

SUMMARY
PINK1 and Parkin are established mediators of mitophagy, the selective removal of damaged mitochondria by autophagy. PINK1 and Parkin have been proposed to act as tumor suppressors, as loss-of-function mutations are correlated with enhanced tumorigenesis. However, it is unclear how PINK1 and Parkin act in coordination during mitophagy to influence the cell cycle. Here we show that PINK1 and Parkin genetically interact with proteins involved in cell cycle regulation, and loss of PINK1 and Parkin accelerates cell growth. PINK1- and Parkin-mediated activation of TBK1 at the mitochondria during mitophagy leads to a block in mitosis due to the sequestration of TBK1 from its physiological role at centrosomes during mitosis. Our study supports a diverse role for the far-reaching, regulatory effects of mitochondrial quality control in cellular homeostasis and demonstrates that the PINK1/Parkin pathway genetically interacts with the cell cycle, providing a framework for understanding the molecular basis linking PINK1 and Parkin to mitosis.
Graphical Abstract
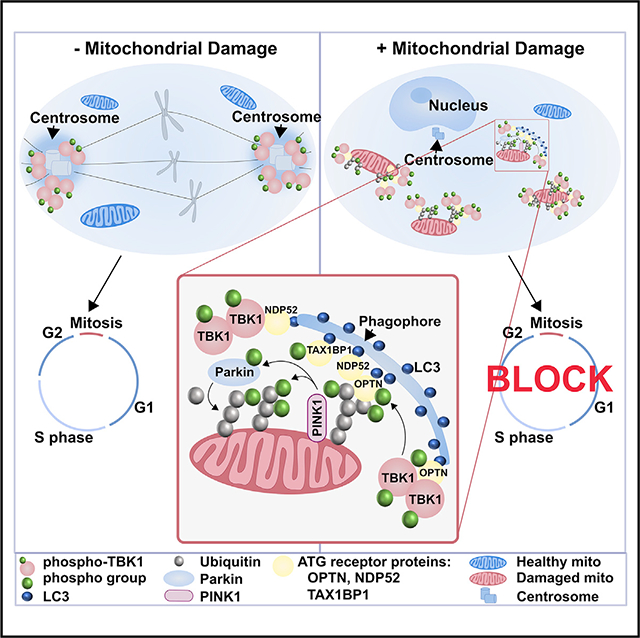
In Brief
Sarraf et al. use mouse and fly genetics to discover that PINK1 and Parkin influence cell cycle progression. Mitophagy and mitosis independently activate TBK1 at damaged mitochondria and centrosomes, respectively, influencing whether the cell will address mitochondrial quality control or progress with proliferation.
INTRODUCTION
Parkin and PINK1 promote the removal of dysfunctional mitochondria, a process termed mitophagy, by specifically targeting damaged mitochondria for lysosomal degradation (Pickrell and Youle, 2015). Loss-of-function mutations and deletions in PINK1 and Parkin have been associated with multiple forms of cancer, indicating that both proteins are possible tumor suppressors. Pathogenic PINK1 germline variants predispose individuals to high-risk neuroblastomas (Pugh et al., 2013). PARK2, the gene that encodes Parkin, resides within a common fragile site at chromosome 6q26 (Mitsui et al., 2010). Parkin mutations and deletions have also been identified in sporadic colorectal cancer, glioblastomas, and other cancer types (Poulogiannis et al., 2010; Veeriah et al., 2010). Previous literature has proposed mechanisms by which PINK1 and Parkin independently influence the cell cycle, but these studies did not consider mitophagy (Bernardini et al., 2017). PINK1’s known roles include activation of inactive cytosolic Parkin upon mitochondrial damage, while in the absence of mitochondrial damage, PINK1 is constitutively degraded and thus unable to be contribute to cellular processes occurring in the absence of mitochondrial damage (Pickrell and Youle, 2015). Therefore, we hypothesized that both PINK1- and Parkin-mediated mitophagy must influence cell cycle progression, albeit directly or indirectly.
During mitophagy, PINK1 and Parkin trigger the translocation and activation of Tank Binding Kinase 1 (TBK1) to damaged mitochondria (Heo et al., 2015; Lazarou et al., 2015; Moore and Holzbaur, 2016; Richter et al., 2016). TBK1 phosphorylates autophagy receptors at the mitochondria to promote binding between the developing autophagosome and damaged mitochondria (Heo et al., 2015; Lazarou et al., 2015; Moore and Holzbaur, 2016; Richter et al., 2016). Our data demonstrate that TBK1 acts at both the damaged mitochondria during mitophagy and at centrosomes, where it is activated and necessary during mitosis. We show that both PINK1 and Parkin genetically interact with tumor-suppressing genes and uncover that PINK1/Parkin-mediated activation of TBK1 causes its sequestration at damaged mitochondria, thus blocking TBK1’s ability to participate in cell cycle progression.
RESULTS
PINK1 and Parkin Genetically Interact with DNA Damage Cell Cycle Checkpoint Proteins
Ataxia telangiectasia mutated (ATM) is a serine threonine kinase activated in response to DNA damage, hypoxia, and oxidative stress that results in the phosphorylation of hundreds of targets involved in cell-cycle arrest, DNA repair, and apoptosis and senescence (Maréchal and Zou, 2013). Several reports also support the notion that loss of ATM leads to mitochondrial dysfunction (Eaton et al., 2007; Schroeder et al., 2013; Valentin-Vega et al., 2012). This observation led us to hypothesize that ATM and Parkin may function together in regulating mitochondrial responses to stress. To further explore this possibility, we undertook a genetic approach crossing ATM-knockout (KO) and Parkin-KO mice to investigate the effect of loss of ATM on Parkin-mediated phenotypes.
Parkin-KO mice are viable, with no overt phenotypes, neuro-degeneration, or reduced lifespan (Goldberg et al., 2003). ATM-KO mice are also born viable, but half of the mice develop thymic lymphomas after 3 months of age (Barlow et al., 1996). Because both of these genetic mutant murine lines are healthy at birth, it was unanticipated that mice with a combinatory loss of ATM and Parkin were found at sub-Mendelian ratios, indicating a synthetic lethality (Table 1; Figure S1). In order to more readily explore genetic interactions of the ATM and Parkin pathways, we confirmed this finding in Drosophila.
Table 1.
PINK1 and Parkin Genetically Interact with DNA Damage Cell Cycle Checkpoint Proteins in Mouse
| ATM Genotype | Parkin Genotype | Actual Number of Mice | Actual Frequency (%) | Expected Frequency (%) |
|---|---|---|---|---|
| Survival of Mice from the ATM+/−Parkin+/− Cross | ||||
| +/+ | +/+ | 11 | 6.70 | 6.25 |
| +/+ | +/− | 8 | 4.87 | 6.25 |
| +/+ | −/− | 7 | 4.27 | 6.25 |
| +/− | +/+ | 22 | 13.41 | 12.5 |
| +/− | +/− | 55 | 33.54 | 25 |
| +/− | −/− | 29 | 29.87 | 12.5 |
| −/− | +/+ | 4 | 2.43 | 6.25 |
| −/− | +/− | 8 | 4.87 | 6.25 |
| −/− | −/− | 0 | 0 | 6.25**** |
| Survival of Mice from the ATM+/−Parkin+/− × ATM+/−Parkin KO Cross | ||||
| +/+ | +/+ | 4 | 2.79 | 12.5 |
| +/+ | +/− | 7 | 4.89 | 12.5 |
| +/− | −/− | 58 | 40.56 | 25 |
| +/− | +/+ | 68 | 47.55 | 25 |
| −/− | +/− | 5 | 3.49 | 12.5 |
| −/− | −/− | 1 | .07 | 12.5**** |
| Survival of Mice from the ATM+/− Cross | ||||
| ATM genotype | ||||
| +/+ | 10 | 27.03 | 25 | |
| +/− | 20 | 54.05 | 50 | |
| −/− | 7 | 18.92 | 25 | |
Table showing the breeding crosses and genotyped progeny from ATM+/−Parkin+/− (top) and ATM+/−Parkin+/− × ATM+/−Parkin KO mice (middle) cross. The progeny from ATM+/− crosses were also examined (bottom). ATM+/−Parkin+/− cross, ****p < 0.0001; ATM+/−Parkin+/− × ATM ± Parkin KO cross, ****p < 0.0001. ATM+/− cross, not significant.
We crossed the park25 mutant fly with a combination of ATM mutant fly lines harboring various point mutations (Table 2). Flies homozygous for the STOP codon mutations (ATM3/ATM3 and ATM6/ATM6) are lethal (Pedersen et al., 2010). Hence, we made combinatory fly lines to disrupt ATM. As in the mouse, these crosses resulted in a synthetic lethality, resulting in fewer double-mutant pupae hatching when both alleles were mutated (Table 2). PINK1 and Parkin reside in the same pathway to control mitophagy and were also shown with epistasis experiments in Drosophila (Clark et al., 2006; Park et al., 2006; Poole et al., 2008). This interaction with ATM extended to PINK1 as heteroallelic combinations of Pink1B9 ATM flies also eclosed below the expected number of progeny (Table 2).
Table 2.
PINK1 and Parkin Genetically Interact with DNA Damage Cell Cycle Checkpoint Proteins in Fly
| Genotype | Eclosion (%) |
|---|---|
| park25 | 100% |
| park25, ATM3/+ | 100% |
| park25, ATM6/+ | 100% |
| park25, ATM8/+ | 100% |
| ATM3/6 | 29%**** |
| ATM3/8 | 100% |
| ATM6/8 | 72%**** |
| park25, ATM3/6 | 1.5%**** |
| park25, ATM3/s | 35.9%**** |
| park25, ATM6/8 | 65.4%*** |
| Pink1B9 | 100% |
| Pink1B9;;ATM6/+ | 88.2% |
| Pink1B9;;ATM8/+ | 54.2%**** |
| Pink1B9;;ATM6/8 | 34%**** |
| Mei-41[D12];;park25/+ | 100% |
| Mei-41[D12]/+;;park25/+ | 100% |
| Mei-41[D12]/+;;park25 | 60.5%**** |
| Mei-41[D12];;park25 | 46.6%**** |
| hs-G4; park25/+ | 100% |
| hs-G4; park25 | 100% |
| hs-G4 > stg-HA; park25/+ | 100% |
| hs-G4 > stg-HA; park25 | 52.5%**** |
| hs-G4; park25/+ | 100% |
| hs-G4; park25 | 100% |
| hs-G4 > stgmKEN.md; park25/+ | 100% |
| hs-G4 > stgmKEN.md; park25 | 44.5%**** |
| Ik2[1]/CyO | 100% |
| Ik2[1]/ Ik2[1] | 3.55%**** |
| Ik2[1 / Ik2[1]; park25/+ | 44%**** |
| Ik2[1]/ Ik2[1]; park25 | 66.15**** |
Table summarizing the ATM mutant flies used in this study. Percentage of flies eclosed for each genotype from park25 self-crosses; ATM3/TM3 × ATM6/TM3; ATM3/TM3 × ATM6, park25/TM3; ATM3, park25/TM3 × ATM6, park25/TM3; ATM8/TM3 × ATM3/TM3; ATM8, park25/TM3 × ATM3, park25/TM3; ATM8, park25/TM3 × ATM3/TM3; ATM8, park25/TM3 × ATM6/TM3; ATM8/TM3 × ATM6/TM3, and ATM8, park25/TM3 × ATM6, park25/TM3. Percentage of flies eclosed from each genotype from FM7 × Pink1B9/FM7 self-crosses, Pink1B9/FM7 × ATM6/TM3, Pink1B9/FM7 × ATM8/TM3, and Pink1B9/FM7; ATM6/TM3 × Pink1B9/FM7; ATM8/TM3b. Percentage of flies eclosed for each genotype from Mei-41[D-12]/FM7; park25/TM3 self-cross. Percentage of flies eclosed for each genotype from UAS-stg-HA/CyO; park25/TM3 × hs-G4/CyO; park25/TM3 and UAS-HA-stgmKEN.md/CyO; park25/TM3 × hs-G4/CyO; park25/TM3. Percentage of flies eclosed for each genotype from ik2[1]/CyO (#5322) and ik2[1]/CyO; park25/TM6B self-crosses. At least 300 flies were screened from crosses.
p > 0.001
p > 0.0001.
The Loss of ATM Does Not Affect PINK1 Accumulation or Parkin Translocation upon Mitochondrial Damage
The HCT116 cell line expresses endogenous ATM and Parkin, maintains an intact p53 response, and has been extensively used to study Parkin-mediated mitophagy (Sarraf et al., 2013; Yamano et al., 2014). To test whether ATM and PINK1/Parkin were directly interacting, we generated ATM-KO HCT116 cells using CRISPR (Table S1). Using this cell line, we probed the effects of ATM loss on PINK1/Parkin in the context of mitochondrial dysfunction.
Mitochondrial damage induced by an antimycin A and oligomycin A cocktail (OA) caused Parkin translocation and PINK1 accumulation as expected (Lazarou et al., 2015) and was completely ATM independent (Figures S2A–S2C). Mitochondrial damage did not induce autophosphorylation and activation of ATM (Figure S2C). These results were confirmed in healthy human and ATM patient fibroblast lines (Figure S2D). In conclusion, Parkin-mediated mitophagy was uninhibited in the absence of ATM, and the loss of ATM was not sufficient to trigger Parkin translocation without exogenous mitochondrial damage.
Given that ATM had no role upstream of Parkin, we examined events downstream of PINK1 accumulation and Parkin translocation. In cells stably expressing tagged Parkin, prolonged treatment with OA showed that the presence or absence of ATM had no effect on the degradation of Mfn2, a Parkin substrate (Sarraf et al., 2013) (Figure S2E). The accumulation of p62, an autophagy receptor protein, and the lipidation of the autophagosomal protein LC3 occurred with OA treatment, indicating normal progression of mitophagy in the absence of ATM (Figure S2F). The loss of inner membrane mitochondrial proteins (COXII and Tim23) and absence of GRP75, a mitochondrial matrix protein, suggested completion of mitophagy in both cell lines with prolonged OA treatment (Figure S2G); thus, loss of ATM did not influence Parkin-mediated mitophagy.
We then tested whether the loss of PINK1 or Parkin affected nuclear DNA damage signaling. We confirmed using western blot that our ATM-KO HCT116 cells displayed a typical response to etoposide (Figure S3A). We generated Parkin-KO and PINK1-KO HCT116 (Table S1; Figures S3B and S3C) to compare genotypes. We used a homologous recombination (HR) reporter construct (DR-GFP) and a non-homologous end- joining (NHEJ) reporter construct (EJGFP) to test whether DNA damage repair was inhibited when the PINK1-Parkin pathway was disrupted (Figure S3D). In brief, DR-GFP or EJGFP cannot express GFP fluorescence unless an SceI restriction site is recombined through HR or NHEJ (Bennardo et al., 2008; Pierce et al., 1999) (Figure S3D). We found that PINK1 and Parkin did not have an impact on HR (Figure S3E). The loss of ATM in wild-type cells resulted in deficient HR (Kass et al., 2013), which we used as a positive control (Figure S3E). We also checked NHEJ, which occurs at a higher rate than HR (Mao et al., 2008). Interestingly, we found that NHEJ did appear slightly impaired in PINK1-KO compared with wild-type (WT) and Parkin-KO cells, but not to the extent seen in ATM-KO cells (Figure S3F). This did not appear to be dependent on ATM signaling or p53 accumulation (Figure S3G).
To determine the specificity of the genetic interaction between ATM and PINK1 and Parkin, we crossed ATR mutant flies with Parkin mutant flies. ATR is a PI3K in the same family as ATM that responds to single-strand breaks by phosphorylating multiple protein targets involved in overlapping pathways with those triggered by ATM activation (Figure 1A) (Maréchal and Zou, 2013). Unlike ATM, ATR has not been associated in the literature with mitochondrial dysfunction. Interestingly, ATR and Parkin double mutants also caused a synthetic lethality (Table 2). Mitochondrial-localized PINK1 and Parkin are unlikely to directly interact with nuclear ATM and ATR. Furthermore, we did not observe robust evidence that PINK1 and Parkin indirectly affected ATM or vice versa (Figures S2 and S3); therefore, we felt that PINK1/Parkin were most likely influencing a common pathway downstream of ATM/ATR.
Figure 1. CDK1, CDK2, and CDC25 Interact with and Rescue PINK1 and Parkin Thorax Indentations.

(A) Cartoon of proteins downstream of ATM and ATR that were selected in the targeted RNAi fly screen to cross with Pink1B9 or park25 flies driven under the systemic da-G4 or hs-G4, respectively.
(B) Percentage of flies with the presence or absence of thorax indentations in the Pink1B9 background expressing UAS-stgRNAi (#29556) under the da-G4 driver. Two hundred to 300 flies were screened from cross.
(C) Percentage of flies with the presence or absence of thorax indentations in the park25 background expressing UAS-CDK2RNAi (#36128) under the hs-G4 driver.
(D) Percentage of flies with the presence or absence of thorax indentations in the Pink1B9 background expressing UAS-CDK2RNAi (#36128) under the da-G4 driver.
(E) Percentage of flies with the presence or absence of thorax indentations in the park25 background expressing UAS-CDK1RNAi (#36117) under the hs-G4 driver.
(F) Percentage of flies with the presence or absence of thorax indentations in the Pink1B9 background expressing UAS-CDK1RNAi (#36117) (G) UAS-CDK2RNAi (#41898) and (H) UAS-CDK2RNAi (#35267) under the da-G4 driver. (C–F) At least 200–300 flies were screened from crosses.
CDK1, CDK2, and CDC25 Interact with and Rescue PINK1 and Parkin Thorax Indentations
We screened 14 RNAi fly lines targeted to genes functioning downstream of ATM and ATR to test whether PINK1/Parkin interacted with cell cycle regulation (Table S2; Figure 1A). park25 mutant flies display flight muscle degeneration, causing thorax indentations and dopaminergic neuron loss perfectly phenocopying mutant Pink1B9 flies (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). We scored flies both for survival and for thoracic indentations to identify any genetic interactions with the PINK1/Parkin pathway.
The RNAi fly strain targeting String (stg), the CDC25 human ortholog, partially rescued the PINK1 mutant fly thorax indentation phenotype (Figure 1B). CDC25 removes phosphate residues on active sites of cyclin-dependent kinase (CDK) complexes responsible for cell cycle progression (Lim and Kaldis, 2013). CDK1 and CDK2 are major kinases that drive the cell cycle by binding to cyclin proteins that flux during G1/S and G2/M transitions (Lim and Kaldis, 2013). In a Parkin mutant background, overexpression of stg or a non-degradable stg mutant allele (HA-stgmKEN.md) resulted in lethality (Table 2).
Further validating the connection to the cell cycle, our screen also identified CDK1 and CDK2 as genetic interactors of both PINK1 and Parkin. Knockdown (KD) of CDK1 and CDK2 using multiple different RNAi fly lines ameliorated both PINK1 and Parkin mutant thorax phenotypes (Figures 1C–1H; Tables S2 and S3). These data support the notion of a genetic interaction between components of the cell cycle machinery and the PINK1/Parkin pathway.
The Loss of Endogenous PINK1/Parkin Interferes with Cell Growth
We further explored the effect of PINK1 and Parkin on cell growth by examining primary cell lines lacking PINK1 or Parkin. Using Parkin-KO mouse embryonic fibroblasts (MEFs), we measured cell growth. In comparison with control MEFs, we found that proliferation was accelerated in Parkin-KO MEF lines (Figure 2A) consistent with an increase in the number of Parkin-KO cells at S and M phases (Figures 2B and 2C). Additionally, we examined a PINK1 mutant human fibroblast line and found a trend toward accelerated growth compared with healthy control fibroblast lines, which also exhibited a similar increase in cells entering S and M phases of the cell cycle (Figures 2D–2F). Our data are consistent with other observations reporting that cell division was increased after the loss of PINK1 and Parkin (Agnihotri et al., 2016; Gong et al., 2014; Requejo-Aguilar et al., 2014).
Figure 2. Loss of Endogenous PINK1 and Parkin Disrupts Cell Growth and Cell Cycle.

(A) Normalized change in manual cell counts for primary MEF lines.
(B and E) Percentage of MEFs (B) and patient fibroblasts (E) positive for BrdU.
(C and F) Percentage of MEFs (C) and patient fibroblasts (F) with Aurora B accumulated in the nucleus.
(D) Normalized luminescence measurements for primary patient cell lines.
In (A) and (D), error bars indicate ±SD; n = 2 independent experiments. In (B), (C), (E), and (F), error bars indicate ±SD; n = 3 independent experiments.
TBK1 Is Activated During and Necessary for Mitosis
TBK1’s role in innate immunity is extensively studied because it is required to stimulate the interferon response (Clément et al., 2008). Our work and that of others previously revealed that TBK1 activation at damaged mitochondria promoted efficient mitophagy dependent on PINK1 and Parkin activity (Heo et al., 2015; Lazarou et al., 2015; Moore and Holzbaur, 2016; Richter et al., 2016) (Figure S4). However, an additional role for TBK1 in mitosis has also been described (Onorati et al., 2016; Pillai et al., 2015). Because it represents a potential link between both Parkin-mediated mitophagy and the cell cycle, we tested whether TBK1 was active and necessary for mitosis.
We found that TBK1 was phosphorylated and active during all stages of mitosis at the centrosome and midbody (Figure 3A). This activation was not apparent by western blotting in an asynchronous population, but upon synchronization of cells at G2/M phase, p-TBK1 levels were strikingly increased (Figures 3B and 3C), leading to the question of whether TBK1 was necessary for the G2/M transition. Either pharmacologically inhibiting TBK1 kinase activity or genetic KO of TBK1 (Table S1) resulted in a growth defect (Figures 3D–3F); moreover, we observed decreased numbers of cells in M phase in an asynchronous population (Figure 3G). Upon rescue of the TBK1-KO line, we were able to revert the growth phenotype (Figures 3H and 3I). There was an increased presence of polyploidy and multinucleate cells, presumably harboring cytokinetic defects, in the TBK1-KO cells (Figure 3J).
Figure 3. TBK1 Is Activated During and Necessary for Mitosis.

(A) Confocal images of YFP-Parkin-expressing (green) HeLa cells demonstrating p-TBK1 (orange) localization throughout the different stages of mitosis. Cells were counterstained with DAPI (blue) and α-tubulin (red). Scale bar, 10 μM.
(B and C) Western blot analysis of HeLa cells either in asynchronized growth (B) or synchronized at G2/M with nocodazole and released into normal growth media
(C). β-actin was used as a loading control.
(D and E) Normalized luminescence measurements for HeLa cells or (D) treated with 1 μM BX-795 (E) or two independent TBK1-KO clones. Error bars indicate ±SD for technical replicates.
(F) Western blot analysis of TBK1-KO clones. β-actin was used as a loading control.
(G) Mitotic cell counts from an asynchronous population of wild-type or TBK1-KO clones. Error bars indicate ±SD; n = 3 independent experiments.
(H) Normalized luminescence measurements for HeLa cells, TBK1-KO clone 2, or TBK1 rescue line. Error bars indicate ±SD for technical replicates.
(I) Western blot analysis of HeLa cells and TBK1 rescue line in asynchronous and G2/M synchronization conditions. β-actin was used as a loading control.
(J) Confocal images of HeLa cells or TBK1-KO cells in asynchronous conditions. DAPI (blue) was used as a nuclear counterstain, and the cytoskeleton was identified by alpha-tubulin staining (red). Scale bar, 20 μm.
Activated TBK1 Is Sequestered from the Centrosomes to Damaged Mitochondria during Parkin-Mediated Mitophagy
These data suggest that TBK1 plays an important physiological role in promoting cell cycle progression, and because of TBK1’s additional role in promoting mitophagy at damaged mitochondria, we hypothesized that during mitochondrial damage, active TBK1 was sequestered from centrosomes to mitochondria, resulting in cell-cycle arrest. TBK1’s interaction with PINK1/Parkin is not direct; however, TBK1 translocation to damaged mitochondria and activation is dependent upon the activation of PINK1 and Parkin (Lazarou et al., 2015). In cells either expressing or lacking Parkin, we synchronized at G1/S followed by release into either normal growth media or OA (Figure 4A). By 9 h post-release in normal growth media, cells progressed through G2/M regardless of the presence or absence of Parkin (Figures 4B, 4D, 4E). However, in the context of mitochondrial damage, the presence of Parkin inhibited the progression of cells entering the G2/M transition (Figures 4B and 4C) via sequestration of active p-TBK1 from the centrosome to damaged mitochondria (Figures 4B and 4C). Our data suggest that this sequestration blocked the ability of cells to enter mitosis because the status of PINK1 and Parkin had no effect during normal G2/M progression in the absence of mitochondrial damage (Figure 4E).
Figure 4. Activated TBK1 Is Sequestered from the Centrosomes to Damaged Mitochondria.

(A) Confocal images of YFP-Parkin-expressing (green) or wild-type HeLa cells detecting p-TBK1 (red) localization after double thymidine block (DTB) in DMEM or OA-treated media. Cells were counterstained with DAPI (blue) and α-tubulin (gray). White squares point to cells magnified in insets. Bottom: timeline of double thymidine synchronization for G2/M. Scale bar, 10 μM.
(B) Percentage of cells with p-TBK1 localization at centrosome or mitochondria from conditions in (A) after DTB.
(C and D) Percentage of mitotic YFP-Parkin or wild-type cells after DTB release into (C) OA media or (D) normal growth media.
(E) Western blot analysis of HeLa, YFP-Parkin, or PINK1-KO cells either in asynchronous growth or synchronized in G2/M. β-actin was used as a control.
(F) Cartoon of induced dimerization system used to physically tether TBK1 to undamaged mitochondria upon exposure to rapalog. Illustration made in BioRender (biorender.com, with the standard academic license (G) Confocal images of FKBP-GFP-TBK1 (green) in the FRB-BFP-Fis1 (blue) parental cell line in the TBK1-KO background treated with rapalog for 24 h. Cells were counterstained with MitoTracker Red CMXRos (red). Scale bar, 10 μM. Inset scale bar, 2 μM.
(H) Mitotic cell counts from an asynchronous population of cells expressing FKBP-GFP-TBK1 and FRB-BFP-Fis1 in the TBK1-KO background treated with or without rapalog for 24 h.
In (B)–(D) and (H), error bars indicate ±SEM; n = 3 independent experiments.).
Mitochondrial damage did negatively affect cell growth (Figures 4C and 4D) (Mitra et al., 2009), so to further test the idea that TBK1 sequestration at mitochondria causes decreased mitosis, we attempted two approaches. First, we generated cell lines stably expressing HA-FLAG-tagged and untagged TBK1 as well as YFP-Parkin (Figure S5A), hypothesizing that this may allow us to test our model by increasing additional TBK1 availability during mitochondrial dysfunction. However, we found that overexpression of either tagged or untagged TBK1 in a background with endogenous TBK1 caused abnormal activation and localization in the absence of stimuli (Figures S5B and S5C). We next attempted to physically tether TBK1 to mitochondria using the rapalog-induced dimerization system (FRB-FKBP). We stably expressed FRB-BFP-Fis1 in the TBK1-KO cell line and transfected in FKBP-GFP-TBK1 (Figure 4F). In the absence of rapalog, low levels of FKBP-GFP-TBK1 were predominately cytosolic (Figure S5D). However, in the presence of rapalog, FKBP-GFP-TBK1 translocated to mitochondria and colocalized with the membrane potential-dependent dye MitoTracker Red CMXROS (Figure 4G). By physically tethering TBK1 to mitochondria and bypassing exogenous mitochondrial damage, we observed a significant decrease in the number of mitotic cells in asynchronous conditions compared with FKBP-GFP-TBK1-transfected cells without rapalog (Figure 4H).
Drosophila Ortholog ik2 Genetically Interacts with PINK1 and Parkin
In a genome-wide screen for kinases that regulate cell cycle, the TBK1 ortholog in flies (ik2) was discovered to regulate mitosis, and KD of ik2 was found to halt cell cycle progression during mitosis, resulting in cytokinetic defects (Bettencourt-Dias et al., 2004). We decided to test whether ik2 interacted with PINK1 and Parkin genetically. Loss of ik2 on both alleles causes first instar lethality (Shapiro and Anderson, 2006). Unexpectedly, we found that loss of Parkin suppressed the ik2 recessive lethality (Table 2). Upon further examination, we found the loss of ik2 caused a reversal of the thorax indentation phenotype in the Parkin and PINK1 mutant flies (Figures 5A and 5B). These experiments provided additional genetic evidence of PINK1 and Parkin’s interaction with TBK1.
Figure 5. Drosophila Ortholog ik2 Genetically Interacts with PINK1 and Parkin.

(A) Percentage of flies with the presence or absence of thorax indentations in the ik2[1]/CyO; park25/TM6B self-cross. At least 200–300 flies were screened from crosses.
(B) Percentage of flies with the presence or absence of thorax indentations from the FM7/y; ik2/CyO × Pink1B9/Fm7c; pin/Cyo. At least 200–300 flies were screened from crosses.
DISCUSSION
Our work demonstrates a strong genetic interaction between tumor-suppressing genes in both mouse and fly and genes involved in mitophagy, leading us to conclude that the PINK1/Parkin pathway influences the cell cycle. We attribute the ability of PINK1/Parkin-dependent mitophagy to cause the translocation and sequestration of active TBK1 at damaged mitochondria to this genetic interaction. This sequestration of active TBK1 at the mitochondria from its physiological role at the centrosome during mitosis contributes to an arrest in G2/M.
TBK1 has previously been suggested to have tumor-suppressing activities and involvement in cell cycle control. It was first identified in a small interfering RNA (siRNA) screen looking for genes causing synthetic lethality with oncogenic K-Ras mutations (Barbie et al., 2009). Follow up studies reported a more general role for TBK1 in cell cycle, demonstrating its activation during mitosis (Onorati et al., 2016; Pillai et al., 2015). Our data agree with these studies, but it is yet unclear how TBK1 is activated at the centrosome and midbody; previous reports disagree as to the targets that TBK1 phosphorylates (Kim et al., 2013; Pillai et al., 2015). Our data showing abnormal TBK1 activation upon overexpression indicate that TBK1 levels and cellular distribution are tightly regulated during each of its physiological roles: mitosis, innate immunity, and selective autophagy. Additional studies are needed to understand TBK1 regulation during mitosis and how it translocates and is selectively activated at different organelles, a topic currently under investigation. Onorati et al. (2016) reported mitotic arrest in the context of Zika infection and attributed the mechanism to the sequestration of active TBK1 at Zika viral particles. These results are consistent with what we observed during mitophagy. Another intriguing line of future inquiry will be to understand how innate immunity (Xiao et al., 2017) has the potential to suppress tumorigenesis in the context of TBK1.
Mitochondrial damage triggers the activation of p-TBK1 at mitochondria in a PINK1/Parkin-dependent manner (Heo et al., 2015; Lazarou et al., 2015; Moore and Holzbaur, 2016; Richter et al., 2016), and our data suggest that this response arrests the cell cycle. Mitosis and mitophagy are both tightly regulated processes. Mitosis is a highly bioenergetically demanding process that requires stringent regulation of mitochondrial import, morphology, and distribution during cell cycle progression (Harbauer et al., 2014; Kanfer et al., 2015; Kashatus et al., 2011). Our data indicate that cell cycle progression is arrested during mitophagy. If PINK1 and Parkin are absent, the cell cycle continues to progress. We speculate that progression through mitosis before or without removing damaged mitochondria would be detrimental to the daughter cell. Previous observations found that “fit” healthy mitochondria segregate during cell division in stem cells and yeast daughter cells through an undefined mechanism (Higuchi et al., 2013; Katajisto et al., 2015). We propose that during mitophagy and the clearance of these damaged organelles, it is bioenergetically unfavorable to initiate mitosis. A block in mitosis could be a protective mechanism to halt cellular division during active mitochondrial clearance.
Our data do not support a direct interaction between ATM and Parkin-mediated mitophagy. However, we did find that PINK1-KO cells had impaired NHEJ, and it is possible that Parkin may have a link to other DNA damage repair pathways (Sumpter et al., 2016). Sumpter et al. (2016) found that Fanconi anemia complementation group proteins had a distinct role, separate from their function in the DNA damage response, which was required for efficient mitophagy and interacted directly with Parkin. Parkin-null mice have also been found to have increased rates of cancer and tumorigenesis after irradiation or when crossed to a mouse model of colon cancer (Poulogiannis et al., 2010; Zhang et al., 2011). Our findings demonstrate that the DNA damage proteins, ATM and ATR, genetically interact with the PINK1/Parkin pathway through their shared function in influencing the cell cycle. This does not exclude other possible types of interactions with other damage-sensing proteins. There may also be overlap between nuclear and mitochondrial stress signaling. Previously, we reported that mtDNA double-strand break signaling may rely partly on nuclear DNA damage pathways for premature senescence (Pinto et al., 2017).
Chk1 and Chk2 did not revert the Pink1B9 thoracic indentations upon KD (Table S2). Unlike CDC25 and the CDKs, Chk1 and Chk2 mutant Drosophila do not show dysregulation of cell cycle without the presence of DNA-damaging stimuli (Masrouha et al., 2003; Royou et al., 2005). We speculate that this may be why there was no genetic interaction between Chk1/2 and PINK1.
Our genetic data indicate that PINK1 and Parkin interact with the cell cycle by directly interacting with genes influencing cell cycle progression (CDKs and CDC25/stg) or with genes that influence the cell cycle but may have additional roles (ATM and TBK1/ik2). The thorax and flight wing develop from adult muscle precursors and a highly proliferative stem cell pool (Gunage et al., 2014). We evaluated the flies immediately after eclosion, so we did not evaluate a degenerative phenotype upon aging of the post-mitotic thorax muscles but rather a developmental defect that was rescued by the KD of the CDKs required for cell cycle progression (Pines, 1995). The mouse line is a complete KO of ATM and Parkin, while the Drosophila crosses were performed with point mutation mutant lines. ATM-KO mice have less severe phenotypes compared with kinase-dead ATM point-mutant mice, which are embryonic lethal, accounting for possible compensatory mechanisms in null versus kinase-dead mice (Daniel et al., 2012). Therefore, when evaluating the genetic data, we cannot directly compare findings from mouse and Drosophila, only the general outcome. In the Drosophila model, some ATM/Parkin and ATM/PINK1 double-mutant flies survived eclosion and notably exhibited phenotypes of each individual mutant fly, indicating that ATM and PINK1/Parkin are likely not in the same pathway, further implying that the genetic interaction is in parallel ultimately converging upon a common function or outcome involved in cell cycle progression. Experiments performed to check for the presence of an interaction between ATM and mitophagy or mitophagy and DNA damage confirm this.
Our report and other evidence suggest a global response to PINK1/Parkin-mediated mitochondrial clearance. Numerous recent efforts have helped elucidate many of the molecular events that occur at the mitochondria as they pertain to Parkin-dependent mitophagy (Yamano et al., 2016). However, it is becoming apparent that myriad factors contribute to a global cellular response to mitochondrial damage. In a PINK1/Parkin-dependent manner, mitophagy triggers the nuclear translocation of the transcriptional co-activator TFEB, which upregulates genes required for lysosomal biogenesis (Nezich et al., 2015). The C. elegans protein SKN-1 and its human ortholog NRF2, which are transcriptional co-activators involved in mitochondrial biogenesis, are also activated during mitophagy (Palikaras et al., 2015). These data support a global cellular response aimed at arresting cell cycle during mitochondrial quality control.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alicia M. Pickrell, Ph.D. (alicia.pickrell@vt.edu).
The pHAGE untagged TBK1 and pHAGE N-FLAG-HA TBK1 constructs generated in this study have been deposited to Addgene under Cat# 131791 and 131792, respectively (see Key Resource Table). All KO cell lines generated in this study are available by contacting the Lead Contact. All Drosophila lines can be obtained from the Indiana University Bloomington Drosophila Stock Center, except for the park25 null and the Pink1B9 null flies, which were a kind gift from Dr. Leo Pallanck (University of Washington). CRISPR plasmids were generated with targeting sequences from either published sources or advice from colleagues and are available upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-GRP75 | Cell Signaling | Cat#2816; RRID: AB_2120331 |
| rabbit anti-ATM S1981 | Abcam | Cat#ab81292; RRID: AB_1640207 |
| rabbit anti-PINK1 | Cell Signaling | Cat#6946; RRID: AB_11179069 |
| rabbit anti-Histone H3 S10 | Cell Signaling | Cat#53348; RRID: AB_2799431 |
| rabbit anti-Histone H3 T11 | Cell Signaling | Cat#9764; RRID: AB_659964 |
| mouse anti-Parkin (PRK8) | Santa Cruz | Cat#sc-32282; RRID: AB_628104 |
| rabbit anti-GAPDH | Sigma | Cat#G9545; RRID: AB_796208 |
| mouse anti-actin | Sigma | Cat#A2228; RRID: AB_476697 |
| rabbit anti-Mfn2 | in house (RJY) | N/A |
| rabbit anti-TBK1 S172 | Cell Signaling | Cat#5483; RRID: AB_10693472 |
| rabbit anti-TBK1 | Cell Signaling | Cat#3504; RRID: AB_2255663 |
| rabbit anti-LC3 | Cell Signaling | Cat#3868; RRID: AB_2137707 |
| rabbit anti-ATM S1981 | Cell Signaling | Cat#13050; RRID: AB_2798100 |
| mouse anti-COXII | Abcam | Cat#ab110258: RRID: AB_10887758 |
| mouse anti-p53 (DO1) | Santa Cruz | Cat#sc-126; RRID: AB_628082 |
| rabbit anti-ATM | Cell Signaling | Cat#2873; RRID: AB_2062659 |
| guinea pig anti-p62 | Cedarlane now Progen | Cat#GP62-C; RRID: AB_2687531 |
| mouse anti-TIM23 | BD Biosciences | Cat#611222; RRID: AB_398754 |
| mouse anti-BrdU | Sigma | Cat#B8434; RRID: AB_476811 |
| rabbit anti-AIM1/Aurora B | Sigma | Cat#A5102; RRID: AB_476740 |
| mouse anti-alpha-tubulin | Sigma | Cat#T9026; RRID: AB_477593 |
| mouse anti-HA.11 | Covance | Cat#MMS-101R; RRID: AB_291262 |
| rabbit anti-TOM20 | Santa Cruz | Cat#sc-11415; RRID: AB_2207533 |
| mouse anti-ATM | Sigma | Cat#A1106; RRID: AB_796190 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MitoTracker Red CMXRos | Thermo Fisher | Cat#M7512 |
| TO-PRO-3 Iodide | Thermo Fisher | Cat#T3605 |
| Lipofectamine LTX with Plus Reagent | Thermo Fisher | Cat#A12621 |
| XtremeGENE 9 DNA Transfection Reagent | Roche | Cat#XTG9-RO |
| NuPAGE LDS Sample Buffer (4X) | Invitrogen | Cat#NP0007 |
| Oligomycin | EMD Millipore | Cat#495455 |
| Antimycin A | Sigma | Cat#A8674 |
| Etoposide | Sigma | Cat#E1383 |
| BX-795 | Sigma | Cat#204001 |
| Thymidine | Sigma | Cat#T1895 |
| Nocodazole | Sigma | Cat#487928 |
| QVD-Oph | Abcam | Cat#ab141421 |
| A/C Heterodimerizer (rapalog) | Takara | Cat#635056 |
| DL-Dithiothreitol | Sigma | Cat#D0632 |
| SuperSignal West Femto Maximum Sensitivity Substrate | Thermo Fisher | Cat#34095 |
| ECL Prime Western Blotting System | GE Healthcare | Cat#RPN2232 |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | Cat#E2621 |
| Gateway LR Clonase II Enzyme mix | Invitrogen | Cat#11791100 |
| Gateway BP Clonase Enzyme Mix | Invitrogen | Cat#11789013 |
| Puromycin | InvivoGen | Cat#ant-pr-1 |
| Polybrene | Sigma | Cat# TR-1003-G |
| Critical Commercial Assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat#G7570 |
| Deposited Data | ||
| pHAGE-N-FLAG-HA-TBK1 | Addgene | Cat#131791 |
| pHAGE-TBK1-C-FLAG-HA (untagged) | Addgene | Cat#131792 |
| Western blot data | Mendeley Data | https://doi.org/10.17632/789652xcr3.110.17632/789652xcr3.1 |
| Experimental Models: Cell Lines | ||
| HeLa | ATCC® | Cat#CCL-2; subcloned from RJY |
| HCT116 | ATCC® | Cat#CCL-247 |
| Patient Fibroblasts | Coriell | GM05294 |
| Patient Fibroblasts | Coriell | GM02052 |
| Patient Fibroblasts | NINDS depository | ND40066 |
| Patient Fibroblasts | NINDS depository | ND36091 |
| Patient Fibroblasts | NINDS depository | ND34769 |
| HEK293T | ATCC® | Cat#CRL-3216 |
| ATM KO HCT116 | This paper | N/A |
| PINK1 KO HCT116 | This paper | N/A |
| Parkin KO HCT116 | This paper | N/A |
| TBK1 KO HeLa Clone 2 | This paper | N/A |
| TBK1 KO HeLa Clone 4 | This paper | N/A |
| PINK1 KO HeLa | Kane et al., 2014 | N/A |
| TBK1 KO Clone 2 rescued with TBK1 WT HeLa | This paper | N/A |
| FRB-BFP-Fis1 HeLa | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| ATM KO | Jackson Laboratories | B6.129S6-Atmtm1Awb/J |
| Parkin KO | Jackson Laboratories | B6.129S4-Prkntm1Shn/J |
| Recombinant DNA | ||
| hCas9 | Addgene | Cat#41815 |
| pSpCas9(BB)-2A-Puro (PX459) | Addgene | Cat#48139 |
| pBMN-mCherry-Parkin | Addgene | Cat#59419 |
| pBMN-YFP-Parkin | Addgene | Cat#59416 |
| pCBASceI | Addgene | Cat#26477 |
| pimEJ5GFP | Addgene | Cat#44026 |
| pDRGFP | Addgene | Cat#26475 |
| pmCherry-C1 | Clonetech | Cat#632524 |
| pEYFP-C1 | Clonetech | Cat#6005–1 |
| gRNA Cloning Vector | Addgene | Cat#41824 |
| pDONR223 | Invitrogen | No longer available |
| pHAGE-FRB-BFP-Fis1 | Vargas et al., 2019 | N/A |
| pHAGE-FKBP-mEGFP-TBK1 | Vargas et al., 2019 | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
ATM KO mice (ATMtm1Awb/J) were crossed with Parkin KO mice (Park2tm1Shn/J) (strains from Jackson Laboratories). The nuclear background was C57BL/6J (backcrossed at least > F10). ATM+/− mice of mixed sexes were bred until 1 year of age. ATM+/− Parkin+/− or ATM+/− Parkin KO mice of mixed sexes were bred until 1 year of age. Parkin KO MEFs were generated with Parkin KO mice of mixed sexes bred until 1 year of age. Genotyping was performed according to Jackson Laboratories. All mouse procedures were performed according to a protocol approved by the NIH/NINDS Institutional Animal Care and Use Committee. Mice were housed in a virus-antigen-free facility at the NIH Division of Veterinary Resources in a 12 hr light/dark cycle at RT and fed ad libitum with standard rodent diet.
Drosophila
Drosophila strains were maintained on standard cornmeal/molasses medium at 25°C with a 12-hr light-dark cycle. The park25 null and the Pink1B9 null flies were a kind gift from Dr. Leo Pallanck (University of Washington). Fly stocks were obtained from Bloomington Fly Stock Depository (Table S2). Multiple male and virgin female flies were set up for experimental crosses. Flies of both sexes were analyzed for phenotype and eclosion except for PINK1B9 crosses where males were examined. Fly survival and phenotyping for thorax indentation were scored 24 hr after eclosion.
Cell Lines
HCT116 cell lines were maintained in McCoy’s 5A (Modified) Media with 10% FBS. HEK293T and HeLa cell lines were maintained in DMEM high glucose with 10% FBS, 2mM L-Glutamine, 10mM HEPES, 0.1mM non-essential amino acids, and 1mM sodium pyruvate. Cells were routinely tested for mycoplasma contamination. CRISPR KO lines generated are found in (Table S1). The PINK1 KO HeLa cell line was a kind gift from RJY.
Primary Cells
Patient fibroblast lines obtained from the NINDS depository: ND40066, ND36091, ND34769 and from Coriell Institute for Medical Research were as followed: GM05294 and GM02052. Patient fibroblasts and MEF cells were maintained in DMEM high glucose with 10% FBS, 2mM L-Glutamine, 10mM HEPES, 0.1mM non-essential amino acids, and 1mM sodium pyruvate. Cells were routinely tested for mycoplasma contamination. Cells for growth curve experiments were used at matching passage numbers. Pharmacological drugs used in to treat either cell lines or primary cells are as follows: 10 μg/ml oligomycin (EMD Millipore) and 5 μg/ml antimycin A (Sigma), 10 μM etoposide, 1μM BX-795, 100ng/mL nocodazole, 2.5mM thymidine (Sigma), 0.5nM A/C heterodimer (rapalog) (Takara), and 10 μM QVD-OPh (Abcam).
METHOD DETAILS
MEF Generation
Timed pregnant females E12.5–14.5 were sacrificed. Embryos were dissected to remove the cerebrum, internal organs, and limbs. The remaining tissue was digested with 0.5% trypsin-EDTA. MEFs were used between passages 2–4. Each embryo = clone.
Western Blotting
Cells were lysed using 2x LDS buffer containing 50mM DTT and boiled for 15 min. Approximately 50 μg of protein lysate was loaded onto 4%–12% Bis-Tris gels. Proteins were transferred onto PVDF membranes and blocked with 5% non-fat powdered milk. Primary antibodies were incubated 4°C overnight. Antibodies used: PINK1, p-ATM S1981, LC3B, TBK1, p-TBK1 S172, p-Histone H3 S10 and p-Histone H3 T11 (Cell Signaling), pATM S1981, COXII (Abcam), ATM (Cell Signaling or Sigma), Tim23 (BD Biosciences), Mfn2 (in-house), TOM20, Parkin, p53 (Santa Cruz), GAPDH and actin (Sigma), p62 (Cedarlane), and HA.11 (Covance). Secondary HRP-linked antibodies (GE Healthcare) were incubated at RT for 1 hr. Blots were exposed using peroxidase-based ECL (Pierce) detected by a ChemiDoc Imaging System (BioRad).
CRISPR and Knockout Cell Line Generation
CRISPR design was aided by publicly available software provided by MIT www.crispr.mit.edu. CRISPR sequences are found in Table S1. Primers were annealed and cloned into the linearized gRNA vector, which was a gift from George Church (Addgene #41824) using the Gibson Assembly Cloning Kit (NEB). HCT116 or HeLa cells were cotransfected using Lipofectamine LTX (Invitrogen) or X-trem-eGENE9 (Roche) using the above CRISPR plasmid, pEYFP-C1 (Clonetech), and the hCas9 plasmid, which was a gift from George Church (Addgene #41815) or was cloned into SpCas9–2A-Puro, which was a gift from Feng Zhang (Addgene #48139). YFP-positive cells were FACS sorted or selected with 1 μg/mL puromycin and serially diluted for single colony clones. DNA was extracted using the Zymo gDNA Isolation Kit and genotyped using primers (Table S1). PCR products were cloned using CloneJET PCR cloning kit (Invitrogen) according to the manufacturer’s instructions. Clones were sequenced to verify KOs (Table S1).
Synchronizing Cells
To synchronize cells at the G1/S transition, cells were subjected to a double thymidine block (DTB). In brief, cells were exposed to thymidine-containing media for 17 hr, released in fresh media for 8 hr, and returned to thymidine-containing media for another 16 hr. Cells synchronized at G2/M phase were treated for 16 hr with 100ng/mL of nocodazole.
Cloning
pBMN-mCherry-Parkin and pBMN-YFP-Parkin were cloned into BamHI–SalI sites of the pBMN-Z vector to generate stable cells lines as previously described (Yamano et al., 2014) (Addgene # 59419 and #59416). To generate TBK1 rescue lines, TBK1 cDNA was cloned into pDONR223 and transferred into the pHAGE-N’-FLAG-HA-IRES-puro or pHAGE-C’-FLAG-HA-IRES-puro vectors using l recombinase in vitro (Invitrogen). A stop codon inserted at the end of the TBK1 cDNA ensured it was untagged. The FRB-BFP-Fis1 and FKBP-GFP-TBK1 construct cloning was described previously (Vargas et al., 2019) and given as kind gifts from RJY. Stably expressing FRB-BFP-Fis1 cells were sorted via flow cytometry for low BFP expression and evaluated to ensure normal mitochondrial morphology. All constructs were confirmed by DNA sequencing.
Retrovirus or Lentiviral Generation
Retroviral/lentiviral helpers and retroviral/lentiviral constructs were transfected into HEK293T with Lipofectamine LTX at a 1:3 ratio. 24 hr after transfection, media was changed. Infectious media containing live virus was collected 40 hr later and filtered using a 0.45 μM PES membrane. Virus was used to transduce cells with the addition 8 μg/ml of polybrene (Sigma).
Subcloning
TBK1 KO clone 2 was selected to stably reconstitute wild-type TBK1 to generate the rescue line. After lentiviral infection, positive cells resistant to 1 μg/mL puromycin were subcloned from single cell colonies.
Growth Curves
Cell growth was determined by manual cell counting and CellTiter-Glo® Luminescent Cell Viability Assay (Promega). For manual counting, 5 × 103 cells were plated in a 12- well dish or 3 × 104 cells were plated in a 6-well dish and counted every 24 hr period using an automated cell counter (Logos Biosystems). For luminescence experiments, 300 to 600 cells were plated in white-coated 96-well plates and read with a luminescence reader (Victor) according to the manufacturer’s directions every 24 hr. Cells treated with 1 μm BX-795 were plated in the presence of the inhibitor, and the drug was replaced every 48 h.
BrdU Analysis
Cells were incubated with 10 μM BrdU (Sigma) for 1 hr 30 min. Cells were immediately fixed with 4% PFA and incubated with 2N HCl to denature DNA to facilitate antibody penetration. 0.1mM sodium borate buffer was used to neutralize HCl prior to routine ICC. Cells were imaged on a Zeiss LSM 510 confocal. 5 random fields of view were taken for each individual experiment to sample 200–300 cells.
Immunocytochemistry
Cells for imaging were plated in Nunc two-well chamber slides (Thermo Fisher). Cells were fixed with 4% PFA and permeabilized with 0.15% Triton X-100 followed by blocking with 5% BSA. Primary antibodies were incubated 1hr at RT or overnight at 4°C. Antibodies used were as followed: GRP75, p-TBK1S172 (Cell Signaling), TOM20 (Santa Cruz), AIM-1/Aurora B and BrdU (BD Biosciences), α-tubulin (Sigma), and HA.11 (Covance). Secondary AlexaFluor® (Thermo Fisher) conjugated antibodies were incubated for 1 hr RT. Cells were counterstained with 300 nM of DAPI for 5 min (Thermo Fisher). Cells were imaged on a Zeiss LSM 510 confocal. 5 random fields of view were taken for each individual experiment to sample 200–300 cells. Mitotic cells were identified by chromosome condensation and verified by α-tubulin localization/morphology. p-TBK1 localization was determined by its colocalization with either DAPI or TOM20.
FRB-BFP-Fis1 cells were grown on glass coverslips and transfected 24hrs later with FKBP-GFP-TBK1 plasmid using XtremeGene9 (Roche). 24hrs after transfection, media was changed and cells were treated with rapalog for 24hrs. Cells were either stained with 1nM MitoTracker Red CMXROS (ThermoFisher) for 15 minutes and then fixed with 4% PFA or fixed and permeabilized as above. Cells were counterstained with 1nM TO-PRO-3 (ThermoFisher) for 15 minutes. Cells were imaged on a Nikon C2 confocal. 5 random fields of view were taken for each individual experiment to sample 200–300 cells. Mitotic cells were identified by chromosome condensation.
HR and NHEJ Recombination Assay
Cells were transfected using Lipofectamine LTX with: pCDNA mCherry-C1, pDRGFP (Addgene#26475) or pimEJ5GFP (Addgene#44026), and pCBA SceI (Addgene#26477), which were kind gifts from Maria Jasin or Jeremy Stark. Cells were sorted on a FACS analyzer (BD Biosciences) to determine the population of GFP positive cells normalized to mCherry-C1 expression to account for differences in transfection efficiency (Bennardo et al., 2008; Pierce et al., 1999).
QUANTIFICATION AND STATISTICAL ANALYSIS
A Student’s t test was used to determine statistical significance with comparisons between two groups. For comparisons between three or more groups, a one way-ANOVA with Tukey post hoc analysis was used. A chi-square test was used to determine significance for experiments comparing theoretical Mendelian genetic probabilities to experimental probabilities for Drosophila eclosion experiments and mouse genotyping experiments. GraphPad Prism v7 software was used. Error bars ± SD or SEM noted in legend. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
DATA AND CODE AVAILABILITY
The western blot data generated during this study are available at Mendeley Data https://doi.org/10.17632/789652xcr3.110.17632/789652xcr3.1 The pHAGE untagged TBK1 and pHAGE N-FLAG-HA TBK1 constructs generated in this study have been deposited to Addgene under Cat# 131791 and 131792, respectively (see Key Resource Table). Any other raw datasets generated during this study are currently being used for future studies and to obtain grant funding, but data is available upon request.
Supplementary Material
Highlights.
PINK1 and Parkin genetically interact with cell cycle-regulatory proteins
PINK1 and Parkin do not directly signal with the nuclear DNA damage response
Active TBK1 sequestered at damaged mitochondria during mitophagy perturbs mitosis
Fly ortholog of TBK1 rescues PINK1- and Parkin-knockout phenotypes
ACKNOWLEDGMENTS
This work was supported in part by the NIH NINDS intramural program (R.J.Y., S.A.S., D.P.S., C.-H.H., and A.M.P.), departmental startup funds (A.M.P.), the Virginia Tech Open Access Subvention Fund (A.M.P.), and NIGMS Postdoctoral Research Associate Fellowships (A.M.P. and S.A.S.). We would like to thank the NINDS Microscopy Core, NHLBI Flow Cytometry Core, Dr. Chunxin Wang for technical advice and stimulating scientific conversation, and Dr. Katherine Roche for stimulating scientific conversation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.08.085.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Agnihotri S, Golbourn B, Huang X, Remke M, Younger S, Cairns RA, Chalil A, Smith CA, Krumholtz SL, Mackenzie D, et al. (2016). PINK1 is a negative regulator of growth and the Warburg effect in glioblastoma. Cancer Res. 76, 4708–4719. [DOI] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. (2009). Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 462, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, and Wynshaw-Boris A (1996). Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86, 159–171. [DOI] [PubMed] [Google Scholar]
- Bennardo N, Cheng A, Huang N, and Stark JM (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardini JP, Lazarou M, and Dewson G (2017). Parkin and mitophagy in cancer. Oncogene 36, 1315–1327. [DOI] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Giet R, Sinka R, Mazumdar A, Lock WG, Balloux F, Zafiropoulos PJ, Yamaguchi S, Winter S, Carthew RW, et al. (2004). Genome-wide survey of protein kinases required for cell cycle progression. Nature 432, 980–987. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, and Guo M (2006). Drosophila pink1 is required for mitochondrial function and interacts genetically with Parkin. Nature 441, 1162–1166. [DOI] [PubMed] [Google Scholar]
- Clément JF, Meloche S, and Servant MJ (2008). The IKK-related kinases: from innate immunity to oncogenesis. Cell Res. 18, 889–899. [DOI] [PubMed] [Google Scholar]
- Daniel JA, Pellegrini M, Lee BS, Guo Z, Filsuf D, Belkina NV, You Z, Paull TT, Sleckman BP, Feigenbaum L, and Nussenzweig A (2012). Loss of ATM kinase activity leads to embryonic lethality in mice. J. Cell Biol 198, 295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, and Shadel GS (2007). Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Invest 117, 2723–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, et al. (2003). Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem 278, 43628–43635. [DOI] [PubMed] [Google Scholar]
- Gong Y, Zack TI, Morris LG, Lin K, Hukkelhoven E, Raheja R, Tan IL, Turcan S, Veeriah S, Meng S, et al. (2014). Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat. Genet 46, 588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunage RD, Reichert H, and VijayRaghavan K (2014). Identification of a new stem cell population that generates Drosophila flight muscles. eLife 3, e03126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbauer AB, Opalińska M, Gerbeth C, Herman JS, Rao S, Schönfisch B, Guiard B, Schmidt O, Pfanner N, and Meisinger C (2014). Mitochondria. Cell cycle-dependent regulation of mitochondrial preprotein translocase. Science 346, 1109–1113. [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, and Harper JW (2015). The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 60, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi R, Vevea JD, Swayne TC, Chojnowski R, Hill V, Boldogh IR, and Pon LA (2013). Actin dynamics affect mitochondrial quality control and aging in budding yeast. Curr. Biol 23, 2417–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, and Youle RJ (2014). PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J. Cell Biol 205, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanfer G, Courthéoux T, Peterka M, Meier S, Soste M, Melnik A, Reis K, Aspenström P, Peter M, Picotti P, and Kornmann B (2015). Mitotic redistribution of the mitochondrial network by Miro and Cenp-F. Nat. Commun 6, 8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD, and Counter CM (2011). RALA and RALBP1 regulate mitochondrial fission at mitosis. Nat. Cell Biol 13, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass EM, Helgadottir HR, Chen CC, Barbera M, Wang R, Westermark UK, Ludwig T, Moynahan ME, and Jasin M (2013). Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc. Natl. Acad. Sci. U S A 110, 5564–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katajisto P, Döhla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, and Sabatini DM (2015). Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348, 340–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Welsh EA, Oguz U, Fang B, Bai Y, Kinose F, Bronk C, Remsing Rix LL, Beg AA, Rix U, et al. (2013). Dissection of TBK1 signaling via phosphoproteomics in lung cancer cells. Proc. Natl. Acad. Sci. U S A 110, 12414–12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, and Youle RJ (2015). The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, and Kaldis P (2013). Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 140, 3079–3093. [DOI] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, and Gorbunova V (2008). Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst.) 7, 1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal A, and Zou L (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masrouha N, Yang L, Hijal S, Larochelle S, and Suter B (2003). The Drosophila chk2 gene loki is essential for embryonic DNA double-strand-break checkpoints induced in S phase or G2. Genetics 163, 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K, Wunder C, Roysam B, Lin G, and Lippincott-Schwartz J (2009). A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. U S A 106, 11960–11965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui J, Takahashi Y, Goto J, Tomiyama H, Ishikawa S, Yoshino H, Minami N, Smith DI, Lesage S, Aburatani H, et al. (2010). Mechanisms of genomic instabilities underlying two common fragile-site-associated loci, PARK2 and DMD, in germ cell and cancer cell lines. Am. J. Hum. Genet 87, 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AS, and Holzbaur EL (2016). Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. U S A 113, E3349–E3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezich CL, Wang C, Fogel AI, and Youle RJ (2015). MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J. Cell Biol 210, 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onorati M, Li Z, Liu F, Sousa AMM, Nakagawa N, Li M, Dell’Anno MT, Gulden FO, Pochareddy S, Tebbenkamp ATN, et al. (2016). Zika virus disrupts phospho-TBK1 localization and mitosis in human neuroepithelial stem cells and radial glia. Cell Rep. 16, 2576–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, and Tavernarakis N (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, and Chung J (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by Parkin. Nature 441, 1157–1161. [DOI] [PubMed] [Google Scholar]
- Pedersen M, Tiong S, and Campbell SD (2010). Molecular genetic characterization of Drosophila ATM conserved functional domains. Genome 53, 778–786. [DOI] [PubMed] [Google Scholar]
- Pickrell AM, and Youle RJ (2015). The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce AJ, Johnson RD, Thompson LH, and Jasin M (1999). XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 13, 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai S, Nguyen J, Johnson J, Haura E, Coppola D, and Chellappan S (2015). Tank binding kinase 1 is a centrosome-associated kinase necessary for microtubule dynamics and mitosis. Nat. Commun 6, 10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J (1995). Cyclins and cyclin-dependent kinases: a biochemical view. Biochem. J 308, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto M, Pickrell AM, Wang X, Bacman SR, Yu A, Hida A, Dillon LM, Morton PD, Malek TR, Williams SL, and Moraes CT (2017). Transient mitochondrial DNA double strand breaks in mice cause accelerated aging phenotypes in a ROS-dependent but p53/p21-independent manner. Cell Death Differ. 24, 288–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, and Pallanck LJ (2008). The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl. Acad. Sci. U S A 105, 1638–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulogiannis G, McIntyre RE, Dimitriadi M, Apps JR, Wilson CH, Ichimura K, Luo F, Cantley LC, Wyllie AH, Adams DJ, and Arends MJ (2010). PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc. Natl. Acad. Sci. U S A 107, 15145–15150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, et al. (2013). The genetic landscape of high-risk neuroblastoma. Nat. Genet 45, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Requejo-Aguilar R, Lopez-Fabuel I, Fernandez E, Martins LM, Almeida A, and Bolaños JP (2014). PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nat. Commun 5, 4514. [DOI] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, et al. (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. U S A 113, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royou A, Macias H, and Sullivan W (2005). The Drosophila Grp/Chk1 DNA damage checkpoint controls entry into anaphase. Curr. Biol 15, 334–339. [DOI] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, and Harper JW (2013). Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder EA, Raimundo N, and Shadel GS (2013). Epigenetic silencing mediates mitochondria stress-induced longevity. Cell Metab. 17, 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro RS, and Anderson KV (2006). Drosophila Ik2, a member of the I kappa B kinase family, is required for mRNA localization during oogenesis. Development 133, 1467–1475. [DOI] [PubMed] [Google Scholar]
- Sumpter R Jr., Sirasanagandla S, Fernández AF, Wei Y, Dong X, Franco L, Zou Z, Marchal C, Lee MY, Clapp DW, et al. (2016). Fanconi anemia proteins function in mitophagy and immunity. Cell 165, 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentin-Vega YA, Maclean KH, Tait-Mulder J, Milasta S, Steeves M, Dorsey FC, Cleveland JL, Green DR, and Kastan MB (2012). Mitochondrial dysfunction in ataxia-telangiectasia. Blood 119, 1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, and Youle RJ (2019). Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol. Cell 74, 347–362.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, et al. (2010). Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet 42, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Zou Q, Xie X, Liu T, Li HS, Jie Z, Jin J, Hu H, Manyam G, Zhang L, et al. (2017). The kinase TBK1 functions in dendritic cells to regulate T cell homeostasis, autoimmunity, and antitumorimmunity. J. Exp. Med 214, 1493–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Fogel AI, Wang C, van der Bliek AM, and Youle RJ (2014). Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 3, e01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Matsuda N, and Tanaka K (2016). The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep. 17, 300–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, and Lu B (2006). Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc. Natl. Acad. Sci. U S A 103, 10793–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, and Feng Z (2011). Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. U S A 108, 16259–16264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The western blot data generated during this study are available at Mendeley Data https://doi.org/10.17632/789652xcr3.110.17632/789652xcr3.1 The pHAGE untagged TBK1 and pHAGE N-FLAG-HA TBK1 constructs generated in this study have been deposited to Addgene under Cat# 131791 and 131792, respectively (see Key Resource Table). Any other raw datasets generated during this study are currently being used for future studies and to obtain grant funding, but data is available upon request.