodroski J, Finzi A. 2013. The highly conserved layer-3 component of the HIV-1 gp120 inner domain is critical for CD4-required conformational transitions. J Virol 87:2549–2562. doi: 10.1128/JVI.03104-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finzi A, Xiang SH, Pacheco B, Wang L, Haight J, Kassa A, Danek B, Pancera M, Kwong PD, Sodroski J. 2010. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol Cell 37:656–667. doi: 10.1016/j.molcel.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pacheco B, Alsahafi N, Debbeche O, Prevost J, Ding S, Chapleau JP, Herschhorn A, Madani N, Princiotto A, Melillo B, Gu C, Zeng X, Mao Y, Smith AB III, Sodroski J, Finzi A. 2017. Residues in the gp41 ectodomain regulate HIV-1 envelope glycoprotein conformational transitions induced by gp120-directed inhibitors. J Virol 91:e02219-16. doi: 10.1128/JVI.02219-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, Hahn BH, Kappes JC. 2012. Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J Virol 86:2715–2728. doi: 10.1128/JVI.06157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bar KJ, Tsao CY, Iyer SS, Decker JM, Yang Y, Bonsignori M, Chen X, Hwang KK, Montefiori DC, Liao HX, Hraber P, Fischer W, Li H, Wang S, Sterrett S, Keele BF, Ganusov VV, Perelson AS, Korber BT, Georgiev I, McLellan JS, Pavlicek JW, Gao F, Haynes BF, Hahn BH, Kwong PD, Shaw GM. 2012. Early low-titer neutralizing antibodies impede HIV-1 replication and select for virus escape. PLoS Pathog 8:e1002721. doi: 10.1371/journal.ppat.1002721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, Hora B, Berg A, Cai F, Hopper J, Denny TN, Ding H, Ochsenbauer C, Kappes JC, Galimidi RP, West AP Jr, Bjorkman PJ, Wilen CB, Doms RW, O’Brien M, Bhardwaj N, Borrow P, Haynes BF, Muldoon M, Theiler JP, Korber B, Shaw GM, Hahn BH. 2013. Phenotypic properties of transmitted founder HIV-1. Proc Natl Acad Sci U S A 110:6626–6633. doi: 10.1073/pnas.1304288110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fenton-May AE, Dibben O, Emmerich T, Ding H, Pfafferott K, Aasa-Chapman MM, Pellegrino P, Williams I, Cohen MS, Gao F, Shaw GM, Hahn BH, Ochsenbauer C, Kappes JC, Borrow P. 2013. Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology 10:146. doi: 10.1186/1742-4690-10-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ding S, Veillette M, Coutu M, Prevost J, Scharf L, Bjorkman PJ, Ferrari G, Robinson JE, Sturzel C, Hahn BH, Sauter D, Kirchhoff F, Lewis GK, Pazgier M, Finzi A. 2016. A highly conserved residue of the HIV-1 gp120 inner domain is important for antibody-dependent cellular cytotoxicity responses mediated by anti-cluster A antibodies. J Virol 90:2127–2134. doi: 10.1128/JVI.02779-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richard J, Prevost J, Baxter AE, von Bredow B, Ding S, Medjahed H, Delgado GG, Brassard N, Sturzel CM, Kirchhoff F, Hahn BH, Parsons MS, Kaufmann DE, Evans DT, Finzi A. 2018. Uninfected bystander cells impact the measurement of HIV-specific antibody-dependent cellular cytotoxicity responses. mBio 9:e00358-18. doi: 10.1128/mBio.00358-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richard J, Veillette M, Ding S, Zoubchenok D, Alsahafi N, Coutu M, Brassard N, Park J, Courter JR, Melillo B, Smith AB III, Shaw GM, Hahn BH, Sodroski J, Kaufmann DE, Finzi A. 2016. Small CD4 mimetics prevent HIV-1 uninfected bystander CD4 + T cell killing mediated by antibody-dependent cell-mediated cytotoxicity. EBioMedicine 3:122–134. doi: 10.1016/j.ebiom.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Madani N, Princiotto AM, Easterhoff D, Bradley T, Luo K, Williams WB, Liao HX, Moody MA, Phad GE, Vazquez Bernat N, Melillo B, Santra S, Smith AB III, Karlsson Hedestam GB, Haynes B, Sodroski J. 2016. Antibodies elicited by multiple envelope glycoprotein immunogens in primates neutralize primary human immunodeficiency viruses (HIV-1) sensitized by CD4-mimetic compounds. J Virol 90:5031–5046. doi: 10.1128/JVI.03211-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kwong PD, Doyle ML, Casper DJ, Cicala C, Leavitt SA, Majeed S, Steenbeke TD, Venturi M, Chaiken I, Fung M, Katinger H, Parren PW, Robinson J, Van Ryk D, Wang L, Burton DR, Freire E, Wyatt R, Sodroski J, Hendrickson WA, Arthos J. 2002. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420:678–682. doi: 10.1038/nature01188. [DOI] [PubMed] [Google Scholar]
- 51.Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG. 1998. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393:705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 52.Moody MA, Gao F, Gurley TC, Amos JD, Kumar A, Hora B, Marshall DJ, Whitesides JF, Xia SM, Parks R, Lloyd KE, Hwang KK, Lu X, Bonsignori M, Finzi A, Vandergrift NA, Alam SM, Ferrari G, Shen X, Tomaras GD, Kamanga G, Cohen MS, Sam NE, Kapiga S, Gray ES, Tumba NL, Morris L, Zolla-Pazner S, Gorny MK, Mascola JR, Hahn BH, Shaw GM, Sodroski JG, Liao HX, Montefiori DC, Hraber PT, Korber BT, Haynes BF. 2015. Strain-specific V3 and CD4 binding site autologous HIV-1 neutralizing antibodies select neutralization-resistant viruses. Cell Host Microbe 18:354–362. doi: 10.1016/j.chom.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fontaine J, Chagnon-Choquet J, Valcke HS, Poudrier J, Roger M. 2011. High expression levels of B lymphocyte stimulator (BLyS) by dendritic cells correlate with HIV-related B-cell disease progression in humans. Blood 117:145–155. doi: 10.1182/blood-2010-08-301887. [DOI] [PubMed] [Google Scholar]
- 54.Fontaine J, Coutlee F, Tremblay C, Routy JP, Poudrier J, Roger M. 2009. HIV infection affects blood myeloid dendritic cells after successful therapy and despite nonprogressing clinical disease. J Infect Dis 199:1007–1018. doi: 10.1086/597278. [DOI] [PubMed] [Google Scholar]
- 55.International HIV Controllers Study, Pereyra F, Jia X, McLaren PJ, Telenti A, de Bakker PIW, Walker BD, Ripke S, Brumme CJ, Pulit SL, Carrington M, Kadie CM, Carlson JM, Heckerman D, Graham RR, Plenge RM, Deeks SG, Gianniny L, Crawford G, Sullivan J, Gonzalez E, Davies L, Camargo A, Moore JM, Beattie N, Gupta S, Crenshaw A, Burtt NP, Guiducci C, Gupta N, Gao X, Qi Y, Yuki Y, Piechocka-Trocha A, Cutrell E, Rosenberg R, Moss KL, Lemay P, O’Leary J, Schaefer T, Verma P, Toth I, Block B, Baker B, Rothchild A, Lian J, Proudfoot J, Alvino DML, Vine S, Addo MM, et al. 2010. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science 330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kamya P, Boulet S, Tsoukas CM, Routy JP, Thomas R, Cote P, Boulassel MR, Baril JG, Kovacs C, Migueles SA, Connors M, Suscovich TJ, Brander C, Tremblay CL, Bernard N. 2011. Receptor-ligand requirements for increased NK cell polyfunctional potential in slow progressors infected with HIV-1 coexpressing KIR3DL1*h/*y and HLA-B*57. J Virol 85:5949–5960. doi: 10.1128/JVI.02652-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peretz Y, Ndongala ML, Boulet S, Boulassel MR, Rouleau D, Cote P, Longpre D, Routy JP, Falutz J, Tremblay C, Tsoukas CM, Sekaly RP, Bernard NF. 2007. Functional T cell subsets contribute differentially to HIV peptide-specific responses within infected individuals: correlation of these functional T cell subsets with markers of disease progression. Clin Immunol 124:57–68. doi: 10.1016/j.clim.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 58.Alsahafi N, Debbeche O, Sodroski J, Finzi A. 2015. Effects of the I559P gp41 change on the conformation and function of the human immunodeficiency virus (HIV-1) membrane envelope glycoprotein trimer. PLoS One 10:e0122111. doi: 10.1371/journal.pone.0122111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ding S, Gasser R, Gendron-Lepage G, Medjahed H, Tolbert WD, Sodroski J, Pazgier M, Finzi A. 4 September 2019. CD4 incorporation into HIV-1 viral particles exposes envelope epitopes recognized by CD4-induced antibodies. J Virol doi: 10.1128/JVI.01403-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robinson JE, Holton D, Elliott S, Ho DD. 1992. Distinct antigenic sites on HIV gp120 identified by a panel of human monoclonal antibodies. J Cell Biochem Suppl 16E:Q449. [Google Scholar]
- 61.Moore JP, Willey RL, Lewis GK, Robinson J, Sodroski J. 1994. Immunological evidence for interactions between the first, second, and fifth conserved domains of the gp120 surface glycoprotein of human immunodeficiency virus type 1. J Virol 68:6836–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richard J, Veillette M, Batraville LA, Coutu M, Chapleau JP, Bonsignori M, Bernard N, Tremblay C, Roger M, Kaufmann DE, Finzi A. 2014. Flow cytometry-based assay to study HIV-1 gp120 specific antibody-dependent cellular cytotoxicity responses. J Virol Methods 208:107–114. doi: 10.1016/j.jviromet.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 63.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 64.Collaborative Computational Project, Number 4. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 65.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 66.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 67.Davis IW, Murray LW, Richardson JS, Richardson DC. 2004. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res 32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structures were deposited in the Protein Data Bank (PDB) with accession numbers 6ONV [(S)-MCG-III-027-D05], 6ONE [(S)-MCG-III-188-A01], 6ONF [(S)-MCG-III-188-A02], 6ONH [(S)-MCG-IV-031-A05], and 6P9N [(S)-MCG-IV-210].