

Abstract
1-Aminopyridinium ylides are efficient directing groups for palladium-catalyzed β-arylation and alkylation of sp3 C–H bonds in carboxylic acid derivatives. The efficiency of these directing groups depends on the substitution at the pyridine moiety. The unsubstituted pyridine-derived ylides allow functionalization of primary C–H bonds, while methylene groups are unreactive in the absence of external ligands. 4-Pyrrolidinopyridine-containing ylides are capable of C-H functionalization in acyclic methylene groups in the absence of external ligands, thus rivaling the efficiency of the aminoquinoline directing group. Preliminary mechanistic studies have been performed. A cyclopalladated intermediate has been isolated and characterized by X-ray crystallography, and its reactivity was studied.
Graphical abstract

1. INTRODUCTION
Carbon–hydrogen bond activation and functionalization methodology has evolved from organometallic curiosity to applications in the synthesis of complex natural products.1 However, selective functionalization of unactivated (not benzylic or α to a heteroatom) sp3 C–H bonds still presents a challenge. Positional selectivity is usually achieved by employing a directing group that coordinates metal and positions it for cleavage of the desired C–H bond. Both mono and bidentate directing groups have been extensively used for this purpose. Monodentate directing groups can be broadly categorized as X or L type (Figure I). Examples of X-type directing groups, sometimes categorized as weak, include carboxylates 1, polyfluoroanilides 2, and hydroxamic acids 3.2 L-Type monodentate auxiliaries (or strong directing groups) include pyridines, oxazolines, oxime derivatives, and tertiary amines.3 With few exceptions,3a,4 monodentate groups allow functionalization of only primary unactivated sp3 C–H bonds. Secondary C–H bonds in acyclic structures typically react only in the presence of added ligands, and transformations often require high loadings of palladium catalyst as well as forcing conditions. Reactivity in the presence of added external ligands has important practical consequences with respect to asymmetric C–H functionalization, since background reaction leading to erosion of enantioselectivity is suppressed.5
Figure I.

Directing groups for sp3 C–H functionalization.
Bidentate, usually monoanionic, auxiliaries have been categorized as strong directing groups.1e,6 They allow for an efficient functionalization of both primary and secondary alkane C–H bonds. In some cases, even tertiary C–H bonds are reactive.7 The aminoquinoline, picolinamide, and 2-thiomethyaniline auxiliaries were introduced in 2005 and 2010.8 Many similar directing groups have been found subsequently.9 Recently, C–H functionalizations have been rendered catalytic with respect to bidentate auxiliaries, and sometimes the formation of the directing groups can be performed in situ.10 The electron-rich, bidentate, monoanionic auxiliaries facilitate both the C–H activation and functionalization steps, especially if high-valent transition metal intermediates are involved. There are cases, however, where weaker or monodentate coordination may be preferable. For example, development of catalytic, asymmetric transformations requires binding of external ligands to metal. Strong, bidentate auxiliaries may outcompete the chiral ligand, resulting in significant racemic background reaction. After the C–H activation step, the substrate coordinates to metal as a tridentate ligand, saturating its coordination sites (14). The decreased number of open sites at the metal and the stability of the intermediate palladacycles may interfere with the subsequent C–H functionalization step. Consequently, monodentate auxiliaries possessing efficiency of bidentate directing groups would have advantages in C–H bond functionalization, perhaps allowing for mild C–H functionalization and strong influence of added ligands.
In 1974, McWhinnie and co-workers showed that benzoylated 1-aminopyridine ylides can be cyclometalated by palladium, rhodium, and platinum complexes.11 In the next 40 years, these ylides were used as substitutes for pyridine oxides, mostly for functionalization of the pyridine ring.12 We speculated that sp3 C–H functionalization could be achieved if aliphatic carboxylic acid amides of 1-aminopyridine were employed. We report here that 1-aminopyridine ylides are the first monodentate directing groups that allow for general functionalization of primary and secondary unactivated sp3 C–H bonds in the absence of external ligands. Properties of these auxiliaries can be easily tuned by substitution at the 4-position of the pyridine moiety. These new directing groups rival aminoquinoline in efficiency and may supplement it in the synthesis of complex natural products and multiple bond functionalization.13
2. RESULTS AND DISCUSSION
2.1. Reaction Optimization.
The initial reaction optimization was based on previous results using monodentate directing groups for C–H functionalization (Table 1).14 However, conditions employing acetic acid solvent resulted in low yield of 17 (entry 1). Performing the reaction in hexafluoroisopropanol resulted in an improved 63% yield (entry 2). Addition of disodium hydrogen phosphate or sodium tetrafluoroborate increased the yield (entries 3 and 4). Further improvement was observed when sodium triflate additive was used (entry 5). An increased amount of NaOTf did not result in higher reaction yield (entry 6). Highest yield was obtained at 90 °C (entry 8), while other temperatures gave lower yields (entries 7 and 9). Aryl bromides and aryl triflates are unreactive under these conditions.
Table 1.
Optimization of Reaction Conditionsa
 | ||||
|---|---|---|---|---|
| entry | T, °C | solvent | additive | yield, % |
| 1 | 100 | AcOH | 40 | |
| 2 | 110 | HFIP | 63 | |
| 3 | 110 | HFIP | Na2HPO4 | 74 |
| 4 | 110 | HFIP | NaBF4 | 80 |
| 5 | 110 | HFIP | NaOTf | 87 |
| 6b | 110 | HFIP | NaOTf | 87 |
| 7 | 100 | HFIP | NaOTf | 87 |
| 8 | 90 | HFIP | NaOTf | 91 |
| 9 | 80 | HFIP | NaOTf | 74 |
Amide 0.15 mmol, 4-iodotoluene 2.5 equiv, AgOAc 1.5 equiv, additive 1.0 equiv. Yields of 17 were determined by 1H NMR analysis using 1,1,2-trichloroethane internal standard.
NaOTf 1.2 equiv. AcOH = acetic acid, HFIP = hexafluoroisopropanol, OTf = triflate.
2.2. Arylation of Primary C—H Bonds.
The arylation of 1-(propionylamino) pyridinium ylide was conducted with a broad range of electron-rich (entries 1–5 and 15), electron poor (entries 6–14), and heterocyclic (entries 15–18) aryl iodides. The products were obtained in good yields (Table 2). This reaction tolerates substituents at either para-or meta-positions of the aryl iodides. No arylation products were observed if ortho-substituted aryl iodides were employed, consistent with results obtained with aminoquinoline amides. The reaction can accommodate a range of functional groups, such as methoxy (entry 2), alkyl (entries 1 and 3), bromo (entry 7), ether (entries 2, 5, and 15), ester (entry 6), trifluoromethyl (entries 9, 10, and 13), fluoro (entries 11 and, 14), chloro (entry 14), and enolizable ketone (entry 17). Interestingly, even an aldehyde moiety is tolerated (entries 12 and 16). Some heterocycles, such as dihydrobenzofuran (entry, 15), furan (entry 16), thiophene (entry 17), and protected indole (entry 18), are also compatible with reaction conditions. Introduction of strongly electron-withdrawing substituents decreases reaction yields (entries 8 and 9).
Table 2.
Arylation Scope with Respect to Aryl Iodidesa
 | |||
|---|---|---|---|
| entry | ArI | product | yield, % |
| 1 |  |
 |
81 |
| 2 |  |
 |
65 |
| 3 |  |
 |
80 |
| 4 |  |
 |
77 |
| 5 |  |
 |
62 |
| 6 |  |
 |
76 |
| 7 |  |
 |
72 |
| 8 |  |
 |
53 |
| 9 |  |
 |
40 |
| 10 |  |
 |
75 |
| 11 |  |
 |
66 |
| 12 |  |
 |
67 |
| 13 |  |
 |
75 |
| 14 |  |
 |
84 |
| 15 |  |
 |
52 |
| 16 |  |
 |
84 |
| 17 |  |
 |
86 |
| 18 | 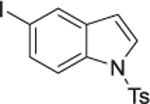 |
 |
35 |
Amide 0.5 mmol, hexafluoroisopropanol 3.5 mL. Yields are iso;ated yields. Py = 1-pyridyl. Please see Supporting Information for details.
Next, we investigated the possibility of room-temperature reaction and low catalyst loadings (Scheme 1). Arylation of ylide 16 was successful by using 1 mol % Pd(OAc)2 catalyst if an acetyl-protected aminoethyl phenyl thioether was added.2e Room-temperature arylation was also successful in the presence of this ligand.
Scheme 1.

Reactions with Low Catalyst Loading and at Room Temperature
Reaction scope with respect to amide coupling component was investigated subsequently (Table 3). The arylation proceeded exclusively at the methyl group for the 2-methylbutyric acid derivative (entry 1). The phenyl-substituted compound afforded product in 91% yield (entry 2). Arylation of isobutyric acid amide gave a mixture of diarylation and monoarylation products in 58% and 37% yields, respectively (entry 3). The reaction is compatible with trifluoromethyl and methoxy substitution on the amide (entries 4 and 6). More hindered substrates, such as cyclohexyl-substituted amide, are reactive as well, giving product in 83% yield (entry 5). Interestingly, the phthaloyl-protected alanine derivative affords arylation product in a reasonable yield, and no loss of enantiomeric excess was observed (entry 7). No arylation of secondary C(sp3)–H bonds was observed when the reaction was carried out under the standard conditions.
Table 3.
Arylation Scope with Respect to Amidesa



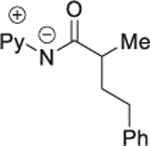


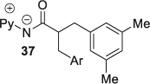








Amide 0.5 mmol, hexafluoroisopropanol 3.5 mL. Yields are isolated yields. Please see Supporting Information for details.
Ar = 3,5-Me2C6H3. Monoarylation product also isolated (37% yield).
Arylation of starting material (92% ee) gave 41 (92% ee).
2.3. Arylation of Secondary C–H Bonds.
Yu has shown that addition of mono-N-protected amino acid ligands allows palladium-catalyzed arylation of secondary, unactivated sp3 C–H bonds even when weak directing groups are used.5a Inspired by this precedent, we investigated the effect of mono-N-protected amino acid ligands on secondary C(sp3)–H arylation. After considerable optimization, it was discovered that addition of N-acetyl-L-phenyalanine ligand afforded optimal reactivity. The results of secondary C(sp3)–H arylation are presented in Table 4. A higher loading of palladium acetate and use of silver trifluoroacetate additive is required to obtain good yields of products. The butanoic acid derivative afforded product in 67% isolated yield (entry 1). Cyclic substrates can be arylated as well. Cyclohexanecarboxylic acid amide gave two monoarylation diastereoisomers in 92% yield with a crude diastereomer ratio of 4:1 (entry 2). The cyclopentanecarboxylic acid derivative is monoarylated in 85% yield (entry 3), while cyclobutanecarboxylic acid amide reacts to produce diarylated product in 82% yield (entry 4). Interestingly, β-methylene C(sp3)-H bond arylation of a protected amino acid occurs in reasonable yield (entry 5), and single product diastereomer was obtained, consistent with results obtained with an aminoquinoline directing group.15
Table 4.
N-Acetyl-L-phenyalanine-Enabled Secondary C(sp3)−H Arylationa











Amide 0.5 mmol, hexafluoroisopropanol 3.5 mL. Yields are isolated yields. Please see Supporting Information for details. Ar = 4-FC6H4.
Enantiomeric excess: 25%.
Cis/trans 4.6/1.
Enantiomeric excess: 33% for the cis isomer.
Enantiomeric excess: 23%.
As shown above, unsubstituted iminopyridinium ylide auxiliary is about as efficient in directing C–H bond functionalization as Yu’s perfluoroarylamides,5a requiring added ligands to effect arylation of methylene groups. Assuming a catalytic cycle similar to that proposed for aminoquinoline amides,8b,15 it is likely that a more electron-rich substrate/ligand will facilitate oxidation, forming Pd(III) (or Pd(IV)) species, irrespective of whether the reaction proceeds via Pd(IV) or dimeric Pd(III) complexes.16,17 Furthermore, while the electronic effect of coordinated ligand/substrate on the C–H activation step is unclear, in some cases strong σ-donor ligands allow for efficient cyclometalation.18 Consequently, a strongly donating auxiliary may increase the efficiency of C–H arylation. Functionalization of 1-aminopyridine ylide butyramides possessing an electron-donor substituent was investigated (Scheme 2). As predicted, strongly donating pyrrolidinopyridine-substituted ylide 50 gave the highest product yield, while methoxy-and dimethylamino-substituted 48 and 49 afforded lower conversions. Pyridine derivative 47 did not react under these conditions. The reactivities track the corresponding pyridine catalytic efficiencies in alcohol acylation reactions.19
Scheme 2.

Arylation of Substituted Ylides
After a short optimization, the scope of acylated 4-pyrrolidinopyridine ylide arylation was investigated (Table 5). The butyryl derivative was arylated in a 68% yield (entry 1). Alkyl groups possessing a phenyl (entry 2), cyclohexyl (entry 3), or branched chains (entry 4) were arylated in good yields. For some substrates, increasing the palladium acetate loading to 10 mol % gives higher yields (entries 2 and 4). Cyclic structures, such as ylides derived from cyclohexane-and cyclobutanecarboxylic acids, are arylated in good yields (entries 5 and 6). In those cases, cis-products are obtained either predominately (entries 5 and 7) or exclusively (entry 6). Amide-containing substrates also react, albeit at higher palladium loading (entry 7). Fluoro-and trifluoromethyl functionalities are compatible with reaction conditions, and products were isolated in acceptable yields (entries 8 and 9).
Table 5.
Secondary C(sp3)−H Arylation without Added External Ligandsa















Amide 0.5 mmol, hexafluoroisopropanol 3.5 mL. Yields are isolated yields. Please see Supporting Information for details. Ar = 4-FC6H4.
Ten mol % Pd(OAc)2.
Cis/trans 5/1.
Diastereomer ratio: 1.6/1; 36 h, 110 °C.
36 h, 110 °C.
Diastereomer ratio: 1.5/1; 36 h, 110 °C. PPY = 4-pyrrolidino-1-pyridyl.
2.4. Alkylation of C–H Bonds.
Alkylation of C–H bonds was also successful (Scheme 3). Reactions require use of dibenzyl phosphate silver salt additive.20a Ethyl iodoacetate reacts with a propionic acid derivative to give an 82% yield of 62. Similarly, reactions with trifluoromethyl-and alkyl-substituted ylides afford products in acceptable to good yields (63 to 65). Benzyl ester gives alkylation product 66 in a reduced 14% yield. Methyl iodide is a competent alkylation agent, and 67 was obtained in 51% yield.
Scheme 3.

Alkylation of C−H Bonds
2.5. Directing Group Removal.
The pyridinium moiety can be easily removed through zinc-mediated N–N bond cleavage. In the case of a 4-pyrrolidinopyridinium auxiliary, magnesium was used instead of zinc (Scheme 4). Additionally, the directing group can be cleaved by Lewis acid promoted methanolysis with no erosion of enantiomeric excess (41 to 70).20b
Scheme 4.

Directing Group Removal
2.6. Mechanistic Considerations.
A presumed C–H functionalization intermediate 72 was formed by reaction of amide 71 with palladium acetate in acetonitrile, isolated, and fully characterized (Scheme 5). It exists as a green-yellow solid that is soluble in hydroxylic solvents, is insoluble in acetonitrile, and decomposes in dichloromethane, forming a chloro-bridged complex. The structure of 72 was verified by single-crystal X-ray diffraction analysis. The ORTEP diagram of 72 is shown in Figure 2. Complex 72 crystallizes in monoclinic space group C2/c and exists as a dimer in the solid state with a Pd-Pd distance of 2.8821(3) Å, which is shorter than the van der Waals radii sum of 3.26 A, signifying a Pd-Pd bonding interaction.21 The palladium–C(alkyl) bond length is 2.007(2) Å, while the palladium–N(amide) distance is 1.984(2) Å. The N(1)–Pd(l)–C(1) angle is 80.17(21)°, showing distortion from an idealized square-planar geometry at the palladium center. It appears that no transition metal complexes of acylated 1-aminopyridine ylides have been crystallographically characterized.22
Scheme 5.

Cyclometalated Intermediate
Figure 2.

ORTEP view of 72. Selected interatomic distances (Å) and angles (deg): Pd1-N1 1.984(2); Pd1-C1 2.007(2); Pd1-Pd1 2.8821(3); N1-N2 1.415(3); N1-Pd1-C1 80.17(10); C1-Pd1-O2 94.77(10).
Our attempts to oxidize 72 to a Pd(III) or Pd(IV) complex with a range of oxidants such as halogens and peroxides were not successful, and ligand loss from palladium was observed. Reactions with 4-fluorophenyl iodide were more informative. Complex 72 did not react with aryl iodide in methanol at 25 °C on a time scale of days. However, reactions in hexafluoroisopropanol were successful (Table 6).
Table 6.
Reactivity of 72 with Aryl Iodidea
 | |||||
|---|---|---|---|---|---|
| Entry | solvent | additive | 73 (%) | 74 (%) | 75 (%) |
| 1 | HFIP | none | 44 | 21 | 0 |
| 2 | HFIP | AgOCOCF3 | 16 | 38 | 11 |
| 3 | HFIP | NaOCOCF3 | 36 | 25 | 2 |
| 4 | HFIP | NaOTf | 39 | 24 | 2 |
| 5 | MeOH | AgOCOCF3 | 0 | 0 | 0 |
| 6 | HFIP | CH3CN AgOCOCF3 | 0 | 0 | 0 |
| 7b | HFIP | none | 44 | 28 | 5 |
| 8b | HFIP | AgOCOCF3 | 11 | 41 | 26 |
Time: 1 h, trifluoroacetate or triflate additive: 1.2 equiv, acetonitrile additive: 7.5 equiv. Yield determined by NMR with 1,1,2,2-tetrachloroethane internal standard.
Time: 24 h.
Reaction in hexafluoroisopropanol solvent for one hour gave a mixture of mono-and diarylation products (entry 1). After 24 h, some triarylation product was observed (entry 7). Addition of silver trifluoroacetate increased the rate of arylation (entry 2), while sodium trifluoroacetate and triflate had little effect on the reaction (entries 3 and 4). No arylation was observed in methanol solvent even in the presence of silver trifluoroacetate (entry 5). Addition of acetonitrile shut down the reaction completely (entry 6). Acetonitrile presumably breaks up the dimeric structure of 72. After 24 h, reaction with added silver salt gave a mixture of arylation products (entry 8). Furthermore, it appears that cyclometalation is irreversible (Scheme 6). No H/D exchange in the methyl group was observed if amide 76 was heated under the catalytic conditions with added acetic acid-d4. If amide 76 was arylated in HFIP-d2, deuterium was not incorporated in the methylene group of the recovered starting material. This is distinct from the corresponding reactivity of aminoquinoline amides, where H/D exchange is observed.8b,15 Furthermore, C–H functionalizations directed by bidentate, monoanionic auxiliaries proceed in coordinating solvents such as acetonitrile.8b While extensive mechanistic speculations are premature at this point, pyridine ylide-directed C–H functionalizations may proceed via dimeric species, as evidenced by the shutting down of the reaction when coordinating solvents are added.23
Scheme 6.

H/D Exchange Experiments
3. SUMMARY
We report here that 1-aminopyridinium ylides are efficient directing groups for palladium-catalyzed β-arylation and alkylation of sp3 C–H bonds in carboxylic acid derivatives. The unsubstituted pyridine-derived ylides can direct function-alization of primary C–H bonds. Addition of external ligands allows for functionalization of methylene groups. Directing group efficiency is improved if electron-donor substituents are introduced at the 4-position of the pyridine ring. Thus, 4-pyrrolidinopyridine-containing ylides are capable of C–H functionalization of acyclic methylene groups in the absence of external ligands. Preliminary mechanistic studies have been performed. A cyclopalladated intermediate was isolated, characterized by X-ray crystallography, and its reactions were studied. Aminopyridine ylide directing groups may find extensive use in new methodology development as well as in synthesis of natural products, supplementing current applications of bidentate, monoanionic auxiliaries.
4. EXPERIMENTAL SECTION
4.1. General Procedure for the Arylation Directed by a 1-Amino-4-pyrrolidinopyridine Auxiliary.
A 8-dram vial equipped with a magnetic stir-bar was charged with amide (0.5 mmol), 1-fluoro-4-iodobenzene (278 mg, 1.25 mmol), Pd(OAc)2 (5.7 mg, 5 mol %), AgOCOCF3 (133 mg, 1.2 equiv), NaOTf (86 mg, 1.0 equiv), and hexafluoroisopropanol (3.5 mL). The mixture was stirred at room temperature for 5 min, covered with aluminum foil, and then heated at 90 °C for 24 h. After completion, the reaction was cooled to room temperature and diluted with a CH2 Cl2/MeOH mixture (5 mL, 9:1). The suspension was filtered through a pad of Celite, and the solid phase was washed with dichloromethane (2 × 20 mL). The filtrate was concentrated under reduced pressure. The residue was subjected to column chromatography on silica gel using an appropriate eluent. After purification, the product was dried under reduced pressure.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Welch Foundation (Chair No. E-0044) and NIGMS (Grant No. R01GM077635) for supporting this research. We are grateful to Dr. Xiqu Wang for collecting and solving the X-ray structure of 72 and to Dr. Rana M. Kashif Khan for initial experiments with a 1-aminopyridine auxiliary.
Footnotes
Supporting Information
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b06643.
Crystallographic data for 72 (CIF)
Detailed experimental procedures and characterization data for new compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1) (a).Chu JCK; Rovis T. Complementary Strategies for Directed C(sp3)-H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem. Int. Ed 2018, 57, 62–101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Parasram M; Gevorgyan V. Silicon-Tethered Strategies for C-H Functionalization Reactions. Acc. Chem. Res 2017, 50, 2038–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Labinger JA Platinum-Catalyzed C-H Functionalization. Chem. Rev 2017, 117, 8483–8496. [DOI] [PubMed] [Google Scholar]; (d) Rej S; Chatani N. Rhodium-Catalyzed C(sp2)-or C(sp3) -H Bond Functionalization Assisted by Removable Directing Groups. Angew. Chem., Int. Ed 2019, 58, 8304–8329. [DOI] [PubMed] [Google Scholar]; (e) He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Palladium-Catalyzed Transformations of Alkyl C-H Bonds. Chem. Rev 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) He G; Wang B; Nack WA; Chen G. Syntheses and Transformations of α-Amino Acids via Palladium-Catalyzed Auxiliary-Directed sp3 C-H Functionalization. Acc. Chem. Res 2016, 49, 635–645. [DOI] [PubMed] [Google Scholar]; (g) Hartwig JF Evolution of C-H Bond Functionalization from Methane to Methodology. J. Am. Chem. Soc 2016,138, 2–24. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Wang W; Lorion MM; Shah J; Kapdi AR; Ackermann L. Late-stage peptide diversification by position-selective C-H activation. Angew. Chem. Int. Ed 2018, 57, 14700–14717. [DOI] [PubMed] [Google Scholar]; (i) Xu Y; Dong G. sp3 C-H activation via exo-type directing groups. Chem. Sci 2018, 9, 1424–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Baudoin O. Ring Construction by Palladium(0)-Catalyzed C(sp3)-H Activation. Acc. Chem. Res 2017, 50, 1114–1123. [DOI] [PubMed] [Google Scholar]
- (2) (a).Giri R; Maugel N; Li J-J; Wang D-H; Breazzano SP; Saunders LB; Yu J-Q Palladium-Catalyzed Methylation and Arylation of sp2 and sp3 C-H Bonds in Simple Carboxylic Acids. J. Am. Chem. Soc 2007, 129, 3510–3511. [DOI] [PubMed] [Google Scholar]; (b) Wasa M; Engle KM; Yu J-Q Pd(0)/PR3-Catalyzed Intermolecular Arylation of sp3 C-H Bonds. J. Am. Chem. Soc 2009, 131, 9886–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang D-H; Wasa M; Giri R; Yu J-Q Pd(II)-Catalyzed Cross-Coupling of sp3 C-H Bonds with sp2 and sp3 Boronic Acids Using Air as the Oxidant. J. Am. Chem. Soc 2008, 130, 7190–7191. [DOI] [PubMed] [Google Scholar]; (d) Kapoor M; Liu D; Young MC Carbon Dioxide-Mediated C(sp3)-H Arylation of Amine Substrates. J. Am. Chem. Soc 2018, 140, 6818–6822. [DOI] [PubMed] [Google Scholar]; (e) Zhuang Z; Yu C-B; Chen G; Wu Q-F; Hsiao Y; Joe CL; Qiao JX; Poss MA; Yu J-Q Ligand-Enabled β-C(sp3)-H Olefination of Free Carboxylic Acids. J. Am. Chem. Soc 2018, 140, 10363–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3) (a).Desai LV; Hull KL; Sanford MS Palladium-Catalyzed Oxygenation of Unactivated sp3 C-H Bonds. J. Am. Chem. Soc 2004, 126, 9542–9543. [DOI] [PubMed] [Google Scholar]; (b) Giri R; Chen X; Yu J-Q Palladium-Catalyzed Asymmetric Iodination of Unactivated C-H Bonds under Mild Conditions. Angew. Chem. Int. Ed 2005, 44, 2112–2115. [DOI] [PubMed] [Google Scholar]; (c) Chen X; Goodhue CE; Yu J-Q Palladium-Catalyzed Alkylation of sp2 and sp3 C-H Bonds with Methylboroxine and Alkylboronic Acids: Two Distinct C-H Activation Pathways. J. Am. Chem. Soc 2006, 128, 12634–12635. [DOI] [PubMed] [Google Scholar]; (d) Stowers KJ; Fortner KC; Sanford MS Aerobic Pd-Catalyzed sp3 C-H Olefination: A Route to Both N-Heterocyclic Scaffolds and Alkenes. J. Am. Chem. Soc 2011, 133, 6541–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) He C; Gaunt MJ Ligand-Enabled Catalytic C-H Arylation of Aliphatic Amines by a Four-Membered-Ring Cyclopalladation Pathway. Angew. Chem. Int. Ed 2015, 54, 15840–15844. [DOI] [PubMed] [Google Scholar]; (f) Calleja J; Pla D; Gorman TW; Domingo V; Haffemayer B; Gaunt MJ A Steric Tethering Approach Enables Palladium-Catalysed C-H Activation of Primary Amino Alcohols. Nat. Chem 2015, 7, 1009–1016. [DOI] [PubMed] [Google Scholar]; (g) Desai LV; Stowers KJ; Sanford MS Insights into Directing Group Ability in Palladium-Catalyzed C-H Bond Functionalization. J. Am. Chem. Soc 2008, 130, 13285–13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4) (a).Cabrera-Pardo JR; Trowbridge A; Nappi M; Ozaki K; Gaunt MJ Selective Palladium(II)-Catalyzed Carbonylation of Methylene β-C–H Bonds in Aliphatic Amines. Angew. Chem. Int. Ed 2017, 56, 11958–11962. [DOI] [PubMed] [Google Scholar]; (b) Gulia N; Daugulis O. Palladium-Catalyzed Pyrazole-Directed sp3 C-H Bond Arylation for the Synthesis of beta-Phenethylamines. Angew. Chem. Int. Ed 2017, 56, 3630–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5) (a).Saint-Denis TG; Zhu R-Y; Chen G; Wu Q-F; Yu J-Q Enantioselective C(sp3)-H bond activation by chiral transition metal catalysts. Science 2018, 359, eaao4798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Newton CG; Wang S-G; Oliveira CC; Cramer N. Catalytic Enantioselective Transformations Involving C-H Bond Cleavage by Transition-Metal Complexes. Chem. Rev 2017, 117, 8908–8976. [DOI] [PubMed] [Google Scholar]; (c) Chu L; Xiao K-J; Yu J-Q Room-temperature enantioselective C-H iodination via kinetic resolution. Science 2014, 346, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Daugulis O; Roane J; Tran LD Bidentate, Monoanionic Auxiliary-Directed Functionalization of Carbon-Hydrogen Bonds. Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rouquet G; Chatani N. Catalytic functionalization of C(sp2)-H and C(sp3)-H bonds by using bidentate directing groups. Angew. Chem. Int. Ed 2013, 52, 11726–11743. [DOI] [PubMed] [Google Scholar]
- (7) (a).Hoshiya N; Kobayashi T; Arisawa M; Shuto S. Palladium-Catalyzed Arylation of Cyclopropanes via Directing Group-Mediated C(sp3)-H Bond Activation To Construct Quaternary Carbon Centers: Synthesis of cis-and trans-1,1,2-Trisubstituted Chiral Cyclopropanes. Org. Lett 2013, 15, 6202–6205. [DOI] [PubMed] [Google Scholar]; (b) Parella R; Arulananda Babu S. Palladium-catalyzed double activation and arylation of 2° and 3° C(sp3)-H bonds of the norbornane system: formation of a C-C bond at the bridgehead carbon and bridgehead quaternary stereocenter. Synlett 2014, 25, 1395–1402. [Google Scholar]
- (8) (a).Zaitsev VG; Shabashov D; Daugulis O. Highly Regioselective Arylation of sp3 C-H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc 2005, 127, 13154–13155. [DOI] [PubMed] [Google Scholar]; (b) Shabashov D; Daugulis O. Auxiliary-Assisted Palladium-Catalyzed Arylation and Alkylation of sp2 and sp3 Carbon-Hydrogen Bonds. J. Am. Chem. Soc 2010, 132, 3965–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9) (a).Chen F-J; Zhao S; Hu F; Chen K; Zhang Q; Zhang S-Q; Shi B-F Pd(II)-Catalyzed Alkoxylation of Unactivated C(sp3)-H and C(sp2)-H Bonds Using a Removable Directing Group: Efficient Synthesis of Alkyl Ethers. Chem. Sci 2013, 4, 4187–4192. [Google Scholar]; (b) Gu Q; Al Mamari HH; Graczyk K; Diers E; Ackermann L. Iron-Catalyzed C(sp2)-H and C(sp3)-H Arylation by Triazole Assistance. Angew. Chem., Int. Ed 2014, 53, 3868–3871. [DOI] [PubMed] [Google Scholar]; (c) Poveda A; Alonso I; Fernández-Ibáñez MA Experimental and Computational Studies on the Mechanism of the Pd-Catalyzed C(sp3)-H γ-Arylation of Amino Acid Derivatives Assisted by the 2-Pyridylsulfonyl Group. Chem. Sci 2014, 5, 3873–3882. [Google Scholar]; (d) Ye X; He Z; Ahmed T; Weise K; Akhmedov NG; Petersen JL; Shi X. 1,2,3-Triazoles as Versatile Directing Group for Selective sp2 and sp3 C-H Activation: Cyclization vs Substitution. Chem. Sci 2013, 4, 3712–3716. [Google Scholar]; (e) Fan M; Ma D. Palladium-Catalyzed Direct Functionalization of 2-Aminobutanoic Acid Derivatives: Application of a Convenient and Versatile Auxiliary. Angew. Chem. Int. Ed 2013, 52, 12152–12155. [DOI] [PubMed] [Google Scholar]; (f) Wang C; Chen C; Zhang J; Han J; Wang Q; Guo K; Liu P; Guan M; Yao Y; Zhao Y. Easily accessible auxiliary for palladium-catalyzed intramolecular amination of C(sp2)-H and C(sp3)-H bonds at δ-and ε-positions. Angew. Chem. Int. Ed 2014, 53, 9884–9888. [DOI] [PubMed] [Google Scholar]; (g) Topczewski JT; Cabrera PJ; Saper NI; Sanford MS Palladium-Catalysed Transannular C–H Functionalization of Alicyclic Amines. Nature 2016, 531, 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Hao X-Q; Chen L-J; Ren B; Li L-Y; Yang X-Y; Gong J-F; Niu J-L; Song M-P Copper-Mediated Direct Aryloxylation of Benzamides Assisted by an N,O-Bidentate Directing Group. Org. Lett 2014, 16, 1104–1107. [DOI] [PubMed] [Google Scholar]
- (10) (a).Zhang F-L; Hong K; Li T-J; Park H; Yu J-Q Functionalization of C(sp3)-H bonds using a transient directing group. Science 2016, 351, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu Y; Young MC; Wang C; Magness DM; Dong G. Catalytic C(sp3)-H Arylation of Free Primary Amines with an exo Directing Group Generated In Situ. Angew. Chem., Int. Ed 2016, 55, 9084–9087. [DOI] [PubMed] [Google Scholar]; (c) Yang K; Li Q; Liu Y; Li G; Ge H. Catalytic C-H Arylation of Aliphatic Aldehydes Enabled by a Transient Ligand. J. Am. Chem. Soc 2016, 138, 12775—12778. [DOI] [PubMed] [Google Scholar]; (d) St John-Campbell S; White AJP; Bull JA Single operation palladium catalysed C(sp3) -H functionalisation of tertiary aldehydes: investigations into transient imine directing groups. Chem. Sci 2017, 8, 4840–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chen Y-Q; Wang Z; Wu Y; Wisniewski SR; Qiao JX; Ewing WR; Eastgate MD; Yu J-Q Overcoming the Limitations of γ-and δ-C-H Arylation of Amines through Ligand Development. J. Am. Chem. Soc 2018, 140, 17884—17894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Dias SA; Downs AW; McWhinnie W. R Metal-ylide complexes. I. Metallation reaction of N-(1-pyridinio) benzamidate and related compounds with palladium(II), platinum(II), rhodium-(III), and iridium(III). J. Chem. Soc. Dalton Trans. 1975, 162–71. [Google Scholar]
- (12) (a).Qiu G; Kuang Y; Wu J. N-Imide Ylide-Based Reactions: C-H Functionalization, Nucleophilic Addition and Cycloaddition. Adv. Synth. Catal 2014, 356, 3483–3504. [Google Scholar]; (b) Larivée A; Mousseau JJ; Charette AB Palladium-Catalyzed Direct C—H Arylation of N-Iminopyridinium Ylides: Application to the Synthesis of (±)-Anaba-sine. J. Am. Chem. Soc 2008, 130, 52–54. [DOI] [PubMed] [Google Scholar]; (c) Mousseau JJ; Larivée A; Charette AB Palladium-Catalyzed Benzylic C—H Insertion of 2-Substituted N-Iminopyridinium Ylides. Org. Lett 2008, 10, 1641 — 1643. [DOI] [PubMed] [Google Scholar]
- (13) (a).Beck JC; Lacker CR; Chapman LM; Reisman SE A modular approach to prepare enantioenriched cyclobutanes: synthesis of (+)-rumphellaone A. Chem. Sci 2019, 10, 2315–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kinsinger T; Kazmaier U. C-H functionalization of N-methylated amino acids and peptides as tool in natural product synthesis: Synthesis of Abyssenine A and Mucronine E. Org. Lett 2018, 20, 7726–7730. [DOI] [PubMed] [Google Scholar]; (c) Maetani M; Zoller J; Melillo B; Verho O; Kato N; Pu J; Comer E; Schreiber SL Synthesis of a Bicyclic Azetidine with In Vivo Antimalarial Activity Enabled by Stereo-specific, Directed C(sp3) -H Arylation. J. Am. Chem. Soc 2017, 139, 11300—11306. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chapman LM; Beck JC; Wu L; Reisman SE Enantioselective Total Synthesis of (+)-Psiguadial B. J. Am. Chem. Soc 2016, 138, 9803–9806. [DOI] [PubMed] [Google Scholar]; (e) Feng Y; Chen G. Total Synthesis of Celogentin C by Stereo-Selective C-H Activation. Angew. Chem., Int. Ed 2010, 49, 958–961. [DOI] [PubMed] [Google Scholar]; (f) Gutekunst WR; Baran PS Total Synthesis and Structural Revision of the Piperarborenines via Sequential Cyclobutane C-H Arylation. J. Am. Chem. Soc 2011, 133, 19076—19079. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ting CP; Maimone TJ C-H Bond Arylation in the Synthesis of Aryltetralin Lignans: A Short Total Synthesis of Podophyllotoxin. Angew. Chem. Int. Ed 2014, 53, 3115–3119. [DOI] [PubMed] [Google Scholar]; (h) Liu Z; Wang Y; Wang Z; Zeng T; Liu P; Engle KM Catalytic Intermolecular Carboamination of Unactivated Alkenes via Directed Aminopalladation. J. Am. Chem. Soc 2017, 139, 11261—11270. [DOI] [PubMed] [Google Scholar]
- (14).Shabashov D; Daugulis O. Catalytic Coupling of C-H and C-I Bonds Using Pyridine As a Directing Group. Org. Lett 2005, 7, 3657–3659. [DOI] [PubMed] [Google Scholar]
- (15).Tran LD; Daugulis O. Nonnatural Amino Acid Synthesis by Carbon-Hydrogen Bond Functionalization Methodology. Angew. Chem., Int. Ed 2012, 51, 5188–5191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16) (a).Powers DC; Geibel MAL; Klein JEMN; Ritter T. Bimetallic Palladium Catalysis: Direct Observation of Pd(III)—Pd(III) Intermediates. J. Am. Chem. Soc 2009, 131, 17050—17051. [DOI] [PubMed] [Google Scholar]; (b) Deprez NR; Sanford MS Synthetic and Mechanistic Studies of Pd-Catalyzed C—H Arylation with Diaryliodonium Salts: Evidence for a Bimetallic High Oxidation State Pd Intermediate. J. Am. Chem. Soc 2009, 131, 11234—11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vicente J; Arcas A; Juliá-Hernández F; Bautista D. Synthesis of a palladium(IV) complex by oxidative addition of an aryl halide to palladium(II) and its use as precatalyst in a C-C coupling reaction. Angew. Chem. Int. Ed 2011, 50, 6896–6899. [DOI] [PubMed] [Google Scholar]
- (18) (a).Bowen RJ; Fernandes MA; Layh M. Synthesis and Crystal Structures of Novel Lithium-and Palladium-1-azaallyls. J. Organomet. Chem 2004, 689, 1230–1237. [Google Scholar]; (b) Ren Z; Dong G. Direct Observation of C-H Cyclopalladation at Tertiary Positions Enabled by an Exo-Directing Group. Organometallics 2016, 35, 1057–1059. [Google Scholar]
- (19).Scriven EFV 4-Dialkylaminopyridines: super acylation and alkylation catalysts. Chem. Soc. Rev 1983, 12, 129–61. [Google Scholar]
- (20) (a).Zhang S-Y; He G; Nack WA; Zhao Y; Li Q; Chen G. Palladium-Catalyzed Picolinamide-Directed Alkylation of Unactivated C(sp3)-H Bonds with Alkyl Iodides. J. Am. Chem. Soc 2013, 135, 2124–2127. [DOI] [PubMed] [Google Scholar]; (b) Miltsov S; Rivera L; Encinas C; Alonso J. Boron trifluoride—methanol complex—mild and powerful reagent for deprotection of labile acetylated amines. Tetrahedron Lett. 2003, 44, 2301–2303. [Google Scholar]
- (21).Bondi A. van der Waals Volumes and Radii. J. Phys. Chem 1964, 68, 441–451. [Google Scholar]
- (22).A Zn complex: Molina A; de las Heras MA; Martinez Y; Vaquero JJ; Navio JLG; Alvarez-Builla J; Gomez-Sal P; Torres R. Synthesis and structure of complexes of acyl N-aminides with zinc(II) salts. Tetrahedron 1997, 53, 6411–6420. [Google Scholar]
- (23).Complex 72 shows the presence of several rapidly interconverting species in hexafluoropropanol-d2 (1H NMR analysis, ambient temperature). Cooling to 0 °C did not simplify the spectra, and lower temperatures could not be achieved, as HFIP crystallizes at —4 °C. Addition of 15 equiv of CD3CN simplified the spectrum, presumably due to formation of the acetonitrile complex.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.