

Abstract
Malignant gliomas are glial-derived, primary brain tumors that carry poor prognosis. Existing therapeutics are largely ineffective and dramatically affect quality of life. The standard of care details a taxing combination of surgical resection, radiation of the resection cavity, and temozolomide (TMZ) chemotherapy, with treatment extending life by only an average of months (Maher et al., 2001, Stupp et al., 2005). Despite scientific and technological advancement, surgery remains the most important treatment modality. Therapeutic obstacles include xenobiotic protection conveyed by the blood-brain barrier (Zhang et al., 2015), invasiveness and therapeutic resistance of tumor cell populations (Bao et al., 2006), and distinctive attributes of secondary glioma occurrence (Ohgaki and Kleihues, 2013). While these brain malignancies can be classified by grade or grouped by molecular subclass, each tumor presents itself as its own complication. Based on all of these obstacles, new therapeutic approaches are urgently needed. These will likely emerge from numerous exciting studies of glioma biology that are ongoing and reviewed here. These show unexpected roles for ion channels, amino-acid transporters, and connexin gap junctions in supporting the invasive growth of gliomas. These studies have identified a number of proteins that may be targeted for therapy in the future.
Keywords: glioma, classification, mutation, therapeutics, anti-invasive strategies
• What are gliomas?
Gliomas are highly aggressive, primary cancers arising from cells within the central nervous system (CNS). Tumor location and disease progression may manifest in symptoms varying from unexplained headaches, personality changes, visual field defects, motor deficiencies and even seizures. Gliomas display a heterogeneity in their anatomical location, with 40% presenting in the frontal lobe (Figure 1A) (Larjavaara et al., 2007). Gliomas are relatively rare, occurring in every 5 out of 100,000 adults per year (Ostrom et al., 2014). Out of the 20 of 100,000 people diagnosed with primary brain tumors per year, gliomas account for 27% of all brain tumors and 80% of all malignant brain tumors (Ostrom et al., 2015). Glioma occurrence is stratified throughout different stages of life, accounting for 47% of primary brain tumors in children ages 0–19 years but has a mean age of diagnosis at 62 years of age in adults (Ostrom et al., 2015, Arora et al., 2009). Incidence rates may also differ slightly based on subtype, tumor grade, and any inconsistencies in glioma definition during diagnosis. Unlike tumors to the periphery of the CNS that can be more easily biopsied and delineated, brain tumors have historically posed challenges in their classification and treatment.
Figure 1:
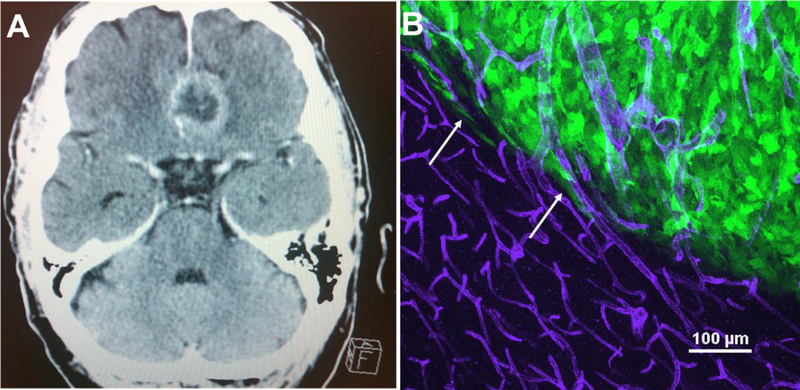
(A) A T2-weighted MRI of a GBM patient displays the tumor mass residing in the frontal lobe of the brain. This region is where most GBMs can be visualized through MRI. (B) Glioma cells migrate away from the main tumor core using white matter tracts and the vasculature of the brain. Mouse models of glioma such as human glioma D54- eGFP cells transplanted into SCID mice nicely recapitulate this biology observed in humans. As seen in this maximum intensity project of a GFP+ tumor that was intracranially implanted in an immunocompromised mouse, GFP+ tumor cells branch out of the tumor core (white arrows) and attach to CD31 + blood vessels (purple) outside of the tumor core. Of note is the difference in vessel morphology, as angiogenesis that has occurred in the tumor mass because of a hypoxic environment, results in larger, abnormal, and leaky vessels.
• Glioma classification and management
Pioneering neurosurgeons Percival Bailey and Harvey Cushing performed the first histological classification of brain tumors in 1926, whereby abnormal features were demarcated with the use of light microscopy (Bailey and Cushing, 1926). Since then, histological observation has persisted for brain tumor pathological diagnosis. Common pathological features of CNS tumors include nuclear atypia, increased proliferation, vascular growth, and the potential for pseudopalisading necrosis (Huse and Holland, 2010). The specific presence of certain pathological combinations also predicts disease progression and life expectancy. In vivo mouse models of glioma such as human glioma D54-eGFP cells transplanted into SCID mice recapitulate a multitude of these processes seen in human brain cancer, including vascular (Figure 1B) and white matter routes of invasion (Cuddapah et al., 2014).
While glioma diagnosis still relies on histology, the World Health Organization (WHO) further refined glioma classification. In 1979, the WHO established a scale for degrees of malignancy, whereby the WHO I-IV rating reflects anticipated tumor behavior, invasiveness, and predicted life expectancy (Huse and Holland, 2010). Glioblastoma (GBM) is the most malignant form (grade IV) of glioma, accounts for 55% of all gliomas, and has a dismal 5-year survival rate of <5% (Ostrom et al., 2014) (Ostrom et al., 2015).
The most recent 2016 CNS WHO update integrates the phenotypic and more recently identified genotypic classifications for a more objective diagnoses within the heterogeneous glioma patient population (Louis et al., 2016). While these integrations may prove to be efficient diagnostic criteria, genotype data could help refine diagnosis over pure histological observation based on one portion of the tumor.
The first line of treatment almost always involves surgical tumor resection. Tumor surgery was pioneered by Harvey Cushing, also known as the father of neurosurgery, who introduced antiseptic and hemostatic operative techniques that reduced infection and blood loss (Ballance, 1921, Mark C. Preul, 2005). Radiation therapy was added during the 1960– 1970’s and, based on a 1998 randomized study of 95 patients, it appears effective. Surgical resection combined with 50.4 Gy total postoperative radiotherapy allowed for fewer recurrences in patients than those that received surgery alone (Patchell et al., 1998). The combination of surgery and radiation extends patient survival from 6 months to 12.1 months post treatment (Wilson et al., 2014). All the while, the era of chemotherapy had commenced in the 1940’s when Louis Goodman and Alfred Gilman pioneered the use of nitrogen mustard to treat a patient with non-Hodgkin’s lymphoma (Chabner and Roberts, 2005). Clinical studies using nitrosourea chemotherapy for glioma varied with patient numbers, posing limits in the conclusions of treatment efficacy. Until the early 2000’s, the benefits of adjuvant chemotherapy postradiation treatment remained under debate (Fine et al., 1993, Wilson et al., 2014). A 2004 clinical trial of patients receiving concurrent temozolomide (TMZ) treatment with postoperative radiation had an increased, albeit still low median survival of 14.6 months versus the 12.1 month median survival for those receiving radiotherapy alone (Stupp et al., 2005). This so called “Stupp protocol” has since been deemed the standard of care in glioma therapy.
• What is the clinical challenge?
While surgery remains the most important therapeutic option for gliomas, complete surgical resection is essentially unattainable as brain tumors infiltrate the normal brain by migrating diffusely away from the main tumor core. Chemotherapeutics have shown limited success, partially due to lack of efficacy, specificity, and partial access to the brain. The current chemotherapy of choice, TMZ, is also the only approved medication in the standard of care regimen, with adequate permeability across the blood-brain barrier (BBB) unlike other therapies that are easily excluded by BBB transport proteins (Deeken and Löscher, 2007). While abnormal vascularization (Figure 1B) and BBB disruption at the primary tumor site exists, infiltrative satellite tumors are found away from the main tumor mass, posing a greater challenge for chemotherapy delivery (Juillerat-Jeanneret, 2008). Furthermore, these invasive cells express multi-drug resistant transporters such as ABCG2 and are more resistant to chemotherapy, the last effort in the standard of care regimen (Bao et al., 2006, Bleau et al., 2009, Natarajan et al., 2012).
• How can we do better?
To develop truly specific therapies, it is essential to gain an improved understanding of genetic changes that occur specifically in glioma compared to their non-malignant counterparts. Furthermore, it is crucial to understand how such changes affect function at cellular and molecular levels and how this alters the tumor’s interaction with the surrounding, normal brain. This approach has been termed a “neurocentric” approach before as its focus on the specific biology of the tumor is in the context of the brain rather than an “oncogenic” approach that focuses on well known genetic changes shared with most other cancers. Major strides between both approaches have been made and are discussed in more detail below.
Genetic classifications:
Since the 2007 CNS WHO update, a genetic basis of glioma has been defined by the The Cancer Genome Atlas (TCGA) Network study (McLendon R, 2008). An additional molecular stratification of GBM identified expression-based subtypes as Proneural, Neural, Classical, and Mesenchymal, also providing a framework for how responsive these different subtypes may be to the same standard of care provided to every glioma patient (Verhaak et al., 2010). These subtypes, named for their specific gene signatures, possess different mutation frequencies in cell-cycle regulation (TP53 and RB1), proliferation (PTEN, PIK3R1, PIK3CA, NF1, IDH1), and tyrosine kinase receptor activation (EGFR, ERBB2, EGFRvIII, and PDGFRA) genes that were identified in the 2008 TCGA study (Verhaak et al., 2010, McLendon R, 2008). Genetic dysregulation in the TP53, RB1, and PTEN signaling axes have a synergistic effect on high-grade glioma development versus when each pathway is individually disrupted (Chow et al.,2011). While mouse models have shown the cooperativity of these pathways in glioma progression, these tyrosine kinase and cell-cycle regulators are commonly mutated in other cancers, providing a core requirement for GBMs but a lack of personalized approach to studying the disease (Chalhoub and Baker, 2009, Sherr and McCormick, 2002).
Cell of Origin:
Important advancements have been made in understanding the cell in which cancer causing mutations occur, otherwise known as the the glioma cell of origin. As most differentiated brain cells remain in a non-proliferative state, the notion of cancer-prone, growth-competent brain cells seems incongruous. However, the discovery of neural stem cell niches in the subventricular and subgranular zones gives notion that proliferative brain cells could be the culprit for neoplastic transformation (Eriksson et al., 1998, Sanai et al., 2004). In fact, glioma stem cells isolated from primary tumors stay undifferentiated and akin to their original tumor mass in serum free, growth factor-containing media (Lee et al., 2006). Furthermore, as few as 100 CD133+ primary glioma stem cells are enough to initiate tumor development versus 106 CD133 − glioma cells when intracranially implanted into NOD-SCID mice (Singh et al., 2004). Neural stem cells also possess many behavioral characteristics and immature expression profiles intrinsic to gliomas (Sanai et al., 2005). In the Maturation-Arrest Theory, glial progenitor cells accumulate mutations that promote proliferation but not maturation into differentiated CNS cell types (Sanai et al., 2005). There is also an oligodendrocyte progenitor pool of NG2+cells within the CNS that has a greater capacity to proliferate versus astrocyte progenitors, suggesting a more tumorigenic niche for oligodendrocyte progenitors (Zerlin et al., 2004, Canoll and Goldman, 2008). However, when astrocytes and progenitor cells were given the appropriate set of driver mutations in genes such as those found in the 2008 TCGA study, tumors were found in and out of proliferative zones (Chow et al., 2011, McLendon R, 2008). Whether from a mutated neural stem cell or differentiated glia, it could be fathomed that cancers derived from these particular cells should also harbor antigens of the cell from which the tumor originates.
However, the cell of mutation may not be the cell of origin. Gliomas are considered to be derived from astrocytes, as the differentiated markers glial fibrillary associated protein (GFAP) and S100β are also present in glioma cells (Jones et al., 1981, Wang et al., 2013). Additionally, S100β has a proliferative effect on glioma cells and astrocytes but not neurons in vitro, suggesting a glial-specific, mitogenic effect of S100β (Selinfreund et al., 1991). Cells could potentially obtain a mutational signature and therefore be predestined to become oncogenic.
It is also possible that mutations may de-differentiate the cell of mutation to another cellular destiny. When infected with lentivirus over-expressing oncogenic Ras and shRNA against p53, GFAP-Cre mice developed gliomas whereby expression of the differentiated astrocyte marker GFAP decreased over time while conversely increasing expression of the progenitor marker nestin (Friedmann-Morvinski et al., 2012). Gliomas also release extracellular factors into their environment, having the potential to transform the proper cell of origin. Transplantation and in vitro studies with embryonic conditioned media also suggest a diffusible, inducing factor can de-differentiate astrocytes into radial glia (Maher et al., 2001, Hunter and Hatten, 1995). Furthermore, glia comprise most resident cells of the brain, and while differentiated, cells such as Ng2+ glia are still mitotic, it is possible that these cells could acquire mutations during cell division (Ge et al., 2009). Over all, the heterogeneity of gliomas indicates that both progenitors and differentiated glia may be at play, making the cell of origin hard to define (Sanai et al., 2005, Friedmann-Morvinski et al., 2012).
Interestingly, at present, none of this knowledge translates into patient treatment and all glioma patients receive the same standard of care. Nevertheless, genetic biomarkers that can predict prognosis or treatment response have been uncovered. For example, mutations in the Krebs cycle protein isocitrate dehydrogenase (IDH), have been identified as early biomarkers in gliomagenesis as well as a distinguishing factor between primary and secondary gliomas (Thon et al., 2013). The biomarker with the largest clinical fingerprint thus far has been the promoter for the DNA repair enzyme methylguanine methyltransferase (MGMT). MGMT is crucial for genome stability because it repairs naturally occurring DNA lesions from errors during DNA replication and transcription. In certain patients, the MGMT promoter is silenced via methylation and therefore TMZ treatment is more efficient because MGMT is inactivated. Studies have confirmed that the epigenetic inactivation of the MGMT promoter is a predictive value in response to TMZ in combination with radiation therapy (Stupp et al., 2007, Hegi et al., 2005).
While significant effort is being made to identify frequently mutated genes amidst the mutational heterogeneity of tumors, each glioma presents itself as a unique challenge. Furthermore, these distinct mutations could exist in part due to the specific environment of the individual from which the tumor developed. In fact, studies analyzing larger samples sizes of whole-exome sequencing data suspecting to distinguish true cancer-driver genes instead gathered a longer list of more implausible and false-positive “hits” (Lawrence et al., 2013). Understanding the array of mutations driving GBM provides a detailed microscopic view of this disease but overall, these malignant tumors share a very macroscopic, invasive phenotype.
• Potential for new therapeutic strategies: Anti-invasive strategies
The above studies indicate that our ability to genotype gliomas is advancing rapidly but development of novel treatments are at a standstill. This may be misleading because while no discovery has changed the standard of care yet, there are a number of compounds that have been studied in preclinical and small clinical trials.
Unlike other cancers of the body, brain tumors rarely cross into the bloodstream and metastasize outside of the CNS. However, brain tumor cells are notorious for forming satellite tumors as they invade throughout the tight extracellular CNS space. Ahead of his time in the 1930’s, pathologist Dr. Hans Scherer identified migratory brain tumor cells in the perivascular, perifasicular, and parenchymal spaces (Scherer, 1938). His insight into these structures suggested that rather than being defined based on tumor type, a glioma cell’s morphology was due to the space in which it was confined. Invasion throughout the extracellular space poses quite a feat for tumor cells, as these spaces are smaller than the average cell nucleus. A common mean for glioma cell travel within the CNS space is distributing matrix-degrading enzymes that break down the extracellular milieu surrounding basement membranes and their adjacent cells (Cuddapah et al., 2014). Outside of factors that gliomas release, recent studies have interrogated endogenous proteins important for glioma infiltration of the CNS that may prove useful as future drug targets.
• Anti-invasive strategies: Ion channels and volume changes
The endogenous balance of K+ and Cl− ions play a substantial role not only in regular physiology, but also in tumor invasion. An electrochemical K+ gradient exists between normal prokaryotic cells and the surrounding extracellular space, whereby K+ is favored to leave the cell, however, Cl− normally remains in an electrochemical equilibrium (Sontheimer, 2008). While it was hypothesized some time ago, studies have now affirmed preservation of these sizeable intracellular ionic gradients helps favor salt efflux and reduce intracellular water during tumor invasion. Furthermore, over-expression of aquaporin water channels AQP1 and AQP4 increases water permeability from glioma cells (McCoy and Sontheimer, 2007). During invasion, movement of K+ and Cl− through ion channels removes all unbound cytoplasmic water molecules, allowing tumor cells to decrease their volume by 30–35% and occupy tight extracellular spaces in vitro and in vivo (Watkins and Sontheimer, 2011, Sontheimer, 2008). Misappropriation of ion channels and transporters are important players for establishing the driving force behind this process.
Astrocytes express Kir4.1, a glial ion channel which normally buffers K+ release from neurons, but is mislocalized to the nucleus in glioma cells (Olsen and Sontheimer, 2004). While gliomas express a non-functional Kir4.1, over-expression of Kir4.1 in glioma cells causes growth arrest (Higashimori and Sontheimer, 2007). K+ efflux is maintained in glioma cells by the expression of Ca2+ activated big conductance (BK) K+ channels (Sontheimer, 2008). Glioma cells express a specific 34AA variant of these channels, deemed gBK, which is also highly expressed in glioma cells versus normal brain, making it a promising therapeutic target (Liu et al., 2002). gBK have a higher sensitivity to changes in intracellular Ca2+ than the regular BK channel, highlighting the important role of Ca2+ as an effector in glioma biology (Ransom et al., 2002). Indeed, Bradykinin is believed to be orchestrating Ca2+ increase, transiently activating gBK currents and this activation is diminished in the presence of the Ca2+ chelator BAPTA-AM (Ransom and Sontheimer, 2001). Directed movement of glioma cells to the perivascular space is facilitated by the presence of gBK channels and directed chemotactically by bradykinin signaling. Bradykinin, while central for Ca2+ levels, is also important in the vasculature, a favored route for invasive tumor cells. Glioma cells express bradykinin receptors, mediating intracellular Ca2+ oscillations, and enhanced glioma migration (Montana and Sontheimer, 2011). Bradykinin acts as a chemoattractant during invasion, so that genetic and chemical blockade of bradykinin deters glioma cells from migrating to blood vessels (Montana and Sontheimer, 2011).
Unusually high intracellular Cl− concentrations are maintained in glioma due to pronounced expression of the NKCC1 Cl− transporter (Ernest et al., 2005). Voltage-gated Cl− channels, CIC-2 and CIC-3, are also highly expressed in gliomas and upon blockage, reduce the inward currents in these cells (Olsen et al., 2003). These Cl− channels are important pathways that can efflux Cl− from invading gliomas. Furthermore, ClC-3 co-localizes with and is regulated via Ca2+/calmodulin-dependent protein kinase II (CaMKII) (Cuddapah and Sontheimer, 2010). Infusion of CaMKII via patch pipette, robustly enhanced the ClC-3 Cl− current in glioma cells and this conductance was reversed with addition of the CaMKII inhibitor AIP (Cuddapah and Sontheimer, 2010). Targeting these ClC channels has shown great promise in human patients. Chlorotoxin (Cltx), a Cl− channel-specific peptide purified from scorpion venom, binds within acute biopsies from GBM patients (Sontheimer, 2008). Cltx impedes migration in slice invasion assays through a complex formed between MMP-2 and ClC-3, endocytosing the ClC-3 channel (Soroceanu et al., 1999, McFerrin and Sontheimer, 2006). TM-601, a synthetic version of Cltx, passed Phase I/II clinical trials for the use in glioma treatment, highlighting the promise of targeting ion channels in tumor invasion.
• Anti-invasive strategies: System XC and Glutamate (Glu) Release in Glioma
It is not uncommon for glioma patients to present with seizures before realizing the deeper truth of their diagnosis. In fact, around 80% of glioma patients present with at least one seizure and 50% develop reoccurring tumor-associated epilepsy. This kind of abnormal brain activity is normally maintained due to an excitation-inhibition in the CNS. Astrocytes sheathing synapses express glutamate transporters, so that they can maintain proper glutamate equilibrium. A disturbance in this balance is common in gliomas and partially due to an over 100-fold higher concentration of the excitatory neurotransmitter Glu (Marcus et al., 2010). One player that may be responsible for these high Glu concentrations within the CNS is a sodium-independent, amino acid antiporter, System XC (SXC).
SXC transports L-cystine into the cell at equimolar exchange for the extracellular release of Glu (Figure 3A). Structurally, SXC exists as a dimer in the plasma membrane, composed of the regulatory CD98 subunit and catalytic xCT subunit, which is encoded by the SLC7A11 gene (Sato et al., 1999). After L-cystine is transported into the cell it becomes reduced to cysteine the precursor for the production of the antioxidant species, glutathione (GSH) (Bridges et al., 2012). The primary role of SXC is to provide cellular antioxidant defense against free radicals, such as those species generated in radiation therapy performed in glioma treatment. Furthermore, the glioma microenvironment is hypoxic and requires the sprouting of new blood vessels to deliver the appropriate nutrients for cellular homeostasis. After chronic decrease in oxygen supply in vitro, GSH was utilized at a higher rate and was accompanied by an increase in L-cystine uptake, demonstrating a need for SXC-mediated GSH production under hypoxic conditions present in the tumor environment (Ogunrinu and Sontheimer, 2010).
Figure 3:
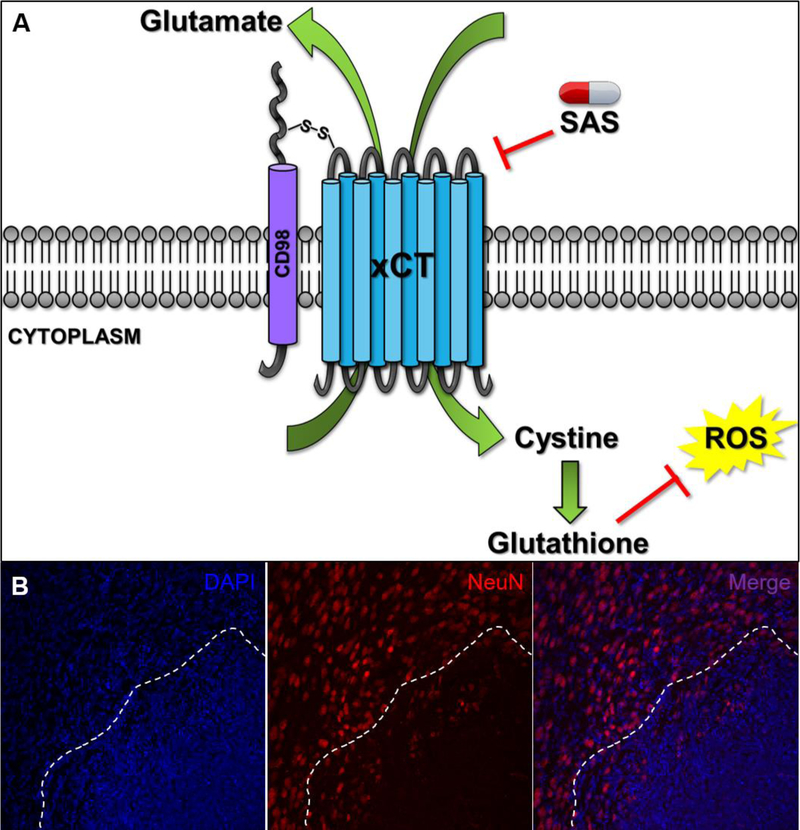
(A) As depicted in this representation, the System XC (SXC) transporter is composed of the 12 transmembrane, xCT catalytic subunit and CD98 regulatory subunit. xCT is responsible for the equimolar exchange of cystine into the cell and glutamate release into the extracellular space. Import of cystine conveys the production of glutathione, an important intracellular antioxidant that helps fight off any reactive oxygen species that may be produced endogenously or by radiation and chemotherapy treatment. Glutamate release out of the glioma cell can cause an excitotoxic build-up to surrounding neurons. Sulfasalazine (SAS) is an FDA-approved inhibitor of SXC and has been used in a small, clinical pilot trial for GBM patients (B) GBM22, a glioma that highly expresses SXC, was implanted into an immunocompromised mouse and allowed to engraft for two weeks. After performing immunohistochemistry for the neuronal marker NeuN, neuronal death can be visualized at the tumor core (as denoted by the nuclear marker DAPI) and beginning surrounding the peritumoral border (dotted line).
While SXC has been implicated in various CNS biology such as neurotransmitter release and oxidative protection, previous studies have demonstrated the role of SXC in glioma malignancy in both continual cells and patient-derived xenolines (Chung et al., 2005, Robert and Sontheimer, 2014). Gliomas express high levels of SXC despite the loss of sodium-dependent Glu transporters, such as EAAT2, resulting in high extracellular concentrations of Glu relative to normal astrocytes (Chung et al., 2005, Ye and Sontheimer, 1999). SXC-mediated Glu release is consequently responsible for peritumoral neuronal death (Figure 3B) and glutamatergic, epileptiform hyperexcitability (Buckingham et al., 2011). Glu is also an important autocrine/paracrine molecule in glioma invasion. Gliomas release enough Glu to activate Ca2+−permeable AMPA-R, causing Ca2+ oscillations in the cell, and subsequent cell migration (Lyons et al., 2007). Furthermore, differences in tumor biology persist based on xCT status and help further stratify patient-derived glioma xenolines (Robert et al., 2015). Gliomas with higher xCT expression present with greater Glu-mediated excitotoxicity, higher seizure frequency, and lower survival rates for animal models and human patients (Robert et al., 2015). Recent studies have also demonstrated physical relationships between SXC and cancer-driving pathways such as EGFR, CD44, and p53 (Tsuchihashi et al., 2016, Ishimoto et al., 2011, Liu et al., 2017).
SXC can be chemically targeted with the FDA-approved drug, sulfasalazine (SAS), which is already used to treat Crohn’s disease. Initial docking studies revealed the postulated SAS binding site on SXC, extending beyond that of Glu (Sontheimer and Bridges, 2012). The promise of SAS in vivo has been validated in animal models demonstrating reduced tumor burden after SAS treatment (Chung et al., 2005, Lyons et al., 2007). In vitro patch-clamp recordings in brain slices bearing gliomas also confirmed Glu-mediated peritumoral hyperexcitability and a reduction upon bath application of SAS (Buckingham et al., 2011). A small, pilot clinical trial also showed a benefit after oral SAS treatment whereby patients saw a decrease in tumor SXC-mediated Glu release as measured by magnetic resonance spectroscopy (Robert et al., 2015). Therefore, SXC expression segregates gliomas into distinct biological phenotypes, understanding SXC function and regulation in glioma could be beneficial for disease therapy, and targeting SXC chemically has proven hopeful for the future of tumor and tumor-associated seizure management.
• Anti-invasive strategies: Network formation
Cell-cell network formation is crucial for glioma cells during invasion. Connexin 43 (Cx43), a structural component of gap junction proteins that form intercellular pores between apposing cell membranes, is widely expressed in astrocytes in the healthy CNS (Rozental et al., 2000). In addition to cellular communication, Cx43 has roles associated with Glu release and Ca2+ propagation, two important effector molecules in glioma as previously discussed (Ye et al., 2003, Toyofuku et al., 1998). When astrocytic tumors expressing Cx43 were implanted into mice, Cx43-expressing glioma cells dispersed over a greater brain volume versus Cx43-mutant gliomas (Lin et al., 2002). While Cx43 expression has been identified as inversely related with glioma grade, this could be due to that the most invasive tumor cells that are not easily resected are not actually surveyed in these expression studies (Soroceanu et al., 2001, Huang et al., 1999). Furthermore, antibody specificity and the isoform of Cx43 analyzed may matter in glioma biology, in that non-phosphorylated Cx43 has been shown in high grade versus low-grade gliomas (Aronica et al., 2001). Phosphorylation by particular non-receptor protein kinases correlates with a disruption in intercellular junctional communication, suggesting non-phosphorylated Cx43 could be advantageous in glioma invasion (Lampe and Lau, 2004). Furthermore, EGFR is activated in glioma and the downstream MAPK signaling pathway mediates S278/282 phosphorylation of Cx43, disrupting its function (Lampe and Lau, 2004, McLendon R, 2008).
A capacity for glioma invasion has recently been explained by a “syncytium” of tumor cells that are connected via microtubules filled with Cx43 pores (Osswald et al., 2015). This “neighborhood” of glioma cells were also highly resistant to radiation therapy, could stabilize Ca2+ distribution, and protected one another after toxic laser exposure (Figure 4) (Osswald et al., 2015). Such a community of glioma cells has the capability to maintain an invasive network, whereby molecules can be dispersed and sacrifices can be made so cells within the network can still survive. Recent studies have shown promising results with Cx43 as a potential drug target in glioma therapy. Cx43 levels in glioma seem to be inversely related to TMZ resistance and upon addition of a Cx43 C-terminal mimetic peptide, MGMT-deficient, TMZ-resistant glioma cells became sensitized to TMZ treatment (Sheng et al., 2015). This study provides evidence that Cx43 may be used as a biomarker for gliomas that are resistant to TMZ treatment and that upon Cx43 inhibition, glioma cells may respond better to the currently available standard of care regimen. However, caution should be made when designing drug target inhibitors against Cx43, as Cx43 is highly important in heart physiology and expressed elsewhere in the body. GAP-43, a protein important for neural growth cone formation during neuron migration, was identified as another important component of the microtubule glioma syncytium that was resistant to radiation (Osswald et al., 2015, Denny, 2006). Because this was studied in the context of adult glioma and its involvement in development, GAP-43 may therefore be an additional, promising target in anti-invasive strategies.
Figure 4:

(With permission from Sontheimer, Nature, 2015) Osswald et al., 2015 identified a “syncytium” of glioma cells that are resistant to radiation therapy. The cellular mechanism behind these networks was due in part to Cx43 pores. Cx43 is a protein important in the formation of gap junction channels between cell membranes and is normally expressed in astrocytes. However, as seen in these cell-cell contacts, Cx43 is harnessed to form microtubule highways to help spread effector molecules like Ca2+ and even provide a damaged cell with its own nucleus.
• Conclusions
Glioma is a devastating CNS disease that claims the lives of a majority of those afflicted. Surgical removal is still one of the most pertinent options for disease management, yet the average survival with even aggressive care is typically less than 2 years. One of the major challenges is the resiliency of satellite tumor cells migrating from the tumor core. These “stem-like” tumor cells tend to escape the defined area of post-resection radiation and are more resistant to chemotherapy. In an era where technology and knowledge is at the forefront, we must do a better job at treating this disease. Both oncogenic and neurogenic approaches identifying disease-driving mutations that are distinct from non-pathological cells of the CNS have exciting translational potential. However, the more we learn about gliomas as either a cancer or as a CNS pathology, we must not forget that each tumor presents as its own obstacle.
Therefore, targets against tumor invasiveness prove promising for the future of glioma treatment. Gliomas share an invasive phenotype that has been more recently dissected in the history of glioma biology. Ion channels expressed on glioma cells play an important role in maintaining an ideal gradient for ion and water flow during invasion.
Movement of K+ and Cl− down these established concentration gradients provides glioma cells with the impressive ability to change their volume in tight extracellular CNS spaces. A subset of gliomas also highly express a Glu/cystine antiporter, SXC, which creates an excitotoxic build-up of Glu in the peritumoral space. The death of these surrounding neurons provides extra space for tumor cells to invade. Additionally, the equimolar exchange of Glu for cystine provides the cell with a rate-limiting reagent in the assembly of the antioxidant, GSH. Reactive oxygen species conveyed by glioma therapies are therefore futile in the presence of SXC-mediated GSH production in high SXC-expressing gliomas. Furthermore, studies have investigated the role of network formation in gliomas that are resistant to the standard of care. Gap junction protein Cx43 as well as GAP-43 are crucial in maintaining this fighting syncytium. By targeting these more resistant, invasive subset of glioma cells, therapies may have a larger grasp on the tumor cell population that may exist among all patients, no matter what specific mutations they may harbor.
Figure 2:

Glioma cells express different K+ and Cl− ion channels to mediate ion flow during tumor invasion. The flow of salts across the membrane is critical for cytoplasmic water to leave the cell as it is invading tight extracellular spaces, sometimes smaller than the average nucleus. A few of these players include gBK, NKCC1, and ClC-3 channels as well as Bradykinin signaling. In vivo and in vitro studies have shown that inhibition of these channels or Ca2+ related mediation, impedes glioma invasion. More specifically, the ClC-3 channel blocker Chlorotoxin has made successful progress in PhaseI/II clinical trials as an anti-invasive strategy for glioma treatment.
Highlights:
Glioma classifications, etiology, and disease management are discussed.
A major clinical challenge in glioma treatment is tumor invasion.
Invasion involves volume changes, glutamate release, and network formation.
Anti-invasive strategies may lead to new therapies that are urgently needed.
Acknowledgments
This work was supported by the National Institutes of Health [5R01NS052634-11 &7R01 NS036692-16]
References
- ARONICA E, GORTER JA, JANSEN GH, LEENSTRA S, YANKAYA B. & TROOST D. 2001. Expression of connexin 43 and connexin 32 gap-junction proteins in epilepsy-associated brain tumors and in the perilesional epileptic cortex. Acta Neuropathologica, 101, 449–459. [DOI] [PubMed] [Google Scholar]
- ARORA RS, ALSTON RD, EDEN TOB, ESTLIN EJ, MORAN A. & BIRCH JM 2009. Age-incidence patterns of primary CNS tumors in children, adolescents, and adults in England. Neuro-Oncology, 11, 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAILEY P. & CUSHING H. 1926. A classification of the tumours of the glioma group on a histogenetic basis, with a correlated study of prognosis. [Google Scholar]
- BALLANCE C. 1921. THE HISTORY OF BRAIN SURGERY. British Medical Journal, 2, 1041–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAO S, WU Q, MCLENDON RE, HAO Y, SHI Q, HJELMELAND AB, DEWHIRST MW, BIGNER DD & RICH JN 2006. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature, 444, 756–60. [DOI] [PubMed] [Google Scholar]
- BLEAU AM, HUSE JT & HOLLAND EC 2009. The ABCG2 resistance network of glioblastoma. Cell Cycle, 8, 2936–44. [PubMed] [Google Scholar]
- BRIDGES RJ, NATALE NR & PATEL SA 2012. System xc(−) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol, 165, 20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUCKINGHAM SC, CAMPBELL SL, HAAS BR, MONTANA V, ROBEL S, OGUNRINU T. & SONTHEIMER H. 2011. Glutamate release by primary brain tumors induces epileptic activity. Nat Med, 17, 1269–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANOLL P. & GOLDMAN JE 2008. The interface between glial progenitors and gliomas. Acta neuropathologica, 116, 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHABNER BA & ROBERTS TG 2005. Chemotherapy and the war on cancer. Nat Rev Cancer, 5, 65–72. [DOI] [PubMed] [Google Scholar]
- CHALHOUB N. & BAKER SJ 2009. PTEN and the PI3-Kinase Pathway in Cancer. Annual review of pathology, 4, 127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOW LM, ENDERSBY R, ZHU X, RANKIN S, QU C, ZHANG J, BRONISCER A, ELLISON DW & BAKER SJ 2011. Cooperativity within and among Pten, p53, and Rb pathways induces high grade astrocytoma in adult brain. Cancer Cell, 19, 305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHUNG WJ, LYONS SA, NELSON GM, HAMZA H, GLADSON CL, GILLESPIE GY & SONTHEIMER H. 2005. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci, 25, 7101–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUDDAPAH VA, ROBEL S, WATKINS S. & SONTHEIMER H. 2014. A neurocentric perspective on glioma invasion. Nat Rev Neurosci, 15, 455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUDDAPAH VA & SONTHEIMER H. 2010. Molecular Interaction and Functional Regulation of ClC-3 by Ca(2+)/Calmodulin-dependent Protein Kinase II (CaMKII) in Human Malignant Glioma. The Journal of Biological Chemistry, 285, 11188–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEEKEN JF & LÖSCHER W. 2007. The Blood-Brain Barrier and Cancer: Transporters, Treatment, and Trojan Horses. Clinical Cancer Research, 13, 1663–1674. [DOI] [PubMed] [Google Scholar]
- DENNY JB 2006. Molecular mechanisms, biological actions, and neuropharmacology of the growth- associated protein GAP-43. Curr Neuropharmacol, 4, 293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ERIKSSON PS, PERFILIEVA E, BJORK-ERIKSSON T, ALBORN A-M, NORDBORG C, PETERSON DA & GAGE FH 1998. Neurogenesis in the adult human hippocampus. Nat Med, 4, 1313–1317. [DOI] [PubMed] [Google Scholar]
- ERNEST NJ, WEAVER AK, VAN DUYN LB & SONTHEIMER HW 2005. Relative contribution of chloride channels and transporters to regulatory volume decrease in human glioma cells. Am J Physiol Cell Physiol, 288, C1451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FINE HA, DEAR KB, LOEFFLER JS, BLACK PM & CANELLOS GP 1993. Meta-analysis of radiation therapy with and without adjuvant chemotherapy for malignant gliomas in adults. Cancer, 71, 2585–97. [DOI] [PubMed] [Google Scholar]
- FRIEDMANN-MORVINSKI D, BUSHONG EA, KE E, SODA Y, MARUMOTO T, SINGER O, ELLISMAN MH & VERMA IM 2012. Dedifferentiation of Neurons and Astrocytes by Oncogenes Can Induce Gliomas in Mice. Science, 338, 1080–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GE W-P, ZHOU W, LUO Q, JAN LY & JAN YN 2009. Dividing glial cells maintain differentiated properties including complex morphology and functional synapses. Proceedings of the National Academy of Sciences, 106, 328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEGI ME, DISERENS A-C, GORLIA T, HAMOU M-F, DE TRIBOLET N, WELLER M, KROS JM, HAINFELLNER JA, MASON W, MARIANI L, BROMBERG JEC, HAU P, MIRIMANOFF RO, CAIRNCROSS JG, JANZER RC & STUPP R. 2005. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. New England Journal of Medicine, 352, 997–1003. [DOI] [PubMed] [Google Scholar]
- HIGASHIMORI H. & SONTHEIMER H. 2007. Role of Kir4.1 channels in growth control of glia. Glia, 55, 1668–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG R-P, HOSSAIN MZ, SEHGAL A. & BOYNTON AL 1999. Reduced connexin43 expression in high-grade human brain glioma cells. Journal of Surgical Oncology, 70, 21–24. [DOI] [PubMed] [Google Scholar]
- HUNTER KE & HATTEN ME 1995. Radial glial cell transformation to astrocytes is bidirectional: regulation by a diffusible factor in embryonic forebrain. Proceedings of the National Academy of Sciences, 92, 2061–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUSE JT & HOLLAND EC 2010. Targeting brain cancer: advances in the molecular pathology of malignant glioma and medulloblastoma. Nat Rev Cancer, 10, 319–331. [DOI] [PubMed] [Google Scholar]
- ISHIMOTO T, NAGANO O, YAE T, TAMADA M, MOTOHARA T, OSHIMA H, OSHIMA M, IKEDA T, ASABA R, YAGI H, MASUKO T, SHIMIZU T, ISHIKAWA T, KAI K, TAKAHASHI E, IMAMURA Y, BABA Y, OHMURA M, SUEMATSU M, BABA H. & SAYA H. 2011. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(−) and thereby promotes tumor growth. Cancer Cell, 19, 387–400. [DOI] [PubMed] [Google Scholar]
- JONES TR, BIGNER SH, SCHOLD SC JR., ENG LF & BIGNER DD 1981. Anaplastic human gliomas grown in athymic mice. Morphology and glial fibrillary acidic protein expression. Am J Pathol, 105, 316–27. [PMC free article] [PubMed] [Google Scholar]
- JUILLERAT-JEANNERET L. 2008. The targeted delivery of cancer drugs across the blood-brain barrier: chemical modifications of drugs or drug-nanoparticles? Drug Discov Today, 13, 1099–106. [DOI] [PubMed] [Google Scholar]
- LAMPE PD & LAU AF 2004. The effects of connexin phosphorylation on gap junctional communication. Int J Biochem Cell Biol, 36, 1171–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LARJAVAARA S, MANTYLA R, SALMINEN T, HAAPASALO H, RAITANEN J, JAASKELAINEN J. & AUVINEN A. 2007. Incidence of gliomas by anatomic location. Neuro Oncol, 9, 319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAWRENCE MS, STOJANOV P, POLAK P, KRYUKOV GV, CIBULSKIS K, SIVACHENKO A, CARTER SL, STEWART C, MERMEL CH, ROBERTS SA, KIEZUN A, HAMMERMAN PS, MCKENNA A, DRIER Y, ZOU L, RAMOS AH, PUGH TJ, STRANSKY N, HELMAN E, KIM J, SOUGNEZ C, AMBROGIO L, NICKERSON E, SHEFLER E, CORTÉS ML, AUCLAIR D, SAKSENA G, VOET D, NOBLE M, DICARA D, LIN P, LICHTENSTEIN L, HEIMAN DI, FENNELL T, IMIELINSKI M, HERNANDEZ B, HODIS E, BACA S, DULAK AM, LOHR J, LANDAU D-A, WU CJ, MELENDEZ-ZAJGLA J, HIDALGO-MIRANDA A, KOREN A, MCCARROLL SA, MORA J, CROMPTON B, ONOFRIO R, PARKIN M, WINCKLER W, ARDLIE K, GABRIEL SB, ROBERTS CWM, BIEGEL JA, STEGMAIER K, BASS AJ, GARRAWAY LA, MEYERSON M, GOLUB TR, GORDENIN DA, SUNYAEV S, LANDER ES & GETZ G. 2013. Mutational heterogeneity in cancer and the search for new cancer genes. Nature, 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE J, KOTLIAROVA S, KOTLIAROV Y, LI A, SU Q, DONIN NM, PASTORINO S, PUROW BW, CHRISTOPHER N, ZHANG W, PARK JK & FINE HA 2006. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell, 9, 391–403. [DOI] [PubMed] [Google Scholar]
- LIN JH-C, TAKANO T, COTRINA ML, ARCUINO G, KANG J, LIU S, GAO Q, JIANG L, LI F, LICHTENBERG-FRATE H, HAUBRICH S, WILLECKE K, GOLDMAN SA & NEDERGAARD M. 2002. Connexin 43 Enhances the Adhesivity and Mediates the Invasion of Malignant Glioma Cells. The Journal of Neuroscience, 22, 4302–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU DS, DUONG CP, HAUPT S, MONTGOMERY KG, HOUSE CM, AZAR WJ, PEARSON HB, FISHER OM, READ M, GUERRA GR, HAUPT Y, CULLINANE C, WIMAN KG, ABRAHMSEN L, PHILLIPS WA & CLEMONS NJ 2017. Inhibiting the system xC-/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun, 8, 14844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU X, CHANG Y, REINHART PH, SONTHEIMER H. & CHANG Y. 2002. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J Neurosci, 22, 1840–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOUIS DN, PERRY A, REIFENBERGER G, VON DEIMLING A, FIGARELLA-BRANGER D, CAVENEE WK, OHGAKI H, WIESTLER OD, KLEIHUES P. & ELLISON DW 2016. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathologica, 131, 803–820. [DOI] [PubMed] [Google Scholar]
- LYONS SA, JOON CHUNG W, WEAVER AK, OGUNRINU T. & SONTHEIMER H. 2007. Autocrine Glutamate Signaling Promotes Glioma Cell Invasion. Cancer research, 67, 9463–9471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAHER EA, FURNARI FB, BACHOO RM, ROWITCH DH, LOUIS DN, CAVENEE WK & DEPINHO RA 2001. Malignant glioma: genetics and biology of a grave matter. Genes Dev, 15, 1311–33. [DOI] [PubMed] [Google Scholar]
- MARCUS HJ, CARPENTER KL, PRICE SJ & HUTCHINSON PJ 2010. In vivo assessment of high grade glioma biochemistry using microdialysis: a study of energy-related molecules, growth factors and cytokines. J Neurooncol, 97, 11–23. [DOI] [PubMed] [Google Scholar]
- PREUL MARK C. 2005. History of brain tumor surgery. Neurosurgical Focus, 18, 1–1. [DOI] [PubMed] [Google Scholar]
- MCCOY E. & SONTHEIMER H. 2007. Expression and Function of Water Channels (Aquaporins) in Migrating Malignant Astrocytes. Glia, 55, 1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCFERRIN MB & SONTHEIMER H. 2006. A role for ion channels in glioma cell invasion. Neuron glia biology, 2, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCLENDON R FA, BIGNER D, VAN MEIR EG, BRAT DJ, MASTROGIANAKIS GM, OLSON JJ, MIKKELSEN T, LEHMAN N, ALDAPE K, YUNG WK, BOGLER O, WEINSTEIN JN, VANDENBERG S, BERGER M, PRADOS M, MUZNY D, MORGAN M, SCHERER S, SABO A, NAZARETH L, LEWIS L, HALL O, ZHU Y, REN Y, ALVI O, YAO J, HAWES A, JHANGIANI S, FOWLER G, SAN LUCAS A, KOVAR C, CREE A, DINH H, SANTIBANEZ J, JOSHI V, GONZALEZ-GARAY ML, MILLER CA, MILOSAVLJEVIC A, DONEHOWER L, WHEELER DA, GIBBS RA, CIBULSKIS K, SOUGNEZ C, FENNELL T, MAHAN S, WILKINSON J, ZIAUGRA L, ONOFRIO R, BLOOM T, NICOL R, ARDLIE K, BALDWIN J, GABRIEL S, LANDER ES, DING L, FULTON RS, MCLELLAN MD, WALLIS J, LARSON DE, SHI X, ABBOTT R, FULTON L, CHEN K, KOBOLDT DC, WENDL MC, MEYER R, TANG Y, LIN L, OSBORNE JR, DUNFORD-SHORE BH, MINER TL, DELEHAUNTY K, MARKOVIC C, SWIFT G, COURTNEY W, POHL C, ABBOTT S, HAWKINS A, LEONG S, HAIPEK C, SCHMIDT H, WIECHERT M, VICKERY T, SCOTT S, DOOLING DJ, CHINWALLA A, WEINSTOCK GM, MARDIS ER, WILSON RK, GETZ G, WINCKLER W, VERHAAK RG, LAWRENCE MS, O’KELLY M, ROBINSON J, ALEXE G, BEROUKHIM R, CARTER S, CHIANG D, GOULD J, GUPTA S, KORN J, MERMEL C, MESIROV J, MONTI S, NGUYEN H, PARKIN M, REICH M, STRANSKY N, WEIR BA, GARRAWAY L, GOLUB T, MEYERSON M, CHIN L, PROTOPOPOV A, ZHANG J, PERNA I, ARONSON S, SATHIAMOORTHY N, REN G, YAO J, WIEDEMEYER WR, KIM H, KONG SW, XIAO Y, KOHANE IS, SEIDMAN J, PARK PJ, KUCHERLAPATI R, LAIRD PW, COPE L, HERMAN JG, WEISENBERGER DJ, PAN F, VAN DEN BERG D, VAN NESTE L, YI JM, SCHUEBEL KE, BAYLIN SB, ABSHER DM, LI JZ, SOUTHWICK A, BRADY S, AGGARWAL A, CHUNG T, SHERLOCK G, BROOKS JD, MYERS RM, SPELLMAN PT, PURDOM E, JAKKULA LR, LAPUK AV, MARR H, DORTON S, CHOI YG, HAN J, RAY A, WANG V, DURINCK S, ROBINSON M, WANG NJ, VRANIZAN K, PENG V, VAN NAME E,FONTENAY GV, NGAI J, CONBOY JG, PARVIN B, FEILER HS, SPEED TP, GRAY JW, BRENNAN C, SOCCI ND, OLSHEN A, TAYLOR BS, LASH A, SCHULTZ N, REVA B, ANTIPIN Y, STUKALOV A, GROSS B,CERAMI E, WANG WQ, QIN LX, SESHAN VE, VILLAFANIA L, CAVATORE M, BORSU L, VIALE A, GERALD W, SANDER C, LADANYI M, PEROU CM, HAYES DN, TOPAL MD, HOADLEY KA, QI Y, BALU S, SHI Y, WU J, PENNY R, BITTNER M, SHELTON T, LENKIEWICZ E, MORRIS S, BEASLEY D, SANDERS S, KAHN A, SFEIR R, CHEN J, NASSAU D, FENG L, HICKEY E, BARKER A, GERHARD DS, VOCKLEY J, COMPTON C, VAUGHT J, FIELDING P, FERGUSON ML, SCHAEFER C, ZHANG J, MADHAVAN S, BUETOW KH, COLLINS F, GOOD P, GUYER M, OZENBERGER B, PETERSON J, THOMSON E. 2008. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature, 455, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MONTANA V. & SONTHEIMER H. 2011. Bradykinin Promotes the Chemotactic Invasion of Primary Brain Tumors. The Journal of Neuroscience, 31, 4858–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NATARAJAN K, XIE Y, BAER MR & ROSS DD 2012. Role of Breast Cancer Resistance Protein (BCRP/ABCG2) in Cancer Drug Resistance. Biochemical Pharmacology, 83, 1084–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OGUNRINU TA & SONTHEIMER H. 2010. Hypoxia increases the dependence of glioma cells on glutathione. J Biol Chem, 285, 37716–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OHGAKI H. & KLEIHUES P. 2013. The definition of primary and secondary glioblastoma. Clin Cancer Res, 19, 764–72. [DOI] [PubMed] [Google Scholar]
- OLSEN ML, SCHADE S, LYONS SA, AMARAL MD & SONTHEIMER H. 2003. Expression of Voltage- Gated Chloride Channels in Human Glioma Cells. The Journal of Neuroscience, 23, 5572–5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLSEN ML & SONTHEIMER H. 2004. Mislocalization of Kir channels in malignant glia. Glia, 46, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSSWALD M, JUNG E, SAHM F, SOLECKI G, VENKATARAMANI V, BLAES J, WEIL S, HORSTMANN H, WIESTLER B, SYED M, HUANG L, RATLIFF M, KARIMIAN JAZI K, KURZ FT, SCHMENGER T, LEMKE D, GÖMMEL M, PAULI M, LIAO Y, HÄRING P, PUSCH S, HERL V, STEINHÄUSER C, KRUNIC D, JARAHIAN M, MILETIC H, BERGHOFF AS, GRIESBECK O, KALAMAKIS G, GARASCHUK O, PREUSSER M, WEISS S, LIU H, HEILAND S, PLATTEN M, HUBER PE, KUNER T, VON DEIMLING A, WICK W. & WINKLER F. 2015. Brain tumour cells interconnect to a functional and resistant network. Nature, 528, 93–98. [DOI] [PubMed] [Google Scholar]
- OSTROM QT, BAUCHET L, DAVIS FG, DELTOUR I, FISHER JL, LANGER CE, PEKMEZCI M, SCHWARTZBAUM JA, TURNER MC, WALSH KM, WRENSCH MR & BARNHOLTZ-SLOAN JS 2014. The epidemiology of glioma in adults: a “state of the science” review. Neuro-Oncology, 16, 896–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSTROM QT, GITTLEMAN H, FULOP J, LIU M, BLANDA R, KROMER C, WOLINSKY Y, KRUCHKO C. & BARNHOLTZ-SLOAN JS 2015. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-Oncology, 17, iv1- iv62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATCHELL RA, TIBBS PA, REGINE WF & et al. 1998. Postoperative radiotherapy in the treatment of single metastases to the brain: A randomized trial. JAMA, 280, 1485–1489. [DOI] [PubMed] [Google Scholar]
- RANSOM CB, LIU X. & SONTHEIMER H. 2002. BK channels in human glioma cells have enhanced calcium sensitivity. Glia, 38, 281–91. [DOI] [PubMed] [Google Scholar]
- RANSOM CB & SONTHEIMER H. 2001. BK Channels in Human Glioma Cells. Journal of Neurophysiology, 85, 790–803. [DOI] [PubMed] [Google Scholar]
- ROBERT SM, BUCKINGHAM SC, CAMPBELL SL, ROBEL S, HOLT KT, OGUNRINU-BABARINDE T, WARREN PP, WHITE DM, REID MA, ESCHBACHER JM, BERENS ME, LAHTI AC, NABORS LB & SONTHEIMER H. 2015. SLC7A11 expression is associated with seizures and predicts poor survival in patients with malignant glioma. Sci Transl Med, 7, 289ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERT SM & SONTHEIMER H. 2014. Glutamate transporters in the biology of malignant gliomas. Cell Mol Life Sci, 71, 1839–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROZENTAL R, GIAUME C & SPRAY DC 2000. Gap junctions in the nervous system. Brain Research Reviews, 32, 11–15. [DOI] [PubMed] [Google Scholar]
- SANAI N, ALVAREZ-BUYLLA A. & BERGER MS 2005. Neural Stem Cells and the Origin of Gliomas. New England Journal of Medicine, 353, 811–822. [DOI] [PubMed] [Google Scholar]
- SANAI N, TRAMONTIN AD, QUINONES-HINOJOSA A, BARBARO NM, GUPTA N, KUNWAR S, LAWTON MT, MCDERMOTT MW, PARSA AT, MANUEL-GARCIA VERDUGO J, BERGER MS & ALVAREZ-BUYLLA A. 2004. Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature, 427, 740–4. [DOI] [PubMed] [Google Scholar]
- SATO H, TAMBA M, ISHII T. & BANNAI S. 1999. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem, 274, 11455–8. [DOI] [PubMed] [Google Scholar]
- SCHERER HJ 1938. Structural Development in Gliomas. The American Journal of Cancer, 34, 333. [Google Scholar]
- SELINFREUND RH, BARGER SW, PLEDGER WJ & VAN ELDIK LJ 1991. Neurotrophic protein S100 beta stimulates glial cell proliferation. Proc Natl Acad Sci U S A, 88, 3554–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHENG Z, MURPHY SF, VARGHESE RT, LAMOUILLE S, GUO S, PRIDHAM KJ, KANABUR P, OSIMANI AM, SHARMA S, JOURDAN J, RODGERS CM, SIMONDS GR & GOURDIE RG 2015. Connexin 43 inhibition sensitizes chemoresistant glioblastoma cells to temozolomide. Cancer Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHERR CJ & MCCORMICK F. 2002. The RB and p53 pathways in cancer. Cancer Cell, 2, 103–112. [DOI] [PubMed] [Google Scholar]
- SINGH SK, HAWKINS C, CLARKE ID, SQUIRE JA, BAYANI J, HIDE T, HENKELMAN RM, CUSIMANO MD & DIRKS PB 2004. Identification of human brain tumour initiating cells. Nature, 432, 396–401. [DOI] [PubMed] [Google Scholar]
- SONTHEIMER H. 2008. An unexpected role for ion channels in brain tumor metastasis. Exp Biol Med (Maywood), 233, 779–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONTHEIMER H. & BRIDGES RJ 2012. Sulfasalazine for brain cancer fits. Expert Opin Investig Drugs, 21, 575–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOROCEANU L, MANNING TJ JR. & SONTHEIMER H. 1999. Modulation of glioma cell migration and invasion using Cl(−) and K(+) ion channel blockers. J Neurosci, 19, 5942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOROCEANU L, MANNING TJ JR. & SONTHEIMER H. 2001. Reduced expression of connexin-43 and functional gap junction coupling in human gliomas. Glia, 33, 107–17. [DOI] [PubMed] [Google Scholar]
- STUPP R, HEGI ME, GILBERT MR & CHAKRAVARTI A. 2007. Chemoradiotherapy in malignant glioma: standard of care and future directions. J Clin Oncol, 25, 4127–36. [DOI] [PubMed] [Google Scholar]
- STUPP R, MASON WP, VAN DEN BENT MJ, WELLER M, FISHER B, TAPHOORN MJ, BELANGER K, BRANDES AA, MAROSI C, BOGDAHN U, CURSCHMANN J, JANZER RC, LUDWIN SK, GORLIA T, ALLGEIER A, LACOMBE D, CAIRNCROSS JG, EISENHAUER E, MIRIMANOFF RO, EUROPEAN ORGANISATION FOR, R., TREATMENT OF CANCER BRAIN, T., RADIOTHERAPY, G. & NATIONAL CANCER INSTITUTE OF CANADA CLINICAL TRIALS, G. 2005. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med, 352, 987–96. [DOI] [PubMed] [Google Scholar]
- THON N, KRETH S. & KRETH F-W 2013. Personalized treatment strategies in glioblastoma: MGMT promoter methylation status. OncoTargets and therapy, 6, 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOYOFUKU T, YABUKI M, OTSU K, KUZUYA T, HORI M. & TADA M. 1998. Intercellular calcium signaling via gap junction in connexin-43-transfected cells. J Biol Chem, 273, 1519–28. [DOI] [PubMed] [Google Scholar]
- TSUCHIHASHI K, OKAZAKI S, OHMURA M, ISHIKAWA M, SAMPETREAN O, ONISHI N, WAKIMOTO H, YOSHIKAWA M, SEISHIMA R, IWASAKI Y, MORIKAWA T, ABE S, TAKAO A, SHIMIZU M, MASUKO T, NAGANE M, FURNARI FB, AKIYAMA T, SUEMATSU M, BABA E, AKASHI K, SAYA H. & NAGANO O. 2016. The EGF Receptor Promotes the Malignant Potential of Glioma by Regulating Amino Acid Transport System xc(−). Cancer Res, 76, 2954–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERHAAK RGW, HOADLEY KA, PURDOM E, WANG V, QI Y, WILKERSON MD, MILLER CR, DING L, GOLUB T, MESIROV JP, ALEXE G, LAWRENCE M, O’KELLY M, TAMAYO P, WEIR A, GABRIE S, WINCKLER W, GUPTA S, JAKKULA L, FEILER HS, HODGSON JG, JAMES CD, SARKARIA JN, BRENNAN C, KAHN A, SPELLMAN PT, WILSON RK, SPEED TP, GRAY JW, MEYERSON M, GETZ G, PEROU CM, HAYES DN & THE CANCER GENOME ATLAS RESEARCH, N. 2010. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer cell, 17, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG H, ZHANG L, ZHANG IY, CHEN X, DA FONSECA A, WU S, REN H, BADIE S, SADEGHI S, OUYANG M, WARDEN CD & BADIE B. 2013. S100B Promotes Glioma Growth through Chemoattraction of Myeloid-Derived Macrophages. Clinical cancer research : an official journal of the American Association for Cancer Research, 19, 3764–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATKINS S. & SONTHEIMER H. 2011. Hydrodynamic Cellular Volume Changes Enable Glioma Cell Invasion. The Journal of Neuroscience, 31, 17250–17259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILSON TA, KARAJANNIS MA & HARTER DH 2014. Glioblastoma multiforme: State of the art and future therapeutics. Surg Neurol Int, 5, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE Z-C, WYETH MS, BALTAN-TEKKOK S. & RANSOM BR 2003. Functional Hemichannels in Astrocytes: A Novel Mechanism of Glutamate Release. The Journal of Neuroscience, 23, 3588–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YE ZC & SONTHEIMER H. 1999. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res, 59, 4383–91. [PubMed] [Google Scholar]
- ZERLIN M, MILOSEVIC A. & GOLDMAN JE 2004. Glial progenitors of the neonatal subventricular zone differentiate asynchronously, leading to spatial dispersion of glial clones and to the persistence of immature glia in the adult mammalian CNS. Dev Biol, 270, 200–13. [DOI] [PubMed] [Google Scholar]
- ZHANG F, XU CL & LIU CM 2015. Drug delivery strategies to enhance the permeability of the blood-brain barrier for treatment of glioma. Drug Des Devel Ther, 9, 2089–100. [DOI] [PMC free article] [PubMed] [Google Scholar]