

Seizure-related apnea is common and can be lethal. Its mechanisms however remain unclear and preventive strategies are lacking. We postulate that brainstem spreading depolarization (SD), previously associated with lethal seizures in animal models, initiates apnea upon invasion of brainstem respiratory centers.
Keywords: apnea, brainstem, channelopathy, oxygen, SUDEP, transgenic model
Abstract
Seizure-related apnea is common and can be lethal. Its mechanisms however remain unclear and preventive strategies are lacking. We postulate that brainstem spreading depolarization (SD), previously associated with lethal seizures in animal models, initiates apnea upon invasion of brainstem respiratory centers. To study this, we assessed effects of brainstem seizures on brainstem function and respiration in male and female mice carrying a homozygous S218L missense mutation that leads to gain-of-function of voltage-gated CaV2.1 Ca2+ channels and high risk for fatal seizures. Recordings of brainstem DC potential and neuronal activity, cardiorespiratory activity and local tissue oxygen were performed in freely behaving animals. Brainstem SD occurred during all spontaneous fatal seizures and, unexpectedly, during a subset of nonfatal seizures. Seizure-related SDs in the ventrolateral medulla correlated with respiratory suppression. Seizures induced by stimulation of the inferior colliculus could evoke SD that spread in a rostrocaudal direction, preceding local tissue hypoxia and apnea, indicating that invasion of SD into medullary respiratory centers initiated apnea and hypoxia rather than vice versa. Fatal outcome was prevented by timely resuscitation. Moreover, NMDA receptor antagonists MK-801 and memantine prevented seizure-related SD and apnea, which supports brainstem SD as a prerequisite for brainstem seizure-related apnea in this animal model and has translational value for developing strategies that prevent fatal ictal apnea.
SIGNIFICANCE STATEMENT Apnea during and following seizures is common, but also likely implicated in sudden unexpected death in epilepsy (SUDEP). This underlines the need to understand mechanisms for potentially lethal seizure-related apnea. In the present work we show, in freely behaving SUDEP-prone transgenic mice, that apnea is induced when spontaneous brainstem seizure-related spreading depolarization (SD) reaches respiratory nuclei in the ventrolateral medulla. We show that brainstem seizure-related medullary SD is followed by local hypoxia and recovers during nonfatal seizures, but not during fatal events. NMDA receptor antagonists prevented medullary SD and apnea, which may be of translational value.
Introduction
Seizure-related apnea occurs commonly in patients with epilepsy and can result in severe hypoxemia (Nashef et al., 1996; Bruno et al., 2018; Vilella et al., 2019). If prolonged, apnea may be fatal, as such apnea preceded cardiac arrest in rarely monitored cases of “sudden unexpected death in epilepsy” (SUDEP; Ryvlin et al., 2013), a fatal complication of epilepsy affecting patients of all ages (Massey et al., 2014; Devinsky et al., 2016; Harden et al., 2017). Seizure-related apnea, however, has a benign outcome in the large majority of cases, yet given its presumed role in SUDEP there is an urgent need to understand mechanisms of potentially lethal respiratory suppression. Neuropathological and structural neuroimaging changes (Mueller et al., 2014, 2018; Patodia et al., 2018) in brainstem regions of SUDEP cases suggest seizure-related brainstem dysfunction as a possible critical factor leading to fatal apnea.
In recent years, brainstem spreading depolarization (SD) has been linked to SUDEP based on studies in various transgenic mouse models. A negative DC-potential shift in the dorsal brainstem was observed indicating brainstem SD (Aiba and Noebels, 2015; Aiba et al., 2016). In these studies, SD appeared to follow upon severe respiratory compromise, thereby providing an explanation for the absence of autoresuscitation following apnea (Aiba and Noebels, 2015). Brainstem SD was also demonstrated for cortically induced and spontaneous fatal seizures in mice harboring the homozygous S218L gain-of-function missense mutation in the Cacna1a gene that encodes the α1A subunit of voltage-gated CaV2.1 Ca2+ channels (Loonen et al., 2019). The absence of cortical seizure activity in this model suggests an important role for brainstem circuitry in the genesis of seizures. However, it remains an enigma whether changes in breathing rhythm result in hypoxia and trigger brainstem SD, or vice versa, which is key when considering strategies for (preventative) interventions of seizure-related apnea.
Breathing rhythm is generated by the ventral respiratory column, located in the ventrolateral medulla (VLM; Smith et al., 2013; Del Negro et al., 2018). The pre-Bötzinger complex (pre-BötC), a subregion of the VLM, specifically generates the inspiratory phase that is crucial for respiratory function (Gray et al., 2001; McKay et al., 2005; Feldman and Del Negro, 2006; Del Negro et al., 2018). Rapid suppression of glutamatergic pre-BötC neurons induces fatal apnea (Tan et al., 2008). We postulate that seizure-induced brainstem SD can evoke apnea because of prolonged neuronal suppression in the VLM. To test this, we studied brainstem neuronal activity and cardiorespiratory function during spontaneous seizures and seizures induced by stimulation of the inferior colliculus in freely behaving homozygous Cacna1aS218L mice. Reversibility of brainstem SD was evaluated by mechanical ventilation during apnea. Because brainstem SD commonly preceded apnea and local hypoxia, we also tested whether increasing the SD threshold by NMDA antagonism could prevent lethality.
Materials and Methods
Animals.
Missense mutation S218L was introduced in the mouse Cacna1a gene, generating Cacna1aS218L mice, using a gene-targeting approach (van den Maagdenberg et al., 2010). Mice were backcrossed to C57BL/6J for at least 10 generations and both male and female mice, age 3–6 months, were used in experiments. Standard housing conditions and a 12 h light/dark cycle with food and water available ad libitum were applied. Experiments were approved by local and national ethical committees in accordance with recommendations of the European Communities Council Directive (2010/63/EU) and performed in accordance with ARRIVE guidelines.
Surgery for recordings of spontaneous seizures.
Cacna1aS218L mice (15 males and 17 females) were implanted with either 7 (n = 11) or 16 (n = 21) electrodes (single or paired 75 μm platinum/iridium; PT6718, Advent Research Materials) under isoflurane anesthesia (induction 4%; maintenance 1.5%). Electrode configurations were as follows (coordinates relative to bregma; in mm anterior, lateral, and ventral, respectively): for 7-channel recordings (E363/0 socket contacts and MS373 pedestal; Plastics One), electrodes were implanted in right primary visual cortex (V1; −3.5/2.0/0.5; unipolar) and oral pontine reticular nucleus (PnO; −4.8/0.8/3.7; unipolar or bipolar) and VLM (−6.7/1.3/4.1; unipolar or bipolar); for 16-channel recordings (#0489 pin receptacles and #831 18-pin pedestal; Mill-Max), unipolar electrodes were implanted in right V1 and bilaterally in primary motor cortex (+1.5/1.8/0.6), hippocampus (−2.2/2.0/1.3), and amygdala (−1.7/2.9/4.0), with bipolar electrodes in right inferior colliculus (IC; −5.0/1.0/0.6), PnO, and VLM. Electrodes inserted bilaterally above the cerebellum served as reference and ground. For ECG, electrodes were placed in the flank. For respiratory recordings (n = 13), a thermistor probe (MEAS-G22K7MCD419, Measurement Specialties) was inserted above the epithelium of the anterior nasal cavity as described previously (McAfee et al., 2016). The pedestal was secured to the skull using dental cement (DiaDent Europe). In a subset of animals (n = 14), cerebral blood flow (CBF) was measured using an optic fiber (200 μm; CFM12L02, Thorlabs) placed on the dura overlaying V1. Postoperative analgesia was supplied with carprofen and buprenorphine (5 mg/kg and 0.1 mg/kg respectively, s.c.). Electrode configurations were first tested in wild-type mice for a 2 week period.
Surgery for recordings of induced seizures.
For IC stimulation in homozygous Cacna1aS218L and wild-type animals, bipolar electrodes were randomly implanted in the left or right IC. Two electrode configurations were used, with (1) bipolar electrodes bilaterally in VLM, or (2) electrodes implanted unilaterally in VLM (bipolar) and the rostral parvocellular reticular nucleus (PCRt-r) and caudal parvocellular reticular nucleus PCRt-c (unipolar; PCRt-r: −5.5/1.4/3.6; PCRt-c: −6.1/1.3/4.0) to create an array of brainstem electrodes ipsilateral to the stimulated IC. A unipolar electrode in right V1 and a nasal thermistor were implanted as described in the previous section. Two to 4 d after surgery, a 50 Hz train of 1 ms bipolar pulses was delivered through the IC electrode for 2 s. Threshold for seizure-related behavior, which consistently commenced with wild running, was determined for a range of intensities (30, 50, 100, 200, 500 μA) with 10 min between subsequent pulse trains. The threshold current was delivered twice daily for 3 d, but limited to one stimulation in case of a generalized tonic-clonic seizure. In a separate group of Cacna1aS218L mice, local oxygen partial pressure (pO2) was measured using a technique involving oxygen quenching of fluorescence generated in a platinum-based dye in the tip of an optic fiber (Oxylite, Oxford Optronics). This allows for absolute measurements of pO2 without consuming oxygen (Griffiths and Robinson, 1999). Probes (230 μm), pre-calibrated by the manufacturer and indicated with a T90 response time (i.e., time to reach 90% of the actual pO2 change) of <20 s, were inserted rostral of the VLM electrode (−6.2/1.3/3.8), replacing the PCRt-c electrode. To study the locality of changes in pO2, a subset of mice was implanted with a second oxygen probe in V1, anterior to the V1 electrode (−3.0/2.0/0.5).
Surgery for recordings of cortically induced SD.
For cortical SD induction, a subset of homozygous Cacna1aS218L mice was implanted with a bipolar electrode in V1 and unipolar electrodes in the primary somatosensory cortex (S1; −0.5/2.0/0.5) and M1. Directly adjacent to the S1 electrode, an oxygen probe was implanted. Cathodal stimulation (100 μA for 1 s) was administered once daily for 1 or 2 d and reliably resulted in cortical SD.
Mechanical ventilation and pharmacology.
Post-ictal respiratory support (150 breaths/min for 20–50 s, 250 μl per breath) was administered via the nostrils by a polyethylene tube connected to a mechanical ventilator (Mini-Vent, Harvard Apparatus). In a sham group, the same procedure was performed with the ventilator switched off.
To test the effects of NMDA receptor antagonists on seizure behavior and outcome, mice that showed brainstem SD in response to initial IC stimulation were used. Vehicle, MK-801 (1 mg/kg bodyweight) or memantine hydrochloride (10 mg/kg bodyweight; both from Sigma-Aldrich) were injected intraperitoneally 30 min before IC stimulation by an experimenter blinded to the treatment.
Histology.
Surviving animals were killed by CO2. Electrolytic lesions were made at the tip of brain electrodes (10 μA anodal current) in all animals to enable confirmation of electrode positions. After transcardial perfusion (for killed animals), brains were postfixed in 4% paraformaldehyde for 2.5 h at room temperature, or for 24 h at 4°C (for spontaneously deceased animals), and sucrose-processed. Coronal brain sections (40 μm) were made using a sliding microtome (Leica) and Nissl-stained to confirm electrode positions.
Data acquisition and analyses.
Immediately following surgery, animals were connected to a commutator (ACO32 for 16-channel recordings, Tucker-Davis Technologies; a custom-made system was used for 7-channel recordings). Data were amplified and digitized as described previously (Houben et al., 2017). Recordings of pO2 were acquired at 1 Hz.
SD was defined as a transient negative DC-shift with amplitude >5 mV measured with a delay at two recording locations, or limited to one location if accompanied by depression of local multiunit activity (MUA). Video EEG recordings during these events and the 24 h preceding the fatal seizure were analyzed for seizures, which were scored using the Racine scale (Racine, 1972). In retrospect, all fatal seizures were of Stage 5 and hence Stage 5 nonfatal seizures were used for comparative analyses. Single seizures or seizure clusters lasting >30 min were regarded as status epilepticus (Trinka et al., 2015) and, as a SUDEP exclusion criterion (Nashef et al., 2012), excluded from further analyses.
Electrophysiological data were analyzed using custom-written MATLAB (MathWorks) scripts. For electrocorticogram (ECoG) power analyses, AC-signals were artifact-rejected, digitally low-pass filtered (Chebyshev IIR eighth-order filter) and downsampled to 500 Hz. Total ECoG power (1–100 Hz) was calculated using a fast Fourier transform. For MUA, the root mean square was computed over 10 or 100 ms. Peak detection was used to determine instantaneous heart rate and respiratory rate from ECG and thermistor recordings respectively. For heart rate variability (HRV) analyses, standard deviation of all normal ECG R-R intervals (SDNN) was calculated (Thireau et al., 2008). Pre-ictal and post-ictal parameters were calculated over 5 min periods. Longer-term heart rhythm changes were not analyzed, because HRV is profoundly increased during the first week following surgery (Thireau et al., 2008). Peri-ictal CBF, MUA, and ECoG power were normalized by activity levels during a 1 min period 5 min pre-ictal. For ECoG and CBF analyses, seizures associated with cortical SD were excluded because of local SD-related suppression.
Experimental design and statistical analysis.
In total, data from 104 mice were included: recordings from spontaneous seizures were obtained from 32 homozygous Cacna1aS218L mice, while seizures were induced in 68 mice (3 wild-type and 65 homozygous Cacna1aS218L mice), including 12 mice for mechanical ventilation and 31 mice for pharmacological experiments. A maximum of three seizures were induced in untreated animals, whereas one seizure was induced for experiments involving respiratory or pharmacological intervention. Cortical SD induction was performed in four homozygous Cacna1aS218L mice.
Statistical testing was performed using MATLAB or GraphPad Prism. Data are represented as mean ± SEM in text and time series graphs, total range for box-and-whisker graphs, and mean ± standard deviation for graphs showing individual data points. Data are compared using two-tailed paired or unpaired Student's t tests, or one-way ANOVA with Dunnett's post hoc test for multiple comparisons, unless indicated otherwise. p < 0.05 was considered significant.
Results
Brainstem SD occurs during seizures in freely behaving Cacna1aS218L mice
Over a 2 week recording period, 63% (20/32) of implanted Cacna1aS218L mice died after a mean survival period of 2.7 ± 0.5 d (range: 8 h–9 d). High mortality in the days following surgery in this model was earlier described to be seizure-related (Loonen et al., 2019). Indeed, mice died following a Racine Stage 5 seizure (n = 15; 8 males and 7 females) or status epilepticus (n = 5; 2 males and 3 females). All fatal seizures were accompanied by SDs in brainstem areas including the PnO (11/11 recordings) and VLM (13/13 recordings; Fig. 1A–D) and occasionally IC (3/7 recordings). In contrast, SDs were observed in forebrain areas (neocortex, hippocampus, and amygdala) in only 7/15 fatal seizures and during status epilepticus (5/5). All brainstem SDs occurred during seizure behavior, with behavioral arrest starting 84 ± 17 s (range: 15–160) after onset of VLM SD and 67 ± 18 s (range: 4–142) after PnO SD.
Figure 1.

Brainstem SD and cardiorespiratory dysfunction during a spontaneous fatal and nonfatal seizure in a Cacna1aS218L mutant. A, Top and sagittal view of experimental approach for chronic brainstem recordings. B, Nissl staining showing a lesion marking the electrode tip in the VLM (arrow). C, Schematic of VLM electrode positions (gray dots) in 11 Cacna1aS218L mice that died following a spontaneous fatal seizure with VLM SD. NA, Nucleus accumbens; NTS, nucleus tractus solitarius; OI, oliva inferior; Sp5I, spinal trigeminal nucleus pars interpolaris; XII, nucleus hypoglossus. D, Example of a fatal seizure showing brainstem SD, evidenced by a DC shift (arrowheads) in the PnO and VLM. Note that the SDs occurred during seizure behavior, and were associated with a transient increase in MUA followed by suppression. HR, heart rate; RR, respiratory rate; RMS, root mean square; V1, primary visual cortex). E, Example of a nonfatal seizure that was associated with brainstem SD, after which DC-potential and MUA spontaneously recovered. Note that in both fatal and nonfatal examples, respiratory activity decreased shortly after onset of VLM SD (dashed line).
To assess whether brainstem SD was unique for fatal events, we inspected recordings of the 24 h preceding the fatal events. Of 64 nonfatal Stage 5 seizures (n = 12/15 animals; range: 1–12 per animal), 19% were accompanied by brainstem SD (n = 12 in 7 animals; Fig. 1E). The duration of seizures accompanied by brainstem SD was similar to that of seizures without brainstem SD and fatal seizures (185 ± 24, 170 ± 23, and 194 ± 25 s, respectively; p = 0.525; ANOVA). Brainstem SD was never observed during seizures of lower Racine stage or during inter-ictal periods.
Prolonged SD in the ventrolateral medulla is associated with lethal outcome
In all fatal and in most (10/12) nonfatal seizures, parallel DC recordings in the PnO and VLM demonstrated SD in both brainstem areas. During two nonfatal seizures, only PnO SD occurred, suggesting that during nonfatal seizures SD in the brainstem remains more local than for fatal seizures. Delay between SD onset in PnO and VLM did not indicate evidence for a consistent spreading pattern during nonfatal or fatal seizures: delay was variable and SD could be first detected in either of the two areas (Fig. 2A), indicating that the origin of SD may vary for spontaneous seizures. Duration of VLM SD, as measured from SD onset to partial (20%) recovery of DC-potential, was longer for fatal compared with nonfatal seizures (Fig. 2B).
Figure 2.
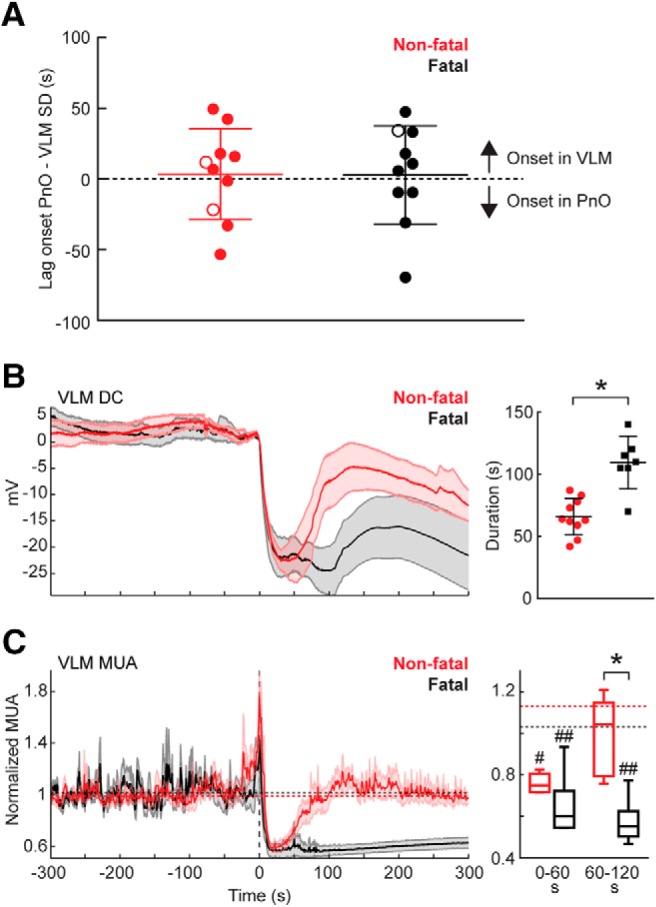
Prolonged medullary neuronal depression following fatal SD in Cacna1aS218L mice. A, Brainstem SD was observed first in either the VLM (positive values) or in the PnO (negative values) during nonfatal (n = 10) and fatal (n = 10) seizures. This variability was observed between animals and within animals, as illustrated by the data points from a single animal with a fatal and two nonfatal seizures associated with brainstem SD (open symbols). B, Averaged DC-recordings in VLM showing SD (left; onset at time = 0 s), indicating earlier recovery for nonfatal (n = 10) compared with fatal (n = 10) seizures (right; 66 ± 15 s vs 109 ± 21 s, t(15) = 5.03, *p = 0.0001, unpaired t test). Note that DC-recordings from three fatal seizures were excluded due to absence of (partial) recovery. C, For fatal versus nonfatal seizures, VLM MUA was reduced after SD onset in both groups (right; n = 5 and n = 6; baseline indicated by dashed lines; #p = 0.009, ##p = 0.0003, and ##p = 0.0004, respectively, ANOVA with Dunnett's test), but then recovered only in nonfatal cases (t(9) = 4.62, *p = 0.001, unpaired t test).
Seizures with brainstem SD confer decreased survival and are followed by ECoG suppression and increased heart rate variability
The time toward a fatal seizure was decreased following nonfatal seizures accompanied by brainstem SD compared with other Stage 5 seizures (4.6 ± 1.1 and 6.3 ± 1.3 h, respectively; t(11) = 2.55, p = 0.031, paired t test). Behaviorally, the majority of nonfatal seizures with brainstem SD terminated with a hindlimb clonus (10/12; all in seizures with VLM SD), a finding previously reported for fatal seizures in Cacna1aS218L mice (Loonen et al., 2019). Onset of hindlimb clonus and VLM SD were closely aligned (Fig. 3A). Pairwise comparisons of pre-ictal and post-ictal parameters of Stage 5 seizures with and without brainstem SD revealed that V1 ECoG was suppressed during the post-ictal period specifically for seizures with brainstem SD, whereas no differences in cortical CBF was observed (Fig. 3B). Post-ictal HRV was increased for seizures with brainstem SD, whereas heart rate remained constant (Table 1). In addition, skipped heart beats occurred more often in the post-ictal period for seizures with brainstem SD (example in Fig. 3C,D) compared with seizures without brainstem SD (Table 1).
Figure 3.
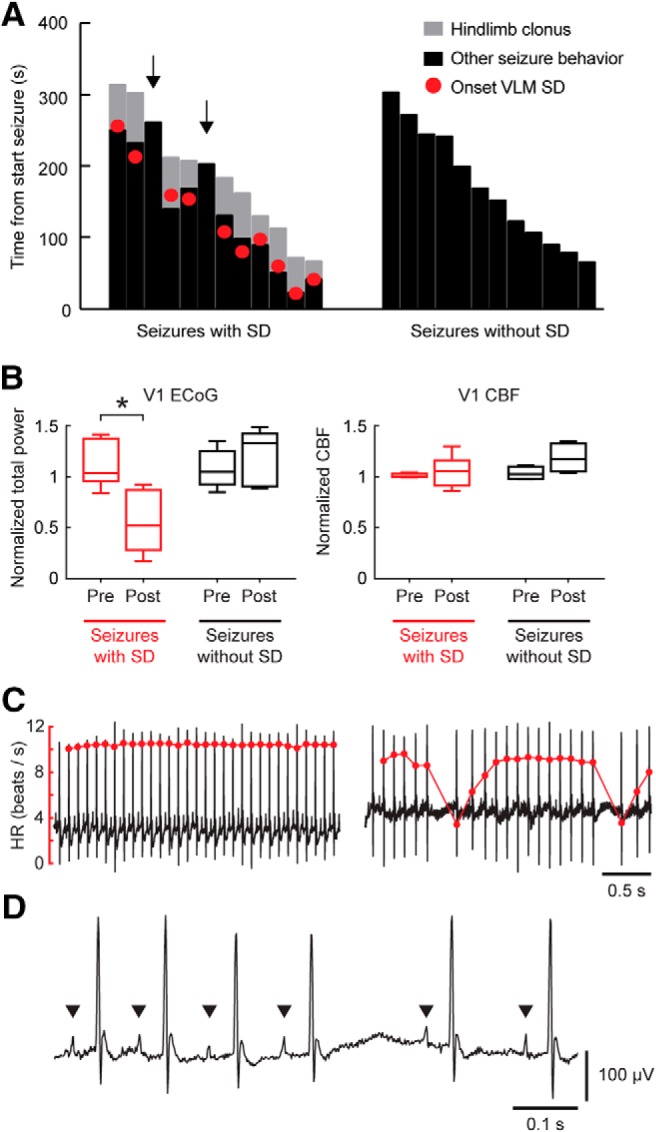
Seizure behavior and post-ictal dynamics indicate brainstem SD during nonfatal seizures in Cacna1aS218L mice. A, Duration of hindlimb clonus and other seizure behavior for seizures with and without brainstem SD, indicated from onset of seizure behavior (at time = 0 s). Hindlimb clonus only occurred during seizures with SD in VLM, and started closely to SD onset. Note that hindlimb clonus did not occur during seizures without brainstem SD and seizures with SD restricted to the PnO (n = 2; arrows). B, ECoG power was significantly suppressed during the post-ictal period for seizures with brainstem SD (t(7) = 5.74, *p = 0.001, paired t test), whereas no difference was present for seizures without brainstem SD. CBF was not significantly different (V1, primary visual cortex). C, Examples of ECG signal (black) and heart rate (HR; red) during a pre-ictal (left) and post-ictal (right) period of a nonfatal seizure with brainstem SD, showing bradyarrhythmias. D, Example of post-ictal ECG signal showing P waves (indicated by arrowheads) and their absence during a prolonged R-R pause.
Table 1.
Post-ictal changes in heart rhythm after brainstem SD
|
n |
Heart rate, beats/s |
Heart rate variability (SDNN), ms |
Skipped heart beats, total count |
|||||
|---|---|---|---|---|---|---|---|---|
| Mice | Seizures | Pre-ictal | Post-ictal | Pre-ictal | Post-ictal | Pre-ictal | Post-ictal | |
| Seizures with brainstem SD | 6 | 9 | 8.12 ± 0.53 | 7.39 ± 0.34 | 22.8 ± 4.8 | 45.1 ± 7.0* | 6.50 ± 4.13 | 65.5 ± 28.5** |
| Seizures without brainstem SD | 6 | 9 | 8.37 ± 0.33 | 8.44 ± 0.49 | 20.2 ± 4.4 | 22.0 ± 3.6 | 6.61 ± 2.72 | 6.13 ± 2.80 |
*p = 0.016,
**p = 0.008 for pre-ictal versus post-ictal via Wilcoxon test.
SDNN, Standard deviation of all normal ECG R-R intervals.
Seizure-related bradypnea correlates with SD in the ventrolateral medulla
Respiratory activity was recorded in a subset of mice with a spontaneous fatal seizure (n = 6). In four mice, eight nonfatal seizures with brainstem SD occurred: two with SD restricted to the PnO and six with SD occurring in both PnO and VLM. All seizures with SD in the VLM were associated with bradypnea (defined as <60 breaths/min), in contrast to the cases with SD limited to PnO (Fig. 4). Dynamics of VLM SD more closely paralleled changes in respiratory rate compared with PnO SD (Fig. 5A,B). In a case with strong coupling of oscillatory VLM MUA and breathing rhythm, VLM SD induced immediate cessation of this oscillatory pattern coinciding with bradypnea onset (Fig. 5C). In contrast to the profound respiratory effects, prolonged bradycardia was not observed for nonfatal seizures with VLM SD, whereas it occurred after ∼1 min following fatal apnea for fatal seizures (Fig. 5D,E).
Figure 4.
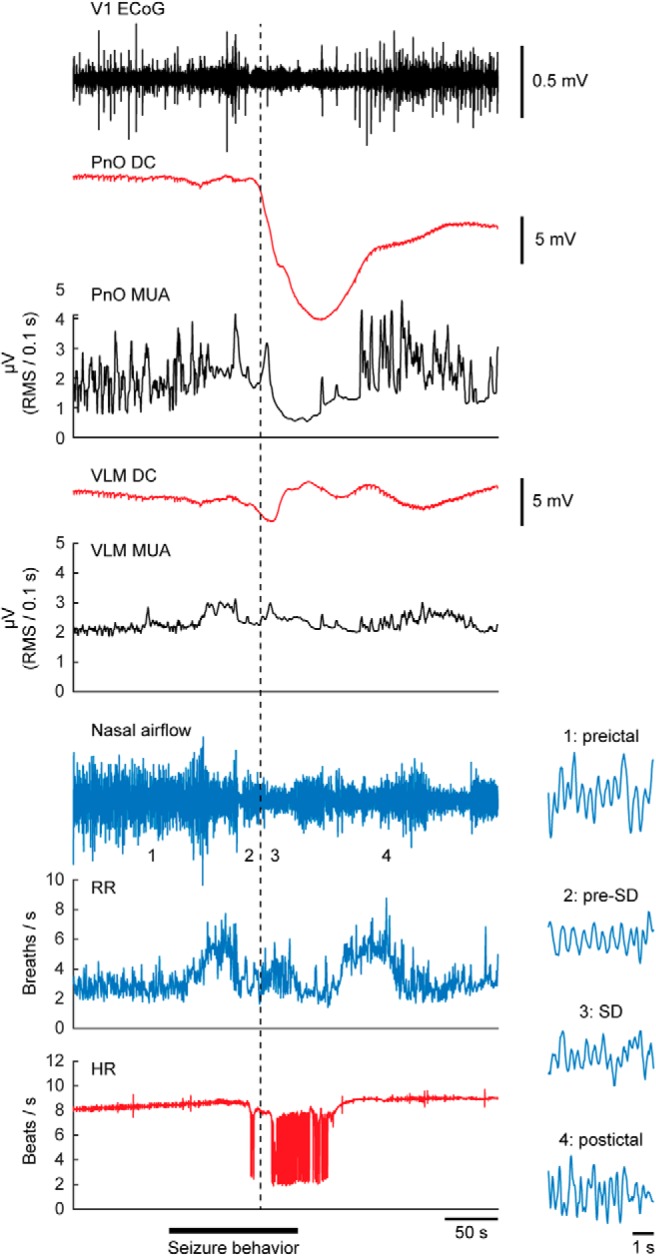
Isolated SD in the rostral brainstem during a spontaneous nonfatal seizure. Example of a nonfatal seizure with SD (dashed line) in the PnO without evidence of ipsilateral SD in the VLM. Note that no bradypnea occurred, whereas bradyarrhythmias were observed during and following PnO SD. Insets show details of nasal airflow at time points 1–4, indicating no disruption of breathing.
Figure 5.
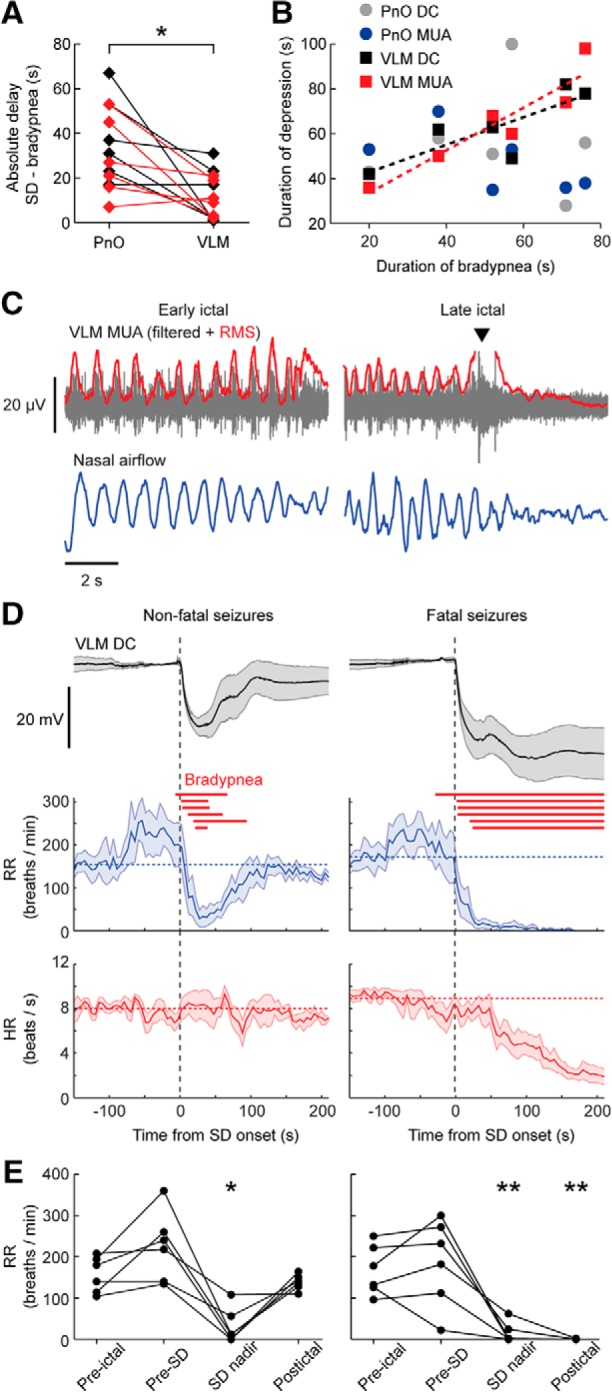
SD in the VLM is strongly correlated with seizure-related respiratory suppression in Cacna1aS218L mice. A, Absolute time delay between onset of bradypnea and SD in the PnO and VLM for nonfatal (red; n = 6) and fatal (black; n = 6) seizures, showing that VLM SD occurred significantly closer to onset of bradypnea (t(11) = 3.64, *p = 0.004, paired t test). B, Duration of brainstem DC and MUA depression plotted against duration of bradypnea. Significant correlation was observed only for VLM DC and MUA (dashed lines; R2 = 0.671, p = 0.046 and R2 = 0.873, p = 0.006, respectively, linear regression). C, Disruption of phase-coupled MUA in the ventrolateral medulla and breathing during a fatal seizure. In this example, VLM MUA was phase-coupled with nasal airflow. Local MUA (RMS, Root mean square) peaked during onset of VLM SD (arrowhead), followed by suppression which coincided with cessation of breathing activity. D, VLM DC-potential, respiratory rate (RR) and heart rate (HR) synchronized at onset of nonfatal (left) and fatal (right) VLM SD (at time = 0 s). Red lines indicate bradypnea duration for single cases. Horizontal dashed lines indicate RR and HR during a 60 s pre-ictal period. E, RR was significantly suppressed during SD nadir, but only for fatal seizures suppression continued into the post-ictal period (30 s after behavioral arrest; *p = 0.002, **p < 0.001, ANOVA with Dunnett's test).
IC stimulation induces brainstem seizures and SD invasion of the ventrolateral medulla coinciding with bradypnea onset
Seizure behavior and absence of cortical epileptiform activity in Cacna1aS218L mice suggest an important role for brainstem networks (Loonen et al., 2019). We therefore tested whether stimulation of the IC, a component of the brainstem seizure network (McCown et al., 1984; Faingold, 2012), could evoke similar seizure behavior but in a more predictable manner than seen with spontaneous brainstem seizures. Unilateral electrical IC stimulation (Fig. 6A) induced a short-lasting running fit (1–3 s) in wild-type mice (n = 3), whereas all Cacna1aS218L mice (n = 12) showed a Stage 5 seizure already during threshold testing. Thresholds for running fits and/or behavioral seizure activity were lower in Cacna1aS218L than wild-type mice [52 μA (range 30–100) vs 267 μA (range 100–500) respectively; p = 0.005, Mann–Whitney test]. In total, 27 seizures were induced in Cacna1aS218L mice, of which 20 were nonfatal and 7 fatal. Stimulation resulted in Stage 5 seizures that were of comparable duration as spontaneous seizures (133 ± 11, 162 ± 6, and 135 ± 3 s for nonfatal seizures without and with brainstem SD and fatal seizures, respectively), and never associated with cortical SD or epileptiform events.
Figure 6.
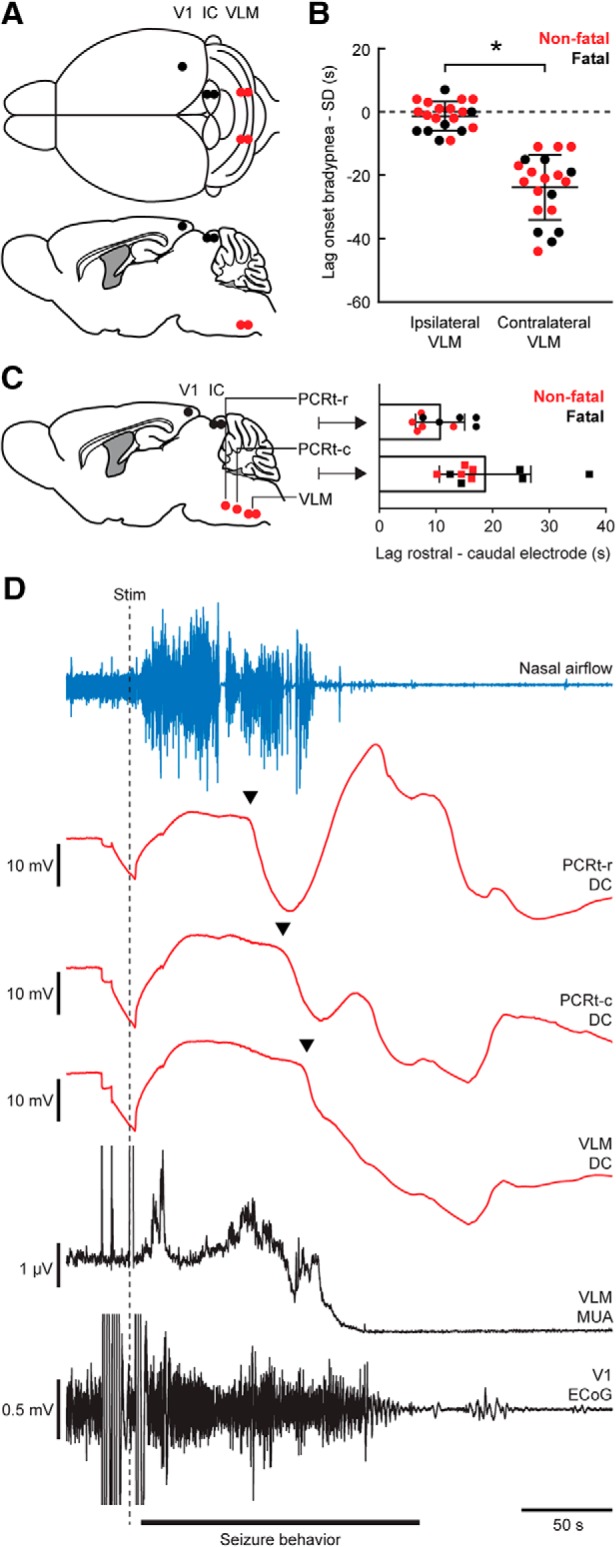
Stimulation of the IC induces early ipsilateral brainstem SD that propagates from rostral to caudal in Cacna1aS218L mice. A, Top and sagittal view of experimental approach for IC stimulation in freely behaving mice (V1, primary visual cortex). B, Time lag in onset of bradypnea (dotted line) and SD in VLM for nonfatal (red) and fatal (black) seizures. Negative values indicate bradypnea preceding SD. Ipsilateral VLM SD always preceded contralateral SD (t(19) = 9.08, *p < 0.0001, paired t test), regardless of laterality (n = 11 for left IC, n = 9 for right IC). C, Brainstem array recordings showed SD propagation from the PCRt to VLM for all seizures induced by IC stimulation. Time lag between SDs was shorter for the rostral (PCRt-r vs PCRt-c) compared with the caudal (PCRt-c vs VLM) electrodes (t(9) = 2.75, p = 0.023, paired t test). D, Example recording of a fatal seizure following IC stimulation (dashed line) indicating sequential SDs (arrowheads) in the rostral (top) to caudal (bottom) brainstem electrodes.
Bilateral VLM recordings in Cacna1aS218L mice revealed SD during all 7 fatal and 13/20 nonfatal seizures, with a delay of 106 ± 3 and 104 ± 7 s after stimulation, respectively (t(19) = 0.33, p = 0.763, unpaired t test). SD never occurred in wild-type mice. Notably, we observed VLM SD first at the site ipsilateral to stimulation, coinciding with bradypnea onset (Fig. 6B), although breathing slowed down already seconds preceding VLM SD. The consistent SD delay between left and right VLM following stimulation contrasts the inconsistent delay between PnO and VLM SD during spontaneous seizures, possibly reflecting lack of a local seizure focus for spontaneous events. Hence, we examined the possibility of spread in a separate group of Cacna1aS218L mice with electrodes in the VLM and the PCRt-r and PCRt-c (Fig. 6C). In all animals stimulated (n = 8; 10 seizures with brainstem SD, of which 5 were fatal), we observed SD first in PCRt-r, followed by PCRt-c and VLM (Fig. 6C,D). This indicates that SD spread following IC stimulation is predictable, but variable during spontaneous seizures, in Cacna1aS218L mice.
Medullary SD is followed by local hypoxia
Ongoing breathing activity does not rule out impaired oxygenation, which can induce brainstem SD (Funke et al., 2009; Richter et al., 2010). We therefore determined in situ brainstem pO2 immediately rostral to the VLM electrode (n = 6 animals; Fig. 7A,B). Following IC stimulation, 7/12 seizures were associated with SD in PCRt-r and VLM of which 4 were fatal. Baseline pO2 in the rostral VLM was 34.2 ± 3.1 mmHg and remained within the normoxic range of 20–40 mmHg during nonfatal seizures without brainstem SD (Fig. 7D). During seizures with brainstem SD, however, severe local hypoxia (pO2 < 10 mmHg) occurred that extended into the post-ictal period for both nonfatal and fatal seizures (pO2 nadir: 1.8 ± 0.9 mmHg and 0 ± 0 mmHg, respectively; Fig. 7E,F). Hypoxia lasted 34–74 s for nonfatal cases (n = 3) and was followed by a period of hyperoxia (Fig. 7G). No pO2 recovery occurred in fatal cases (Fig. 7H). SD in PCRt-r always preceded severe local hypoxia by 29 ± 3.1 s, whereas VLM SD preceded hypoxia in the majority of seizures (5/7; Fig. 7I) by 5.4 ± 3.2 s.
Figure 7.

Seizure-induced brainstem SD precedes local severe hypoxia in Cacna1aS218L mice. A, Top and sagittal view of experimental approach for parallel brainstem pO2 (red) and DC-recordings. B, Nissl staining showing the track of an oxygen-sensing probe close to the VLM. C, Example inter-ictal VLM pO2 recordings for three Cacna1aS218L mice, which remained within the normoxic range of 20–40 mmHg. D–F, Example recordings during termination of IC-induced seizures, including a nonfatal seizure not showing sudden changes in brainstem DC-potential (D) and a nonfatal (E) and fatal seizure (F) during which brainstem SD (arrowheads) occurred. Recordings of local VLM pO2 (bottom) indicate severe hypoxia (pO2 <10 mmHg; shaded red) during brainstem SD. G, Expanded timescale of pO2 measurements during the nonfatal seizure presented in E, showing rebound hyperoxia for minutes following initial severe hypoxia (red dotted line). H, Average pO2 during VLM SD (onset at time = 0 s; n = 3 nonfatal and n = 4 fatal seizures). Recovery from severe hypoxia (red dotted line) occurred only for nonfatal seizures. I, Time lag between onset of SD in PCRt-r and VLM and severe hypoxia. All PCRt-r SDs and the majority of VLM SDs preceded hypoxia.
To confirm that the early hypoxia observed in the brainstem was a local effect, we measured pO2 simultaneously in both V1 cortex and VLM in a subset of animals. Baseline cortical pO2 was similar to baseline pO2 levels observed in VLM (32.3 ± 2.5 mmHg). During fatal seizures (n = 5) severe hypoxia in VLM occurred in close association with VLM SD and preceded severe cortical hypoxia by 25–38 s (Fig. 8A). In a separate group of Cacna1aS218L mice (n = 4), cortical pO2 dynamics during electrically induced cortical SD (Fig. 8B) confirmed that the pO2 monitoring system was capable of detecting shifts in pO2 coinciding with SD, with severe hypoxia detected after 7–16 s. Furthermore, pO2 decreases where more abrupt during induced cortical SDs and SDs in the VLM associated with evoked fatal seizures compared with cortical recordings during fatal seizures not associated with cortical SD (Fig. 8C). Finally, cortical DC shifts did occur >60 s after a fatal seizure and long after severe hypoxia had commenced, indicative of anoxic depolarization (AD; Fig. 8D). These data indicate that the pO2 dynamics measured in the brainstem reflect pO2 changes induced by local SD, which are distinct from pO2 dynamics preceding AD.
Figure 8.

Oxygen dynamics in the ventrolateral medulla indicate that hypoxia is induced by brainstem SD in Cacna1aS218L mice. A, Example of a fatal seizure with parallel pO2 measurements in the VLM (blue) and primary visual cortex (V1; red). In this example, severe hypoxia (dashed line) in the VLM preceded cortical hypoxia by 25 s. B, Schematic (left) showing experimental approach for cortical stimulation in freely behaving mice. M1, Primary motor cortex; S1, primary somatosensory cortex. Cathodal stimulation in the primary visual cortex (V1 stim) resulted in a sequential DC shift in S1 and M1. SD in S1 coincided with a drop in pO2 (right; arrowheads indicate SD onset), followed by severe hypoxia (10 mmHg, dashed horizontal line) 15 s after SD onset in this example. C, Maximum pO2 slope measured in 5 s bins indicating a more abrupt decrease in pO2 for DC shifts during cortically induced SD (black; 6 SDs in n = 4 mice, 1–2 SDs per mouse) and VLM SD (blue) compared with cortical pO2 during fatal seizures (red; t(9) = 7.69 and t(8) = 7.88, respectively, *p < 0.0001, unpaired t tests). D, For cortical recordings, severe hypoxia occurred closer to DC shifts associated with cortically induced SD compared with DC shifts associated with anoxia following fatal seizures (t(9) = 14.4, *p < 0.0001, unpaired t test).
Fatality following brainstem SD can be prevented by timely respiratory resuscitation
In previous experiments, MUA recovery following VLM SD was observed after 26 ± 8 s (range 15–54) and 44 ± 5 s (range: 29–62) for spontaneous and induced nonfatal seizures, respectively. We therefore considered VLM SD without MUA recovery within 60 s fatal, and initiated mechanical ventilation >60 s after SD onset. This led to successful resuscitation in 7/9 animals (65–79 s for successful attempts; 70 and 76 s for unsuccessful attempts). During resuscitation, DC-potential recovery in the VLM was observed, that always preceded the first gasp (Fig. 9A). Bradypnea duration in the seven resuscitated animals was 132 ± 10 s (range: 104–180), significantly longer than in animals with spontaneous recovery [n = 12; 75 ± 5 s (range: 75–102); t(17) = 6.21, p < 0.0001, unpaired t test]. Breathing did not recover in animals receiving sham ventilation (n = 3; 61, 72 and 80 s after SD onset) or mechanical ventilation initiated >80 s after bradypnea onset (n = 3; 81, 91, and 102 s after SD onset).
Figure 9.
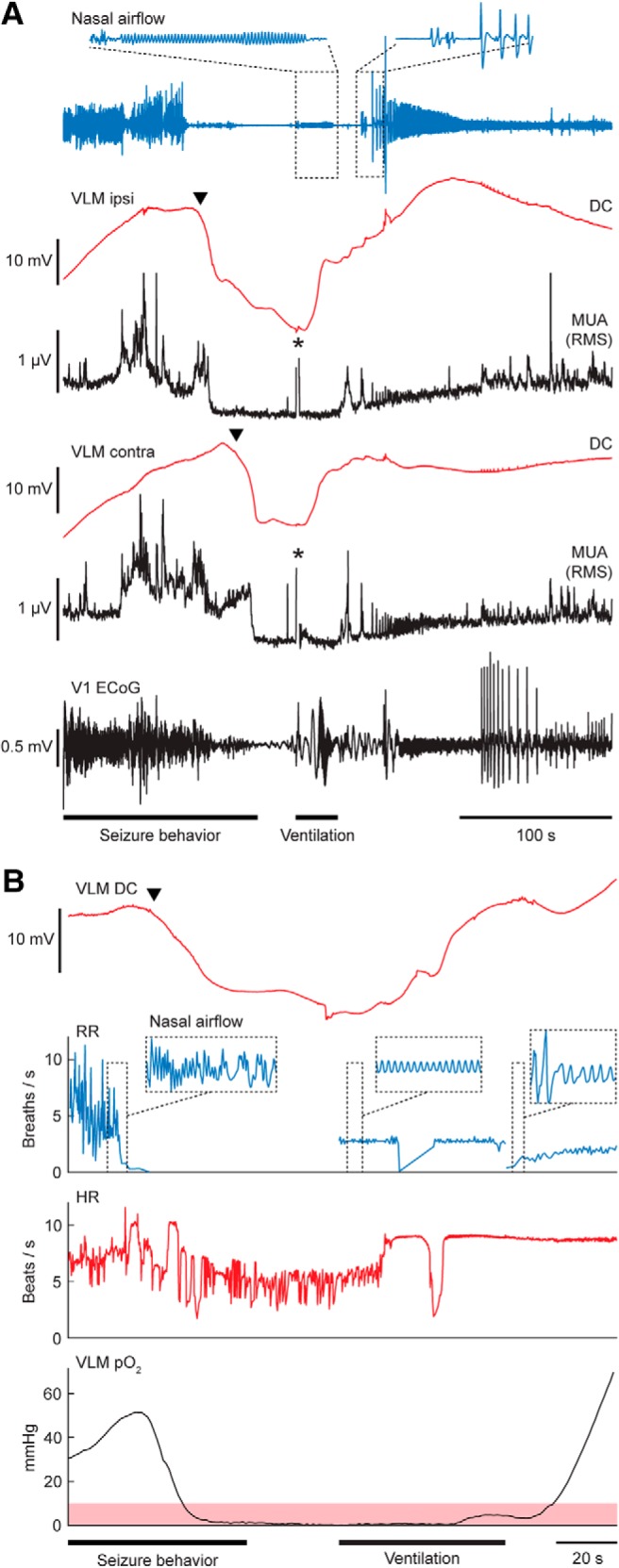
Respiratory resuscitation following SD in VLM prevents fatal outcome in Cacna1aS218L mice. A, Example of a successful resuscitation attempt after bilateral VLM SD (arrowheads) following IC stimulation. Mechanical ventilation was commenced 70 s after VLM SD onset in this case, in absence of spontaneous DC and MUA recovery. Effective delivery of air was confirmed by a regular oscillatory nasal airflow signal (inset, top left), and was always accompanied by early recovery of VLM DC-potential, followed by MUA recovery. Respiratory activity invariably resumed with gasping (inset, top right). Asterisks denote artifacts due to positioning of the animal for mechanical ventilation. RMS, Rroot mean square; V1, primary visual cortex). B, Example of heart rate (HR) and local pO2 changes during mechanical ventilation after VLM SD (arrowhead), showing that recovery of HR and VLM DC precedes recovery of local pO2. Note that effective ventilation was unintendedly interrupted in this case, as indicated by the transient drop in respiratory rate (RR). Severe hypoxia (pO2 <10 mmHg) is indicated in shaded red.
Finally, three mice implanted with a brainstem oxygen-sensing probe were mechanically ventilated at 62–65 s following VLM SD onset. In all animals, an increase in heart rate, recovery of VLM DC-potential and spontaneous breathing activity preceded recovery from severe hypoxia (Fig. 9B). In compliance with respiratory data, hypoxic burden, measured by the area <10 mmHg (Farrell et al., 2016), was greater in resuscitated animals than in animals with spontaneous recovery (752–1580 mmHg·s and 151–523 mmHg·s, respectively; both n = 3).
NMDA receptor antagonists prevent seizure-induced brainstem SD and fatality
In vitro and in vivo studies showed that blockade of NMDA receptors by MK-801 inhibits locally induced brainstem SD (Richter et al., 2008; Aiba and Noebels, 2015). We tested whether seizure-induced brainstem SD and respiratory compromise can be prevented by treatment with MK-801 (1 mg/kg) preceding IC stimulation. Regardless of treatment, all animals showed a Stage 5 seizure. However, MK-801 prevented brainstem SD, bradypnea, and fatal outcome (Fig. 10A,B). Upon recovery of seizures, however, mice demonstrated loss of postural control and hyperactive limb movements for up to 2 h following MK-801 injection. We therefore also tested the effects of memantine, a clinically well tolerated NMDA receptor antagonist (Bullock, 2006; Winblad et al., 2007). Memantine (10 mg/kg) prevented mortality and, in all but one mouse, brainstem SD and bradypnea (Fig. 10A,B), and did not result in overt peri-ictal behavioral changes. Absence of hindlimb clonus, that specifically occurred during termination of spontaneous and induced seizures displaying VLM SD, accounted for a shorter seizure duration for memantine-treated, but not MK-801-treated, mice (Fig. 10C).
Figure 10.
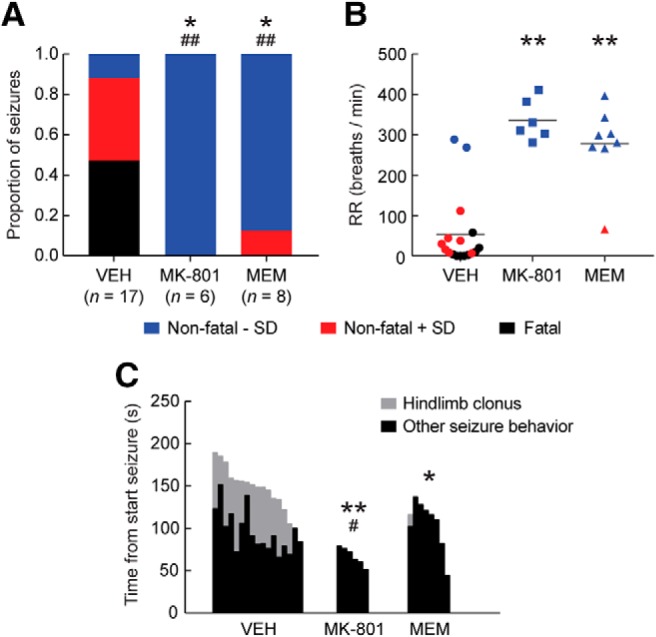
NMDA receptor antagonists prevent seizure-induced medullary SD, respiratory suppression and fatal outcome in Cacna1aS218L mice. Cacna1aS218L mice received acute intraperitoneal treatment with vehicle (VEH), MK-801 (1 mg/kg), or memantine (MEM; 10 mg/kg) 30 min before an IC stimulation-induced seizure. A, Proportion of seizures with fatal or nonfatal outcome, with or without SD in the VLM. Note that all fatal seizures were associated with VLM SD. Animals pretreated with MK-801 or MEM had seizures that were significantly less likely fatal (*p < 0.05, χ2 test) and less likely associated with VLM SD (##p < 0.001, χ2 test). B, Mean respiratory rate (RR) during the last minute of seizure behavior, indicating respiratory suppression during seizures with VLM SD (red and black). RR was significantly greater for animals pretreated with MK-801 or MEM (**p < 0.0001, ANOVA with Dunnett's test). C, Seizures were significantly shorter for animals pretreated with MK-801 or MEM (**p = 0.001 and *p = 0.013, respectively, ANOVA with Dunnett's test). When excluding hindlimb clonus (gray), MK-801 pretreatment resulted in shorter seizures, whereas MEM did not (#p = 0.045 and p = 0.755, respectively, ANOVA with Dunnett's test). Hindlimb clonus only occurred for seizures associated with VLM SD (VEH: n = 15; MEM: n = 1).
Discussion
Here we show that SD causes respiratory compromise and apnea upon invasion of the brainstem VLM during spontaneous and induced seizures in Cacna1aS218L mice. Incidence, onset, and recovery of SD in the VLM were strongly related to apnea, and SD was not caused by local hypoxia but instead initiated such hypoxia. We confirmed that putative fatal SD and subsequent hypoxia were reversible by resuscitation. Furthermore, prevention of SD initiation by NMDA receptor antagonists confirmed that SD is a prerequisite for seizure-related apnea in this animal model.
Cacna1aS218L mice have been proposed as a SUDEP model as homozygotes present spontaneous nonfatal and fatal tonic-clonic seizures at different ages (Loonen et al., 2019). Here we show that prolonged seizure-related apnea frequently occurs in this genetic mouse model. Data from rare monitored SUDEP cases indicate that apnea precedes cardiac arrest (Ryvlin et al., 2013). In patients with implanted electrodes aberrant activity in the amygdala, either caused by a seizure or electrical stimulation, was associated with apnea (Dlouhy et al., 2015). Although this revealed a potential mechanism of seizure-related apnea, breathing resumed before seizure termination, suggesting that other or additional mechanisms are involved in SUDEP as apnea in the post-ictal period appears dangerous (Ryvlin et al., 2013; Vilella et al., 2019).
In the present study, recording electrodes were located in or near the ventral respiratory column, at the level of the pre-BötC. Pre-BötC neurons are essential for breathing rhythm (Del Negro et al., 2018) and the respiratory response to hypoxia (Gray et al., 2001; McKay et al., 2005). Rapid suppression of glutamatergic pre-BötC neurons induces lethal apnea (Tan et al., 2008), whereas unilateral stimulation of inhibitory pre-BötC populations is sufficient to induce apnea during the stimulation period (Sherman et al., 2015). Our data indicate that neuronal suppression in the VLM following SD induces apnea, and subsequently hypoxia: (1) VLM SD occurred specifically during seizures with bradypnea; (2) bradypnea did not occur during seizures with SD limited to the rostral brainstem (PnO); (3) the timing of bradypnea correlated well with neuronal suppression in the VLM, but not with neuronal suppression in the PnO; and (4) brainstem hypoxia occurred in association with brainstem SD and preceded cortical hypoxia. Variations in onset of VLM SD relative to bradypnea may be explained by small deviations in electrode position (∼200 μm), because SD propagation is known to be especially slow in brainstem areas (Richter et al., 2008).
Our findings corroborate previous studies that reported brainstem SD in different SUDEP animal models (Aiba and Noebels, 2015; Aiba et al., 2016; Loonen et al., 2019). Based on DC-potential recordings in the dorsal medulla, it was proposed that brainstem SD occurred in the post-ictal period, secondary to ictal hypoxia as fatal apnea was already present upon detection of brainstem SD (Aiba and Noebels, 2015; Aiba et al., 2016). Whereas this provides a rationale for the absence of successful autoresuscitation efforts during fatal seizures in SUDEP mouse models (Kim et al., 2018; Loonen et al., 2019), it does not explain the onset of apnea. In the present study, brainstem SD preceded apnea, which may be explained by the different location of the brainstem recording electrode. Previously, this electrode was positioned in the dorsocaudal medulla that is primarily involved in cardiovascular function (Aiba and Noebels, 2015; Aiba et al., 2016), whereas we placed the electrode in the ventrolateral medulla that is primarily involved in respiratory function. As such, VLM SD may be the underlying mechanism for both the onset of apnea and the absence of autoresuscitation efforts.
Our data indicate that hypoxia is a consequence, rather than a cause, of seizure-related brainstem SD: (1) SD was detected rostral and immediately caudal from the brainstem oxygen probe at respectively ∼30 and ∼5 s preceding local hypoxia, whereas we found that AD occurred after >60 s following severe hypoxia, in line with a previous study showing AD in the hyperexcitable brainstem after ∼60 s (Richter et al., 2010); (2) pO2 decreases were more abrupt in brainstem than in cortex in the absence of cortical SD, but similar to pO2 decreases after cortical SD, which may be explained by increased local metabolic demand during SD (Takano et al., 2007); and (3) NMDA antagonists prevented seizure-induced brainstem SD, as shown for locally induced SD (Richter et al., 2008; Aiba and Noebels, 2015), but do not delay cortical AD (Lauritzen and Hansen, 1992). Together, this indicates that brainstem hyperexcitability rather than hypoxia initiates seizure-related brainstem SD.
Differences in seizure networks between SUDEP animal models may be crucial in determining the events leading to death. The seizure phenotype in Cacna1aS218L mice suggests an important role for brainstem networks (Loonen et al., 2019), confirmed here by the similar seizure phenotype following IC stimulation. Surgery reduced survival which may be because of an increased risk of SD following tissue trauma, although implanted animals show similar seizures as naive animals (Loonen et al., 2019). Seizures associated with cortical epileptiform activity were prolonged (lasting >30 min), whereas we observed forebrain SDs only during half of the fatal seizures, supporting the premise that increased brainstem network excitability is necessary and sufficient for the (lethal) seizures observed in Cacna1aS218L mice. Although brainstem networks have not been extensively studied for seizures in other models, decreased thresholds for SD implying hyperexcitable brainstem networks were found in multiple SUDEP animal models (Aiba and Noebels, 2015; Aiba et al., 2016). Endogenous brainstem activity was sufficient for hyperthermia- and kainate-induced convulsive seizures in infant rats after pre-collicular transection (Pospelov et al., 2016). Furthermore, in a mouse model of acquired temporal lobe epilepsy, increased excitability was found in the nucleus tractus solitarius (Derera et al., 2017). These preclinical studies implicate brainstem hyperexcitability in SUDEP pathophysiology. However, to generalize our findings, studies in other models are needed that investigate VLM SD in relation to respiratory suppression and hypoxia.
Longer VLM SD duration for fatal seizures was associated with prolonged suppression of local neuronal activity and breathing. Resuscitation resulted in sequential recovery of heart rate, VLM DC-potential, VLM neuronal activity, and breathing activity. Interestingly, VLM DC-potential always recovered before VLM pO2, suggesting that perfusion pressure rather than tissue oxygenation is crucial for initial recovery of brainstem SD, as reported previously for cortical SD (Sukhotinsky et al., 2010). Resuscitation was initiated in a time window that indicated fatal outcome. Earlier studies initiated resuscitation after a few seconds of apnea (Faingold et al., 2010; Kim et al., 2018) or directly after seizure induction (Buchanan et al., 2014). Our data extend these findings and suggest that ventilatory support is sufficient to recover brainstem control of respiratory function during prolonged apnea.
Only nonfatal seizures with VLM SD were associated with apnea. We are unaware of descriptions of prolonged apnea during and following nonfatal seizures in other animal models, although in clinical epilepsy hypoxemia is common (Bruno et al., 2018). Other than respiratory effects, nonfatal seizures with brainstem SD could be discriminated from other nonfatal seizures by post-ictal ECoG suppression and increased HRV. Because nonfatal seizures with brainstem SD were associated with a shorter time toward a fatal seizure, these post-ictal dynamics may be of clinical interest. Increases in pre-ictal and inter-ictal HRV were found in a patient 1 d before a cluster of seizures leading to SUDEP (Jeppesen et al., 2014). These and other potential biomarkers, including postconvulsive central apnea (Vilella et al., 2019) and imaging data implicating atrophy in the mesencephalon and medulla oblongata (Mueller et al., 2014, 2018), may improve SUDEP risk assessment.
Our data showed that hindlimb clonus was specific for seizures with VLM SD and displayed strong overlap in timing with SD. This suggests that hindlimb clonus can be the result of SD-induced motor system activation, for example via reticulospinal pathways (Hirsch et al., 2006). In SUDEP cases, apnea and asystole appear to occur post-ictally (Ryvlin et al., 2013). The obvious difference in brainstem dimensions in humans versus mice and the slow propagation of SD complicates translation of the observed motor effects to patients. SD-induced behavior and cardiorespiratory collapse may thus have different temporal sequences across species.
Treatment with NMDA receptor antagonists MK-801 or memantine did not affect seizure stage, yet total seizure duration was significantly decreased. Although this decrease was largely attributed to the absence of SD-induced hindlimb clonus, duration per se may be a poor predictor of seizure outcome. Indeed, we found no differences in duration of spontaneous seizures with and without brainstem SD in Cacna1aS218L mice. Clinical data in fact indicate the contrary, because post-ictal central apnea is more common for shorter seizures (Vilella et al., 2019). Activation of NMDA receptors by glutamate following postsynaptic depolarization is importantly implicated in SD (Dreier, 2011). Treatment with MK-801 or memantine largely abolished VLM SD and the associated apnea, thereby preventing fatal outcome. These findings suggest the potential of an NMDA receptor antagonist as preventative treatment option in individuals with a high SUDEP risk. Importantly, clinical evidence for seizure-related brainstem SD is crucial before considering such an approach.
Footnotes
This work was supported by the Dutch National Epilepsy Foundation (2017-10; R.D.T., A.M.J.M.v.d.M., E.A.T.), EU IAPP Program “BRAINPATH” (612360; A.M.J.M.v.d.M., E.A.T.), EU Marie Curie Career Integration Grant (294233; E.A.T.), and a CURE SUDEP research award (280560; R.D.T., A.M.J.M.v.d.M., E.A.T.). We thank L. Broos, P. de Brouwer and D. van Abswoude for assistance with histology and data analysis.
The authors declare no competing financial interests.
References
- Aiba I, Noebels JL (2015) Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med 7:282ra46. 10.1126/scitranslmed.aaa4050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiba I, Wehrens XH, Noebels JL (2016) Leaky RyR2 channels unleash a brainstem spreading depolarization mechanism of sudden cardiac death. Proc Natl Acad Sci U S A 113:E4895–4903. 10.1073/pnas.1605216113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno E, Maira G, Biondi A, Richardson MP; RADAR-CNS Consortium (2018) Ictal hypoxemia: a systematic review and meta-analysis. Seizure 63:7–13. 10.1016/j.seizure.2018.10.011 [DOI] [PubMed] [Google Scholar]
- Buchanan GF, Murray NM, Hajek MA, Richerson GB (2014) Serotonin neurones have anti-convulsant effects and reduce seizure-induced mortality. J Physiol 592:4395–4410. 10.1113/jphysiol.2014.277574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock R. (2006) Efficacy and safety of memantine in moderate-to-severe Alzheimer disease: the evidence to date. Alzheimer Dis Assoc Disord 20:23–29. 10.1097/01.wad.0000201847.29836.a5 [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Funk GD, Feldman JL (2018) Breathing matters. Nat Rev Neurosci 19:351–367. 10.1038/s41583-018-0003-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derera ID, Delisle BP, Smith BN (2017) Functional neuroplasticity in the nucleus tractus solitarius and increased risk of sudden death in mice with acquired temporal lobe epilepsy. eNeuro 4:ENEURO.0319–17.2017. 10.1523/ENEURO.0319-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devinsky O, Hesdorffer DC, Thurman DJ, Lhatoo S, Richerson G (2016) Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 15:1075–1088. 10.1016/S1474-4422(16)30158-2 [DOI] [PubMed] [Google Scholar]
- Dlouhy BJ, Gehlbach BK, Kreple CJ, Kawasaki H, Oya H, Buzza C, Granner MA, Welsh MJ, Howard MA, Wemmie JA, Richerson GB (2015) Breathing inhibited when seizures spread to the amygdala and upon amygdala stimulation. J Neurosci 35:10281–10289. 10.1523/JNEUROSCI.0888-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier JP. (2011) The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17:439–447. 10.1038/nm.2333 [DOI] [PubMed] [Google Scholar]
- Faingold CL. (2012) Brainstem Networks: reticulo-cortical synchronization in generalized convulsive seizures. In: Jasper's basic mechanisms of the epilepsies, Ed 4 (Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, eds). New York: Oxford UP. [PubMed] [Google Scholar]
- Faingold CL, Randall M, Tupal S (2010) DBA/1 mice exhibit chronic susceptibility to audiogenic seizures followed by sudden death associated with respiratory arrest. Epilepsy Behav 17:436–440. 10.1016/j.yebeh.2010.02.007 [DOI] [PubMed] [Google Scholar]
- Farrell JS, Gaxiola-Valdez I, Wolff MD, David LS, Dika HI, Geeraert BL, Wang XR, Singh S, Spanswick SC, Dunn JF, Antle MC, Federico P, Teskey GC (2016) Postictal behavioural impairments are due to a severe prolonged hypoperfusion/hypoxia event that is COX-2 dependent. eLife 5:e19352. 10.7554/eLife.19352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman JL, Del Negro CA (2006) Looking for inspiration: new perspectives on respiratory rhythm. Nat Rev Neurosci 7:232–242. 10.1038/nrn1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funke F, Kron M, Dutschmann M, Müller M (2009) Infant brain stem is prone to the generation of spreading depression during severe hypoxia. J Neurophysiol 101:2395–2410. 10.1152/jn.91260.2008 [DOI] [PubMed] [Google Scholar]
- Gray PA, Janczewski WA, Mellen N, McCrimmon DR, Feldman JL (2001) Normal breathing requires preBotzinger complex neurokinin-1 receptor-expressing neurons. Nat Neurosci 4:927–930. 10.1038/nn0901-927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths JR, Robinson SP (1999) The OxyLite: a fibre-optic oxygen sensor. Br J Radiol 72:627–630. 10.1259/bjr.72.859.10624317 [DOI] [PubMed] [Google Scholar]
- Harden C, Tomson T, Gloss D, Buchhalter J, Cross JH, Donner E, French JA, Gil-Nagel A, Hesdorffer DC, Smithson WH, Spitz MC, Walczak TS, Sander JW, Ryvlin P (2017) Practice guideline summary: sudden unexpected death in epilepsy incidence rates and risk factors: report of the guideline development, dissemination, and implementation subcommittee of the am academy of neurology and the am epilepsy society. Neurology 88:1674–1680. 10.1212/WNL.0000000000003685 [DOI] [PubMed] [Google Scholar]
- Hirsch E, Andermann F, Chauvel P, Engel J, da Silva FL, Luders H (2006) Generalized seizures: from clinical phenomenology to underlying systems and networks. Montrouge: John Libbey Eurotext. [Google Scholar]
- Houben T, Loonen IC, Baca SM, Schenke M, Meijer JH, Ferrari MD, Terwindt GM, Voskuyl RA, Charles A, van den Maagdenberg AM, Tolner EA (2017) Optogenetic induction of cortical spreading depression in anesthetized and freely behaving mice. J Cereb Blood Flow Metab 37:1641–1655. 10.1177/0271678X16645113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeppesen J, Fuglsang-Frederiksen A, Brugada R, Pedersen B, Rubboli G, Johansen P, Beniczky S (2014) Heart rate variability analysis indicates preictal parasympathetic overdrive preceding seizure-induced cardiac dysrhythmias leading to sudden unexpected death in a patient with epilepsy. Epilepsia 55:e67–71. 10.1111/epi.12614 [DOI] [PubMed] [Google Scholar]
- Kim Y, Bravo E, Thirnbeck CK, Smith-Mellecker LA, Kim SH, Gehlbach BK, Laux LC, Zhou X, Nordli DR Jr, Richerson GB (2018) Severe peri-ictal respiratory dysfunction is common in Dravet syndrome. J Clin Invest 128:1141–1153. 10.1172/JCI94999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritzen M, Hansen AJ (1992) The effect of glutamate receptor blockade on anoxic depolarization and cortical spreading depression. J Cereb Blood Flow Metab 12:223–229. 10.1038/jcbfm.1992.32 [DOI] [PubMed] [Google Scholar]
- Loonen ICM, Jansen NA, Cain SM, Schenke M, Voskuyl RA, Yung AC, Bohnet B, Kozlowski P, Thijs RD, Ferrari MD, Snutch TP, van den Maagdenberg AMJM, Tolner EA (2019) Brainstem spreading depolarization and cortical dynamics during fatal seizures in Cacna1a S218L mice. Brain 142:412–425. 10.1093/brain/awy325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massey CA, Sowers LP, Dlouhy BJ, Richerson GB (2014) Mechanisms of sudden unexpected death in epilepsy: the pathway to prevention. Nat Rev Neurol 10:271–282. 10.1038/nrneurol.2014.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAfee SS, Ogg MC, Ross JM, Liu Y, Fletcher ML, Heck DH (2016) Minimally invasive highly precise monitoring of respiratory rhythm in the mouse using an epithelial temperature probe. J Neurosci Methods 263:89–94. 10.1016/j.jneumeth.2016.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCown TJ, Greenwood RS, Frye GD, Breese GR (1984) Electrically elicited seizures from the inferior colliculus: a potential site for the genesis of epilepsy? Exp Neurol 86:527–542. 10.1016/0014-4886(84)90087-6 [DOI] [PubMed] [Google Scholar]
- McKay LC, Janczewski WA, Feldman JL (2005) Sleep-disordered breathing after targeted ablation of preBotzinger complex neurons. Nat Neurosci 8:1142–1144. 10.1038/nn1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SG, Bateman LM, Laxer KD (2014) Evidence for brainstem network disruption in temporal lobe epilepsy and sudden unexplained death in epilepsy. Neuroimage Clin 5:208–216. 10.1016/j.nicl.2014.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SG, Nei M, Bateman LM, Knowlton R, Laxer KD, Friedman D, Devinsky O, Goldman AM (2018) Brainstem network disruption: a pathway to sudden unexplained death in epilepsy? Hum Brain Mapp 39:4820–4830. 10.1002/hbm.24325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashef L, Walker F, Allen P, Sander JW, Shorvon SD, Fish DR (1996) Apnoea and bradycardia during epileptic seizures: relation to sudden death in epilepsy. J Neurol Neurosurg Psychiatry 60:297–300. 10.1136/jnnp.60.3.297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nashef L, So EL, Ryvlin P, Tomson T (2012) Unifying the definitions of sudden unexpected death in epilepsy. Epilepsia 53:227–233. 10.1111/j.1528-1167.2011.03358.x [DOI] [PubMed] [Google Scholar]
- Patodia S, Somani A, O'Hare M, Venkateswaran R, Liu J, Michalak Z, Ellis M, Scheffer IE, Diehl B, Sisodiya SM, Thom M (2018) The ventrolateral medulla and medullary raphe in sudden unexpected death in epilepsy. Brain 141:1719–1733. 10.1093/brain/awy078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pospelov AS, Yukin AY, Blumberg MS, Puskarjov M, Kaila K (2016) Forebrain-independent generation of hyperthermic convulsions in infant rats. Epilepsia 57:e1–6. 10.1111/epi.13230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ. (1972) Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281–294. 10.1016/0013-4694(72)90177-0 [DOI] [PubMed] [Google Scholar]
- Richter F, Bauer R, Lehmenkühler A, Schaible HG (2008) Spreading depression in the brainstem of the adult rat: electrophysiological parameters and influences on regional brainstem blood flow. J Cereb Blood Flow Metab 28:984–994. 10.1038/sj.jcbfm.9600594 [DOI] [PubMed] [Google Scholar]
- Richter F, Bauer R, Lehmenkühler A, Schaible HG (2010) The relationship between sudden severe hypoxia and ischemia-associated spreading depolarization in adult rat brainstem in vivo. Exp Neurol 224:146–154. 10.1016/j.expneurol.2010.03.004 [DOI] [PubMed] [Google Scholar]
- Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A, Boon P, Crespel A, Dworetzky BA, Høgenhaven H, Lerche H, Maillard L, Malter MP, Marchal C, Murthy JM, Nitsche M, Pataraia E, Rabben T, Rheims S, Sadzot B, et al. (2013) Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): a retrospective study. Lancet Neurol 12:966–977. 10.1016/S1474-4422(13)70214-X [DOI] [PubMed] [Google Scholar]
- Sherman D, Worrell JW, Cui Y, Feldman JL (2015) Optogenetic perturbation of preBotzinger complex inhibitory neurons modulates respiratory pattern. Nat Neurosci 18:408–414. 10.1038/nn.3938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Abdala AP, Borgmann A, Rybak IA, Paton JF (2013) Brainstem respiratory networks: building blocks and microcircuits. Trends Neurosci 36:152–162. 10.1016/j.tins.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukhotinsky I, Yaseen MA, Sakadzić S, Ruvinskaya S, Sims JR, Boas DA, Moskowitz MA, Ayata C (2010) Perfusion pressure-dependent recovery of cortical spreading depression is independent of tissue oxygenation over a wide physiologic range. J Cereb Blood Flow Metab 30:1168–1177. 10.1038/jcbfm.2009.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Lovatt D, Hansen AJ, Kasischke KA, Nedergaard M (2007) Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci 10:754–762. 10.1038/nn1902 [DOI] [PubMed] [Google Scholar]
- Tan W, Janczewski WA, Yang P, Shao XM, Callaway EM, Feldman JL (2008) Silencing preBotzinger complex somatostatin-expressing neurons induces persistent apnea in awake rat. Nat Neurosci 11:538–540. 10.1038/nn.2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thireau J, Zhang BL, Poisson D, Babuty D (2008) Heart rate variability in mice: a theoretical and practical guide. Exp Physiol 93:83–94. 10.1113/expphysiol.2007.040733 [DOI] [PubMed] [Google Scholar]
- Trinka E, Cock H, Hesdorffer D, Rossetti AO, Scheffer IE, Shinnar S, Shorvon S, Lowenstein DH (2015) A definition and classification of status epilepticus: report of the ILAE task force on classification of status epilepticus. Epilepsia 56:1515–1523. 10.1111/epi.13121 [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, Barrett CF, Gherardini L, van de Ven RC, Todorov B, Broos LA, Tottene A, Gao Z, Fodor M, De Zeeuw CI, Frants RR, Plesnila N, Plomp JJ, Pietrobon D, Ferrari MD (2010) High cortical spreading depression susceptibility and migraine-associated symptoms in Cav2.1 S218L mice. Ann Neurol 67:85–98. 10.1002/ana.21815 [DOI] [PubMed] [Google Scholar]
- Vilella L, Lacuey N, Hampson JP, Rani MRS, Sainju RK, Friedman D, Nei M, Strohl K, Scott C, Gehlbach BK, Zonjy B, Hupp NJ, Zaremba A, Shafiabadi N, Zhao X, Reick-Mitrisin V, Schuele S, Ogren J, Harper RM, Diehl B, et al. (2019) Postconvulsive central apnea as a biomarker for sudden unexpected death in epilepsy (SUDEP). Neurology 92:e171–e182. 10.1212/WNL.0000000000006785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winblad B, Jones RW, Wirth Y, Stöffler A, Möbius HJ (2007) Memantine in moderate to severe Alzheimer's disease: a meta-analysis of randomised clinical trials. Dement Geriatr Cogn Disord 24:20–27. 10.1159/000102568 [DOI] [PubMed] [Google Scholar]