

Abstract
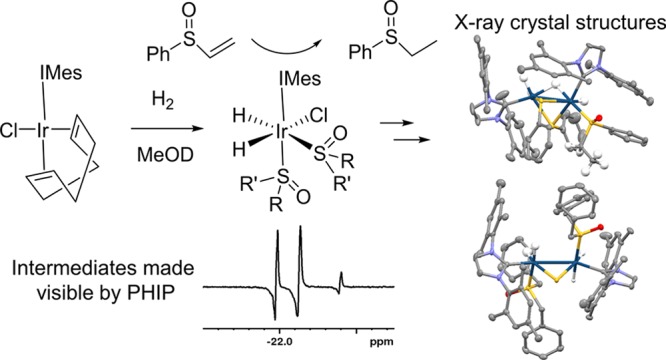
Vinyl sulfoxides are an important functional group used in a wide range of organic transformations. Here, we use [IrCl(COD)(IMes)] where IMes = 1,3-bis(2,4,6-trimethyl-phenyl)imidazole-2-ylidene and COD = cis,cis-1,5-cyclooctadiene to rapidly hydrogenate phenylvinylsulfoxide. We use para-hydrogen-induced hyperpolarization (PHIP) to follow this reaction with [IrCl(H)2(IMes)(S(O)(Ph)(Et))2] dominating in the later stages. Decomposition to form the reduced C–S bond cleavage product [Ir2(H)3(κ2-H)(κ2-SPh)2(IMes)2(S(Et)(Ph)O)] limits turnover. The related product [Ir2(H)4(κ2-S)(IMes)2(S(O)(CH2Ph)2)2] is formed from dibenzylsulfoxide, demonstrating the wider utility of this transformation.
Sulfoxides are present in many organic compounds that find uses as laboratory solvents, pharmaceutical drugs,1 synthetic agents,2 and ligands for metal binding.3 This key functional group is of great interest in asymmetric synthesis due to the chirality of the central sulfur atom.4 The preparation of chiral sulfoxides in an enantiomerically pure form is possible due to the large barrier to inversion of the tetrahedral sulfur atom.5 Vinyl sulfoxides contain conjugated S=O and alkene groups, which can be used in a wide range of organic transformations. Examples include uses as acetylene equivalents in cycloaddition reactions and as Michael acceptors.6 The hydrogenation of vinylsulfoxides, either by transition metal catalysts or free radicals, has also been reported.7,8 Indeed, when chiral hydrogenation catalysts are used, hydrogenated sulfoxides can be prepared in enantiomeric excess.8 These metal-mediated hydrogenations are often facilitated by initial sulfoxide coordination to an active catalyst that in many cases involves rhodium. The mechanisms of these Rh-catalyzed hydrogenation processes have been investigated using NMR spectroscopy and/or high level density functional theory calculations.7−9
Reaction mechanism and low concentration intermediates can also be powerfully studied using a technique called para-hydrogen-induced polarization (PHIP).10,11 Short lived or low concentration species cannot routinely be observed by NMR spectroscopy due to its inherent low sensitivity. However, the PHIP approach creates the necessary non-Boltzmann population distributions within a target molecule by employing para-hydrogen (p-H2), a singlet spin isomer of dihydrogen. The NMR signals derived from these p-H2 hydrogenated products can be up to 3 orders of magnitude larger than those when recorded under Boltzmann conditions.10,11 PHIP belongs to a larger family of hyperpolarization techniques that include dynamic nuclear polarization (DNP) and spin exchange optical pumping (SEOP), which have now produced hyperpolarized molecules suitable for detection in vivo.12,13 Although PHIP has also produced hyperpolarized biomolecules for in vivo detection14,15 it is more commonly used to probe reaction mechanisms, with the sensitivity gains it provides having allowed low concentration catalytic intermediates in Rh-catalyzed alkene hydrogenation reactions to be detected.16−19 H2 activation by frustrated Lewis pairs reflects another area of successful study using PHIP.20,21
Signal amplification by reversible exchange (SABRE) is a p-H2-based hyperpolarization method that does not involve the direct hydrogenation of a target molecule.22 Instead, it uses an oxidative addition reaction to break the symmetry of p-H2. Magnetization can then be catalytically transferred from p-H2-derived hydride ligands to a ligated target molecule at low (∼mT) field through the formation of a temporary J-coupled network.23 This approach and its derivatives, SABRE-SHEATH and SABRE-relay, have led to the polarization of a wide range of molecules and functional groups.24,25 SABRE has been used to make low concentration species visible either in mixtures26 or in chemical reactions.27 Hence, SABRE has been used to track and gain mechanistic insight into chemical reactivity.28
In this communication, we use the typical SABRE precatalyst [IrCl(COD)(IMes)] (where IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene and COD = cis,cis-1,5-cyclooctadiene), which has been used to derive high signal gains for N-donor atoms22,29 to hydrogenate phenylvinylsulfoxide. We then use p-H2 to study this hydrogenation reaction and find that [IrCl(H)2(IMes)(sulfoxide)2]-type intermediates can be made visible to NMR. We use 2D NMR techniques to study and characterize the detected complexes and those created when closely related dibenzylsulfoxide is studied. Notably, X-ray diffraction is used to identify two iridium dimers that are formed by C–S bond activation and account for the catalyst deactivations that are associated with these reactions.
Examination of a methanol-d4 solution containing [IrCl(COD)(IMes)] (1) and phenylvinylsulfoxide (A) (4 equiv) at 245 K yields in the first instance 1H NMR resonances corresponding to the two starting materials. There is no visible evidence to suggest that sulfoxide A can displace the chloride ligand of 1 to form [Ir(COD)(IMes)(A)]Cl as is typically seen with many N-donor reagents.22,30 Upon addition of 3 bar H2 to this solution, hydride-containing complexes yielding resonances at δ −13.32, −18.48, and δ −15.75, −21.99 rapidly form, even at 245 K. These low intensity hydride-containing products are unstable, and their proportion decreases when the reaction time is increased. Notably, the hydride resonances at δ −13.32, −18.48 are consistent with the formation of a species of the type [IrCl(H)2(COD)(IMes)], 2, which results from direct hydrogen addition to 1, and their appearance is independent of sulfoxide ligand. Over time, the resonances corresponding to A decrease and are eventually replaced by those of phenylethylsulfoxide, B, as shown in Figure 1. NMR spectroscopy at 245 K alongside mass spectrometry was used to confirm its identity (see the Supporting Information). The formation of B is readily evident from the large chemical shift difference that exists between the associated alkene protons of A at δ 5.97, 6.18, and 6.79, which decrease in intensity, and those of the hydrogenated product B at δ 1.19, 2.89, and 3.09, which grow in intensity. We note that the two CH2 protons in phenylethylsulfoxide, as denoted by the green square in Figure 1, are diastereotopic. Under these conditions, the slow hydrogenation of A is indicated (Figure 1a) such that after 50 min a turnover frequency of 1.01 × 10–4 s–1 can be estimated. Slow hydrogenation is a consequence of the low reaction temperature (245 K). The formation of B is unsurprising as the hydrogenation of this vinylsulfoxide has been reported in the literature.7,8 However, many reported procedures use rhodium catalysts.7,8 Here, we use cheaper iridium-based catalysts to achieve the hydrogenation of a vinylsulfoxide. As a control, we note that a methanol-d4 solution of A is stable under 3 bar H2 at room temperature overnight.
Figure 1.
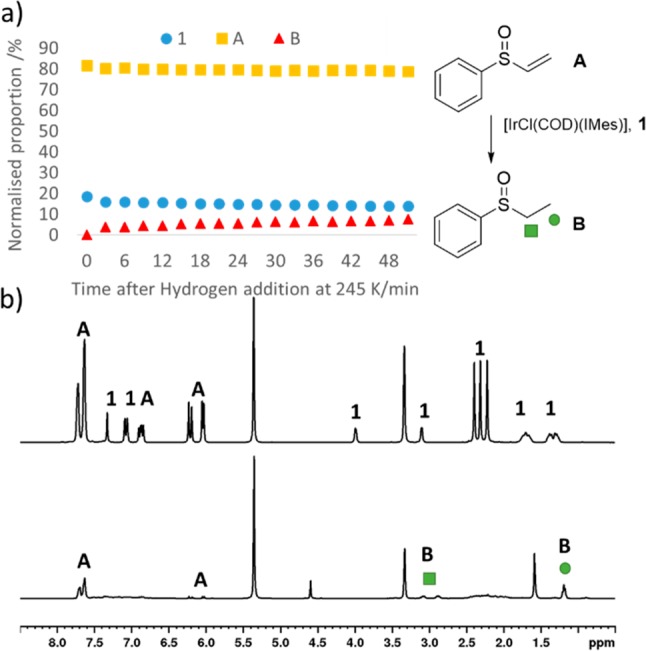
(a) Hydrogenation of A to B catalyzed by 1 can be monitored at 245 K using 1H NMR spectroscopy. (b) 1H NMR spectrum of A and 1 at 245 K prior to H2 addition (top) and the corresponding spectrum after the solution has been left under 3 bar H2 at 245 K for 1 h before increasing to 298 K.
We use the PHIP approach to study the hydrogenation of A by 1 in more detail. A similar methanol-d4 solution of 1 containing 4 equiv of A was therefore exposed to 3 bar p-H2 and immediately shaken for 10 s at 65 G before being placed into a 9.4 T spectrometer. Upon 1H NMR detection at 298 K, hyperpolarized resonances for the CH2 and CH3 groups of B were observed with an ALTADENA-type appearance,31 as shown in Figure 2a. Upon ejecting this sample, immediately adding fresh p-H2, and shaking it for 10 s at 65 G before re-examination, the intensity of these hyperpolarized responses is seen to dramatically decrease. These effects were, however, no longer visible when this sequence of steps is repeated again. This is reflective of the speed of initial hydrogenation, which rapidly consumes both H2 and A; it is so fast that a PHIP response is not seen in a multiple scan measurement. When the hydride region of these single-scan 1H NMR spectra are examined more closely, a pair of strongly hyperpolarized hydride resonances are observed at δ −16.09 and −22.44, which increase in intensity when fresh p-H2 is shaken (see Supporting Information). The species giving rise to these resonances is not visible in the corresponding 1H NMR spectrum recorded when signal strengths are derived from Boltzmann conditions. The complexity of the hydride region of these NMR spectra also increases when monitored after subsequent additions of fresh p-H2.
Figure 2.
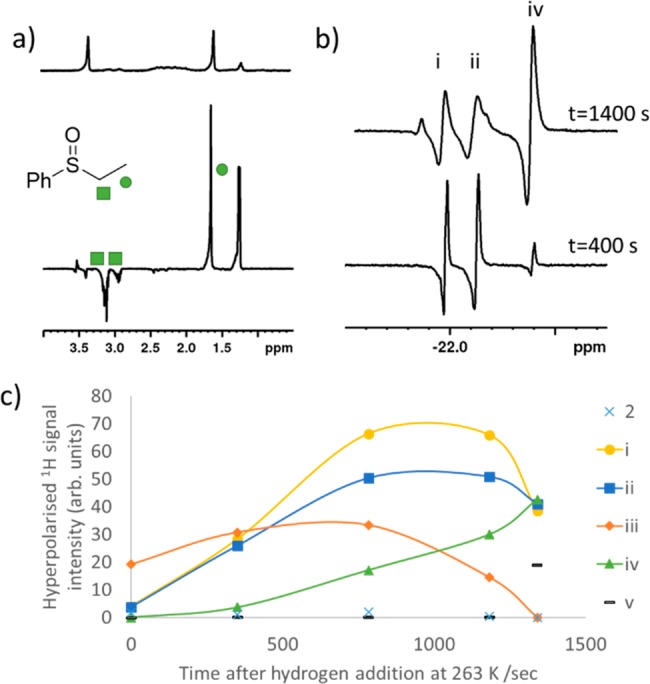
(a) Thermal (top) and hyperpolarized (bottom) 1H spectra recorded using 45° pulses of a solution of 1 and A shaken for 10 s at 65 G immediately after the addition of 3 bar p-H2 at 298 K. (b) Partial hyperpolarized 1H spectra recorded using 45° pulses of a solution of 1 and A shaken for 10 s at 65 G ∼400 and ∼1400 s after initial p-H2 addition at 263 K. Full spectra are given in the Supporting Information. (c) Change in hyperpolarized signals of these hydride-containing species with successive fresh p-H2 shaking can be monitored at 263 K; even though the presented experimental data represent a single sample and, therefore, a series of single-point measurements, we expect them to exhibit a 5% error based on other studies and note the same trends were reproduced by other samples.
We can gain a clearer view of these changes when a fresh solution is monitored in a series of single-scan hyperpolarized 1H NMR spectra at 263 K. The associated measurements start by observing a pair of hydride signals corresponding to 2, which are accompanied by additional sets at δ −15.83, −21.94 (i), δ −15.93, −22.10 (ii), and δ −29.22, −29.58 (iii). After repeating the fresh p-H2 shaking and observation process several times, these resonances are found to decrease in intensity and are replaced by others at δ −16.20, −22.37 (iv) and δ −26.87, −31.28 (v), as shown in Figure 2b,c. The hydride resonances for (iii) and (v) compare well with those previously reported for methanol or water-containing species,32 whereas those of (i), (ii), and (iv) correlate with the chemical shifts previously reported for [IrCl(H)2(DMSO)2(IMes)].33 We therefore expect that these signals arise from similar complexes, which in this case are likely to be of the form [IrCl(H)2(A)2(IMes)] (i) [IrCl(H)2(A)(B)(IMes)] (ii), and [IrCl(H)2(B)2(IMes)] (iv). Based on these observations, we suggest a mechanism for the iridium-catalyzed hydrogenation of A as detailed in Figure 3. It starts with the formation of [IrCl(H)2(IMes)(S(Ο)(CH=CH2)(Ph))2], which is followed by [IrCl(H)2(IMes)(S(Ο)(CH=CH2)(Ph))(S(O)(Et)(Ph))] on the basis of rapid ligand exchange trans to hydride prior to [IrCl(H)2(IMes)(S(O)(Et)(Ph))2].
Figure 3.

Proposed reaction scheme that allows the hydrogenation of vinylsulfoxide A. 4 is a decomposition product observed by X-ray diffraction.
To confirm this behavior, we also investigated dibenzylsulfoxide, C, as a control that does not contain an unsaturated vinyl group. When 3 bar H2 was added to a methanol-d4 solution of 1 and C (4 equiv relative to iridium), the associated reaction proceeds to form a major product that yields 1H NMR signals for two coupled hydride ligands at δ −15.78 and −20.89 (2JHH = 6 Hz). The formation of this species at 245 K was monitored by recording a series of 1H NMR spectra, and the reaction time course is shown in the Supporting Information. NMR characterization of this complex at 245 K reveals it to be [IrCl(H)2(C)2(IMes)], 3. It yields four pairs of diastereotopic CH2 protons due to bound C, as detailed in the Supporting Information. These groups show both COSY and NOESY connections to resonances within the phenyl group of the sulfoxide. NOESY measurements confirmed which resonances arise from the sulfoxide ligand that lies trans to the hydride on the basis of a connection to a carbene ligand CH3 resonance. Full NMR characterization data for 3 are given in the Supporting Information. Its hydride chemical shifts at δ −15.78 and −20.89 are indicative of chemical environments trans to chloride and S-donor ligands, respectively, and correlate well with those of similar complexes.33 We note that addition of A (4 equiv) to a solution of preformed 3 also results in the formation of B, which confirms the lability of the sulfoxide ligands in these complexes necessary for catalysis.
Many of the mechanistic studies that focus on Rh-catalyzed hydrogenation propose intermediates that involve sulfoxide coordination through oxygen rather than sulfur.7,8 In this novel iridium-based system, we suggest that the primary mode of sulfoxide coordination is through the sulfur lone pair. The S-donor binding modes of these sulfoxides in similar iridium(III) complexes have been determined based on the characteristic sulfoxide S–C chemical shift change upon binding.33
When solutions that contain 1 and A in methanol-d4 are left under 3 bar H2 at 278 K, the growth of single crystals was observed. Unfortunately, subsequent X-ray diffraction revealed they contain [Ir2(H)3(κ2-H)(κ2-SPh)2(IMes)2(S(O)(Et)(Ph))] (4), the structure of which is shown in Figure 4 rather than the mononuclear species shown in Figure 3 This iridium(III)-based dimer contains a phenylethylsulfoxide ligand on one center that supports the hydrogenation of A. This ligand is coordinated through a sulfur lone pair rather than oxygen in accordance with the shorter Ir–S bond distance (2.262 Å compared to a 3.248 Å Ir–O distance). Both iridium centers exhibit distorted octahedral geometries, with an Ir–Ir bond distance of 2.798 Å, comparing well to literature values of 2.826,34 2.823,35 2.734,36 2.642,37 and 2.607 Å,37 which suggests there is a metal–metal single bond. Interestingly, the observation of two bridging thiolate ligands confirms that S–Cα bond breaking occurs within the hydrogenated product. The activation of S–C bonds38−40 and subsequent formation of S-bridged iridium dimers in hydrodesulfurization processes has been reported previously.40 In this case, mass spectrometry and NMR confirmed that S–C bond cleavage is limited to the metal products, and there is no evidence for thiol buildup in solution. Hence, 4 is a minor decomposition product that forms slowly.
Figure 4.
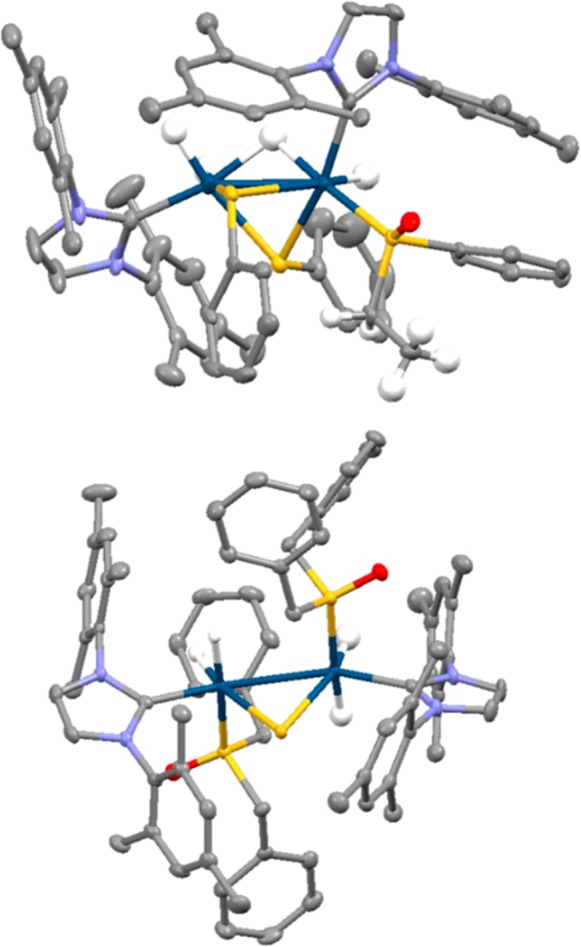
Structures of single crystals of (a) [Ir2(H)3(κ2-H)(κ2-SPh)2(IMes)2(S(O)(Et)(Ph))], 4, and (b) [Ir2(H)4(κ2-S)(IMes)2(S(O)(CH2Ph)2)2], 5, determined from X-ray crystallography. Solvents of crystallization and selected hydrogen atoms have been omitted for clarity.
A related S–C bond cleavage step occurs over long reaction times when solutions of 1 and C in methanol-d4 are left under 3 bar H2 at 278 K. In this case, the associated reaction results in the growth of [Ir2(H)4(κ2-S)(IMes)2(S(O)(CH2Ph)2)2] (5) crystals, the structure of which is also shown in Figure 4. Shorter Ir–S bond distances (2.298 and 2.296 Å) compared to Ir–O distances (3.280 and 3.264 Å) are again seen that indicate sulfur binding. The slightly longer Ir–Ir bond distance of 2.902 Å suggests a weaker metal–metal interaction.
These sulfoxide C–S bond activations are expected to account for catalyst deactivation and the reducing turnover with time. We note that only trace amounts of 4 and 5 crystallized, and that these reflect only some of the many species present in the system at this stage of reaction.
When 4 equiv of A is used relative to 1, 70% of A is hydrogenated after 12 h under 3 bar H2 at 298 K. However, a significant amount of vinylsulfoxide is hydrogenated in the initial stages of the reaction: 30% is converted in the first 15 min, which corresponds to a turnover of 1.2 × 10–3 s–1 in this time period. This suggests that catalyst deactivation caused by S–C bond activation and subsequent dimer formation in these systems is rapid. The initial turnover frequency is comparable to those reported for the hydrogenation of vinylsulfoxide using other Rh-based systems (between 1.4 × 10–3 and 2.2 × 10–2 s–1 in MeOD using 5 bar H2).8 Although these turnovers are not improved by increasing the ratio of A to catalyst (see Supporting Information), in related Rh-based systems, they can be improved by up to 2 orders of magnitude by variation of solvent and the catalyst’s ligands. When hydrogenating a sample of A with a 0.04 mol % catalyst loading relative to substrate at 298 K with 3 bar H2, we calculate a turnover number of 48, which is comparable to those reported with Rh systems (30–70).8 We expect that similar variations in this iridium-based system, including working at higher H2 pressures, higher temperatures, and variation of solvent, could increase these turnovers and turnover numbers.
In conclusion, we have demonstrated that precatalyst [IrCl(COD)(IMes)] is active for the hydrogenation of phenylvinylsulfoxide. This ligand hydrogenation reaction leads to the conversion of initially formed [IrCl(H)2(A)2(IMes)] into [IrCl(H)2(A)(B)(IMes)] and [IrCl(H)2(B)2(IMes)]. We expect that such products are important species in the catalytic hydrogenation of vinylsulfoxides and have observed some of these intermediates using PHIP. However, none of these complexes exhibit long-term stability, with S–C bond cleavage leading to the iridium dimer [Ir2(H)3(κ2-H)(κ2-SPh)2(IMes)2(S(O)(Et)(Ph))]. This lack of S–C bond stability is also evident in the chemistry of dibenzylsulfoxide, which leads first to [IrCl(H)2(sulfoxide)2(IMes)] and then slowly the iridium dimer, [Ir2(H)4(κ2-S)(IMes)2(S(O)(CH2Ph)2)2]. Interestingly, our catalytic system involves S-donor sulfoxide coordination rather than an O-donor. The N-heterocyclic (NHC) ligands used in our catalytic system reflect a structurally diverse range of ligands that are commonly used in SABRE41,42 and other organocatalysis reactions.43 Chiral NHCs can be prepared,44 and the use of such ligands for the enantioselective hydrogenation of sulfoxides may reflect an exciting future application of this work.8
Acknowledgments
We thank Dr. Peter J. Rayner for helpful and insightful discussions, Dr. Victoria Annis for the synthesis of [IrCl(COD)(IMes)], and Dr. R. O. John for experimental assistance. Financial support from the Wellcome Trust (Grant Nos. 092506 and 098335), the MRC (MR/M008991/1), and the EPSRC (B.J.T. studentship) is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.organomet.9b00610. NMR data for this paper can be found at DOI 10.15124/b11b7146-b95d-4aca-bf39-f6fcc9421035.
Experimental details, NMR characterization data, crystallographic data and reaction monitoring (PDF)
Accession Codes
CCDC 1949692–1949693 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- Cotton H.; Elebring T.; Larsson M.; Li L.; Sörensen H.; von Unge S. Asymmetric synthesis of esomeprazole. Tetrahedron: Asymmetry 2000, 11 (18), 3819–3825. 10.1016/S0957-4166(00)00352-9. [DOI] [Google Scholar]
- Carreño M. C. Applications of sulfoxides to asymmetric synthesis of biologically active compounds. Chem. Rev. 1995, 95 (6), 1717–1760. 10.1021/cr00038a002. [DOI] [Google Scholar]
- Cotton F.; Francis R. Sulfoxides as ligands. I. A preliminary survey of methyl sulfoxide complexes. J. Am. Chem. Soc. 1960, 82 (12), 2986–2991. 10.1021/ja01497a003. [DOI] [Google Scholar]
- Trost B. M.; Rao M. Development of chiral sulfoxide ligands for asymmetric catalysis. Angew. Chem., Int. Ed. 2015, 54 (17), 5026–5043. 10.1002/anie.201411073. [DOI] [PubMed] [Google Scholar]
- Fernandez I.; Khiar N. Recent developments in the synthesis and utilization of chiral sulfoxides. Chem. Rev. 2003, 103 (9), 3651–3706. 10.1021/cr990372u. [DOI] [PubMed] [Google Scholar]
- Sipos G.; Drinkel E. E.; Dorta R. The emergence of sulfoxides as efficient ligands in transition metal catalysis. Chem. Soc. Rev. 2015, 44 (11), 3834–3860. 10.1039/C4CS00524D. [DOI] [PubMed] [Google Scholar]
- Ando D.; Bevan C.; Brown J. M.; Price D. W. Contrasting pathways for the directed homogeneous hydrogenation of vinyl sulfoxides and vinyl sulfones. J. Chem. Soc., Chem. Commun. 1992, 8, 592–594. 10.1039/c39920000592. [DOI] [Google Scholar]
- Lao J. R.; Fernández-Pérez H.; Vidal-Ferran A. Hydrogenative Kinetic Resolution of Vinyl Sulfoxides. Org. Lett. 2015, 17 (16), 4114–4117. 10.1021/acs.orglett.5b02139. [DOI] [PubMed] [Google Scholar]
- Dornan P. K.; Kou K. G.; Houk K.; Dong V. M. Dynamic Kinetic Resolution of Allylic Sulfoxides by Rh-Catalyzed Hydrogenation: A Combined Theoretical and Experimental Mechanistic Study. J. Am. Chem. Soc. 2014, 136 (1), 291–298. 10.1021/ja409824b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenschmid T. C.; Kirss R. U.; Deutsch P. P.; Hommeltoft S. I.; Eisenberg R.; Bargon J.; Lawler R. G.; Balch A. L. Para hydrogen induced polarization in hydrogenation reactions. J. Am. Chem. Soc. 1987, 109 (26), 8089–8091. 10.1021/ja00260a026. [DOI] [Google Scholar]
- Bowers C. R.; Weitekamp D. P. Parahydrogen and synthesis allow dramatically enhanced nuclear alignment. J. Am. Chem. Soc. 1987, 109 (18), 5541–5542. 10.1021/ja00252a049. [DOI] [Google Scholar]
- Patz S.; Hersman F. W.; Muradian I.; Hrovat M. I.; Ruset I. C.; Ketel S.; Jacobson F.; Topulos G. P.; Hatabu H.; Butler J. P. Hyperpolarized 129Xe MRI: a viable functional lung imaging modality?. Eur. J. Radiol. 2007, 64 (3), 335–344. 10.1016/j.ejrad.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson S. J.; Kurhanewicz J.; Vigneron D. B.; Larson P. E. Z.; Harzstark A. L.; Ferrone M.; van Criekinge M.; Chang J. W.; Bok R.; Park I.; et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1–13C] pyruvate. Sci. Transl. Med. 2013, 5 (198), 198ra108. 10.1126/scitranslmed.3006070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacharias N. M.; Chan H. R.; Sailasuta N.; Ross B. D.; Bhattacharya P. Real-time molecular imaging of tricarboxylic acid cycle metabolism in vivo by hyperpolarized 1–13C diethyl succinate. J. Am. Chem. Soc. 2012, 134 (2), 934–943. 10.1021/ja2040865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya P.; Chekmenev E. Y.; Perman W. H.; Harris K. C.; Lin A. P.; Norton V. A.; Tan C. T.; Ross B. D.; Weitekamp D. P. Towards hyperpolarized 13C-succinate imaging of brain cancer. J. Magn. Reson. 2007, 186 (1), 150–155. 10.1016/j.jmr.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübler P.; Giernoth R.; Kümmerle G.; Bargon J. Investigating the kinetics of homogeneous hydrogenation reactions using PHIP NMR spectroscopy. J. Am. Chem. Soc. 1999, 121 (22), 5311–5318. 10.1021/ja984353y. [DOI] [Google Scholar]
- Giernoth R.; Heinrich H.; Adams N. J.; Deeth R. J.; Bargon J.; Brown J. M. PHIP detection of a transient rhodium dihydride intermediate in the homogeneous hydrogenation of dehydroamino acids. J. Am. Chem. Soc. 2000, 122 (49), 12381–12382. 10.1021/ja002516o. [DOI] [Google Scholar]
- Ahlquist M.; Gustafsson M.; Karlsson M.; Thaning M.; Axelsson O.; Wendt O. F. Rhodium (I) hydrogenation in water: Kinetic studies and the detection of an intermediate using 13C {1H} PHIP NMR spectroscopy. Inorg. Chim. Acta 2007, 360 (5), 1621–1627. 10.1016/j.ica.2006.08.042. [DOI] [Google Scholar]
- Duckett S. B.; Newell C. L.; Eisenberg R. Observation of new intermediates in hydrogenation catalyzed by wilkinson’s catalyst, RhCl (PPh3) 3, using parahydrogen-induced polarization. J. Am. Chem. Soc. 1994, 116 (23), 10548–10556. 10.1021/ja00102a023. [DOI] [Google Scholar]
- Sorochkina K.; Zhivonitko V. V.; Chernichenko K.; Telkki V.-V.; Repo T.; Koptyug I. V. Spontaneous 15N Nuclear Spin Hyperpolarization in Metal-Free Activation of Parahydrogen by Molecular Tweezers. J. Phys. Chem. Lett. 2018, 9 (4), 903–907. 10.1021/acs.jpclett.7b03433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivonitko V. V.; Sorochkina K.; Chernichenko K.; Kótai B.; Földes T.; Pápai I.; Telkki V.-V.; Repo T.; Koptyug I. Nuclear spin hyperpolarization with ansa-aminoboranes: a metal-free perspective for parahydrogen-induced polarization. Phys. Chem. Chem. Phys. 2016, 18 (40), 27784–27795. 10.1039/C6CP05211H. [DOI] [PubMed] [Google Scholar]
- Adams R. W.; Aguilar J. A.; Atkinson K. D.; Cowley M. J.; Elliott P. I.; Duckett S. B.; Green G. G.; Khazal I. G.; López-Serrano J.; Williamson D. C. Reversible interactions with para-hydrogen enhance NMR sensitivity by polarization transfer. Science 2009, 323 (5922), 1708–1711. 10.1126/science.1168877. [DOI] [PubMed] [Google Scholar]
- Adams R. W.; Duckett S. B.; Green R. A.; Williamson D. C.; Green G. G. R. A theoretical basis for spontaneous polarization transfer in non-hydrogenative para hydrogen-induced polarization. J. Chem. Phys. 2009, 131 (19), 194505. 10.1063/1.3254386. [DOI] [PubMed] [Google Scholar]
- Iali W.; Rayner P. J.; Duckett S. B. Using parahydrogen to hyperpolarize amines, amides, carboxylic acids, alcohols, phosphates, and carbonates. Sci. Adv. 2018, 4 (1), eaao6250 10.1126/sciadv.aao6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong M. L.; Theis T.; Coffey A. M.; Shchepin R. V.; Waddell K. W.; Shi F.; Goodson B. M.; Warren W. S.; Chekmenev E. Y. 15N hyperpolarization by reversible exchange using SABRE-SHEATH. J. Phys. Chem. C 2015, 119 (16), 8786–8797. 10.1021/acs.jpcc.5b01799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshuis N.; van Weerdenburg B. J. A.; Feiters M. C.; Rutjes F. P. J. T.; Wijmenga S. S.; Tessari M. Quantitative trace analysis of complex mixtures using SABRE hyperpolarization. Angew. Chem., Int. Ed. 2015, 54 (5), 1481–1484. 10.1002/anie.201409795. [DOI] [PubMed] [Google Scholar]
- Procacci B.; Aguiar P. M.; Halse M. E.; Perutz R. N.; Duckett S. B. Photochemical pump and NMR probe to monitor the formation and kinetics of hyperpolarized metal dihydrides. Chem. Sci. 2016, 7 (12), 7087–7093. 10.1039/C6SC01956K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova O.; Richardson P. M.; Parrott A. J.; Nordon A.; Halse M. E.; Duckett S. B. Reaction monitoring using SABRE-hyperpolarized benchtop (1 T) NMR spectroscopy. Anal. Chem. 2019, 91 (10), 6695–6701. 10.1021/acs.analchem.9b00729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong M. L.; Theis T.; Coffey A. M.; Shchepin R. V.; Waddell K. W.; Shi F.; Goodson B. M.; Warren W. S.; Chekmenev E. Y. N-15 Hyperpolarization by Reversible Exchange Using SABRE-SHEATH. J. Phys. Chem. C 2015, 119 (16), 8786–8797. 10.1021/acs.jpcc.5b01799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleby K. M.; Mewis R. E.; Olaru A. M.; Green G. G. R.; Fairlamb I. J. S.; Duckett S. B. Investigating pyridazine and phthalazine exchange in a series of iridium complexes in order to define their role in the catalytic transfer of magnetisation from para-hydrogen. Chem. Sci. 2015, 6 (7), 3981–3993. 10.1039/C5SC00756A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pravica M. G.; Weitekamp D. P. Net NMR alignment by adiabatic transport of parahydrogen addition products to high magnetic field. Chem. Phys. Lett. 1988, 145 (4), 255–258. 10.1016/0009-2614(88)80002-2. [DOI] [Google Scholar]
- Knecht S.; Hadjiali S.; Barskiy D. A.; Pines A.; Sauer G.; Kiryutin A. S.; Ivanov K. L.; Yurkovskaya A. V.; Buntkowsky G. Indirect Detection of Short-lived Hydride Intermediates of Iridium N-Heterocyclic Carbene Complexes via Chemical Exchange Saturation Transfer (CEST) Spectroscopy. J. Phys. Chem. C 2019, 123 (26), 16288–16293. 10.1021/acs.jpcc.9b04179. [DOI] [Google Scholar]
- Iali W.; Roy S. S.; Tickner B. J.; Ahwal F.; Kennerley A. J.; Duckett S. B. Hyperpolarizing Pyruvate through Signal Amplification By Reversible Exchange (SABRE). Angew. Chem. 2019, 131 (130), 10377–10381. 10.1002/ange.201905483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen P. G.; Anderson J. E.; Bailey O. H.; Tamres M.; Bayón J. C. A novel metal-metal bonded iridium (II) dimer. J. Am. Chem. Soc. 1985, 107 (1), 279–281. 10.1021/ja00287a066. [DOI] [Google Scholar]
- Gilbert T. M.; Hollander F. J.; Bergman R. G. (Pentamethylcyclopentadienyl) iridium polyhydride complexes: synthesis of intermediates in the mechanism of formation of (pentamethylcyclopentadienyl) iridium tetrahydride and the preparation of several iridium (V) compounds. J. Am. Chem. Soc. 1985, 107 (12), 3508–3516. 10.1021/ja00298a018. [DOI] [Google Scholar]
- Jones W. D.; Chin R. M. Hydrodesulfurization of thiophene to butadiene and butane by a homogeneous iridium complex. J. Am. Chem. Soc. 1994, 116 (1), 198–203. 10.1021/ja00080a022. [DOI] [Google Scholar]
- Li M.-L.; Yang S.; Su X.-C.; Wu H.-L.; Yang L.-L.; Zhu S.-F.; Zhou Q.-L. Mechanism Studies of Ir-Catalyzed Asymmetric Hydrogenation of Unsaturated Carboxylic Acids. J. Am. Chem. Soc. 2017, 139 (1), 541–547. 10.1021/jacs.6b11655. [DOI] [PubMed] [Google Scholar]
- Wang L.; He W.; Yu Z. Transition-metal mediated carbon–sulfur bond activation and transformations. Chem. Soc. Rev. 2013, 42 (2), 599–621. 10.1039/C2CS35323G. [DOI] [PubMed] [Google Scholar]
- Luh T.-Y.; Ni Z.-J. Transition-metal-mediated CS bond cleavage reactions. Synthesis 1990, 1990 (02), 89–103. 10.1055/s-1990-26798. [DOI] [Google Scholar]
- O’Connor J. M.; Bunker K. D.; Rheingold A. L.; Zakharov L. Sulfoxide Carbon– Sulfur Bond Activation. J. Am. Chem. Soc. 2005, 127 (12), 4180–4181. 10.1021/ja050160g. [DOI] [PubMed] [Google Scholar]
- Fekete M.; Bayfield O.; Duckett S. B.; Hart S.; Mewis R. E.; Pridmore N.; Rayner P. J.; Whitwood A. Iridium (III) Hydrido N-heterocyclic carbene–phosphine complexes as catalysts in magnetization transfer reactions. Inorg. Chem. 2013, 52 (23), 13453–13461. 10.1021/ic401783c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weerdenburg B. J.; Eshuis N.; Tessari M.; Rutjes F. P.; Feiters M. C. Application of the π-accepting ability parameter of N-heterocyclic carbene ligands in iridium complexes for signal amplification by reversible exchange (SABRE). Dalton Trans 2015, 44 (35), 15387–15390. 10.1039/C5DT02340H. [DOI] [PubMed] [Google Scholar]
- Marion N.; Díez-González S.; Nolan S. P. N-heterocyclic carbenes as organocatalysts. Angew. Chem., Int. Ed. 2007, 46 (17), 2988–3000. 10.1002/anie.200603380. [DOI] [PubMed] [Google Scholar]
- Gade L. H.; Bellemin-Laponnaz S.. Chiral N-heterocyclic carbenes as stereodirecting ligands in asymmetric catalysis. N-Heterocyclic Carbenes in Transition Metal Catalysis; Springer, 2006; pp 117–157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.