

Previous studies have confirmed that DDX21 is vital for the regulation of various aspects of RNA virus replication. Our research is the first report on the role of DDX21 in HCMV DNA virus replication. We identified that DDX21 knockdown affected HCMV growth and viral late gene transcription. In order to elucidate how DDX21 regulated this transcription, we applied DNA-RNA immunoprecipitation by using the DNA-RNA hybrid-specific S9.6 antibody to test whether more R-loops accumulated on the viral late gene. Consistent with our expectation, more R-loops were detected on the viral late gene at late HCMV infection time points, which demonstrated that the accumulation of R-loops caused by DDX21 knockdown prevented viral late gene transcription and consequently impaired HCMV replication. These results reveal that DDX21 plays an important role in regulating HCMV replication and also provide a basis for investigating the role of DDX21 in regulating other DNA viruses.
KEYWORDS: DDX21, human cytomegalovirus, viral late gene transcription, R-loops, DNA-RNA immunoprecipitation, DDX21
ABSTRACT
DDX21 regulates the biogenesis of rRNA and transcription of ribonucleoprotein genes. Recently, it has been reported that DDX21 regulates the growth of some RNA viruses through various mechanisms, such as inhibiting viral genome replication, suppressing virion assembly and release, and modulating antiviral immune responses (Chen et al., Cell Host Microbe 15:484–493, 2014, https://doi.org/10.1016/j.chom.2014.03.002; Dong et al., Biophys Res Commun, 473:648–653, 2016, https://doi.org/10.1016/j.bbrc.2016.03.120; and Watanabe et al., PLoS Pathog 5:e1000654, 2009, https://doi.org/10.1371/journal.ppat.1000654). The relationship between DDX21 and DNA viruses has not yet been explored. In this study, we used human cytomegalovirus (HCMV), a large human DNA virus, to investigate the potential role of DDX21 in DNA virus replication. We found that HCMV infection prevented the repression of DDX21 at protein and mRNA levels. Knockdown of DDX21 inhibited HCMV growth in human fibroblast cells (MRC5). Immunofluorescence and quantitative PCR (qPCR) results showed that knockdown of DDX21 did not affect viral DNA replication or the formation of the viral replication compartment but did significantly inhibit viral late gene transcription. Some studies have reported that DDX21 knockdown promotes the accumulation of R-loops that could restrain RNA polymerase II elongation and inhibit the transcription of certain genes. Thus, we used the DNA-RNA hybrid-specific S9.6 antibody to stain R-loops and observed that more R-loops formed in DDX21-knockdown cells than in control cells. Moreover, an DNA-RNA immunoprecipitation assay showed that more R-loops accumulated on a viral late gene in DDX21-knockdown cells. Altogether, these results suggest that DDX21 knockdown promotes the accumulation of R-loops, which prevents viral late gene transcription and consequently results in the suppression of HCMV growth. This finding provides new insight into the relationship between DDX21 and DNA virus replication.
IMPORTANCE Previous studies have confirmed that DDX21 is vital for the regulation of various aspects of RNA virus replication. Our research is the first report on the role of DDX21 in HCMV DNA virus replication. We identified that DDX21 knockdown affected HCMV growth and viral late gene transcription. In order to elucidate how DDX21 regulated this transcription, we applied DNA-RNA immunoprecipitation by using the DNA-RNA hybrid-specific S9.6 antibody to test whether more R-loops accumulated on the viral late gene. Consistent with our expectation, more R-loops were detected on the viral late gene at late HCMV infection time points, which demonstrated that the accumulation of R-loops caused by DDX21 knockdown prevented viral late gene transcription and consequently impaired HCMV replication. These results reveal that DDX21 plays an important role in regulating HCMV replication and also provide a basis for investigating the role of DDX21 in regulating other DNA viruses.
INTRODUCTION
Human cytomegalovirus (HCMV) is a slow-growing betaherpesvirus. Its genetic material is double-stranded DNA, and DNA replication occurs in the host cell nucleus (1). During cellular infection, HCMV can establish a suitable environment for long-term latent infection (2, 3). HCMV infection is generally asymptomatic in healthy individuals. However, it can lead to severe infectious complications and even death in immunocompromised individuals such as AIDS patients, organ transplant recipients, or human immunodeficiency carriers (4–6). HCMV is also a serious infectious pathogen causing congenital diseases in newborns. For example, pregnant women carrying this virus have substantially increased risks of giving birth to deaf, blind, or mentally deficient babies (3, 7, 8). HCMV has a very large genome (230 kb) carrying about 192 open reading frames, which makes it quite difficult to explore its underlying pathogenic mechanisms. Limited knowledge exists regarding the regulatory mechanisms underlying interactions between HCMV and its host, and no relevant vaccine has been developed. Thus, it is necessary to explore the relationship between HCMV and various vital host factors, which could increase the understanding of HCMV and facilitate the development of relevant vaccines or small molecule drugs for the timely and effective prevention of HCMV infection.
DEAD-box RNA helicases are a family of multifunctional enzymes that regulate various aspects of RNA metabolism, including transcription, processing, and modification. In some circumstances, these proteins may also participate in microRNA processing, RNA nuclear export and RNA degradation (9, 10). DDX21 is a nucleolar protein belonging to the DEAD-box RNA helicase family. It contains a highly conserved helicase domain, and its helicase activity is involved in the relocalization of DDX21 from the nucleolus to the nucleoplasm. It has been reported that DDX21 can bind to RNA polymerase I (Pol I) or Pol II to regulate multiple stages of RNA biogenesis in human cells. For example, in the nucleolus, DDX21 binds ribosomal DNA and regulates Pol I to promote pre-rRNA transcription, processing, and modification. In the nucleoplasm, it occupies the promoters of Pol II-transcribed genes and controls the transcription of these genes (9, 11–13). Recently, some studies have reported that DDX21 is vital for regulating the replication of certain RNA viruses. For instance, the host DDX21 RNA helicase restricts influenza A virus by binding PB1, inhibiting polymerase assembly and resulting in reduced viral RNA and protein synthesis (14). At early stages of dengue virus infection, DDX21 was found to transfer from the nucleus to cytoplasm, which activates innate immune responses and subsequently inhibits dengue virus replication (15). Borna disease virus (BDV) is a negative-strand RNA virus, and DDX21 was reported to regulate BDV replication by controlling its polycistronic mRNA translation (16). In addition, DDX21 also associates with other members of the DExD/H-box helicase family to regulate immune responses. DExD/H-box helicases belong to the large SF2 helicase superfamily. Within the DExD/H-box family, helicases are further distinguished based on the amino acid sequence of the eponymous conserved helicase motif II (DEAD, DEAH, DExH, and DExD helicases). For example, DDX1, DDX21, and DHX36 can form a complex to regulate immune responses induced by interferon, and depletion of any member of the complex could inhibit these immune responses upon treatment with poly(I⋅C), influenza virus, or reovirus (17). According to these reports, DDX21 can regulate many cellular reactions during virus infection and plays an important role in virus replication. However, these reports have only investigated RNA viruses, and the relationship between DDX21 and DNA virus has not been explored. Thus, it is very important to explore whether DDX21 affects DNA virus replication and the underlying mechanism.
Here, we used HCMV to infect human fibroblast cells (MRC5) to detect the functional role of DDX21 during virus replication. Our results showed that DDX21 mRNA and protein levels gradually decreased in mock-infected cells but maintained high-level expression in HCMV-infected cells. In addition, knockdown of DDX21 inhibited HCMV growth and viral late gene expression. Subsequent results showed that DDX21 knockdown prevented HCMV viral late gene transcription. It has been reported that DDX21 knockdown results in the accumulation of R-loops, and our immunofluorescence assay confirmed this results. R-loops are three-stranded nucleic acid structures consisting of a DNA-RNA hybrid and a displaced single-stranded DNA (18). We applied a DNA-RNA immunoprecipitation (DRIP) assay developed by Song et al. (19) to detect whether more R-loops accumulated on viral late gene. This method involves precipitating DNA-RNA hybrids with the DNA-RNA hybrid-specific S9.6 antibody and monitoring coprecipitated gene sequence by quantitative PCR (qPCR) and has been used in some published research and proved to be an effective method (19–21). By using this DRIP method, we found that higher levels of UL32 were detected in shDDX21-3-expressing cells than that of shC-expressing cells without treatment of RNase H. These results suggested that more R-loops formed on the viral late gene UL32 in DDX21-knockdown cells than in control cells, which may restrain viral late gene transcription and subsequently inhibit HCMV replication. These findings imply that DDX21 plays an important role in regulating HCMV growth.
RESULTS
HCMV infection prevents the repression of DDX21 at protein and mRNA levels.
To explore the potential role of DDX21 in regulating HCMV replication, we first examined DDX21 expression in human embryonic lung fibroblasts (MRC5 cells) with or without HCMV infection. As shown in Fig. 1A, the protein levels of DDX21 gradually decreased in mock-infected MRC5 cells and fell to their lowest level at 72 h. However, DDX21 protein levels in HCMV-infected cells maintained higher levels than in mock-infected cells at late infection times, especially at 72 h postinfection (hpi), although no difference in DDX21 protein levels was observed at 24 hpi. Next, we wanted to determine whether higher DDX21 gene expression in infected cells was caused by increased mRNA transcriptional levels, and we therefore performed quantitative reverse transcription-PCR (RT-qPCR) to detect mRNA levels of DDX21 at three time points with or without HCMV infection. As shown in Fig. 1B, the mRNA levels of DDX21 were gradually reduced in mock-infected cells. In infected cells, DDX21 mRNA levels were much higher than in mock-infected cells, especially at 72 hpi. These results suggested that HCMV infection prevented the repression of DDX21 at protein and mRNA levels.
FIG 1.
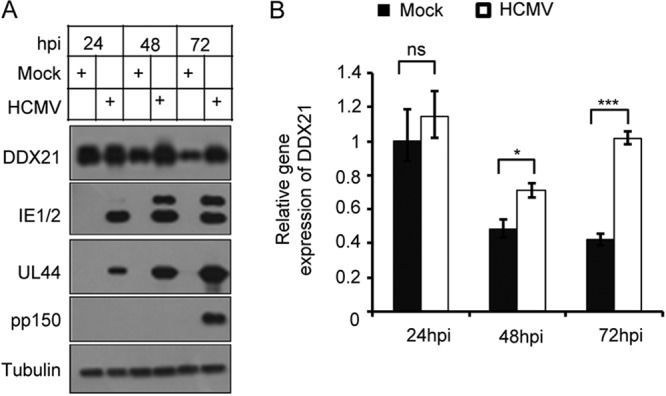
HCMV infection prevents the repression of DDX21 at protein and mRNA levels. (A) MRC5 cells were mock infected or infected with HCMV at an MOI of 1. Cell lysates were collected at the indicated time points postinfection. The protein levels of DDX21 and the viral proteins IE1/2 (immediate early), UL44 (early), and pp150 (late, encoded by UL32) were analyzed by Western blot analysis, and tubulin was used as the loading control. (B) DDX21 mRNA transcriptional levels were higher in HCMV-infected cells than in mock-infected cells at the late stage of infection. MRC5 cells were mock infected or infected with HCMV at an MOI of 1. The total RNA was collected at the indicated times after infection. The mRNA levels of DDX21 were quantified by RT-qPCR. The relative gene expression levels were normalized against that of GAPDH, and the normalized gene expression in mock-infected cells at 24 h was set equal to 1. *, P < 0.05; ***, P < 0.001; ns, not significant.
DDX21 translocates from the nucleolus to the nucleoplasm during HCMV infection.
Almost all members of the DEAD-box RNA helicase family have highly conserved helicase motifs that possess various activities, including ATP binding, ATP hydrolysis, nucleic acid binding and RNA unwinding (17, 22, 23). DDX21 contains this helicase domain, and thus it also has these activities. Although DDX21 is known to be a nucleolar protein, it can also alter its localization upon certain types of stimulation. For example, DDX21 was found to translocate from the nucleus to the cytoplasm during dengue virus infection (15). In addition, DDX21 was reported to translocate from the nucleolus to the nucleoplasm to regulate some genes’ transcription (12). In order to test whether the localization of DDX21 was altered during HCMV infection, we examined the distribution of DDX21 protein in MRC5 cells by confocal microscopy. Nucleolin is a major protein component and a commonly used marker of nucleoli. As shown in Fig. 2A, confocal immunofluorescence analysis showed that DDX21 was located in the nucleolus and colocalized with nucleolin in mock-infected cells. During infection, both DDX21 and nucleolin translocated from the nucleolus to the nucleoplasm, and HCMV infection did not alter the colocalization of DDX21 with nucleolin. The green fluorescent protein (GFP) signal indicated infected cells. In Fig. 2B, we confirmed the results in Fig. 2A in a large representative collection of cells and observed that DDX21 could translocate from the nucleolus to the nucleoplasm during HCMV infection. In Fig. 2C, we used HCMV UL44 as a marker of the viral replication compartment (vRC), and colocalization of DDX21 with UL44 could be observed at 24 hpi. In contrast, at late times of infection and especially at 72 hpi, these two proteins were adjacent to each other, but complete colocalization was not observed. It has been reported that nucleolin can translocate from the nucleolus to the nucleoplasm during HCMV infection. In addition, B. L. Strang and B. J. Bender (24–26) have reported that nucleolin associates with HCMV UL44 in infected cells and is required for viral DNA synthesis and that colocalization of nucleolin with UL44 occurs at the periphery of the viral replication compartment. Our results suggested that DDX21 translocated from the nucleolus to the nucleoplasm in HCMV-infected cells, along with nucleolin. According to our immunofluorescence results and previously reported studies, we speculate that the colocalization of DDX21 with UL44 may occur at the periphery of the viral replication compartment, similar to that observed for nucleolin with UL44.
FIG 2.

DDX21 translocates from the nucleus to the nucleoplasm during HCMV infection. (A) Immunofluorescence analysis of DDX21 localization during HCMV infection. MRC5 cells were mock infected or infected with HCMV at an MOI of 0.1. Cells were fixed at the indicated times after infection and stained with antibodies against nucleolin protein and DDX21 protein. (B) An experiment similar to that described in panel A was performed. Cells were fixed at the indicated times after infection and stained with DDX21 antibody, except the quantity of cells was in a large representative collection. The quantification of nucleoplasmic localization of DDX21 is shown as a graph. (C) An experiment similar to that described in panel A was performed, except that the cells were stained with DDX21 and HCMV UL44 antibodies.
shRNA knockdown of DDX21 inhibits HCMV replication.
Some studies have reported that DDX21 regulates the growth of several RNA viruses (14–16), and thus we sought to determine whether DDX21 also impacted HCMV viral replication. To test this hypothesis, we used two different short hairpin RNA (shRNA) constructs to knock down DDX21. As shown in Fig. 3A, both constructs suppressed the expression of DDX21, and shDDX21-3 had better knockdown efficiency than shDDX21-1. We then conducted viral growth analysis in the presence or absence of DDX21. As shown in Fig. 3B and C, knockdown of DDX21 resulted in reduced viral replication in both shDDX21-1- and shDDX21-3-expressing cells in comparison to that in control shRNA-expressing cells. Consistent with Fig. 3A, shDDX21-3 more significantly inhibited HCMV growth than shDDX21-1, demonstrating that DDX21 regulated HCMV growth and DDX21 knockdown suppressed viral replication. In Fig. 3D, we tested whether these two shRNAs targeting DDX21 could maintain their knockdown efficiency at late time of infection, and our immunoblot results showed that DDX21 expression was reduced by these two shRNAs even at 3 and 6 days postinfection (dpi). In addition, we also tested whether coexpression of these two shRNAs could have better knockdown efficiency, but our results showed that combined expression did not perform better than single expression alone (Fig. 3E).
FIG 3.

shRNA knockdown of DDX21 inhibits HCMV replication. (A) Knockdown efficiency of shRNAs targeting DDX21 genes. Cells were transduced with lentivirus vectors expressing shC, shDDX21-1, or shDDX21-3. Cell lysates were collected at 48 h postransduction, and the protein levels of DDX21 were determined by Western blot analysis. (B) Growth analysis of HCMV infection in cells expressing shC, shDDX21-1, or shDDX21-3. The cells were infected at an MOI of 1, the cell-free viruses in the supernatant were collected at the indicated times after infection, and the titer was determined using a TCID50 assay. Statistical analysis showed that DDX21 knockdown inhibited HCMV replication. ***, P < 0.001. (C) Growth analysis of HCMV infection in cells expressing shC, shDDX21-1, or shDDX21-3 at an MOI of 0.1. The experimental procedures used were similar to those described for panel B. A statistical analysis showed that DDX21 knockdown inhibited HCMV replication. ***, P < 0.001. (D) Knockdown efficiency of these two shRNAs in cells expressing shC, shDDX21-1, or shDDX21-3 at 3 and 6 dpi. The cells were infected at an MOI of 1, the cell lysates were collected at indicated time points, and the protein levels of DDX21 were detected by Western blot analysis. (E) Detection of knockdown efficiency of coexpression of these two shRNAs. Cells were transduced with lentivirus vectors expressing shC, shDDX21-1, shDDX21-3, or both shDDX21-1 and shDDX21-3. Cell lysates were collected at 48 h postransduction, and the protein levels of DDX21 were determined by Western blot analysis.
Suppression of DDX21 gene expression does not affect vRC formation or viral DNA synthesis.
To elucidate which stage of HCMV replication is regulated by DDX21, we first determined whether the process of viral DNA replication was impacted. Then, we tested whether the vRC formation was prevented. As shown in Fig. 4A, we observed that the vRC could form normally despite the knockdown of DDX21, suggesting that its formation was not impaired in DDX21-knockdown cells compared to control cells. We then measured the viral genome copy number at different time points of infection by qPCR, and the results showed that knockdown of DDX21 had no distinct effect on HCMV viral DNA synthesis, which suggested that DDX21 knockdown did not impair viral DNA replication. (Fig. 4B).
FIG 4.

Suppression of DDX21 gene expression does not affect vRC formation or viral DNA synthesis. (A) MRC5 cells transduced with lentivirus respectively expressing shC, shDDX21-1, or shDDX21-3 were infected with HCMV at an MOI of 0.1. Cells were fixed at 72 hpi and stained with antibody against UL44, which is a protein known for localization of the vRC (viral replication compartment). (B) Knockdown of DDX21 had no effect on viral DNA synthesis. MRC5 cells respectively expressing shC, shDDX21-1, or shDDX21-3 were infected with HCMV at an MOI of 1. Intracellular DNA was extracted at the indicated times after infection. Viral DNA levels were quantified by qPCR, and the normalized gene is actin. The result for the DNA sample from shC-expressing cells collected at 2 h was set equal to 1.
DDX21 knockdown inhibits HCMV viral late gene expression and transcription.
The results presented above showed that HCMV DNA synthesis was not impaired in the presence or absence of DDX21. Thus, we tested whether knockdown of DDX21 affected expression or mRNA transcription of viral genes. We first examined the protein level of several viral genes. As shown in Fig. 5A, the expression levels of immediate early (IE1) and early (UL44) genes in DDX21-knockdown cells were almost equal to those of control cells. In contrast, viral late gene protein (pp150, encoded by UL32) levels were significantly reduced, and the two shRNA constructs maintained high DDX21 knockdown efficiency throughout HCMV infection at the protein and mRNA transcription levels (Fig. 5B), which implied that DDX21 knockdown inhibited HCMV viral late gene expression. The IE1/2 immediate early gene expresses two proteins IE1 (lower) and IE2 (upper), IE2 has a late transcriptional promoter, so we also observed that its protein levels were reduced at late time of infection. We speculate that DDX21 may also affect the splicing of IE1/IE2 at IE times of infection. Next, we tested whether viral late gene transcription was also affected, and we therefore performed RT-qPCR to examine the mRNA transcription levels of these viral genes. As shown in Fig. 5C and D, the mRNA levels of immediate early (IE1) and early (UL44) genes were not affected in DDX21-knockdown cells in comparison to control cells, but viral late gene (UL32) mRNA levels were significantly decreased (Fig. 5E), which was consistent with our expectation. Previous research has reported that DDX21 regulates transcription, processing, and modification of pre-rRNA, and its knockdown inhibits the transcription of pre-rRNA (12, 19, 27). Therefore, we sought to ascertain whether a reduction of pre-rRNA in DDX21-knockdown cells during HCMV infection also could mediate the observed decrease in viral late gene protein levels. However, as shown in Fig. 5F, the mRNA transcription levels of pre-rRNA showed no distinct difference at 72 hpi between DDX21-knockdown cells and control cells. We hypothesize that HCMV infection might initiate a signaling pathway to rescue the decreased transcription of pre-rRNA caused by DDX21 knockdown in order to facilitate viral replication. The data demonstrated that DDX21 knockdown inhibited HCMV viral late gene transcription, a major factor leading to reduced viral late gene expression and HCMV replication.
FIG 5.

DDX21 knockdown inhibits HCMV viral late gene expression and transcription. (A) Knockdown of DDX21 reduced HCMV late gene expression. MRC5 cells transduced with lentivirus respectively expressing shC, shDDX21-1, or shDDX21-3 were infected with HCMV at an MOI of 1. Cell lysates were collected at the indicated times after infection. The protein levels of IE1/2, UL44, pp150 (encoded by UL32) and DDX21 were detected by Western blotting. (B) MRC5 cells transduced with lentivirus respectively expressing shC, shDDX21-1, or shDDX21-3 were infected with HCMV at an MOI of 1. The total RNA was collected at the indicated times after infection. The mRNA levels of DDX21 were quantified by RT-qPCR. The relative gene expression level was normalized against that of GAPDH, and the normalized gene expression in mock-infected cells expressing shC was set equal to 1. ***, P < 0.001. (C to F) Cells expressing shC, shDDX21-1, or shDDX21-3 were infected with HCMV at an MOI of 1. The total RNA was collected at 24 hpi. The mRNA levels of IE1 (C) and UL44 (D) were quantified by RT-qPCR, and the normalized gene expression in shC-expressing cells was set equal to 1. ns, not significant. (E and F) The mRNA levels of UL32 (E) and pre-rRNA (F) from cells at 72 hpi were quantified by RT-qPCR, and the normalized gene expression in shC-expressing cells was set equal to 1. **, P < 0.01; ns, not significant.
DDX21 knockdown results in the accumulation of R-loops on HCMV viral late gene.
Next, we explored the mechanism underlying the decrease in viral late gene transcription upon DDX21 knockdown. Recently, it has been reported that DDX21 could effectively unwind R-loops and prevent their accumulation, and DDX21 knockdown resulted in the generation of more R-loops, leading to major genomic instability and DNA damage (19, 28–30). R-loops are structures that consist of an RNA-DNA hybrid with a displaced single-stranded DNA, which can form naturally during transcription and DNA replication (31, 32). Furthermore, R-loop formation impairs the elongation of polymerase II and thus inhibits transcription of a subset of genes (18, 33, 34). Therefore, we investigated whether DDX21 knockdown led to the accumulation of R-loops on viral late gene, which then caused RNA Pol II pausing and subsequent inhibition of viral late gene transcription. To test our hypothesis, we first used the DNA-RNA hybrid-specific S9.6 antibody to stain R-loops in the presence or absence of DDX21 at 72 hpi. As shown in Fig. 6A, consistent with previous research, we detected an enhanced S9.6 signal in DDX21-knockdown cells compared to control cells whether cells were infected or not, which suggested the formation of more R-loops upon knockdown of DDX21. Next, we applied a DNA-RNA immunoprecipitation (DRIP) assay developed by Song et al. (19) to detect whether more R-loops accumulated on viral late gene. In addition, treatment with RNase H could specifically hydrolyze RNA of the DNA-RNA hybrids, which served as a negative control (19, 20, 34, 35). As shown in Fig. 6B, we observed that knockdown of DDX21 led to increased levels of R-loops on the viral late gene UL32 compared to that in control cells, which suggested that DDX21 knockdown resulted in the accumulation of R-loops on an HCMV viral late gene and subsequent inhibition of its transcription. Moreover, we also found that DDX21 knockdown did not result in the accumulation of R-loops on the viral immediate early gene IE1 (Fig. 6C) and early gene UL44 (Fig. 6D). Altogether, these results demonstrated that the accumulation of R-loops caused by DDX21 knockdown prevented HCMV viral late gene transcription and subsequent protein expression, leading to suppression of HCMV replication.
FIG 6.

DDX21 knockdown results in the accumulation of R-loops on HCMV viral late gene. (A) MRC5 cells were transduced with lentivirus vectors respectively expressing shC, shDDX21-1, or shDDX21-3. After 48 h postransduction, the cells were infected with HCMV at an MOI of 0.1. Cells were fixed at 72 hpi and stained with antibodies against S9.6 (marker of R-loops) and DDX21. A bar graph represents the mean intensity of S9.6 fluorescence in the nuclei of individual cells (n = 20). ***, P < 0.001. (B to D) DRIP qPCR assay monitored R loops on viral late gene UL32 (B), viral immediate early gene IE1 (C), and viral early gene UL44 (D) in cells upon siRNA-mediated depletion of DDX21. The 2–ΔCT value is presented as DRIP (% input): ΔCT = CT DRIP – CT Input; ΔCT (RNase H) = CT DRIP (RNase H) – CT Input (RNase H). *, P < 0.05; ns, not significant.
Overexpression of RNase H could rescue reduced UL32 expression and transcription caused by DDX21 knockdown.
According to the results presented above, we have shown that DDX21 knockdown could result in the accumulation of R-loops on a viral late gene, which likely contributed to the inhibition of its transcription. We wondered whether this inhibition would be reversed if we overexpressed RNase H to destroy R-loops. To test this idea, we constructed a lentiviral vector expressing Flag-tagged RNase H to transduce MRC5 cells. As shown in Fig. 7A, we observed weaker GFP signal in DDX21 specific shRNA (shDDX21-3)-expressing cells compared to control shRNA (shC)-expressing cells, but when we coexpressed RNase H with shDDX21-3, we could observed increased GFP signal that was similar to that in shC-expressing cells, suggesting the virus growth was rescued by RNase H expression. In Fig. 7B and C, our Western blot and qPCR results showed decreased pp150 protein and its mRNA levels in shDDX21-3-expressing cells, whereas coexpression of RNase H partially rescued its mRNA and protein accumulation. Taken together, these results proved that DDX21 knockdown resulted in the accumulation of R-loops, which inhibited HCMV viral late gene transcription and finally prevented viral growth.
FIG 7.

Overexpression of RNase H could rescue reduced UL32 expression and transcription caused by DDX21 knockdown. (A) MRC5 cells expressing shC, shDDX21-3, or both shDDX21-3 and RNase H-Flag were infected with HCMV at an MOI of 1. immunofluorescence analysis yielded images of infected cells at 72 hpi. (B) Decreased pp150 protein levels in shDDX21-3-expressing cells could be rescued upon coexpression of RNase H-Flag. MRC5 cells expressing shC, shDDX21-3, or shDDX21-3 and RNase H-Flag were infected with HCMV at an MOI of 1. Cell lysates were collected at 72 hpi, and the protein levels were detected by Western blot analysis with specific antibodies. (C) Coexpression of RNase H-Flag and shDDX21-3 could rescue decreased UL32 mRNA levels in shDDX21-3-expressing cells. MRC5 cells expressing shC, shDDX21-3, or both shDDX21-3 and RNase H-Flag were infected with HCMV at an MOI of 1. Total RNA was collected at 72 hpi. The mRNA levels of UL32 were quantified by RT-qPCR. The relative gene expression level was normalized against that of GAPDH, and the normalized gene expression in shC-expressing cells was set equal to 1. *, P < 0.05.
DISCUSSION
Recent studies have demonstrated that DDX21 regulates the growth of some RNA viruses. DDX21 promotes transcription, processing, and modification of pre-rRNA and is also involved in regulating various immune responses, which implies that DDX21 plays an important role in host cells. Until now, the effect of DDX21 on DNA virus replication and its role in responding to DNA viral infection have not been reported. Here, we show that the DEAD-box RNA helicase DDX21 inhibits HCMV viral late gene transcription and subsequently prevents viral replication. Recent studies have shown that DDX21 negatively regulates the replication of certain RNA viruses by controlling viral mRNA translation, suppressing viral RNA synthesis, or regulating the activity of specific innate immune responses. Our study reveals that HCMV infection prevented the repression of DDX21 at protein and mRNA levels (Fig. 1) and that the knockdown of DDX21 inhibits viral growth (Fig. 3B and C) . Further experiments showed that decreased viral replication is associated with reduced viral late gene transcription (Fig. 5E), which is caused by the accumulation of R-loops (Fig. 6B).
The formation of R-loops has been linked to regulation of several important biological responses. R-loops have been found to positively or negatively affect the transcription and expression of various genes, and they also mediate replication fork stalling and restrain RNA polymerase II elongation. Recent research has shown that DDX21 knockdown results in the accumulation of R-loops, which leads to stalling of RNA Pol II and subsequently prevents specific genes’ transcription. We validated the increased accumulation of R-loops in DDX21-knockdown cells compared to control cells regardless of whether the cells were infected or not (Fig. 6A). We quantified the abundance of R-loops by calculating the S9.6 staining intensity, and our results showed enhanced S9.6 signals in DDX21-knockdown cells, consistent with previous research.
We used the DRIP method published by Song et al. (19) to detect the abundance of R-loops on selected genes. Several recent studies referring to R-loops have utilized this method to monitor R-loop enrichment on target genes. We used the DNA-RNA hybrid-specific S9.6 antibody to precipitate R-loops and compared the abundance of R-loops on viral genes in the presence or absence of DDX21, and our results showed that more R-loops accumulated on the viral late gene in DDX21-knockdown cells than in control cells (Fig. 6B). Also, overexpression of RNase H with shRNA targeting DDX21 could rescue the repression of viral growth caused by DDX21 knockdown (Fig. 7A). In sum, these data imply that the knockdown of DDX21 leads to the accumulation of R-loops, which prevents viral late gene transcription and subsequently inhibits HCMV replication. As for the early genes, though DDX21 knockdown has no effect on their expression and transcription, it may have some differential effects such as on IE RNA splicing; other roles that DDX21 plays in other viral genes may also need to be explored in the future.
Overall, our study is the first report on the relationship between HCMV and DDX21, and it shows that DDX21 is essential for HCMV replication. This study highlights the need to explore the role of DDX21 in the replication of other DNA viruses. In addition, there are many members within the DEAD-box RNA helicase family, and some of them are multifunctional enzymes such as DDX21; therefore, it is also important to investigate the association between these host proteins and viruses.
MATERIALS AND METHODS
Plasmids and antibodies.
We used plasmid pLKO.1 vector to create two shRNAs targeting DDX21 (shDDX21-1 and shDDX21-3). The shRNA targeting sequences were 5′-GCGGAGTTTCAGTAAAGCATT-3′ (shDDX21-1) and 5′-GCATGAGGAATGGGATTGATA-3′ (shDDX21-3). We used the pLKO.1 vector to construct plasmid RNase H-Flag, and the primer sequences were 5′-TGATGATAAAGTCGACATTGATGCGGCAGCC-3′ and 5′-TCGAGGTCGAGAATTCTTATCGCAGCTTGTAAC-3′.
The primary antibodies used in this study include anti-DDX21 (Proteintech, 10528-1-AP), anti-IE1/2 (a generous gift from Jay Nelson, Oregon Health and Science University), anti-UL44 (Virusys), anti-pp150 (a generous gift from Thomas Shenk, Princeton University), anti-tubulin (Proteintech, 66031-1-Ig), anti-Flag (Proteintech, 20543-1-AP), anti-nucleolin (Santa Cruz Biotechnology, sc-8031), and anti-DNA-RNA Hybrid [S9.6] antibody (Kerafast, ENH001). Alexa Fluor 647-goat anti-rabbit IgG(H+L) secondary antibody and Alexa Fluor 555-goat anti-mouse IgG(H+L) secondary antibody were purchased from Invitrogen.
Cells and viruses.
Human embryonic lung fibroblast (MRC5 cells), primary human foreskin fibroblast and HEK293 cells were cultured in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (FBS). Wild-type HCMV carries the whole genome of HCMV lab strain AD169; HCMV-GFP is a recombinant virus derived from it, but the viral US4-US6 region of HCMV is replaced by the GFP under the control of a simian virus 40 early promoter (36, 37). We used HCMV-GFP to perform our experiments throughout this research; GFP indicates infected cells.
RT-qPCR.
Total RNA was isolated from cells with TRIzol reagent (Invitrogen). Portions (1 μg) of each RNA sample were reverse transcribed by using a PrimeScript RT reagent kit (TaKaRa), and cDNA was quantified with these genes’ specific primer pairs (Table 1). qPCRs were conducted in the ABI7900HT (Applied Biosystems) using a SYBR Premix Ex Taq kit (TaKaRa), and reactions were denatured at 95°C for 30 s, followed by 40 two-step cycles of 95°C for 5 s and 60°C for 30 s. The relative gene expression levels were normalized against that of GAPDH (glyceraldehyde-3-phosphate dehydrogenase).
TABLE 1.
Primers used in qPCR analysis
| Primera | Sequence (5′–3′) |
|---|---|
| DDX21 qR | TCATCAAGGACGCACTATCATCT |
| DDX21 qF | CCTTTCAGGGTGATTTCCCTTT |
| IE1 qF | CAAGTGACCGAGGATTGCAA |
| IE1 qR | CACCATGTCCACTCGAACCTT |
| UL44 qF | ACTGCCGTGCACGTTGCGTA |
| UL44 qR | ACTTGCCGCTGTTCCCGACG |
| pp150 qF | GGTTTCTGGCTCGTGGATGTCG |
| pp150 qR | CACACAACACCGTCGTCCGATTAC |
| pre-rRNA qF | TGTCAGGCGTTCTCGTCTC |
| pre-rRNA qR | AGCACGACGTCACCACATC |
| GAPDH qF | CTGTTGCTGTAGCCAAATTCGT |
| GAPDH qR | ACCCACTCCTCCACCTTTGAC |
| IE genome qF | TCTGCCAGGACATCTTTCTCG |
| IE genome qR | GGAGACCCGCTGTTTCCAG |
| Actin qF | CTCCATCCTGGCCTCGCTGT |
| Actin qR | GCTGTCACCTTCACCGTTCC |
| pp150 DRIP qF | CTCGTAAGCGCGGTCATTCTGG |
| pp150 DRIP qR | GCACACCCTCCAGGTTTTCTTC |
| IE1 DRIP qF | CTTAAGTGAGTTCTGTCGGGTGC |
| IE1 DRIP qR | ATGCAGATCTCCTCAATGCGGC |
| UL44 DRIP qF | GCCGTCGCTTATCTTGCAAACG |
| UL44 DRIP qR | ATGAAATTACCCAGCAGCGGCG |
qF, qPCR forward; qR, qPCR reverse.
Transduction and Western blotting.
HEK293 cells were transfected with 12-μg portions of plasmids of these two DDX21-targeting shRNAs, respectively, together with the 10.68-μg portions of the packaging plasmid 9.2ΔR and 1.32 μg of VSV-G to produce lentivirus stocks. Then, 80% confluent MRC-5 cells were transduced with lentivirus supplemented with 5 μg/ml Polybrene. At 48 h posttransduction, the cells were infected with HCMV and collected at the indicated time points. Samples were washed with ice-cold phosphate-buffered saline (PBS) twice and lysed with 3× sodium dodecyl sulfate (SDS) containing protease inhibitor cocktails (Roche). Protein samples were separated by SDS-PAGE using 10% polyacrylamide gels and then transferred to polyvinylidene difluoride membranes. After incubation with antibodies, the proteins were visualized by using Clarity Western ECL substrate (Bio-Rad).
Immunofluorescence.
Cells grown on glass coverslips in a 24-well plate were fixed in 2% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 (in PBS) for 15 min, and then blocked with 5% FBS (in PBS) for 20 min at room temperature. Primary antibodies were incubated with cells for 1 h and then with Alexa Fluor-conjugated secondary antibodies for 1 h. Next, the cells were counterstained with DAPI (4′,6′-diamidino-2-phenylindole, 1:5,000; Beyotime) in the dark for 10 min in order to visualize the nuclei. Finally, labeled cells were mounted on slides with 8 μl of Prolong Gold antifade reagent (Invitrogen/Molecular Probes) overnight. The next day, we sealed the boundaries of these coverslips with colorless nail oil for 30 min. Images were captured with an Olympus FV1200 confocal laser scanning microscope at ×60 magnification.
Virus growth analysis.
MRC5 cells were seeded in a 12-well plate. After 48 h, the cells were incubated with HCMV at a multiplicity of infection (MOI) of 0.1 or 1 in 300 μl of inoculum. Two hours later, the inoculum was removed, and fresh medium was replenished. At different times postinfection, cell-free media from infected cultures were collected, and the virus titers in the media were determined by a 50% tissue culture infective dose (TCID50) assay in human fibroblast cells.
Viral genome determination.
MRC5 cells seeded in a 12-well plate were infected with wild-type virus at an MOI of 1 and then collected at the indicated time points postinfection. Cells were resuspended in 200 μl of digestion buffer (10 mM Tris-HCl [pH 8.0], 200 mM NaCl, 50 mM EDTA, 1% SDS) and then treated with 50 μg/μl protease K at 55°C overnight. Total DNAs were then isolated with phenol-chloroform and treated with 100 μg/ml RNase A at 37°C for 1 h. Next, the DNAs were extracted once more with phenol-chloroform and precipitated with ethanol, and finally 20 μl of water was added to dissolve the DNAs at 4°C overnight. Viral DNAs were quantified with SYBR Premix Ex Taq (TaKaRa) and primers specific for the HCMV IE gene and the human β-actin gene (Table 1). The accumulation of viral DNA was normalized by dividing the number of IE gene equivalents by the number of β-actin gene equivalents.
DRIP.
The viral genome was extracted as described above. Purified DNA was digested with restriction enzymes—1 μl of HindIII (20 U/μl), 1 μl of EcoRI (20 U/μl), 1 μl of SspI (20 U/μl), 1 μl of XbaI (20 U/μl), and 2 μl of BsrGI (10 U/μl)—in NEB buffer at 37°C overnight. Then, half of the samples were treated with 4 μl of RNase H (5 U/μl; NEB) in a total volume of 200 μl at 37°C for 48 h, which served as a negative control. The other half of the sample was not treated. Digested nucleic acids were cleaned by phenol-chloroform extraction and resuspended in water. Next, 1.5 μg of DNA was immunoprecipitated by using 2.5 μg of S9.6 antibody (ENH001; Kerafast) in binding buffer (10 mM NaPO4 [pH 7.0], 140 mM NaCl, 0.05% Triton X-100) for incubation overnight at 4°C. Then, 25 μl of protein A-magnetic beads (NEB) that was preblocked with 1 mg/ml bovine serum albumin (BSA) and 1 mg/ml herring sperm DNA in binding buffer for 4 to 6 h was used for incubation with nucleic acids at 4°C for 3 h. Isolated samples were washed twice with binding buffer and once with Tris-EDTA buffer for 15 min at 4°C, and then 100 μl of elution buffer (50 mM Tris [pH 8.0], 10 mM EDTA, 0.5% SDS) was added to the beads, followed by heating at 65°C for 15 min. Elution was treated with 2.5 μg of proteinase K for 1 h at 55°C. Nucleic acids were extracted using phenol-chloroform. Nucleic acids digested with restriction enzymes (HindIII, EcoRI, SspI, XbaI, and BsrGI) but without immunoprecipitation using protein A-beads were used as input. The relative occupancy of the coprecipitated gene sequence by the DNA-RNA hybrid-specific S9.6 antibody was examined by qPCR as follows: ΔCT = CT DRIP – CT Input, samples treated with RNase H were used as a negative control [ΔCT (RNase H) = CT DRIP (RNase H) – CT Input (RNase H)], and we used 2–ΔCT to describe the accumulated abundance of R-loops on our detected genes, 2–ΔCT is presented as DRIP (% input). The threshold cycle (CT) values for both Input and DRIP are mean threshold cycles of qPCR performed in duplicate on samples from input and specific immunoprecipitations, respectively. The sequences of the primers are shown in Table 1. A Student t test was used to assess statistical significances.
Quantitative analysis.
Quantification of the S9.6 intensity was performed by using ImageJ software.
ACKNOWLEDGMENTS
We thank all members of the Herpesvirus and Molecular Virology Research Unit for helpful discussions and invaluable suggestions, Jay Nelson for IE1/2 antibody, Thomas Shenk for pp150 antibody, and Bin Li for HEK293T cells. We thank the molecular imaging core facility at the Institut Pasteur of Shanghai for assistance with confocal laser scanning microscopy.
This study was supported by the National Natural Science Foundation of China (grants 81371826 and 81572002 to Z.Q. and grants 31300148 and 31570169 to B.X.), the Ministry of Science and Technology of China (2016YFA0502101), and the Chinese Academy of Sciences “100 Talents” program (Z.Q.). The funders had no role in study design, data collection and analysis, the decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol 20:392–401. doi: 10.1016/j.tim.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins-McMillen D, Buehler J, Peppenelli M, Goodrum F. 2018. Molecular determinants and the regulation of human cytomegalovirus latency and reactivation. Viruses 10:444. doi: 10.3390/v10080444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jean Beltran PM, Cristea IM. 2014. The life cycle and pathogenesis of human cytomegalovirus infection: lessons from proteomics. Expert Rev Proteomics 11:697–711. doi: 10.1586/14789450.2014.971116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christensen-Quick A, Vanpouille C, Lisco A, Gianella S. 2017. Cytomegalovirus and HIV persistence: pouring gas on the fire. AIDS Res Hum Retroviruses 33:S23–S30. doi: 10.1089/aid.2017.0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Görzer I, Guelly C, Trajanoski S, Puchhammer-Stöckl E. 2010. Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J Virol 84:7195–7203. doi: 10.1128/JVI.00475-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson EL, Boggavarapu S, Johnson ES, Lal AA, Agrawal P, Bhaumik SK, Murali-Krishna K, Chakraborty R. 2018. Human cytomegalovirus enhances placental susceptibility and replication of human immunodeficiency virus type 1 (HIV-1), which may facilitate in utero HIV-1 transmission. J Infect Dis 218:1464–1473. doi: 10.1093/infdis/jiy327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munro SC, Hall B, Whybin LR, Leader L, Robertson P, Maine GT, Rawlinson WD. 2005. Diagnosis of and screening for cytomegalovirus infection in pregnant women. J Clin Microbiol 43:4713–4718. doi: 10.1128/JCM.43.9.4713-4718.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buxmann H, Hamprecht K, Meyer-Wittkopf M, Friese K. 2017. Primary human cytomegalovirus (HCMV) infection in pregnancy. Dtsch Arztebl Int doi: 10.3238/arztebl.2017.0045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Linder P, Jankowsky E. 2011. From unwinding to clamping: the DEAD-box RNA helicase family. Nat Rev Mol Cell Biol 12:505–516. doi: 10.1038/nrm3154. [DOI] [PubMed] [Google Scholar]
- 10.Diot C, Fournier G, Dos Santos M, Magnus J, Komarova A, van der Werf S, Munier S, Naffakh N. 2016. Influenza A virus polymerase recruits the RNA helicase DDX19 to promote the nuclear export of viral mRNAs. Sci Rep 6:33763. doi: 10.1038/srep33763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourgeois CF, Mortreux F, Auboeuf D. 2016. The multiple functions of RNA helicases as drivers and regulators of gene expression. Nat Rev Mol Cell Biol 17:426–438. doi: 10.1038/nrm.2016.50. [DOI] [PubMed] [Google Scholar]
- 12.Calo E, Flynn RA, Martin L, Spitale RC, Chang HY, Wysocka J. 2015. RNA helicase DDX21 coordinates transcription and ribosomal RNA processing. Nature 518:249–253. doi: 10.1038/nature13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xing YH, Yao RW, Zhang Y, Guo CJ, Jiang S, Xu G, Dong R, Yang L, Chen LL. 2017. SLERT regulates DDX21 rings associated with Pol I transcription. Cell 169:664–678 e616. doi: 10.1016/j.cell.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 14.Chen G, Liu CH, Zhou L, Krug RM. 2014. Cellular DDX21 RNA helicase inhibits influenza A virus replication but is counteracted by the viral NS1 protein. Cell Host Microbe 15:484–493. doi: 10.1016/j.chom.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong Y, Ye W, Yang J, Han P, Wang Y, Ye C, Weng D, Zhang F, Xu Z, Lei Y. 2016. DDX21 translocates from nucleus to cytoplasm and stimulates the innate immune response due to dengue virus infection. Biochem Biophys Res Commun 473:648–653. doi: 10.1016/j.bbrc.2016.03.120. [DOI] [PubMed] [Google Scholar]
- 16.Watanabe Y, Ohtaki N, Hayashi Y, Ikuta K, Tomonaga K. 2009. Autogenous translational regulation of the Borna disease virus negative control factor X from polycistronic mRNA using host RNA helicases. PLoS Pathog 5:e1000654. doi: 10.1371/journal.ppat.1000654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fullam A, Schroder M. 2013. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim Biophys Acta 1829:854–865. doi: 10.1016/j.bbagrm.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santos-Pereira JM, Aguilera A. 2015. R loops: new modulators of genome dynamics and function. Nat Rev Genet 16:583–597. doi: 10.1038/nrg3961. [DOI] [PubMed] [Google Scholar]
- 19.Song C, Hotz-Wagenblatt A, Voit R, Grummt I. 2017. SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev 31:1370–1381. doi: 10.1101/gad.300624.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sridhara SC, Carvalho S, Grosso AR, Gallego-Paez LM, Carmo-Fonseca M, de Almeida SF. 2017. Transcription dynamics prevent RNA-mediated genomic instability through SRPK2-dependent DDX23 phosphorylation. Cell Rep 18:334–343. doi: 10.1016/j.celrep.2016.12.050. [DOI] [PubMed] [Google Scholar]
- 21.Bhatia V, Barroso SI, Garcia-Rubio ML, Tumini E, Herrera-Moyano E, Aguilera A. 2014. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 511:362–365. doi: 10.1038/nature13374. [DOI] [PubMed] [Google Scholar]
- 22.Jankowsky E. 2011. RNA helicases at work: binding and rearranging. Trends Biochem Sci 36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cordin O, Banroques J, Tanner NK, Linder P. 2006. The DEAD-box protein family of RNA helicases. Gene 367:17–37. doi: 10.1016/j.gene.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 24.Bender BJ, Coen DM, Strang BL. 2014. Dynamic and nucleolin-dependent localization of human cytomegalovirus UL84 to the periphery of viral replication compartments and nucleoli. J Virol 88:11738–11747. doi: 10.1128/JVI.01889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strang BL, Boulant S, Kirchhausen T, Coen DM. 2012. Host cell nucleolin is required to maintain the architecture of human cytomegalovirus replication compartments. mBio 3:e00301-11. doi: 10.1128/mBio.00301-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strang BL, Boulant S, Coen DM. 2010. Nucleolin associates with the human cytomegalovirus DNA polymerase accessory subunit UL44 and is necessary for efficient viral replication. J Virol 84:1771–1784. doi: 10.1128/JVI.01510-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romanova L, Grand A, Zhang L, Rayner S, Katoku-Kikyo N, Kellner S, Kikyo N. 2009. Critical role of nucleostemin in pre-rRNA processing. J Biol Chem 284:4968–4977. doi: 10.1074/jbc.M804594200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA. 2017. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell 170:774–786.e719. doi: 10.1016/j.cell.2017.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costantino L, Koshland D. 2018. Genome-wide map of R-loop-induced damage reveals how a subset of R-loops contributes to genomic instability. Mol Cell 71:487–497.e483. doi: 10.1016/j.molcel.2018.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cristini A, Groh M, Kristiansen MS, Gromak N. 2018. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep 23:1891–1905. doi: 10.1016/j.celrep.2018.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aguilera A, García-Muse T. 2012. R loops: from transcription byproducts to threats to genome stability. Mol Cell 46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 32.Hamperl S, Cimprich KA. 2014. The contribution of cotranscriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst) 19:84–94. doi: 10.1016/j.dnarep.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shivji MKK, Renaudin X, Williams ÇH, Venkitaraman AR. 2018. BRCA2 regulates transcription elongation by RNA polymerase II to prevent R-loop accumulation. Cell Rep 22:1031–1039. doi: 10.1016/j.celrep.2017.12.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Chiang H-C, Wang Y, Zhang C, Smith S, Zhao X, Nair SJ, Michalek J, Jatoi I, Lautner M, Oliver B, Wang H, Petit A, Soler T, Brunet J, Mateo F, Angel Pujana M, Poggi E, Chaldekas K, Isaacs C, Peshkin BN, Ochoa O, Chedin F, Theoharis C, Sun L-Z, Curiel TJ, Elledge R, Jin VX, Hu Y, Li R. 2017. Attenuation of RNA polymerase II pausing mitigates BRCA1-associated R-loop accumulation and tumorigenesis. Nat Commun 8:15908. doi: 10.1038/ncomms15908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao H, Zhu M, Limbo O, Russell P. 2018. RNase H eliminates R-loops that disrupt DNA replication but is nonessential for efficient DSB repair. EMBO Rep 19:e45335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terhune S, Torigoi E, Moorman N, Silva M, Qian Z, Shenk T, Yu D. 2007. Human cytomegalovirus UL38 protein blocks apoptosis. J Virol 81:3109–3123. doi: 10.1128/JVI.02124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J Virol 76:2316–2328. doi: 10.1128/jvi.76.5.2316-2328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]