

Previous studies generated recombinant rotaviruses that express FPs by inserting reporter genes into the NSP1 ORF of genome segment 5. Unfortunately, NSP1 is expressed at low levels in infected cells, making viruses expressing FP-fused NSP1 less than ideal probes of rotavirus biology. Moreover, FPs were inserted into segment 5 in such a way as to compromise NSP1, an interferon antagonist affecting viral growth and pathogenesis. We have identified an alternative approach for generating rotaviruses expressing FPs, one relying on fusing the reporter gene to the NSP3 ORF of genome segment 7. This was accomplished without interrupting any of the viral ORFs, yielding recombinant viruses that likely express the complete set of functional viral proteins. Given that NSP3 is made at moderate levels in infected cells, rotaviruses encoding NSP3-based FPs should be more sensitive probes of viral infection than rotaviruses encoding NSP1-based FPs.
KEYWORDS: rotavirus, reverse genetics, green fluorescent protein, African swine fever virus capping enzyme, viral expression vector
ABSTRACT
Rotavirus is a segmented double-stranded RNA (dsRNA) virus that causes severe gastroenteritis in young children. We have established an efficient simplified rotavirus reverse genetics (RG) system that uses 11 T7 plasmids, each expressing a unique simian SA11 (+)RNA, and a cytomegalovirus support plasmid for the African swine fever virus NP868R capping enzyme. With the NP868R-based system, we generated recombinant rotavirus (rSA11/NSP3-FL-UnaG) with a genetically modified 1.5-kb segment 7 dsRNA encoding full-length nonstructural protein 3 (NSP3) fused to UnaG, a 139-amino-acid green fluorescent protein (FP). Analysis of rSA11/NSP3-FL-UnaG showed that the virus replicated efficiently and was genetically stable over 10 rounds of serial passaging. The NSP3-UnaG fusion product was well expressed in rSA11/NSP3-FL-UnaG-infected cells, reaching levels similar to NSP3 levels in wild-type recombinant SA11-infected cells. Moreover, the NSP3-UnaG protein, like functional wild-type NSP3, formed dimers in vivo. Notably, the NSP3-UnaG protein was readily detected in infected cells via live-cell imaging, with intensity levels ∼3-fold greater than those of the NSP1-UnaG fusion product of rSA11/NSP1-FL-UnaG. Our results indicate that FP-expressing recombinant rotaviruses can be made through manipulation of the segment 7 dsRNA without deletion or interruption of any of the 12 open reading frames (ORFs) of the virus. Because NSP3 is expressed at higher levels than NSP1 in infected cells, rotaviruses expressing NSP3-based FPs may be more sensitive tools for studying rotavirus biology than rotaviruses expressing NSP1-based FPs. This is the first report of a recombinant rotavirus containing a genetically engineered segment 7 dsRNA.
IMPORTANCE Previous studies generated recombinant rotaviruses that express FPs by inserting reporter genes into the NSP1 ORF of genome segment 5. Unfortunately, NSP1 is expressed at low levels in infected cells, making viruses expressing FP-fused NSP1 less than ideal probes of rotavirus biology. Moreover, FPs were inserted into segment 5 in such a way as to compromise NSP1, an interferon antagonist affecting viral growth and pathogenesis. We have identified an alternative approach for generating rotaviruses expressing FPs, one relying on fusing the reporter gene to the NSP3 ORF of genome segment 7. This was accomplished without interrupting any of the viral ORFs, yielding recombinant viruses that likely express the complete set of functional viral proteins. Given that NSP3 is made at moderate levels in infected cells, rotaviruses encoding NSP3-based FPs should be more sensitive probes of viral infection than rotaviruses encoding NSP1-based FPs.
INTRODUCTION
Group A rotavirus (RVA) is a primary cause of acute gastroenteritis in infants and children <5 years of age (1). The RVA genome consists of 11 segments of double-stranded RNA (dsRNA), with a total size of 18.5 kbp, and is contained within a nonenveloped icosahedral capsid composed of three concentric protein layers (2). During replication, the genome segments are transcribed, producing eleven 5′-capped but nonpolyadenylated (+)RNAs (3, 4). The (+)RNAs are generally monocistronic, each with a single open reading frame (ORF) that specifies one of the six structural proteins (VP1 to VP4, VP6, and VP7) or six nonstructural proteins (NSP1 to NSP6) of the virus. The (+)RNAs also serve as templates for the synthesis of dsRNA genome segments (5).
Insights into RVA biology have been severely hampered by the lack of a reverse genetics (RG) system to elucidate details of the replication and pathogenesis mechanisms of the virus. This limitation was recently overcome by Kanai et al. (6), who described the development of a fully plasmid-based RG system that allowed genetic engineering of the prototypic RVA simian SA11 strain. Key to the RG system was cotransfection of baby hamster kidney cells expressing T7 RNA polymerase (BHK-T7 cells) with T7 plasmids directing synthesis of the 11 SA11 (+)RNAs, two cytomegalovirus (CMV) plasmids encoding the vaccinia virus D1L-D12R capping enzyme complex (7), and another CMV plasmid encoding the avian reovirus p10FAST fusion protein (8). Subsequent publications described changes to that RG system that were designed to reduce its complexity and/or to enhance the recovery of recombinant virus. Notably, Komoto et al. (9) showed that recombinant virus could be produced simply by transfecting BHK-T7 cells with 11 SA11 T7 plasmids, with the caveat that plasmids for the viroplasm building blocks (NSP2 and NSP5) (10, 11) be added at levels 3-fold greater than levels of the other plasmids. Of possible significance, the RG system described by Komoto et al. (9) used a set of SA11 T7 plasmids with vector backbones that differed in size and sequence from the SA11 T7 plasmids described by Kanai et al. (6).
Plasmid-based RG systems have been used to modify several SA11 genome segments, with the focus mostly on segment 5, which encodes NSP1 (6, 9, 12). Through insertion of reporter genes into the segment 5 dsRNA, recombinant RVAs that express reporter fluorescent proteins (FPs) (e.g., mCherry and enhanced green FP [EGFP]) have been produced (6, 9, 12); these FP-RVAs are important tools for studying virus replication and pathogenesis via live-cell imaging and other fluorescence-based approaches. Unfortunately, the segment 5 product NSP1 is expressed at low levels in infected cells and is subject to proteasomal degradation, making FP-fused NSP1 proteins less than ideal probes of RVA biology (13). Moreover, FP genes were inserted into the segment 5 dsRNA in such a way as to alter the NSP1 ORF, likely compromising the protein’s function as an interferon antagonist (14–15) and thus affecting the biological properties of the virus.
In this study, we explored an alternative approach for making FP-RVAs, one relying on modification of the genome segment that expresses NSP3 (segment 7), a viral translation enhancer that is expressed at moderate levels in infected cells and may not be required for virus replication (16, 17). In generating recombinant RVAs, we employed a simplified RG system requiring only a single support plasmid, namely, a CMV expression vector for the African swine fever virus (ASFV) NP868R capping enzyme (18). Using the NP868R-based RG system, we produced a novel SA11 strain with modified segment 7 dsRNA that expressed NSP3 fused to the small green FP UnaG. This is the first RVA strain engineered to produce a FP that did not involve deletion or interruption of any of the 12 viral ORFs, thus yielding a recombinant virus that likely expressed a complete set of functional viral proteins.
RESULTS AND DISCUSSION
NP868R-based RG system.
A previous study showed that addition of a CMV plasmid expressing the ASFV NP868R capping enzyme to the mammalian reovirus RG system significantly increased recovery of recombinant virus (19). Based on this finding, we constructed a similar CMV plasmid for NP868R (pCMV/NP868R) and evaluated whether its cotransfection with SA11 pT7 plasmids into BHK-T7 cells was sufficient to allow recovery of recombinant RVAs. In our RG experiments, two different types of pT7 vectors for segment 7 (NSP3) were used, one (pT7/NSP3SA11) designed to introduce a wild-type segment 7 dsRNA into recombinant RVA and the second (pT7/NSP3-FL-UnaG) designed to test the possibility of introducing a modified segment 7 RNA that expressed NSP3 with a fluorescent tag. To construct pT7/NSP3-FL-UnaG, the C terminus of the NSP3 ORF in pT7/NSP3SA11 was fused to the ORF for UnaG, a 139-amino-acid (aa) green FP of the unagi eel that utilizes bilirubin as a fluorophore (20) (Fig. 1). To further ease detection of the protein product of the modified pT7 plasmid, a 3× Flag sequence was inserted between the NSP3 ORF and the UnaG ORF. As a result of the addition of Flag and UnaG sequences, the pT7/NSP3-FL-UnaG vector expresses a 1.6-kb RNA that encodes a 477-aa protein instead of the 1.1-kb RNA and 315-aa protein of pT7/NSP3SA11 (Fig. 1). The NSP3-FL-UnaG fusion protein retained the same RNA-binding domain, coiled-coil dimerization domain, and eIF4G-binding domain present in wild-type NSP3 (21, 22).
FIG 1.
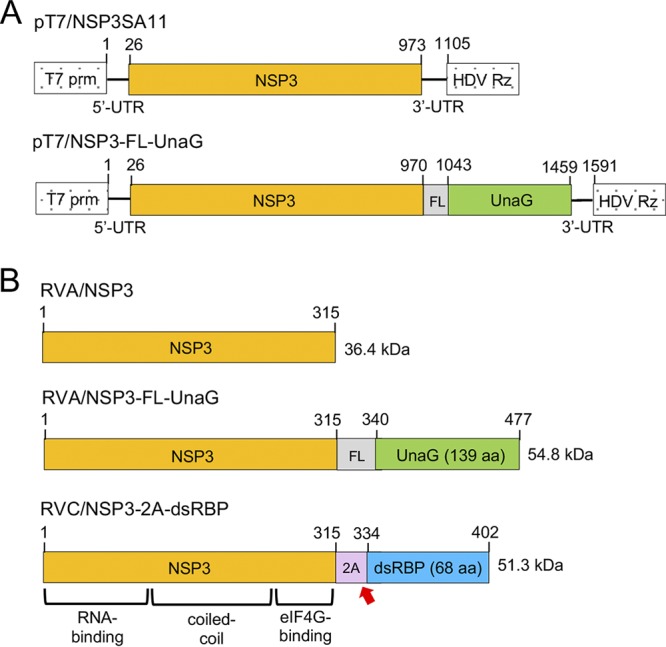
Wild-type and modified NSP3 proteins encoded by pT7 plasmids and rotaviruses. (A) Organization of pT7 plasmids expressing wild-type NSP3 and NSP3-FL/UnaG (+)RNAs, indicating the locations of the T7 promoter (prm) and the hepatitis delta virus (HDV) self-cleaving ribozyme (Rz). Nucleotide positions are labeled. (B) Products of recombinant RVAs expressing wild-type NSP3 and NSP3-FL-UnaG and RVC (Bristol strain) expressing NSP3-2A-dsRBP, including approximate locations of functional domains in NSP3 (21, 22). The red arrow indicates the position of the stop-restart cleavage site in the 2A-like element in the RVC NSP3-2A-dsRBP ORF. Amino acid positions are labeled. FL represents a 3× Flag tag.
Rotavirus RG experiments were performed using stocks of BHK-T7 cells maintained in Glasgow medium enriched with fetal bovine serum (FBS), tryptose-phosphate broth (TPB), and nonessential amino acids (NEAA). Mixtures of RG plasmids were transfected into BHK-T7 cells that were ∼90% confluent and had been seeded into 12-well plates the day before. In our optimized protocol, plasmid mixtures included pCMV/NP868R, 3× levels of pT7/NSP2SA11 and pT7/NSP5SA11, either pT7/NSP3SA11 or pT7/NSP3-FL-UnaG, and the remaining SA11 pT7 plasmids. Transfected BHK-T7 cells were overseeded with MA104 cells to promote amplification of recombinant viruses. At 5 days posttransfection (dpi), the cells were freeze-thawed three times, and large debris was removed by low-speed centrifugation. Recombinant viruses in cell lysates were amplified by passage in MA104 cells, plaque isolated, and amplified again in MA104 cells.
Recombinant SA11 expressing fused NSP3-UnaG.
Analysis of the cell lysates showed that transfection of BHK-T7 cells with RG plasmid mixtures using the optimized protocol supported the generation of recombinant RVAs, including SA11 isolates with a wild-type segment 7 (NSP3) dsRNA (rSA11/wt) or a modified segment 7 RNA (rSA11/NSP3-FL-UnaG) (Fig. 2). The identity of the modified segment 7 dsRNA in rSA11/NSP3-FL-UnaG was verified by gel electrophoresis (Fig. 2A), which revealed that the wild-type 1.1-kb segment 7 dsRNA had been replaced with a segment comigrating with the 1.6-kb segment 5 (NSP1) dsRNA. The authenticity of the segment 7 dsRNA in rSA11/NSP3-FL-UnaG was confirmed by reverse transcription-PCR and sequencing (data not shown). Immunoblot analysis of infected cell lysates with anti-NSP3 and anti-Flag antibodies indicated that segment 7 of rSA11/NSP3-FL-UnaG expressed a protein of the size (55 kDa) expected for NSP3-FL-UnaG and did not express the wild-type 37-kDa NSP3 (Fig. 2B). As a probe of the properties of NSP3-FL-UnaG, we examined whether the protein was able to form dimers in infected cells, as reported previously for wild-type NSP3 (23). Indeed, electrophoretic analysis of rSA11/NSP3-FL-UnaG-infected cell lysates treated with denaturing sample buffer at 25°C showed that the NSP3-FL-UnaG protein migrated as a dimer (Fig. 3), suggesting that the NSP3 coiled-coil dimerization domain retained its function in the fusion product. Under these same electrophoretic conditions, the VP6 inner capsid protein of both rSA11/NSP3-FL-UnaG and rSA11/wt formed trimers that were stable at 25°C (Fig. 3) (24). Quantitation of bands appearing on immunoblots probed with anti-NSP3 and anti-VP6 antibodies indicated that the steady-state level of NSP3-FL-UnaG (normalized to VP6) in rSA11/NSP3-FL-UnaG-infected cells approximated the steady-state level of NSP3 in rSA11/wt-infected cells (Fig. 3). Thus, the segment 7 (+)RNAs of rSA11/NSP3-FL-UnaG and rSA11/wt appear to be translated with nearly equal efficiency.
FIG 2.
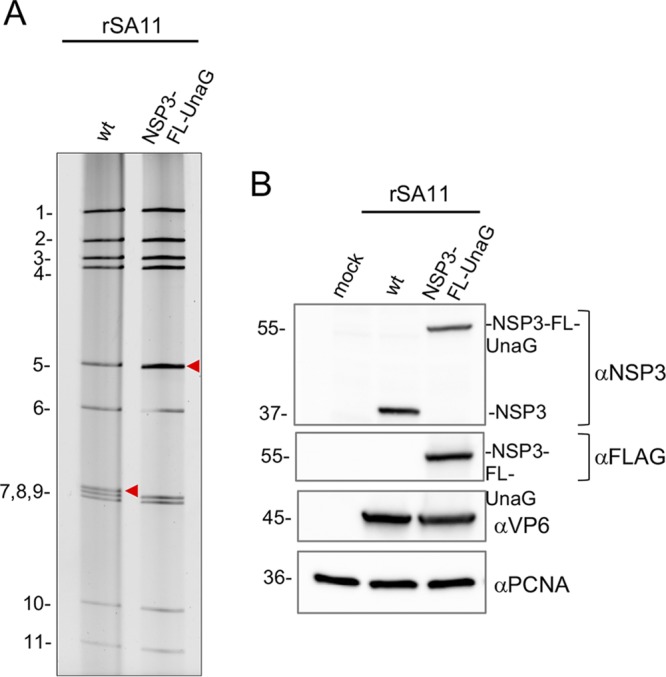
Recovery of recombinant RVA with a modified segment 7 dsRNA that expresses the fused NSP3-FL-UnaG protein. (A) Profiles of the 11 genomic dsRNAs recovered from recombinant SA11 (rSA11) viruses, resolved by PAGE. The red arrowheads note the position of the segment 7 dsRNA. wt, wild-type. (B) Western blot analysis of proteins present at 8 hpi in MA104 cells infected with recombinant viruses.
FIG 3.
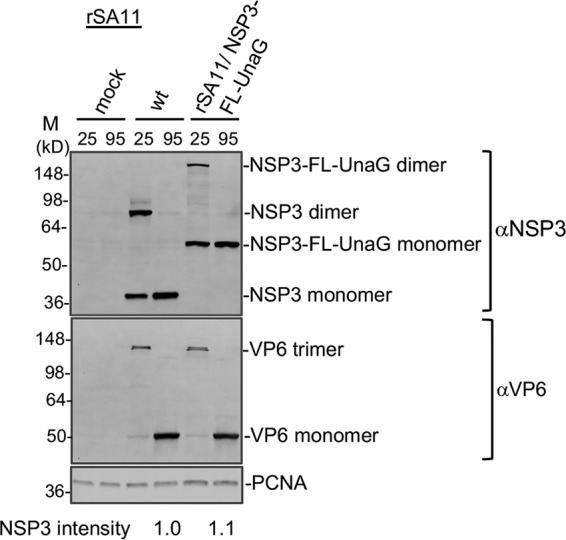
Dimerization of NSP3-FL-UnaG. MA104 cells were mock infected or infected with rSA11/wt (wt) or rSA11/NSP3-FL-UnaG and incubated until 8 hpi, when cells were harvested. Cell lysates were mixed with sample buffer containing sodium dodecyl sulfate and β-mercaptoethanol, incubated for 10 min at either 25°C or 95°C, resolved by electrophoresis on a Novex 8 to 16% polyacrylamide gel, and blotted onto a nitrocellulose membrane. Blots were probed with guinea pig polyclonal anti-NSP3 or anti-VP6 antibodies or with a mouse anti-PCNA monoclonal antibody. Primary antibodies were detected using HRP-conjugated secondary antibodies. Sizes (kilodaltons) of protein markers (M) are indicated. NSP3 band intensities were determined by ImageJ analysis and were normalized to VP6 band intensities.
Plaque analysis showed that rSA11/wt grew to a peak titer in MA104 cells that was ∼2- to 3-fold greater than that of rSA11/NSP3-FL-UnaG (1.6 × 107 to 4.8 × 107 and 0.6 × 107 to 2.0 × 107, respectively) and generated plaques that were ∼2-fold larger (Fig. 4). Ten rounds of serial passaging of rSA11/NSP3-FL-UnaG at low multiplicity of infection (MOI) revealed no difference in the dsRNA profiles of the starting virus and the passage 10 virus, suggesting that the recombinant RVA was genetically stable (Fig. 5). To further test for genetic stability, the segment 6 (VP6) and segment 7 (NSP3) RNAs of three plaque isolates recovered from the passage 10 pool of rSA11/NSP3-FL-UnaG were sequenced. The sequences of segment 6 and segment 7 RNAs were the same as those present in the pT7/VP6SA11 and pT7/NSP3-FL-UnaG plasmids used to make the recombinant viruses. Thus, based on serial passaging and sequencing of P10 viruses, rSA11/NSP3-FL-UnaG appears to stably retain its foreign sequence.
FIG 4.
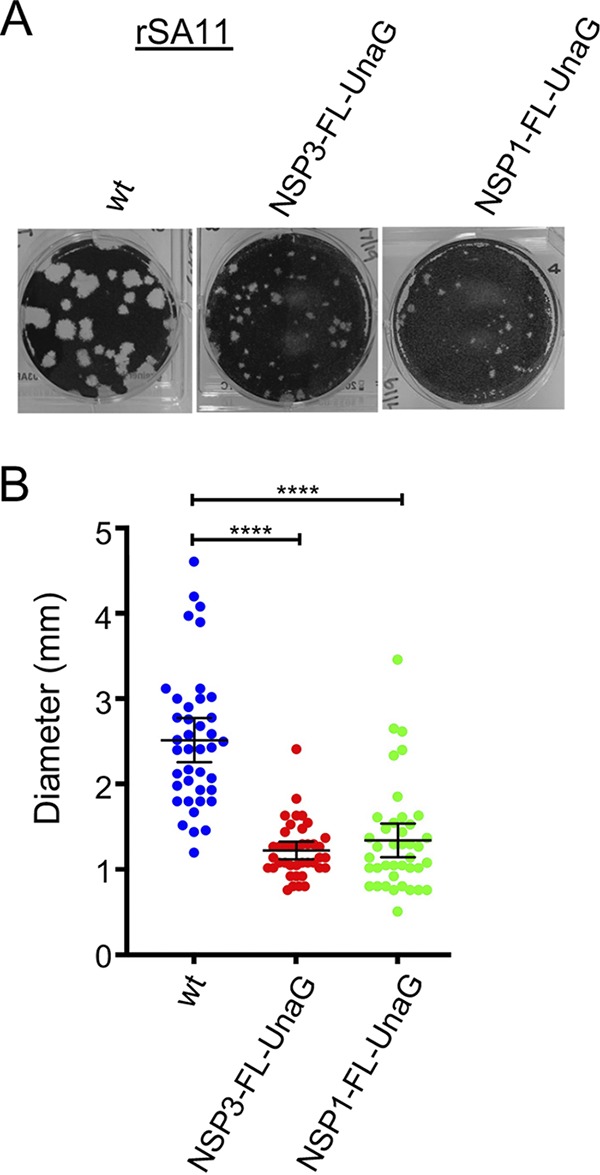
Comparison of plaques formed by wild-type (wt) rSA11 and mutant rSA11 strains containing NSP3-FL-UnaG or NSP1-FL-UnaG sequences. (A) Plaques were generated on MA104 monolayers and detected at 6 dpi by crystal violet staining (39). (B) Sizes of 40 randomly selected plaques from six independent plaque assays were measured, and the means were determined. Mean values and 95% confidence intervals are plotted (black lines). Significance values were calculated using an unpaired Student's t test (GraphPad Prism v8). ****, P < 0.0001.
FIG 5.
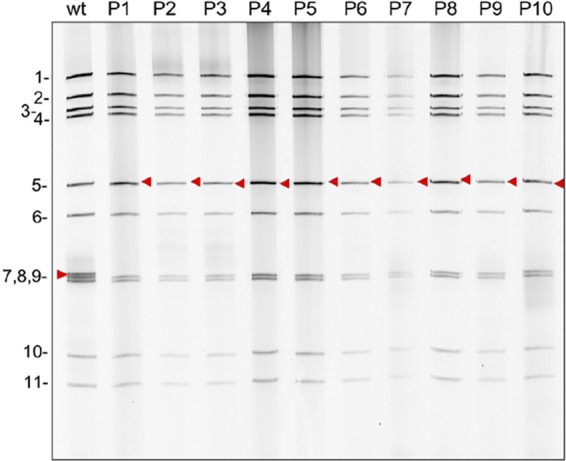
Genetic stability of rSA11/NSP3-FL-UnaG. Virus was serially passaged 10 times (P1 to P10) in MA104 cells, using 1:1,000 dilutions of infected cell lysate as the inoculum. RNA was recovered from infected cell lysates using TRIzol, treated for 30 min with RNase T1 at room temperature, and resolved by electrophoresis on an 8% polyacrylamide gel. RNA was detected by staining the gel with ethidium bromide. The red arrowheads denote the position of the segment 7 (NSP3) RNA. wt, wild-type SA11 rotavirus.
Fluorescence signal of rSA11/NSP3-FL-UnaG.
Examination of MA104 cells infected with rSA11/NSP3-FL-UnaG by live-cell imaging from 0 to 16 h postinfection (hpi) confirmed that UnaG was functional, emitting fluorescent light in a range overlapping that of EGFP (Fig. 6A; also see Movie S1 in the supplemental material) (25). At early times of infection (0 to 8 hpi), the signal was localized predominantly to the cytoplasm. At later times, some UnaG fluorescent signal was also detected in the nucleus (Movie S1). To compare the intensity of the fluorescent signals produced by recombinant viruses expressing UnaG fused to NSP3 versus NSP1, we generated rSA11/NSP1-FL-UnaG (Fig. 7) using the optimized NP868R-based RG protocol. To produce this virus, a FL-UnaG ORF terminating with a stop codon was inserted into the NSP1 ORF of the segment 5 cDNA of pT7/NSP1SA11. As illustrated in Fig. 7, pT7/NSP1-FL-UnaG produces a 2.1-kb RNA that encodes a 575-aa protein instead of the 1.6-kb RNA and 520-aa protein of pT7/NSP1SA11. The segment 5 protein product of pT7/NSP1-FL-UnaG ends with the same Flag-UnaG cassette as the segment 7 protein product of pT7/NSP3-FL-UnaG.
FIG 6.
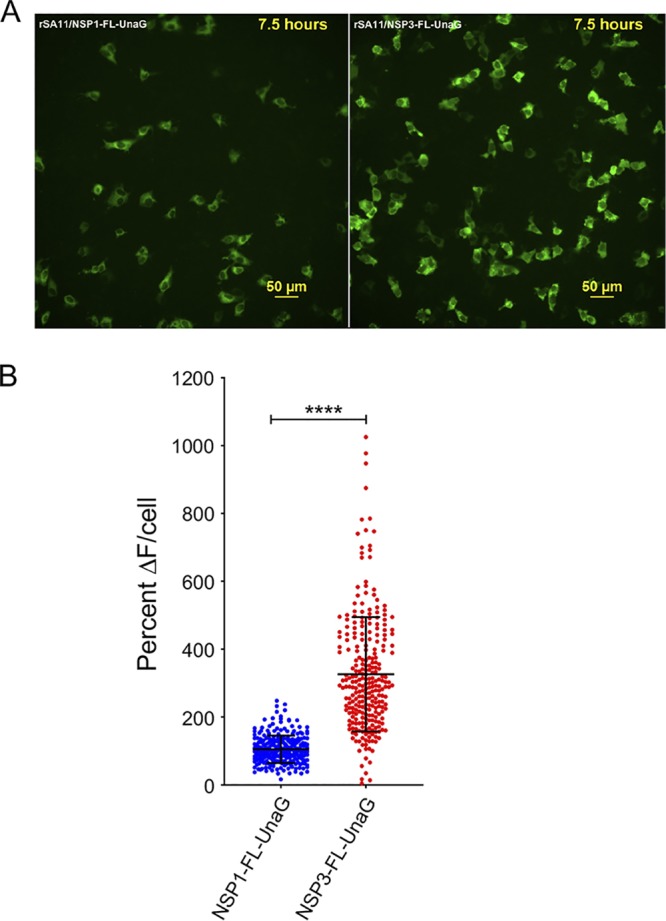
Comparison of UnaG expression by rSA11/NSP3-FL-UnaG versus rSA11/NSP1-FL-UnaG. (A) Images of MA104 cells infected with rSA11/NSP1-FL-UnaG (left panel) and rSA11/NSP3-FL-UnaG (right panel) taken at 7.5 hpi with an epifluorescence Nikon TiE inverted microscope with a 20× Plan Apo (NA, 0.75) differential interference contrast objective. (B) Quantification of the percent change in UnaG fluorescence (ΔF) in MA104 cells infected with rSA11/NSP1-FL-UnaG or rSA11/NSP3-FL-UnaG. Single-cell analysis (n = 270 cells each) was performed to determine the percent ΔF versus baseline. Data are shown as mean ± standard deviation. ****, P < 0.0001.
FIG 7.
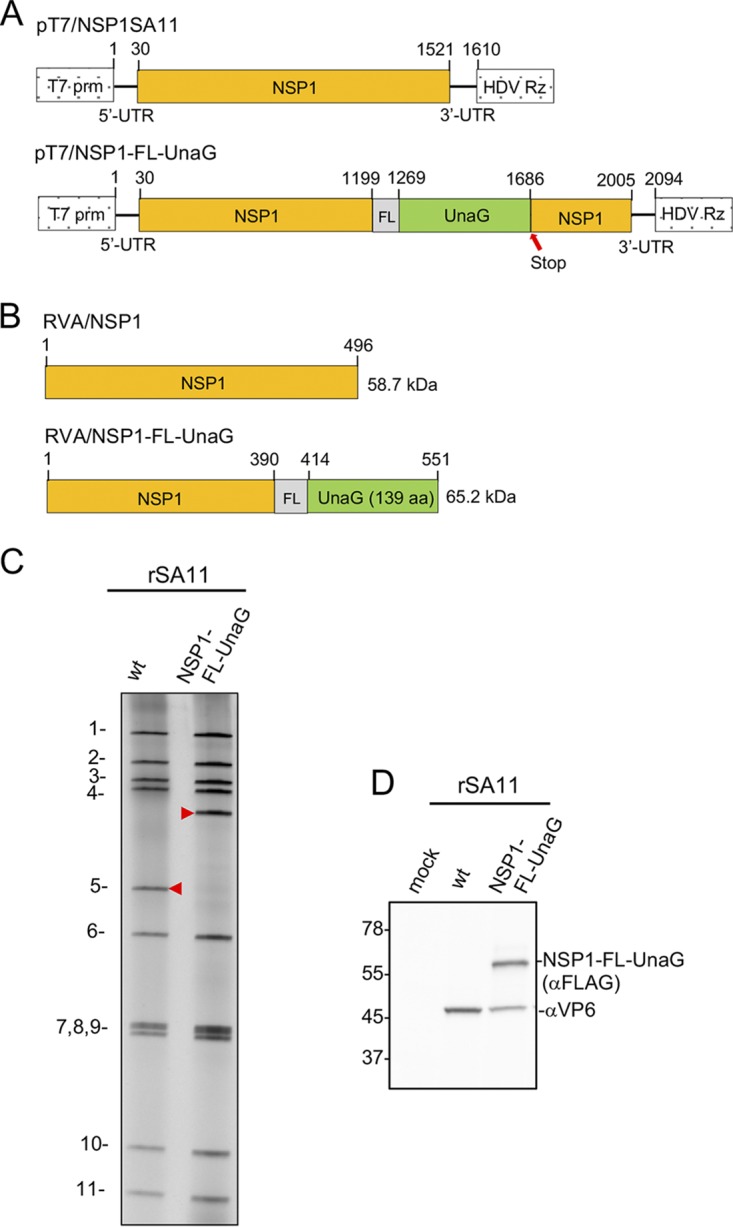
pT7 plasmids and rotaviruses expressing NSP1. (A) Organization of pT7 plasmids expressing wild-type NSP1 and NSP1-FL-UnaG (+)RNAs, indicating the locations of the T7 promoter (prm) and the hepatitis delta virus (HDV) self-cleaving ribozyme (Rz). The red arrow notes the position of the stop codon in the NSP1-FL-UnaG ORF. (B) Products of recombinant RVAs expressing wild-type NSP1 and NSP1-FL-UnaG. (C) Profiles of the 11 genomic dsRNAs recovered from rSA11 viruses, resolved by PAGE. The red arrowheads note the position of the segment 5 dsRNA. wt, wild-type. (D) Western blot analysis of proteins present at 8 hpi in MA104 cells infected with recombinant viruses.
As determined by gel electrophoresis (Fig. 7) and sequencing (data not shown), the genome of rSA11/NSP1-FL-UnaG included the expected 2.1-kb segment 5 dsRNA. Immunoblot analysis of rSA11/NSP1-FL-UnaG-infected cell lysates using anti-Flag antibody indicated that the virus encoded the NSP1-FL-UnaG product. Based on plaque assays, rSA11/NSP1-FL-UnaG grew to a peak titer (0.8 × 107 to 1.0 × 107) that was ∼3-fold less than that of rSA11/wt and produced plaques on MA104 cells that were ∼2-fold smaller than those of rSA11/wt (Fig. 4). The small-plaque phenotype of rSA11/NSP1-FL-NSP1 is similar to the small-plaque phenotype described previously for RVAs encoding truncated or altered NSP1 proteins (26).
Quantification of UnaG fluorescence signals generated by MA104 cells infected with rSA11/NSP3-FL-UnaG were approximately 3-fold greater than those generated by cells infected with rSA11/NSP1-FL-UnaG (Fig. 6B). This result suggests that RVAs expressing fused NSP3-FPs may be more sensitive probes of viral infection than RVAs expressing fused NSP1-FPs.
Stem-loop structure in the 3′ UTR of segment 7.
rSA11/NSP3-FL-UnaG was generated by placing a nonviral 500-bp insert into the segment 7 dsRNA at the junction of the NSP3 ORF and 3′ untranslated region (UTR). Similar recombinant RVAs have been made by inserting nonviral sequences between the NSP2 ORF and 3′ UTR of the segment 8 dsRNA (27). Thus, the junction between the viral ORF and 3′ UTR may represent a site well suited for the introduction of long foreign sequences into RVA genome segments. Interestingly, for RVAs with naturally occurring genome rearrangements, this is the same site in segment 5, 6, 7, 10, and 11 dsRNAs in which viral sequence duplications have been noted to initiate (23, 26, 28–30). The 3′ UTR contains multiple cis-acting signals important for rotavirus replication, including sequences that are recognized by the RVA RNA polymerase VP1 and the translation enhancer NSP3 (21, 31). The fact that well-growing, genetically stable, recombinant RVAs in which a viral ORF has been separated from its 3′ UTR have been recovered indicates that cis-acting signals in the 3′ UTR continue to function although displaced linearly a long distance from the remaining viral sequence of the RNA.
In a previous study (27), an in silico RNA folding analysis (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) was performed to probe how the insertion of sequence duplications and foreign sequences affected the predicted secondary structure of the mutant segment 8 (+)RNAs used in making recombinant RVAs. The results showed that, despite extensive differences in the overall folding predictions for the mutant RNAs, in all cases their 5′ and 3′ UTRs interacted to form stable 5′-3′ panhandles. In addition, the predictions all revealed identical stem-loop structures projecting from the 5′ side of the 5′-3′ panhandle, formed by residues that are highly conserved among RVA segment 8 RNAs. The conservation of the structure and its sequence suggested that the stem-loop may function as a segment-specific packaging signal (27). We performed a similar in silico RNA folding analysis, contrasting the secondary structures predicted for the segment 7 RNAs of rSA11/wt and rSA11/NSP3-FL-UnaG. The results showed that the overall secondary structures predicted for the RNAs differed considerably, with the notable exception that, extending from the 3′ UTR of both RNAs, there was a long (∼70-base) stable stem-loop structure formed by sequences that are highly conserved in RVA segment 7 RNAs (Fig. 8). The stability and location of the stem-loop suggest that this structure may function as a segment-specific packaging signal, in a manner previously proposed for the conserved stem-loop detected in the segment 8 RNA.
FIG 8.

Conservation of a predicted stable stem-loop structure formed by the 3′-UTR sequence of rSA11/wt and rSA11/NSP3-FL-UnaG. Secondary structures associated with minimum free energy were calculated for segment 7 (+)RNAs using RNAfold (http://rna.tbi.univie.ac.at) and were color coded to indicate base-pairing probability (40, 41). Portions of the secondary structures that include the 5′ and 3′ ends of the (+)RNAs (labeled) and the conserved 3′ stem-loops (3′-SL) (boxed) are shown. Also labeled are the start and stop codons (green and red arrowheads, respectively) of both the NSP3 and NSP3-FL-UnaG ORFs.
Summary.
rSA11/NSP3-FL-UnaG is the first recombinant RVA to be described with a modified segment 7 dsRNA. Segment 7 joins segment 4 (VP4) (32, 33), segment 5 (NSP1) (6, 9, 12), segment 8 (NSP2) (27, 31), and segment 11 (NSP5/NSP6) (35) as targets altered by RG and represents only the second RVA segment to be used as a vector for FP expression. Our analysis of rSA11/NSP3-FL-UnaG indicates that it is possible to generate recombinant RVAs that express FPs through their fusion to the C terminus of NSP3. Given that NSP3 is expressed at moderate levels in infected cells, RVAs expressing NSP3-based FPs may be more effective indicators of viral replication in live-cell imaging experiments and other fluorescence-based assay systems than RVAs expressing NSP1-based FPs, since NSP1 is expressed at low levels in vivo (13). Although several recombinant RVAs that express FPs have been described, rSA11/NSP3-FL-UnaG is unique among them, in that none of its ORFs has been deleted or interrupted. Instead, the only impact on rSA11/NSP3-FL-UnaG was to fuse its NSP3 ORF to a FL-UnaG ORF. Importantly, although the NSP3 ORF in RVA strains is not naturally extended and does not encode NSP3 fused to a downstream protein, the NSP3 ORF of group C rotaviruses (RVCs) is extended, encoding an NSP3 protein that is fused to a 2A stop-start translational element (36) and dsRNA-binding protein (dsRBP) (37, 38). Given that RVC segment 6 encodes an NSP3 fusion protein, it seems likely that the NSP3 fusion protein of rSA11/NSP3-FL-UnaG remains functional even when fused to a downstream protein. Interestingly, despite repeated attempts, we were unsuccessful in generating recombinant RVAs using mutated pT7/NSP3SA11 plasmids in which the NSP3 ORF was interrupted through insertion of stop codons (data not shown). This result implies that NSP3 is essential for RVA replication or is required to generate recombinant viruses using the RG system.
Our results suggest that the RVA segment 7 RNA can be reengineered to function as an expression vector of foreign proteins, without compromising the function of any of the viral ORFs. It remains unclear how much foreign sequence can be inserted into the segment 7 RNA, but our analysis thus far indicates that it is possible to generate well-replicating viruses carrying >500 bp of extra sequence. Given the remarkable flexibility in the ability of the rotavirus to accommodate changes in the size and sequence of its RNA noted thus far (6, 9, 12, 33–35), the virus may be a particularly empowering tool for elucidating the shared mechanisms used by the Reoviridae to package and to replicate their genomes.
MATERIALS AND METHODS
Cell culture.
Embryonic monkey kidney cells (MA104) were grown in medium 199 (M199) complete medium (M199 [Lonza] with 1% penicillin-streptomycin [Corning]) containing 5% FBS (Gibco) (39). BHK-T7 cells were a kind gift from Ulla Buchholz and Peter Collins, Laboratory of Infectious Diseases, NIAID, NIH. BHK-T7 cells were grown in Glasgow complete medium (Glasgow minimal essential medium [MEM] [Lonza] with 10% tryptone-peptide broth [Gibco], 1% penicillin-streptomycin, 2% NEAA [Gibco], and 1% glutamine [Gibco]) containing 5% heat-inactivated FBS. Medium used to cultivate BHK-T7 cells was supplemented with 2% G418 (Geneticin) (Invitrogen) every other passage.
Plasmid construction.
RVA (simian SA11 strain) plasmids pT7/VP1SA11, pT7/VP2SA11, pT7/VP3SA11, pT7/VP4SA11, pT7/VP6SA11, pT7/VP7SA11, pT7/NSP1SA11, pT7/NSP2SA11, pT7/NSP3SA11, pT7/NSP4SA11, and pT7/NSP5SA11 were kindly provided by Takeshi Kobayashi (6) through the Addgene plasmid repository (https://www.addgene.org/Takeshi_Kobayashi). To generate the pCMV/NP868R plasmid, a DNA representing the ASFV NP868R ORF (GenBank accession no. NP_042794), bound by upstream XbaI and downstream BamHI sites, was synthesized by GenScript and inserted into the EcoRV site of the pUC57 plasmid. A DNA fragment containing the NP868R ORF was recovered from the plasmid by digestion with NotI and PvuI and ligated into the plasmid pCMV-Script (Agilent Technologies), cut with the same two restriction enzymes. pT7/NSP3-FL-UnaG was constructed by fusing a DNA fragment containing the ORF for 3×FL-UnaG to the 3′ end of the NSP3 ORF in pT7/NSP3SA11 using a TaKaRa In-fusion HD cloning kit. The 3×FL-UnaG fragment was amplified from the reovirus S1 3×FL-UnaG plasmid (19) by using the primer pair 5′-TCATTTGGTTGCGAAGACTACAAAGACCATGACGGTGATTATAAAGA-3′ and 5′-CATGTATCAAAATGGTCATTCTGTGGCCCTTCTGTAGCT-3′, and the pT7/NSP3SA11 plasmid was amplified by using the NSP3 primer pair 5′-CCATTTTGATACATGTTGAACAATCAAATACAGTGT-3′ and 5′-TTCGCAACCAAATGAATATTGATAATTACATCTCTGTATTAAT-3′. pT7/NSP1-FL-UnaG was constructed by inserting the 3×FL-UnaG fragment into the NSP1 ORF of the segment 5 cDNA (position 1199) in pT7/NSP1SA11. For cloning, the pT7/NSP1SA11 plasmid was amplified by using the NSP1 primer pair 5′-TGAAGAAGTGTTTAATCACATGTCGCC-3′ and 5′-TTTGATCCATGTGATTAGTAAACAAACTCCAAA-3′, and the 3×FL-UnaG insert was amplified from pT7/NSP3-R2A-FL-UnaG by using the primer pair 5′-TCACATGGATCAAACCTACAAAGACCATGACGGTGATTATAAAGATCAT-3′ and 5′-TTAAACACTTCTTCATCATTCTGTGGCCCTTCTGTAGC-3′. Transfection-quality plasmids were prepared commercially (Plasmid.com) or by using a QIAprep spin miniprep kit. Primers and sequencing services were provided by Eurofins Genomics. Sequences of recombinant RVAs were determined from cDNAs prepared from viral RNAs using a Superscript III reverse transcriptase kit (Invitrogen).
Optimized RVA RG protocol.
On day 0, freshly confluent monolayers of BHK-T7 cells were disrupted using trypsin-Versene and were resuspended in G418-free Glasgow complete medium containing 5% FBS. Cell numbers were determined with a Nexcelom Cellometer AutoT4 counter. The cells were seeded into 12-well plates in the same medium (2 × 105 to 4 × 105 cells/well). On day 1, plasmid mixtures that contained 0.8 μg each of the 11 RVA pT7 plasmids except for pT7/NSP2SA11 and pT7/NSP5SA11, which were present at 2.4 μg each, were prepared. Included in the plasmid mixtures was 0.8 μg of pCMV/NP868R. The plasmid mixtures were added to 100 μl of prewarmed (37°C) Opti-MEM (Gibco) and mixed by gentle pipetting up and down. Subsequently, 25 μl of TransIT-LTI transfection reagent (Mirus) was added, and the transfection mixtures were gently vortex-mixed and incubated at room temperature for 20 min. During the incubation period, BHK-T7 cells in 12-well plates were washed once with Glasgow complete medium, and then 1 ml of Spinner-modified MEM (Eagle Joklik MEM [Lonza] with 10% tryptose phosphate broth [TBP], 2% NEAA, 1% penicillin-streptomycin, and 1% glutamine) was placed in each well. The transfection mixture was added dropwise to the medium in the wells, and the plates were returned to a 37°C incubator. On day 3, 2 × 105 MA104 cells in 250 μl of M199 complete medium were added to the wells, along with trypsin (porcine pancreatic type IX; Sigma-Aldrich) to a final concentration of 0.5 μg/ml. On day 5, cells in the plates were frozen and thawed three times, and the lysates were placed in 1.5-ml microcentrifuge tubes. After centrifugation at 500 × g for 10 min at 4°C, 300 μl of the supernatant was transferred onto MA104 monolayers in 6-well plates containing 2 ml of M199 complete medium with 0.5 μg/ml trypsin. The plates were incubated at 37°C for 7 days or until complete cytopathic effects were observed. Typically, complete cytopathic effects were noted at 4 to 6 dpi for wells containing replicating RVA.
Analysis of recombinant viruses.
RVAs were propagated in MA104 cells in M199 complete medium containing 0.5 μg/ml trypsin. Viruses were isolated by plaque purification, and titers were determined by a plaque assay or fluorescence focus assay on MA104 cells (39). To determine peak viral titers, virus isolates were grown in parallel on MA104 cell monolayers until cytopathic effects reached 100%. Plaque titers for lysates were calculated from six independent plaque assays. Viral RNAs were recovered from clarified infected-cell lysates by TRIzol extraction, resolved by electrophoresis on Novex 8% polyacrylamide gels (Invitrogen), and detected by staining with ethidium bromide.
For immunoblot assays, proteins in lysates prepared at 8 hpi from MA104 cells infected with RVA at a MOI of 5 were resolved by electrophoresis on Novex 8 to 16% (linear gradient) polyacrylamide gels and transferred to nitrocellulose membranes. After blocking with phosphate-buffered saline containing 5% nonfat dry milk, blots were probed with guinea pig polyclonal anti-NSP3 (lot 55068; 1:2,000) or anti-VP6 (lot 53963; 1:2,000) antiserum (23), mouse monoclonal Flag M2 antibody (product no. F1804 [Sigma]; 1:2,000), or rabbit monoclonal proliferating cell nuclear antigen (PCNA) antibody (product no. 13110S [Cell Signaling Technology]; 1:1,000). Primary antibodies were detected using 1:10,000 dilutions of horseradish peroxidase (HRP)-conjugated secondary antibodies, i.e., horse anti-mouse IgG (Cell Signaling Technology), goat anti-guinea pig IgG (KPL), or goat anti-rabbit IgG (Cell Signaling Technology). Signals were developed using Clarity Western enhanced chemiluminescence (ECL) substrate (Bio-Rad) and were detected using a Bio-Rad ChemiDoc imaging system. ImageJ analysis was used to determine the intensity of bands on immunoblots (https://imagej.net/ImageJ).
Assessment of genetic stability.
MA104 cell monolayers in 6-well plates were infected with recombinant RVA at a MOI of ∼0.1. When cytopathic effects were complete (4 to 5 dpi), the cells were frozen and thawed twice in their medium, and lysates were centrifuged at low speed to remove debris. Virus in clarified lysates was serially passaged 10 times through sequential infection of MA104 cells with 2 μl of lysate combined with 2 ml of fresh M199 complete medium. To analyze viral dsRNA content, 0.6 ml of clarified lysate was incubated with 1 μl of RNase T1 (1,000 U/ml; Fermentas) for 30 min at 37°C, extracted with TRIzol, and pelleted by ethanol precipitation. Purified dsRNA was resolved by electrophoresis on 8% polyacrylamide gels and detected by staining with ethidium bromide. Plaque purification was used to recover six virus isolates from the passage 10 pool of rSA11/NSP3-FL-UnaG. The segment 6 (VP6) and segment 7 (NSP3) RNAs of three isolates were sequenced by Eurofins Genomics.
Live-cell imaging of UnaG fluorescence in infected cells.
MA104 cells were seeded at a density of 25,000 cells per well in a 96-well optical-bottom plate (Greiner Bio-One) and grown for 2 days at 37°C in 5% CO2 before being infected with rSA11/NSP1-FL-UnaG or rSA11/NSP3-FL-NSP3-UnaG at ∼4 fluorescent focus units per cell. After 1 h, the inoculum was removed and 200 μl FluoroBrite (Thermo Fisher Scientific) was added to each well for maintenance medium. The plate was mounted in an Okolab stage-top environmental chamber equilibrated to 37°C in 5% CO2. Wells were imaged with a wide-field epifluorescence Nikon TiE inverted microscope using a Spectra X LED light source (Lumencor) and a 20× Plan Fluor (numerical aperture [NA], 0.45) phase-contrast objective. Fluorescence light images were recorded using an ORCA-Flash 4.0 scientific complementary metal oxide semiconductor (sCMOS) camera (Hamamatsu), and Nikon Elements Advanced Research v4.5 software was used for multipoint position selection, data acquisition, and image analysis. Single cells were selected as regions of interest (ROIs), and fluorescence intensity was measured for the experiment. Fluorescence intensity values were exported to Microsoft Excel and normalized to the initial baseline fluorescence value. The baseline and maximum fluorescence values for each ROI selected were calculated; from this, the change in fluorescence (ΔF) was calculated by subtracting the baseline value from the maximum value. The ΔF was plotted as percent change, and biostatistical analyses were performed using GraphPad Prism v8.1, with results presented as mean ± standard deviation. Comparisons used a nonparametric Mann-Whitney test, and differences were considered statistically significant for P values of <0.05.
Accession number(s).
Nucleotide sequences were deposited in GenBank and have the following accession numbers: pCMV/NP868R, MH212166; pT7/NSP3-FL-UnaG, MK868472; pT7/NSP1-FL-UnaG, MH197081.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to all of the members of the Patton and Danthi laboratories for their support and encouragement on this project. Our thanks also go to Ulla Buckholtz and Peter Collins (NIAID, NIH) for their gift of BHK-T7 cells.
This work was supported by NIH grants R03 AI131072 (J.T.P.), R21 AI144881 (J.T.P.), and R01DK115507 (J.M.H.). J.T.P. was also supported by Indiana University Start-Up Funding and the Lawrence M. Blatt Endowment.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01616-19.
REFERENCES
- 1.Crawford SE, Ramani S, Tate JE, Parashar UD, Svensson L, Hagbom M, Franco MA, Greenberg HB, O'Ryan M, Kang G, Desselberger U, Estes MK. 2017. Rotavirus infection. Nat Rev Dis Primers 3:17083. doi: 10.1038/nrdp.2017.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC. 2011. Atomic model of an infectious rotavirus particle. EMBO J 30:408–416. doi: 10.1038/emboj.2010.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imai M, Akatani K, Ikegami N, Furuichi Y. 1983. Capped and conserved terminal structures in human rotavirus genome double-stranded RNA segments. J Virol 47:125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trask SD, McDonald SM, Patton JT. 2012. Structural insights into the coupling of virion assembly and rotavirus replication. Nat Rev Microbiol 10:165–177. doi: 10.1038/nrmicro2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guglielmi KM, McDonald SM, Patton JT. 2010. Mechanism of intraparticle synthesis of the rotavirus double-stranded RNA genome. J Biol Chem 285:18123–18128. doi: 10.1074/jbc.R110.117671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanai Y, Komoto S, Kawagishi T, Nouda R, Nagasawa N, Onishi M, Matsuura Y, Taniguchi K, Kobayashi T. 2017. Entirely plasmid-based reverse genetics system for rotaviruses. Proc Natl Acad Sci U S A 114:2349–2354. doi: 10.1073/pnas.1618424114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kyrieleis OJ, Chang J, de la Peña M, Shuman S, Cusack S. 2014. Crystal structure of vaccinia virus mRNA capping enzyme provides insights into the mechanism and evolution of the capping apparatus. Structure 22:452–465. doi: 10.1016/j.str.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salsman J, Top D, Boutilier J, Duncan R. 2005. Extensive syncytium formation mediated by the reovirus FAST proteins triggers apoptosis-induced membrane instability. J Virol 79:8090–8100. doi: 10.1128/JVI.79.13.8090-8100.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komoto S, Fukuda S, Ide T, Ito N, Sugiyama M, Yoshikawa T, Murata T, Taniguchi K. 2018. Generation of recombinant rotaviruses expressing fluorescent proteins by using an optimized reverse genetics system. J Virol 92:e00588-18. doi: 10.1128/JVI.00588-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fabbretti E, Afrikanova I, Vascotto F, Burrone OR. 1999. Two non-structural rotavirus proteins, NSP2 and NSP5, form viroplasm-like structures in vivo. J Gen Virol 80:333–339. doi: 10.1099/0022-1317-80-2-333. [DOI] [PubMed] [Google Scholar]
- 11.Eichwald C, Rodriguez JF, Burrone OR. 2004. Characterization of rotavirus NSP2/NSP5 interactions and the dynamics of viroplasm formation. J Gen Virol 85:625–634. doi: 10.1099/vir.0.19611-0. [DOI] [PubMed] [Google Scholar]
- 12.Kanai Y, Kawagishi T, Nouda R, Onishi M, Pannacha P, Nurdin JA, Nomura K, Matsuura Y, Kobayashi T. 2018. Development of stable rotavirus reporter expression systems. J Virol 93:e01774-18. doi: 10.1128/JVI.01774-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martínez-Álvarez L, Piña-Vázquez C, Zarco W, Padilla-Noriega L. 2013. The shift from low to high non-structural protein 1 expression in rotavirus-infected MA-104 cells. Mem Inst Oswaldo Cruz 108:421–428. doi: 10.1590/S0074-0276108042013005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barro M, Patton JT. 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc Natl Acad Sci U S A 102:4114–4119. doi: 10.1073/pnas.0408376102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davis KA, Patton JT. 2017. Shutdown of interferon signaling by a viral-hijacked E3 ubiquitin ligase. Microb Cell 4:387–389. doi: 10.15698/mic2017.11.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montero H, Arias CF, Lopez S. 2006. Rotavirus nonstructural protein NSP3 is not required for viral protein synthesis. J Virol 80:9031–9038. doi: 10.1128/JVI.00437-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gratia M, Sarot E, Vende P, Charpilienne A, Baron CH, Duarte M, Pyronnet S, Poncet D. 2015. Rotavirus NSP3 is a translational surrogate of the poly(A)-binding protein-poly(A) complex. J Virol 89:8773–8782. doi: 10.1128/JVI.01402-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dixon LK, Chapman DA, Netherton CL, Upton C. 2013. African swine fever virus replication and genomics. Virus Res 173:3–14. doi: 10.1016/j.virusres.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 19.Eaton HE, Kobayashi T, Dermody TS, Johnston RN, Jais PH, Shmulevitz M. 2017. African swine fever virus NP868R capping enzyme promotes reovirus rescue during reverse genetics by promoting reovirus protein expression, virion assembly, and RNA incorporation into infectious virions. J Virol 91:e02416-16. doi: 10.1128/JVI.02416-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumagai A, Ando R, Miyatake H, Greimel P, Kobayashi T, Hirabayashi Y, Shimogori T, Miyawaki A. 2013. A bilirubin-inducible fluorescent protein from eel muscle. Cell 153:1602–1611. doi: 10.1016/j.cell.2013.05.038. [DOI] [PubMed] [Google Scholar]
- 21.Deo RC, Groft CM, Rajashankar KR, Burley SK. 2002. Recognition of the rotavirus mRNA 3ʹ consensus by an asymmetric NSP3 homodimer. Cell 108:71–81. doi: 10.1016/s0092-8674(01)00632-8. [DOI] [PubMed] [Google Scholar]
- 22.Groft CM, Burley SK. 2002. Recognition of eIF4G by rotavirus NSP3 reveals a basis for mRNA circularization. Mol Cell 9:1273–1283. doi: 10.1016/s1097-2765(02)00555-5. [DOI] [PubMed] [Google Scholar]
- 23.Arnold MM, Brownback CS, Taraporewala ZF, Patton JT. 2012. Rotavirus variant replicates efficiently although encoding an aberrant NSP3 that fails to induce nuclear localization of poly(A)-binding protein. J Gen Virol 93:1483–1494. doi: 10.1099/vir.0.041830-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clapp LL, Patton JT. 1991. Rotavirus morphogenesis: domains in the major inner capsid protein essential for binding to single-shelled particles and for trimerization. Virology 180:697–708. doi: 10.1016/0042-6822(91)90083-N. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez EA, Campbell RE, Lin JY, Lin MZ, Miyawaki A, Palmer AE, Shu X, Zhang J, Tsien RY. 2017. The growing and glowing toolbox of fluorescent and photoactive proteins. Trends Biochem Sci 42:111–129. doi: 10.1016/j.tibs.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patton JT, Taraporewala Z, Chen D, Chizhikov V, Jones M, Elhelu A, Collins M, Kearney K, Wagner M, Hoshino Y, Gouvea V. 2001. Effect of intragenic rearrangement and changes in the 3ʹ consensus sequence on NSP1 expression and rotavirus replication. J Virol 75:2076–2086. doi: 10.1128/JVI.75.5.2076-2086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navarro A, Trask SD, Patton JT. 2013. Generation of genetically stable recombinant rotaviruses containing novel genome rearrangements and heterologous sequences by reverse genetics. J Virol 87:6211–6220. doi: 10.1128/JVI.00413-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ballard A, McCrae MA, Desselberger U. 1992. Nucleotide sequences of normal and rearranged RNA segments 10 of human rotaviruses. J Gen Virol 73:633–638. doi: 10.1099/0022-1317-73-3-633. [DOI] [PubMed] [Google Scholar]
- 29.Shen S, Burke B, Desselberger U. 1994. Rearrangement of the VP6 gene of a group A rotavirus in combination with a point mutation affecting trimer stability. J Virol 68:1682–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gault E, Schnepf N, Poncet D, Servant A, Teran S, Garbarg-Chenon A. 2001. A human rotavirus with rearranged genes 7 and 11 encodes a modified NSP3 protein and suggests an additional mechanism for gene rearrangement. J Virol 75:7305–7314. doi: 10.1128/JVI.75.16.7305-7314.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tortorici MA, Broering TJ, Nibert ML, Patton JT. 2003. Template recognition and formation of initiation complexes by the replicase of a segmented double-stranded RNA virus. J Biol Chem 278:32673–32682. doi: 10.1074/jbc.M305358200. [DOI] [PubMed] [Google Scholar]
- 32.Johne R, Reetz J, Kaufer BB, Trojnar E. 2016. Generation of an avian-mammalian rotavirus reassortant by using a helper virus-dependent reverse genetics system. J Virol 90:1439–1443. doi: 10.1128/JVI.02730-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohanty SK, Donnelly B, Dupree P, Lobeck I, Mowery S, Meller J, McNeal M, Tiao G. 2017. A point mutation in the rhesus rotavirus VP4 protein generated through a rotavirus reverse genetics system attenuates biliary atresia in the murine model. J Virol 91:e00510-17. doi: 10.1128/JVI.00510-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trask SD, Taraporewala ZF, Boehme KW, Dermody TS, Patton JT. 2010. Dual selection mechanisms drive efficient single-gene reverse genetics for rotavirus. Proc Natl Acad Sci U S A 107:18652–18657. doi: 10.1073/pnas.1011948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Komoto S, Kanai Y, Fukuda S, Kugita M, Kawagishi T, Ito N, Sugiyama M, Matsuura Y, Kobayashi T, Taniguchi K. 2017. Reverse genetics system demonstrates that rotavirus nonstructural protein NSP6 is not essential for viral replication in cell culture. J Virol 91:e00695-17. doi: 10.1128/JVI.00695-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donnelly ML, Hughes LE, Luke G, Mendoza H, ten Dam E, Gani D, Ryan MD. 2001. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J Gen Virol 82:1027–1041. doi: 10.1099/0022-1317-82-5-1027. [DOI] [PubMed] [Google Scholar]
- 37.James VL, Lambden PR, Deng Y, Caul EO, Clarke IN. 1999. Molecular characterization of human group C rotavirus genes 6, 7 and 9. J Gen Virol 80:3181–3187. doi: 10.1099/0022-1317-80-12-3181. [DOI] [PubMed] [Google Scholar]
- 38.Langland JO, Pettiford S, Jiang B, Jacobs BL. 1994. Products of the porcine group C rotavirus NSP3 gene bind specifically to double-stranded RNA and inhibit activation of the interferon induced protein kinase PKR. J Virol 68:3821–3829. doi: 10.1089/jir.1988.8.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arnold M, Patton JT, McDonald SM. 2009. Culturing, storage, and quantification of rotaviruses. Curr Protoc Microbiol Chapter 15:Unit 15C.3. doi: 10.1002/9780471729259.mc15c03s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hofacker IL. 2003. Vienna RNA secondary structure server. Nucleic Acids Res 31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hofacker IL, Fontana W, Stadler PF, Bonhoeffer S, Tacker M, Schuster P. 1994. Fast folding and comparison of RNA secondary structures. Monatsh Chem 125:167–188. doi: 10.1007/BF00818163. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.