

Abstract
Objective
This study measured and analyzed the serum levels of 24‐hydroxycholesterol in patients with probable Alzheimer's disease (AD) and age‐/sex‐matched controls.
Methods
A case‐control study involving 40 AD patients and 40 controls was performed at a tertiary neurological teaching hospital in eastern India. Blood and serum samples were collected for APOE genotyping and 24‐hydroxycholesterol levels, respectively.
Results
Serum 24‐hydroxycholesterol was significantly lower in AD patients compared to controls (median concentration: controls, 47.14 ng/mL (interquartile range, 16.34); AD patients, 32.93 ng/mL (interquartile range, 9.45); P < 0.001) but showed no significant correlation with Mini Mental State Examination (MMSE) score in AD cases (r = −0.169, P = 0.298) or in controls (r = 0.18, P = 0.26). No statistically significant difference was observed between serum 24‐hydroxycholesterol levels of the APOE4‐positive and ‐negative subgroups in AD patients (P = 0.79). Findings were consistent and unchanged even when the ratio of serum 24‐hydroxycholesterol to serum total cholesterol was considered.
Conclusion
The decreased 24‐hydroxycholesterol level in peripheral circulation in AD cases observed in the present study may suggest its role in AD pathogenesis. The lack of a clear correlation between serum levels of 24‐hydroxycholesterol and MMSE score—a surrogate marker of AD severity—raises the question as to whether 24‐hydroxycholesterol level declines with decreasing neuronal mass or whether the steroid continues to play a protective role.
Keywords: 24‐hydroxycholesterol, Alzheimer's dementia, APOE, biomarkers, case‐control, India
1. INTRODUCTION
The pathogenesis of sporadic Alzheimer's disease (AD) is complex and multifactorial with an interplay of genetic and environmental risk factors and metabolic alterations.1 A body of experimental data suggests that cholesterol impacts the processing, trafficking, and fibrillization of amyloid beta peptide (Aβ), which is considered as the primary mediator of AD pathogenesis.2, 3, 4, 5 Epidemiological studies have also indicated that mid‐life hypercholesterolemia is a risk factor for sporadic AD even when corrected for other confounding factors.6 Several genes related to cholesterol metabolism and transport, including APOE, are now known to be associated with AD susceptibility.5, 7 Thus, altered cholesterol homeostasis in the brain has been implicated as a contributor in the pathogenesis of sporadic AD even though the molecular pathways are as yet ill defined.
The brain is enriched in cholesterol, but in the adult the organ has no access to circulating cholesterol in the plasma because the cholesterol‐containing lipoproteins cannot cross the blood‐brain barrier (BBB).5, 8 The cholesterol homeostasis in the brain is, therefore, maintained primarily by the recycling of the existing cholesterol in the organ with some de novo synthesis compensating for the loss by degradation and removal from the brain.5, 8
A group of degradation products of cholesterol in the brain, derived enzymatically or non‐enzymatically, are oxysterols, such as 24‐hydroxycholesterol, 27‐hydroxycholesterol, 7β‐hydroxycholesterol, 7α‐hydroxycholesterol, 4β‐hydroxycholesterol, and 7‐ketocholesterol, and by far the most important of them is 24‐hydroxycholesterol.8, 9 The enzyme 24‐hydroxylase (CYP46A1) converts cholesterol to 24‐hydroxycholesterol and the latter (approximately 0.09 mg/kg/d) readily comes out of the brain through the BBB into peripheral circulation constituting the major pathway of cholesterol removal from the brain.8, 9 The level of 24‐hydroxycholesterol has been estimated in the cerebrospinal fluid (CSF) and plasma in multiple studies in AD patients to determine its suitability as a biomarker for this disease.10, 11, 12, 13, 14 Likewise, many experimental studies have attempted to identify the effects of 24‐hydroxycholesterol on neuronal functions in order to elucidate its possible role in AD pathogenesis.15, 16, 17 In both kinds of studies, however, the results have been somewhat inconsistent, necessitating further investigations and reanalysis of existing data. In the present case‐control study, we measured the serum levels of 24‐hydroxycholesterol in patients with probable AD, analyzed their correlation with degree of dementia and APOE4 positivity, and attempted to understand the biological significance of our results in the light of recent experimental evidence of neuroprotective functions of 24‐hydroxycholesterol.
2. METHODS AND SUBJECTS
2.1. Study design
The current study had a single‐center, case‐control design with probable AD patients constituting the cases and with age‐ and sex‐matched controls.
2.2. Biological sample collection, processing, and storage
Non‐fasting blood samples were collected from all the subjects under study in sterile vials between 12:30 pm and 2:00 pm without any anticoagulant for analysis of serum parameters and in the presence of the anticoagulant EDTA for subsequent isolation of DNA and transported with care to prevent hemolysis. Serum was separated within 1 hour of collection of blood by centrifugation and stored at −20°C in aliquots for further analysis. An aliquot of a fresh sample of serum was immediately analyzed for routine biochemical parameters. For APOE genotyping, the blood sample was collected in the presence of EDTA and the DNA isolation was done within 24 hours of collection, and the purified DNA stored at −40°C.
2.3. Study setting and participants
The patients with probable AD (n = 40) were randomly selected from those visiting the dementia clinic of an apex tertiary care neurological hospital serving a large population in the eastern part of India, between July 2014 and March 2015. Recruitment was stopped on reaching the desired sample size. The patients underwent a preliminary determination of Mini Mental State Examination (MMSE) scores (maximum of 30; cutoff of 26 for dementia). The diagnosis of probable AD was then made using the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition)18 criteria as well as detailed neuropsychological evaluation by a neurologist with extensive expertise in dementia management. The diagnosis was supported by magnetic resonance imaging of the brain, which showed the characteristic pattern of cerebral atrophy seen in AD as determined by an expert radiologist while ruling out other causes, such as cerebrovascular disease. All AD patients at the time of inclusion were on donepezil treatment and a few of the advanced cases were also given memantine. Further, the patients were given intermittent courses of supportive treatment with vitamins, antihypertensives, and so forth. The age‐ and sex‐matched healthy controls (n = 40) were randomly chosen from subjects visiting the free health camps organized by our group. We excluded from our study AD patients with diabetes mellitus, chronic kidney disease, overt cardiac disease, chronic infectious disease, and cancer. All healthy controls underwent neuropsychological evaluation to exclude elements of dementia and informed consent was taken from each control subject to include them in the study. For AD patients, the informed consent was taken either from the patient or the primary caregiver of the patient in cases of advanced stage of the disease. The study was approved by the institutional ethics committee. The recruitment of patients is shown in Figure 1.
Figure 1.
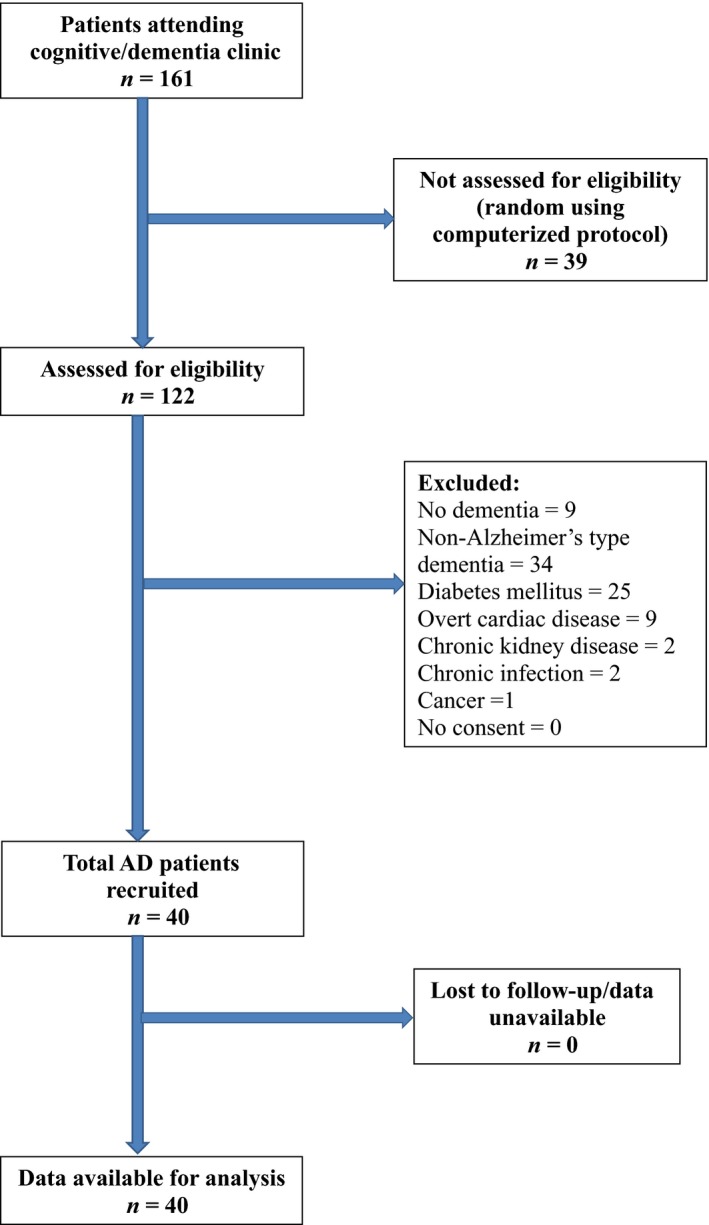
Strengthening the reporting of observational studies in epidemiology flow diagram showing the recruitment of study patients (Alzheimer's disease [AD]). Recruitment was terminated on reaching the predetermined sample size of 40 patients
2.4. Study parameters
The basic demographic data of each participant of the study, including MMSE scores, were collected in predesigned case report form. For all the subjects under this study, non‐fasting serum samples were used for the estimation of 24‐hydroxycholesterol and routine biochemical parameters while the blood samples were utilized for APOE genotyping. All measured values were entered into the case report form.
2.4.1. Estimation of 24‐hydroxycholesterol
The serum level of 24‐hydroxycholesterol was estimated by using a commercial kit (Shanghai Sunred Biological Technology Co. Ltd.) that is based on the principle of double‐antibody sandwich ELISA employing wells precoated with 24‐hydroxycholesterol antibody (capture antibody), biotin‐labeled 24‐hydroxycholesterol antibody (detection antibody), streptavidin‐horseradish peroxidase conjugate, and a chromogenic substrate. A calibration curve of the standards (4‐64 ng/mL) was prepared and absorbance measured at 450 nm on a microplate reader (Tecan, Sunrise). Each sample was analyzed in duplicate and the measurements of all samples were completed in two batches.
2.4.2. APOE genotyping
DNA was extracted from blood collected in EDTA vials using a commercial kit (Qiagen) that utilizes a silica‐based spin‐column. A 245 base‐pair fragment of APOE gene was amplified by touch‐down polymerase chain reaction (PCR) using the appropriate forward and reverse primers in a reaction volume of 50 μL containing 10 mM Tris, 50 mM KCl, dNTPs (each 200 μM), 3 mM MgCl2, 150 nM of each primer, 25 ng of template DNA, 10% dimethyl sulfoxide, 6% glycerol, and 2.5 units of Taq polymerases adopted from a published procedure.19 The amplified PCR product (244 bp) was digested without purification with HhaI for 3 hours at 37°C followed by electrophoresis on 15% polyacrylamide gel, post‐staining with ethidium bromide, and UV visualization to identify the restriction fragments. The different alleles (E2, E3, E4) of APOE produced characteristic restriction fragments and, accordingly, different genotypes (E3/E3, E2/E3, E3/E4, E4/E4) were identified in the controls and AD patients.
2.4.3. Routine biochemical parameters
The routine biochemical parameters of the controls and cases were estimated by a clinical auto‐analyzer (Randox) under internal quality‐control program.
2.5. Sample size estimation
The current study did not aim to validate 24‐hydroxycholesterol as a biomarker of AD but instead focused on studying changing serum levels of this molecule with MMSE scores and APOE genotype in a relatively pure subset of probable AD patients free from chronic inflammatory comorbidities. In view of the limited number of past studies in this area and similar sample sizes employed, a sample size of 40 in each group was considered to be adequate.
2.6. Statistical methods
Mann‐Whitney U test and unpaired t test were used for comparing quantitative variables of a non‐parametric and parametric nature, respectively. To identify the correlation between two variables, Spearman's correlation coefficient was used. A value of P < 0.05 was considered as statistically significant. The statistical analysis was performed by using Graph Pad Prism software, San Diego, CA, USA (version 5, 2007).
The frequencies of E2, E3, and E4 alleles were estimated by gene counting; for example, the frequency of APOE3 = (2 × APOE3/E3 + APOE3/E2 + APOE3/E4)/Total number of APOE alleles. The statistical difference in the distribution of allelic variants in controls versus AD cases was calculated using Fisher's exact test (with Freeman Halton extension).
3. RESULTS
3.1. Participants
The AD patients, randomly selected from those visiting the dementia clinic (n = 40) and in accordance with the inclusion and exclusion criteria, all gave consent for the study. The data from all of these patients as well as from controls were analyzed. No loss of collected biological sample occurred at any stage.
3.2. Descriptive data
The demographic and biochemical profiles of the subjects under study are presented in Table 1. None of the AD patients included in our study had any family history of the disease and the mean (± SD) MMSE score was 12.70 (± 8.03) for AD patients, indicating sampling from different stages of the disease (Table 1). The male:female ratios in the control and AD groups did not differ statistically and there was no significant difference in the age or body mass index between the two groups (Table 1). The routine serum biochemical parameters were within the reference range in controls and AD cases and were not significantly different in the two groups (data not presented) except for the level of serum total cholesterol, which was higher in the AD group with a statistically significant difference. There were no missing data in the study.
Table 1.
Demographic and biochemical profile of AD patients (cases) and controls
| Parameter | Control (n = 40) | AD (n = 40) |
|---|---|---|
| Age (years) | 71.95 ± 6.27 | 70.20 ± 5.17 |
| Sex (M/F) | 22/18 | 25/15 |
| Body mass index (kg/m2) | 22.03 ± 1.39 | 21.66 ± 1.00 |
| MMSE score | 29.05 ± 0.71 | 12.70 ± 8.03* |
| Random blood sugar (mg/dL) | 88.13 ± 11.64 | 89.10 ± 11.36 |
| Total cholesterol (mg/dL) | 175.20 ± 16.88 | 199.90 ± 23.60* |
Abbreviations: AD, Alzheimer's disease; MMSE, Mini Mental State Examination.
All quantitative values expressed as mean ± SD.
Statistically significant difference between control and AD groups; i.e, P < 0.001.
3.3. Main results
3.3.1. Serum levels of 24‐hydroxycholesterol in study population
The data presented in Figure 2 show that the serum level of 24‐hydroxycholesterol was significantly lower in AD patients compared to controls (median 24‐hydroxycholesterol concentration: controls, 47.14 ng/mL [interquartile range, 16.34]; AD patients, 32.93 ng/mL [interquartile range, 9.45]; P < 0.001).
Figure 2.
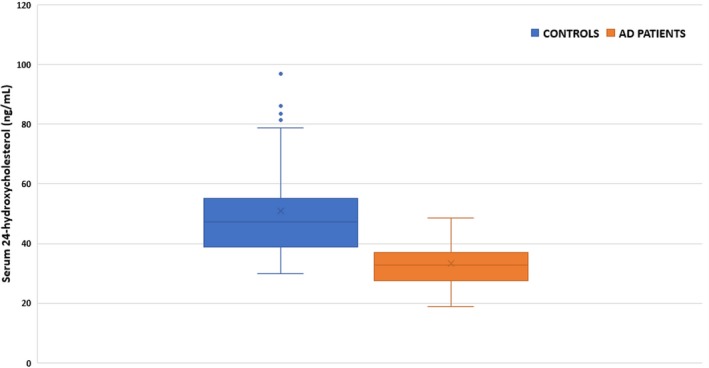
Serum 24‐hydroxycholesterol levels among controls and Alzheimer's disease (AD) patients. Box and whisker plots represent median, first quartile, third quartile, and minimum to maximum range. Median serum 24‐hydroxycholesterol concentration in ng/mL: controls, 47.14 (interquartile range, 16.34); AD patients, 32.93 (interquartile range, 9.45; P < 0.001; Mann‐Whitney U)
3.3.2. Genotypes of patients/controls in study population
Genotypes of patients/controls in the study population are represented in the form of bar charts (Figure 3). The E3/E3 isoform was the most frequent in both AD and control groups (52.5% in AD cases and 87.5% in controls). Table 2 denotes the allelic frequency of the three alleles (E2, E3, E4) in AD patients and controls. The E3 allele was noted to be the most frequent allele in both groups. Fisher's exact text confirmed a significant difference between the study groups (P < 0.05) due to differential distribution of the E4 allele.
Figure 3.
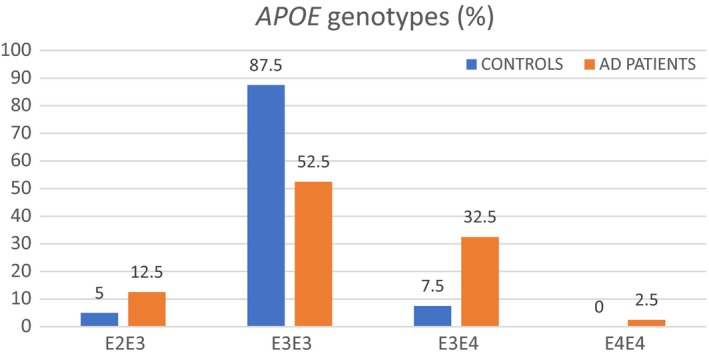
Distribution of different APOE genotypes in Alzheimer's disease (AD) patients and controls, expressed as percentage of total subjects in each group. E3/E3 was the commonest genotype in both groups (52.5% in AD cases and 87.5% in controls)
Table 2.
Allelic frequency of the three alleles (E2, E3, E4) in AD patients and controls
| Allelic frequency (%) | ||
|---|---|---|
| Allele | Frequency in AD | Frequency in controls |
| E2 | 6.25 | 2.50 |
| E3 | 75.00 | 93.75 |
| E4 | 18.75 | 3.75 |
Abbreviation: AD, Alzheimer's disease.
3.3.3. Serum 24‐hydroxycholesterol in study groups
As presented in Figure 4, the serum 24‐hydroxycholesterol level showed no significant correlation with MMSE score in controls (rho = 0.18, P = 0.26; Spearman's) or in AD cases (rho = −0.167, P = 0.302; Spearman's). When serum 24‐hydroxycholesterol levels were compared between the APOE4‐positive and APOE4‐negative subgroups of the AD cases, no statistically significant difference was observed between them (P = 0.79).
Figure 4.
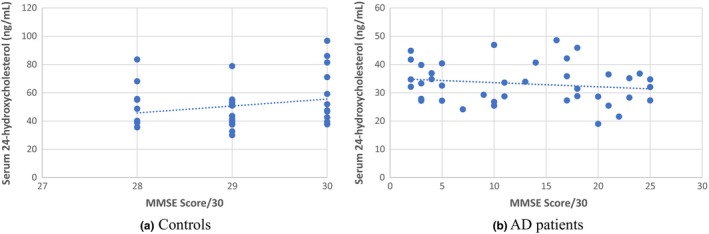
XY scatter plot between Mini Mental State Examination (MMSE) scores and serum levels of 24‐hydroxycholesterol in (a) controls and (b) Alzheimer's disease (AD) patients. Correlation coefficient was insignificant in both groups (rho = 0.18, P = 0.26 in controls; rho = −0.167, P = 0.302 in AD cases)
4. DISCUSSION
4.1. Serum 24‐hydroxycholesterol as an AD biomarker
Apart from the positron emission tomography‐based imaging studies, the only recommended biomarkers of AD are the CSF levels of Aβ42, and total and phosphorylated tau proteins. Many attempts have been made to find a suitable diagnostic or prognostic biomarker for AD in peripheral circulation without any success. Since 24‐hydroxycholesterol is produced almost exclusively in the brain and enters the peripheral circulation through the BBB, it has been considered as a putative biomarker for AD, especially when early reports suggested alterations in CSF and serum levels of this cholesterol derivative in AD. Several case‐control studies have indicated that plasma and CSF levels of 24‐hydroxycholesterol are increased in AD and other types of dementia.10, 11, 12 A meta‐analysis of available data from several sources also suggests an elevated level of 24‐hydroxycholesterol in the CSF in AD.20 However, in contrast to these, there are other reports of decreased levels of 24‐hydroxycholesterol in plasma in AD, which is consistent with autopsy findings of decreased levels of 24‐hydroxycholesterol in various regions of the AD brain compared to that in controls.13, 14, 21, 22 A very elegant recent study measured different oxysterols in post‐mortem AD brains and clearly demonstrated a significant decrease in brain 24‐hydroxycholesterol along with decreased mRNA expression of cholesterol‐24‐hydroxylase at different stages of the disease.9 The conflicting reports related to plasma levels of 24‐hydroxycholesterol in various studies may have arisen because of selection of patients with different degrees of disease severity in different studies, differences in assay procedure, drug history, dietary habits, APOE4 positivity, and male:female ratio of the subjects under study, as well as the presence of other comorbidities in the patients. The different rates of hepatic clearance of 24‐hydroxycholesterol from the peripheral circulation, polymorphism of 24‐hydroxylase, and possibly other confounders may have added to the variability of the published results.10, 21, 22, 23, 24, 25, 26 However, it is well established that 24‐hydroxycholesterol is formed exclusively in the brain and an efflux of this oxysterol occurs from the brain to peripheral circulation. Thus, when the published reports of brain and plasma levels of 24‐hydroxycholesterol in AD are judged in conjunction, a decrease in the level of this oxysterol in peripheral circulation is the most likely event and our present results amply confirm this. However, the decrease in the level of serum 24‐hydroxycholesterol in AD subjects observed in our study is apparently not related to disease severity as measured by MMSE scores. This contradicts the suggestion that 24‐hydroxycholesterol level in CSF or plasma increases in early AD but decreases when the disease becomes severe with loss of neuronal tissue.10, 22 Other confounding factors already mentioned may explain the lack of correlation between MMSE scores and the plasma levels of 24‐hydroxycholesterol in AD in our study, but one important factor in this regard could be the aberrant expression of cholesterol‐24‐hydroxylase in astrocytes in the AD brain as the disease progresses, which may partially compensate for the loss of this enzyme in degenerating neurons.9, 14, 22
Some authors compared ratios of plasma 24‐hydroxycholesterol and total plasma cholesterol in AD and control subjects indicating normalization of 24‐hydroxycholesterol with respect to varying cholesterol concentrations in plasma.13, 23 However, 24‐hydroxycholesterol is exclusively produced in the brain, which has no access to peripheral circulating cholesterol because of the inability of cholesterol‐containing lipoproteins to cross the BBB. Thus, apparently the ratio of 24‐hydroxycholesterol and total cholesterol in peripheral circulation carries no biological significance. Even so, we calculated such ratios in our analysis (results not presented), but this did not affect our conclusions, showing consistency across all statistical calculations compared to when absolute values of serum 24‐hydroxycholesterol concentrations were considered. In this context, a minor observation may be pointed out. A mild but statistically significant elevation of total serum cholesterol was seen in AD subjects compared to controls in the present study. Although the cause is not known, this was consistently found in different series of case‐control studies on AD conducted by our lab and reported earlier.27, 28 However, there is no consensus on this issue in the literature.
Nearly 35% of AD cases in the present study had at least one allele of APOE4, which is lower than that reported in several populations in the United States and Europe but consistent with published data on APOE4 positivity (E3/E4 or E4/E4) in AD patients in Asia.29 More importantly, we have not observed any effect of APOE4 positivity on serum 24‐hydroxycholesterol level in AD subjects and it is important to point out that the effects of APOE4 positivity on serum 24‐hydroxycholesterol levels in AD subjects are not clearly known.10, 11 However, given the fact that 24‐hydroxycholesterol transport from the brain to peripheral circulation across the BBB is not mediated by apolipoprotein E containing lipoprotein, it is not surprising that APOE4 positivity has failed to show any correlation with serum 24‐hydroxycholesterol level in AD subjects in our study.
The current study has included only cases of probable AD who have none of the chronic comorbidities, such as diabetes mellitus, chronic kidney disease, overt cardiac diseases, chronic infectious diseases, and cancer. Furthermore, patients with significant cerebrovascular disease have been excluded based on MRI brain findings. Hence, it is expected that the serum levels of 24‐hydroxycholesterol in the present study would be more representative of the characteristic pattern of AD pathogenesis while being unaffected by other chronic conditions with lipid‐vascular abnormalities. Although we have observed a significant decrease in serum 24‐hydroxycholesterol level in AD patients in comparison to age‐matched controls, its suitability as a disease marker is doubtful. The serum level of this oxysterol is altered in other CNS disorders, which lowers its specificity as a biomarker of AD.13, 24 Further, as is apparent from our study, the level of 24‐hydroxycholesterol does not reflect the clinical severity of the disease. Thus, instead of establishing 24‐hydroxycholesterol as a biomarker of AD, we have thought it prudent to analyze our finding in clinical cases vis‐à‐vis the very interesting experimental results existing in the literature that suggest a protective role of this oxysterol against AD neurodegeneration.
4.2. Serum 24‐hydroxycholesterol in AD pathogenesis: Findings of the current study and how they fit into newer molecular insights
There is a substantial body of evidence that suggests a protective role of 24‐hydroxycholesterol against AD pathogenesis in different experimental animal and cell‐based models. For example, 24‐hydroxycholesterol has been shown to inhibit the production of Aβ in SH‐SY5Y and CHO cell lines stably expressing the human amyloid precursor protein (APP) gene and the phenomenon occurs through impaired trafficking of APP at the endoplasmic reticulum.16 Another report has suggested that in SH‐SY5Y cells, 24‐hydroxycholesterol activates the non‐amyloidogenic pathway of APP processing, inhibits 27‐hydroxycholesterol‐induced increase in Aβ production and increases the expression of the cholesterol transporter ABCA1.17 It is interesting that the disruption of ABCA1 causes increased Aβ deposition in the brains of transgenic APP23 mice carrying Swedish mutation of familial AD in the APP gene.30 In an elegant study in triple transgenic AD mice carrying three mutant genes, including one human APP (hAPP) with Swedish mutation, the ablation of the acylcholesterol acyltransferase (ACAT1) gene has been shown to increase the level of 24‐hydroxycholesterol in the brain associated with considerably decreased accumulation of mutant human APP and its proteolytic products and significant amelioration of cognitive deficits.31 Additionally, in cultured hippocampal neurons from such triple transgenic mice, a decreased production of mutant hAPP has been noted upon exposure to varying concentrations of 24‐hydroxycholesterol.31 Other experimental studies have indicated that 24‐hydroxycholesterol is a modulator of NMDA receptors and hippocampal long‐term potentiation (LTP), and it prevents the LTP deficits induced by NMDA receptor blockers.32 Another interesting report suggests that in SK‐N‐BE cells, 24‐hydroxycholesterol is neuroprotective through upregulation of Sirtuin 1 and inhibition of tau phosphorylation.33 This study has also claimed that the levels of 24‐hydroxycholesterol and cholesterol 24‐hydroxylase are both decreased in the post‐mortem AD brain along with downregulation of Sirtuin 1.33 Thus, the clear majority of experimental data, including some elegant molecular genetic studies, tends to support the protective effect of this compound against AD pathogenesis, although a few studies have indicated the neurotoxic potential of 24‐hydroxycholesterol.21 In brief, the decreased 24‐hydroxycholesterol level in peripheral circulation in AD cases observed in different studies, including the present one, is very much consistent with experimental data and, further, it suggests that the deficiency of this oxysterol has a role in AD pathogenesis. A lack of clear correlation was observed between serum levels of 24‐hydroxycholesterol and MMSE score, a surrogate marker of AD severity or stage of disease. This goes against previously made suggestions that 24‐hydroxycholesterol levels decline with decreasing neuronal mass with advancing disease. Whether the steroid plays a protective role preventing AD and, hence, is universally low in all stages of the disease may be explored in more focused studies.
4.3. Limitations of the study
The small sample size and the lack of molecular validation for the hypothesis raised by the current study are its main limitations. There have been similar studies in the past with similar sample sizes generating disparate results. However, the results of the current study are more in line with recent molecular findings. MMSE scores are not the most reliable marker of stage of AD but in a predominantly poor and rural population, other markers, such as duration of disease, are not reliably determined; hence, these could not be used. Another limitation of this study is the different drug histories of the recruited subjects in the AD and the control groups. We used healthy controls with no history of chronic use of drugs at the point of sample collection. The AD patients, however, were on donepezil or donepezil and memantine in different doses and, in addition, most of them were given various B‐vitamins, anti‐hypertensive agents, statins, and neuropsychiatric drugs as intermittent short or long courses. This may have had a confounding effect on our results and, especially in the context of the current study, it is important to remember that statins have been shown to produce variable effects on serum 24‐hydroxycholesterol level in different studies. However, due to the variability of medication history and the small numbers of patients in each subgroup of the AD case group, not much can be read into the effects of statins on the results.34, 35, 36 The findings of the current study, namely decreased serum 24‐hydroxycholesterol in AD cases, suggest that the deficiency of this oxysterol has a role in AD pathogenesis. Lack of a clear correlation between serum levels of 24‐hydroxycholesterol and MMSE scores raises the question as to whether 24‐hydroxycholesterol level indeed declines with decreasing neuronal mass. It is instead quite possible that the cholesterol metabolite plays a protective role throughout the AD disease course and, hence, decreased levels are uniformly present in all stages of AD. A major advantage of this study is perhaps the fact that a group of AD patients free from chronic inflammatory and vascular comorbidities and significant cerebrovascular disease were included in analysis of 24‐hydroxycholesterol serum levels and the data thus obtained may be more representative of AD cases. Moreover, we have dwelt upon the link between experimental data on AD pathogenesis and our observation in clinical cases of AD.
AUTHOR CONTRIBUTIONS
Debashree Roy (lead), Anindita Banerjee, Atanu Biswas were involved in data collection and analysis and in writing the manuscript. Pallav Sharma was involved in performing review of literature and statistical analysis. Sankha Shubhra Chakrabarti wrote the manuscript and performed the major part of statistical analysis. Sasanka Chakrabarti was the lead in study design, data validation, monitoring of all experimental data, final writing and editing of manuscript, and is the corresponding author.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
ACKNOWLEDGEMENTS
S.S.C. would like to thank the National Programme for Healthcare of the Elderly, Ministry of Health & Family Welfare, Government of India for research support. S.C. would like to thank the Maharshi Markandeshwar (deemed to be) University, Mullana, Haryana for research support.
Roy D, Chakrabarti SS, Banerjee A, Sharma P, Biswas A, Chakrabarti S. Serum 24‐hydroxycholesterol in probable Alzheimer's dementia: Reexploring the significance of a tentative Alzheimer's disease biomarker. Aging Med. 2019;2:74‐81. 10.1002/agm2.12068
REFERENCES
- 1. Chakrabarti S, Khemka VK, Banerjee A, Chatterjee G, Ganguly A, Biswas A. Metabolic risk factors of sporadic Alzheimer's disease: implications in the pathology, pathogenesis and treatment. Aging Dis. 2015;6(4):282‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reiss AB, Voloshyna I. Regulation of cerebral cholesterol metabolism in Alzheimer's disease. J Investig Med. 2012;60(3):576‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of beta‐amyloid in hippocampal neurons. Proc Natl Acad Sci USA. 1998;95(11):6460‐6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang C, Shou Y, Pan J, Du Y, Liu C, Wang H. The relationship between cholesterol level and Alzheimer's disease‐associated APP proteolysis/Aβ metabolism. Nutr Neurosci. 2018;22(7):453‐463. 10.1080/1028415x.2017.1416942 [DOI] [PubMed] [Google Scholar]
- 5. Martín MG, Pfrieger F, Dotti CG. Cholesterol in brain disease: sometimes determinant and frequently implicated. EMBO Rep. 2014;15(10):1036‐1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anstey KJ, Ashby‐Mitchell K, Peters R. Updating the evidence on the association between serum cholesterol and risk of late‐life dementia: review and meta‐analysis. J Alzheimers Dis. 2017;56(1):215‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wollmer MA. Cholesterol‐related genes in Alzheimer's disease. Biochim Biophys Acta. 2010;1801(8):762‐773. [DOI] [PubMed] [Google Scholar]
- 8. Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem. 2009;390(4):287‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Testa G, Staurenghi E, Zerbinati C, et al. Changes in brain oxysterols at different stages of Alzheimer's disease: their involvement in neuroinflammation. Redox Biol. 2016;10:24‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lütjohann D, Papassotiropoulos A, Björkhem I, et al. Plasma 24S‐hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res. 2000;41(2):195‐198. [PubMed] [Google Scholar]
- 11. Papassotiropoulos A, Lütjohann D, Bagli M, et al. 24S‐hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J Psychiatr Res. 2002;36(1):27‐32. [DOI] [PubMed] [Google Scholar]
- 12. Zuliani G, Donnorso MP, Bosi C, et al. Plasma 24S‐hydroxycholesterol levels in elderly subjects with late onset Alzheimer's disease or vascular dementia: a case‐control study. BMC Neurol. 2011;11:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kölsch H, Heun R, Kerksiek A, Bergmann KV, Maier W, Lütjohann D. Altered levels of plasma 24S‐ and 27‐hydroxycholesterol in demented patients. Neurosci Lett. 2004;368(3):303‐308. [DOI] [PubMed] [Google Scholar]
- 14. Solomon A, Leoni V, Kivipelto M, et al. Plasma levels of 24S‐hydroxycholesterol reflect brain volumes in patients without objective cognitive impairment but not in those with Alzheimer's disease. Neurosci Lett. 2009;462(1):89‐93. [DOI] [PubMed] [Google Scholar]
- 15. Yamanaka K, Saito Y, Yamamori T, Urano Y, Noguchi N. 24(S)‐hydroxycholesterol induces neuronal cell death through necroptosis, a form of programmed necrosis. J Biol Chem. 2011;286(28):24666‐24673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Urano Y, Ochiai S, Noguchi N. Suppression of amyloid‐β production by 24S‐hydroxycholesterol via inhibition of intracellular amyloid precursor protein trafficking. FASEB J. 2013;27(10):4305‐4315. [DOI] [PubMed] [Google Scholar]
- 17. Prasanthi JR, Huls A, Thomasson S, Thompson A, Schommer E, Ghribi O. Differential effects of 24‐hydroxycholesterol and 27‐hydroxycholesterol on β‐amyloid precursor protein levels and processing in human neuroblastoma SH‐SY5Y cells. Mol Neurodegener. 2009;4(1):74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. 4th ed Washington, DC: American Psychiatric Association; 1994. [Google Scholar]
- 19. Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31(3):545‐548. [PubMed] [Google Scholar]
- 20. Wang H‐L, Wang Y‐Y, Liu X‐G, et al. Cholesterol, 24‐hydroxycholesterol, and 27‐hydroxycholesterol as surrogate biomarkers in cerebrospinal fluid in mild cognitive impairment and Alzheimer's disease: a meta‐analysis. J Alzheimers Dis. 2016;51(1):45‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Costa AC, Joaquim HPG, Nunes VS, et al. Donepezil effects on cholesterol and oxysterol plasma levels of Alzheimer's disease patients. Eur Arch Psychiatry Clin Neurosci. 2018;268(5):501‐507. [DOI] [PubMed] [Google Scholar]
- 22. Heverin M, Bogdanovic N, Lütjohann D, et al. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer's disease. J Lipid Res. 2004;45(1):186‐193. [DOI] [PubMed] [Google Scholar]
- 23. Vega GL, Weiner M, Kölsch H, et al. The effects of gender and CYP46 and apo E polymorphism on 24S‐hydroxycholesterol levels in Alzheimer's patients treated with statins. Curr Alzheimer Res. 2004;1(1):71‐77. [DOI] [PubMed] [Google Scholar]
- 24. Leoni V, Caccia C. 24S‐hydroxycholesterol in plasma: a marker of cholesterol turnover in neurodegenerative diseases. Biochimie. 2013;95(3):595‐612. [DOI] [PubMed] [Google Scholar]
- 25. Lütjohann D, von Bergmann K. 24S‐hydroxycholesterol: a marker of brain cholesterol metabolism. Pharmacopsychiatry. 2003;36(suppl 2):S102‐S106. [DOI] [PubMed] [Google Scholar]
- 26. Garcia ANM, Muniz MTC, Souza e Silva HR, da Silva HA, Athayde‐Junior L. Cyp46 polymorphisms in Alzheimer's disease: a review. J Mol Neurosci. 2009;39(3):342‐345. [DOI] [PubMed] [Google Scholar]
- 27. Khemka VK, Bagchi D, Bandyopadhyay K, et al. Altered serum levels of adipokines and insulin in probable Alzheimer's disease. J Alzheimers Dis. 2014;41(2):525‐533. [DOI] [PubMed] [Google Scholar]
- 28. Khemka VK, Ganguly A, Bagchi D, et al. Raised serum proinflammatory cytokines in Alzheimer's disease with depression. Aging Dis. 2014;5(3):170‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crean S, Ward A, Mercaldi CJ, et al. Apolipoprotein E ε4 prevalence in Alzheimer's disease patients varies across global populations: a systematic literature review and meta‐analysis. Dement Geriatr Cogn Disord. 2011;31(1):20‐30. [DOI] [PubMed] [Google Scholar]
- 30. Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005;280(52):43224‐43235. [DOI] [PubMed] [Google Scholar]
- 31. Bryleva EY, Rogers MA, Chang CCY, et al. ACAT1 gene ablation increases 24(S)‐hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci USA. 2010;107(7):3081‐3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Paul SM, Doherty JJ, Robichaud AJ, et al. The major brain cholesterol metabolite 24(S)‐hydroxycholesterol is a potent allosteric modulator of N‐methyl‐D‐aspartate receptors. J Neurosci. 2013;33(44):17290‐17300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gamba P, Testa G, Staurenghi E, et al. Neuron survival modulated by 24‐hydroxycholesterol: the role of sirtuin pathway in Alzheimer's disease. Free Radic Biol Med. 2016;96:S48‐S49. [Google Scholar]
- 34. Serrano‐Pozo A, Vega GL, Lütjohann D, et al. Effects of simvastatin on cholesterol metabolism and Alzheimer disease biomarkers. Alzheimer Dis Assoc Disord. 2010;1:220‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thelen KM, Lütjohann D, Vesalainen R, et al. Effect of pravastatin on plasma sterols and oxysterols in men. Eur J Clin Pharmacol. 2006;62(1):9‐14. [DOI] [PubMed] [Google Scholar]
- 36. Dias IHK, Milic I, Lip GYH, Devitt A, Polidori MC, Griffiths HR. Simvastatin reduces circulating oxysterol levels in men with hypercholesterolaemia. Redox Biol. 2018;16:139‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]