

Abstract
To dissect therapeutic mechanisms of transplanted stem cells and develop exosome-based nanotherapeutics in treating autoimmune and neurodegenerative diseases, we assessed the effect of exosomes secreted from human mesenchymal stem cells (MSCs) in treating multiple sclerosis using an experimental autoimmune encephalomyelitis (EAE) mouse model. We found that intravenous administration of exosomes produced by MSCs stimulated by IFNγ (IFNγ-Exo) (i) reduced the mean clinical score of EAE mice compared to PBS control, (ii) reduced demyelination, (iii) decreased neuroinflammation, and (iv) upregulated the number of CD4+CD25+FOXP3+ regulatory T cells (Tregs) within the spinal cords of EAE mice. Co-culture of IFNγ-Exo with activated peripheral blood mononuclear cells (PBMCs) cells in vitro reduced PBMC proliferation and levels of pro-inflammatory Th1 and Th17 cytokines including IL-6, IL-12p70, IL-17AF, and IL-22 yet increased levels of immunosuppressive cytokine indoleamine 2,3-dioxygenase. IFNγ-Exo could also induce Tregs in vitro in a murine splenocyte culture, likely mediated by a third-party accessory cell type. Further, IFNγ-Exo characterization by deep RNA sequencing suggested that IFNγ-Exo contains anti-inflammatory RNAs, where their inactivation partially hindered the exosomes potential to induce Tregs. Furthermore, we found that IFNγ-Exo harbors multiple anti-inflammatory and neuroprotective proteins. These results not only shed light on stem cell therapeutic mechanisms but also provide evidence that MSC-derived exosomes can potentially serve as cell-free therapies in creating a tolerogenic immune response to treat autoimmune and central nervous system disorders.
Keywords: nanotherapeutics, exosomes, extracellular vesicles, mesenchymal stem cells, multiple sclerosis, regulatory T cells, drug delivery
Graphical Abstract
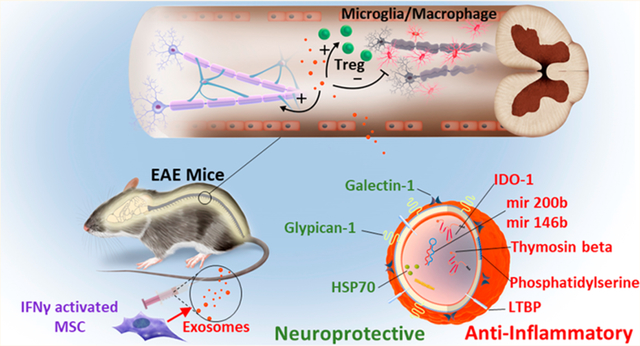
Stem cells hold great promise for treating several devastating diseases. In particular, mesenchymal stem (or stromal) cells (MSCs) are being tested in over 700 clinical trials, including treating autoimmune and neurodegenerative diseases.1,2 Unfortunately, preclinical studies and clinical trials using MSCs have produced mixed outcomes, which is in part because of the limited understanding of their mechanisms of action (MOA).2 One particular puzzle in the MSC field is that following systemic transplantation, MSCs are quickly entrapped in the lung vasculature bed due to their big size, with typically <1% of MSCs reaching and engrafting at the target sites,3 yet therapeutic effects are often observed.4–6 Current paradigm attributes MSC’s therapeutic functions mainly to paracrine factors.1,2 We reasoned that extracellular vesicles (EVs) generated by transplanted cells in situ in vivo could be a mechanism by which stem cells contribute to tissue remodeling and regeneration in distant sites.7 Indeed, several recent studies demonstrated that EVs can modulate the immune system through multiple mechanisms8–11 and MSC-derived EVs possess therapeutic functions.12–18
To develop EVs as cell-free therapeutics and shed light on their potential roles in stem cell’s effects in vivo, in this study we evaluated exosomes released from MSCs (MSC-exosomes) in treating devastating autoimmune and neurodegenerative diseases, specifically, multiple sclerosis (MS). MS is an inflammatory disease of the central nervous system (CNS) that can lead to demyelination, neuronal injury and loss, and eventually neurological disability,19,20 which currently is a significant burden on the healthcare system.21 Specifically, therapeutic effects of exosomes derived from native MSC (Native-Exo) or MSC activated by IFNγ (IFNγ-Exo) in a MS mouse experimental autoimmune encephalomyelitis (EAE) model22 were evaluated, and their main cellular and molecular mechanisms were investigated. We found that systemic injection of IFNγ-Exo resulted in sustained clinical recovery with enhanced improvement in motor skills, reduction in neuroinflammation, and reduced demyelination in EAE mice. In addition, we observed increased numbers of CD4+CD25+FOXP3+ regulatory T cells (Tregs), while a reduced number of total macrophages/microglia and pro-inflammatory T cells within the spinal cords of IFNγ-Exo-treated animals. Additionally, co-culture of IFNγ-Exo with activated peripheral blood mononuclear cells (PBMCs) cells reduced PBMC proliferation and levels of pro-inflammatory Th1 and Th17 cytokines including IL-6, IL-12p70, IL-17AF, and IL-22, yet increased levels of immunosuppressive cytokine indoleamine 2,3-dioxygenase (IDO). IFNγ-Exo could also induce Tregs in vitro in a murine splenocyte culture, likely mediated by a third-party accessory cell type. We further delineate exosome’s molecular mechanisms through deep RNA sequencing and proteomics, which revealed that IFNγ-Exo are enriched in anti-inflammatory and/or neuroprotective RNAs and proteins. In summary, our work helps to understand the therapeutic mechanism of transplanted stem cells and provides therapeutic platforms based on cell-free exosomes to treat MS, and in the future, other autoimmune and neurodegenerative diseases.
RESULTS AND DISCUSSION
Isolation and Characterization of MSC Exosomes.
To isolate exosomes, conditioned media from MSCs cultured for 3 days under IFNγ stimulation or native conditions (without stimulation) went through serial ultracentrifugation, and the exosome pellet was reconstituted in phosphate-buffered saline (PBS) and stored at −80 °C before use (see Materials and Methods section for details). IFNγ, a pleiotropic cytokine that is involved in onset, orchestration, and resolution of adaptive immune and autoimmune responses,23 has recently been reported to promote immunosuppressive effects of MSCs.24 Activation of MSC by IFNγ was confirmed by the upregulation of major histocompatibility complex II (MHCII) and programmed death-ligand 1 (pD-L1) expression on MSCs (Figure S1), which are characteristic MSC markers induced upon activation by pro-inflammatory cytokines.25 Isolated MSC exosomes were characterized using different methods including Western blotting, flow cytometry, electron microscopy, and nanoparticle tracking analysis (NTA). The presence of CD81 and tumor susceptibility gene 101 protein (TSG101), two proteins commonly enriched in exosomes, was confirmed in both IFNγ-Exo and Native-Exo using Western Blotting (Figure S2A). In addition, the absence of Calnexin (an endoplasmic reticulum marker) suggests the MSC exosome pellet does not have contamination from endoplasmic reticulum-derived vesicles (Figure S2A). On the CD63-captured exosomes, the tetraspanins CD63 and CD81 were expressed on exosomes, whereas the CD9 expression was low, as characterized by flow cytometry (Figure S2B). A similar trend of expression for CD63, CD81, and CD9 was observed for MSCs. Stimulation of MSCs with IFNγ resulted in no major differences in expression for the above markers on MSCs and exosomes (Figure S2A and Figure S2B). Transmission electron microscopy (TEM) showed spherically shaped vesicles within the size range of exosomes (Figure S2C). Additionally, the presence of CD63 was confirmed by using an immunogold anti-CD63 antibody (Figure S2C). The sizes of exosomes were characterized by NTA that tracks the dynamic of particle movement and approximates it with Brownian motion to obtain size information. A screenshot of the NTA video is presented in Figure S2D, left panel. The average size of exosomes was determined to be approximately 115 nm in diameter (Figure S2D). Together these data show that our protocol isolated vesicles with the size of exosomes, expressing several of the commonly used exosomal markers.
MSC Exosomes Improve Functional Outcomes in EAE.
We next sought to examine MSC exosome’s therapeutic effect in treating MS in a well-established EAE model. To induce EAE, female C57BL/6J mice were immunized with complete Freund’s adjuvant (CFA), MOG35–55 peptide, and pertussis toxin (injected on days 0 and 2) (Figure 1A). Approximately 15–20 days later, mice displayed the peak of the disease, showing complete paralysis of the tail and hind limbs with flattened posture. Each mouse was graded every other day and assigned a clinical score ranging from 0 to 4, where 0 represents healthy wild-type (WT) mouse and 4 represents a dead mouse (detailed scoring criteria are provided in the Materials and Methods). Exosomes derived from Native-Exo or from IFNγ-Exo were infused intravenously (i.v.), as schematically shown in Figure 1A. A dose of approximately 150 μg (or 1.06 × 109 ± 9.6 × 107 particles per NTA, n = 4), which was derived from 5 to 7 million MSCs, per mouse was used because a lower dose (30 μg) was shown to be less potent (Figure 1B,C) and higher doses than 150 μg are impractical due to the inefficiencies in exosome preparation. While the focus of this study is exosomes, native and IFNγ stimulated MSCs (1 million) were also included as control groups, as we aim to compare exosome’s efficacy to MSCs’. A single injection of IFNγ-Exo (n = 6) at the peak of the disease (day 18) resulted in a mean clinical score of 1.2 ± 0.3 at day 40, which is a significant improvement (p < 0.001) compared with PBS control (n = 6) (mean clinical score of 2.9 ±0.6) (Figure 1B). Native-Exo (n = 6) also ameliorated the disease but to a lesser extent than IFNγ-Exo with the mean clinical score of 2.2 ± 0.5 (p < 0.05). Comparison of exosomes with their MSC counterparts showed similar efficacy in EAE model. IFNγ-treated MSCs (n = 6) showed comparable clinical scores (1.5 ± 0.6; n.s.) to that of IFNγ-Exo (1.2 ± 0.3). Similarly, native MSCs (n = 6) displayed comparable clinical scores (2.1 ±0.4; n.s.) compared to Native-Exo (2.2 ± 0.5). We acknowledge that due to their small size and the enrichment steps, we are able to administer higher dosages of exosomes (based on the number of MSCs from which they were derived) compared to the MSC controls in this set of studies, demonstrating an advantage of exosome-based therapeutics. MSCs exhibited a better efficacy than their equivalent dose of exosome counterparts, probably due to that MSCs may exert additional therapeutic mechanisms (e.g., secreted paracrine factors) beyond exosomes. Despite the dosage difference, the comparability in efficacy between MSCs and their exosomes suggests that exosomes could be considered as a surrogate treatment to the cell therapy.
Figure 1.

MSC-derived exosomes improve clinical scores and associated neurological parameters in EAE mice. (A) Schematic representation of the procedure to induce EAE and the treatment (exosomes or MSCs) that the mice received at day 18, followed by clinical scoring until day 40. (B) Clinical scores of EAE mice after PBS injection, native MSCs, and IFNγ stimulated MSCs and their respective exosomes (n = 6 for all groups); Mann-Whitney t tests were used to determine the p values (*p < 0.05; ***p < 0.001). (C) Dose study was conducted using two doses of exosomes and results compared against the PBS control. Mann-Whitney t tests were used to determine the p values (*p < 0.05; ***p < 0.001). (D) Representative spinal cord sections stained with Luxol fast blue. Dashed lines indicate white matter damage areas. (E) Quantification of demyelinated areas in the different groups. Unpaired t tests were used to determine p values (n = 3; **p < 0.01; ***p < 0.001).
Demyelination Was Reduced in Exosome-Treated EAE Mice.
Demyelination is considered to be one of the hallmarks of MS, which eventually results in clinical symptoms.26 It is also known that clinical symptoms in EAE mice are associated with a demyelination in spinal cord.22 Therefore, to analyze the severity of demyelination of treated mice, spinal cord sections of variously treated mice were analyzed for myelin using Luxol fast blue (LFB). Spinal cord sections from IFNγ-Exo-treated mice (n = 3; >10 sections per animal) showed a reduction in the degree of demyelination (9.5% ± 1.09%; p < 0.001) when compared to PBS control mice (n = 3; 34.4% ± 1.1%) (Figure 1D,E). The degree of demyelination was also reduced in Native-Exo-treated mice (n = 3; 13.7% ± 1.3%; p < 0.001) compared to PBS control and to a lesser extent compared to IFNγ-Exo (Figure 1D,E).
Reduction of Neuroinflammation and Induction of Tolerance in Exosome-Treated EAE Mice.
Early MS lesions are characterized by focal infiltration of monocytes and lymphocytes into regions of the brain and spinal cord.27 For instance, infiltrated macrophages secrete an excessive amount of pro-inflammatory factors including cytokines, nitric oxide, and reactive oxygen species, which can result in neuronal damage.28 To reveal the mechanisms of exosome-mediated effect in neurologic function, we investigated levels of neuroinflammation in exosome-treated mice. Ionized calcium binding adaptor protein (Iba-1) is a marker of macrophages/microglia. Therefore, Iba-1 positive cells were counted in the spinal cords sections in the white and gray matter areas (Figure 2A). IFNγ-Exo (n = 3; 301 ± 23 cells per section; p < 0.001) and Native-Exo (581 ± 21 cells per section; p < 0.01) treated mice showed a significant decrease in macrophage/microglia infiltration compared to the PBS-treated group (n = 3; 940 ± 75 cells per section) (Figure 2B). Note also that exosome treatments transformed the macrophage/microglia morphology from a characteristic swollen truncated amoeboid-like structure that is usually displayed in activated microglia/macrophage29,30 and observed here in the PBS-treated group into a long, ramified morphology with small cell bodies as observed in healthy WT mice (Figure 2A and Figure S3).
Figure 2.

MSC-derived exosome-treated mice showed a reduced number of infiltrated macrophages/microglia and T cells in the spinal cords of EAE mice. (A) Immunohistochemistry of Iba-1, a Ca2+-binding protein indicative of macrophages and microglia, and the nuclear stain DAPI in spinal cord sections of EAE mice (Left and middle panel scale: 400 μm; right panel scale: 80 μm). Yellow inset frames are magnified images showing the morphology of macrophages. (B) Quantification of Iba-1 positive cells from immunohistochemistry images. Iba-1 positive cells were counted in the spinal cords’ sections in both white and gray matter areas (n = 3; **p < 0.01; ***p < 0.001). (C) Representative flow cytometry plots of CD4+ and CD8+ T cells within spinal cords. (D) and (E) Flow cytometry analysis of spinal cords stained for infiltrated CD4+ and CD8+ T-cells, respectively. Unpaired t tests were used to determine p values (n = 3; *p < 0.05; **p < 0.01).
T-cell infiltration into the CNS is another hallmark of neuroinflammation, particularly in MS. Therefore, we also assessed the T-cell infiltration into the spinal cord of EAE mice using flow cytometry. For this set of experiments, whole spinal cords from each animal were excised and strained to harvest the cells. As shown in Figure 2C,D, significant reduction in the number of infiltrated CD4+ (n = 3; 77.3 ± 24 cells per spinal cord; p < 0.01) into the spinal cords of IFNγ-Exo-treated mice was observed compared to PBS control (n = 3; 263.3 ± 38.1 cells per spinal cord). IFNγ-Exo exhibited a superior effect in reducing the number of infiltrated CD4+ compared to Native-Exo group (77.3 ± 24 vs 165.3 ± 12.5 cells per spinal cord; p <0.01). Moreover, infiltration of the CD8+ cells was also dramatically reduced in IFNγ-Exo group (n = 3) compared to PBS (n = 3) control (44 ± 11.5 vs 260 ± 129 cells per spinal cord; p < 0.05) (Figure 2E). In this case, the Native-Exo group (n = 3) exhibited a comparable effect (46 ± 7.5 cells per spinal cord) in CD8+ cell reduction into spinal cords compared to the IFNγ-Exo group. The number of infiltrated CD4+ and CD8+ in the WT animals was almost undetectable.
Regulatory T cells (Tregs) are critical players in the pathogenesis of CNS autoimmune inflammation.31 Recent reports signify that MS is accompanied by impaired maturation or dysfunction of Tregs.32 Treg deletion prompts spontaneous autoimmune disease in mice, while restoring the Treg function prevents development of experimental autoimmune encephalomyelitis, an animal model of MS.33 We then asked whether MSC-derived exosomes could induce Tregs in vivo and also if clinical recovery is associated with the number of Tregs. To this end, EAE was induced in a FOXP3-eGFP “Treg reporter” model. This model labels all Tregs with GFP, therefore eliminating the need for intracellular staining for FOXP3. The mice received IFNγ-Exo, Native-Exo, or PBS as a vehicle control. Immunohistochemistry analyses revealed the presence of Tregs (CD4+FOXP3+) in spinal cords of exosome-treated animals (Figure 3A). Quantification of number of CD4+FOXP3+ cells in spinal cords shows an increase of Tregs in IFNγ-Exo (n = 3, 21 ± 5.2 cells per section; p < 0.01) and Native-Exo-treated mice (n = 3, 9.6 ± 3.7; p < 0.05) compared to PBS controls (n = 3, 2 ± 1) (Figure 3B). In addition, we found that CD4+FOXP3+ are often in direct contact with other CD4+ cells (inset boxes in Figure 3A). Indeed, it has been reported that FOXP3+ Tregs are dynamic and continuously interact with conventional T cells and other immune cells to exert their immunoregulatory function.34 Importantly, we are able to replicate IFNγ-Exo’s efficacy in this FOXP3-eGFP EAE model: IFNγ-Exo-treated mice showed superior therapeutic recovery as measured by clinical scores compared to other groups (Figure 3C). This recovery was similar to what we observed in C57BL/6J mice before (Figure 1B). Flow cytometry analysis of dissociated cells from spinal cords also showed an increase in the frequency of Tregs (CD4+CD25+FOXP3+) in the spinal cords of IFNγ-Exo-treated mice (n = 3, 43.3 ± 12.6%; p < 0.05) compared to Native-Exo-treated mice (n = 3, 28.2 ± 4.2%) and PBS control (n = 3, 26.3 ± 4.4%) (Figure S4A). Further analyses using flow cytometry on spleens did not show any major difference in the number of Tregs between different groups: IFNγ-Exo (n = 3, 15.9 ± 1.5%), native-Exo (n = 3, 15.9 ± 0.6%), and PBS control (16.06 ± 1.8%) (Figure S4B). A similar trend was observed for the number of Tregs analyzed in lymph nodes of IFNγ-Exo (n = 3, 13.36 ± 0.4%), native-Exo (n = 3, 15.7 ± 1.0%), and PBS-treated controls (13.6 ± 0.9%) (Figure S4C). We note that the total numbers of Tregs are similar in the spleen and lymph node between different groups, and it does not necessarily mean that exosomes did not have any effects on Tregs in those tissues, since MOG-specific Tregs might have been increased without affecting the total number of Tregs, which we will investigate in the future work.
Figure 3.

MSC-derived exosomes increase Tregs in the spinal cords of EAE mice. (A) Immunohistochemistry of CD4+FOXP3+ cells and DAPI in spinal cord sections of EAE mice after injection of PBS, Native-Exo, and IFNγ-Exo. Scale bar: 100 μm. (B) Quantification of number of CD4+ FOXP3+ Tregs in immunohistochemistry staining of spinal cords. Unpaired t tests were used to determine p values (n = 3; *p < 0.05; **p < 0.01). (C) Clinical scores of EAE in FOXP3-eGFP Treg reporter C57BL/6 mice after injection of PBS (n = 4), Native-Exo (n = 4), and IFNγ-Exo (n = 8). Mann-Whitney t tests were used to determine the p values (*p < 0.05; **p < 0.01; ***p < 0.001).
Recent evidence has suggested several pro-inflammatory T cell subsets including Th1 and particularly Th17 cells are key drivers in EAE.35,36 We thus sought to characterize the potential effect of our exosomes on Th1 and Th17 responses in the spinal cord. Flow cytometry analyses of mouse spinal cord cells showed that while the percentage of CD4+IFNγ+ T cells (Th1) (n = 3, from 0.38 ± 0.25% in PBS treated to 0.13 ± 0.12% in IFNγ-Exo treated) and CD4+IL-17+ T cells (Th17) (n = 3, from 0.52 ±0.4% in PBS treated to 0.14 ± 0.09% in IFNγ-Exo treated) decrease in the IFNγ-Exo-treated mice, the difference was not significant (Figure S5A,B). Moreover, to measure inflammatory cytokines in spinal cords, a Luminex assay using a Th1/Th2/Th9/Th17/Th22/Treg Cytokine 17-Plex Mouse ProcartaPlex Panel was performed, but we were not able to detect any of these cytokines (data not shown), likely due to that their concentrations are below the limit of detection of our assay, especially following sample processing and dilution steps prior to the Luminex assay.
In our initial study, we chose to inject exosomes on day 18 post-immunization, which is the peak of the disease and therefore represents an established disease stage. We reason that if MSC-derived exosomes work by suppressing the pro-inflammatory cells and cytokines, we might need to intervene at an earlier time point during the inflammatory induction phase in order to produce an optimal therapeutic outcome. Indeed, it has been known that inflammatory T cells infiltrate the CNS and mediate inflammation before the start of EAE clinical symptoms.37–39 Moreover, we hypothesize that increasing the number of exosome injections could further attenuate inflammation. While systematically optimizing treatment timing and the number of injections is beyond the scope of the present work, we performed a pilot study where we transplanted two doses of MSC-derived exosomes at earlier time points on days 12 and 14 post-immunization, which is known to be during the T cell inflammatory induction phase.38 While it exhibited a comparable treatment efficacy with respect to clinical score reduction (Figure S6) and similar trends of total CD4+ T cell reduction and Treg increase in the spinal cord compared to the previous regimen of single injection on day 18, intervention with multiple injections at earlier time points showed a significant reduction of the Th17 cells (Figure S7). Th17 cells have recently been shown to be a key player in EAE.35,36 This data warrants future work to continue to optimize treatment timing and number of injections so we can better understand the evolving molecular and cellular heterogeneity as the disease progresses and eventually stratify patients and treatment regimens to achieve desirable outcomes.40
IFNγ-Exo Suppress T Cell Proliferation and Induce Tregs in vitro.
As we mentioned above, T cells play a key role in inducing and regulating MS pathophysiology, and the development of therapeutics for MS has often focused on targeting factors that mediate T cell function.33 As we observed a reduction of neuroinflammation and an induction of tolerance in exosome-treated EAE mice in vivo, we next sought to further confirm exosome-mediated immunomodulatory effects in vitro. To examine if MSC exosomes suppress T-cell activation ex vivo, carboxyfluorescein succinimidyl ester (CFSE)-labeled human PBMCs were activated with anti-CD3/CD28 Dyna beads (1:1 ratio) and further cultured with or without IFNγ-Exo and Native-Exo. Both Native-Exo and IFNγ-Exo suppressed activation of the gated T cells, with IFNγ-Exo being considerably more suppressive (Figure 4A). These results are consistent with previous reports where the ability of MSC-derived exosomes to suppress T cell activation and proliferation was evaluated.12,15,16 It is well established that MSC can suppress PBMC activation and proliferation in co-cultures mainly through secreted immunomodulatory or immunosuppressive cytokines including IDO, IL-10, PGE2, TSG6, and TGFβ.1,2,41,42 We further sought to elucidate the underlying pathways for the effect of exosomes on the PBMC proliferation. We conducted Luminex and ELISA assays to investigate cytokine profiles in the supernatant of PBMC co-cultures (Figures S8 and S9 and Figure 4B). Interestingly, the IDO level in the PBMC co-culture was significantly increased in the presence of IFNγ-Exo. IDO is an immunosuppressive enzyme that enhances the catabolism of tryptophan to kynurenine; both depletion of tryptophan and accumulation of the toxic kynurenine are known to inhibit T cell proliferation.43 As Th1 and Th17 cells play key roles in the pathology of autoimmune disorders including MS/EAE,35,36 to further probe the MOA of MSC-derived exosomes in suppressing T cell induction to Th1/Th17 subtypes, we analyzed several key representative Th1 and Th17 cytokines in this MSC exosome/PBMC co-culture experiment. We found that in the presence of MSC-derived exosomes, the levels of several pro-inflammatory Th1 and Th17 cytokines including notably IL-6 (Th17 cytokine), IL-12p70 (Th1), IL-17AF (Th17), and IL-22 (Th17) were significantly reduced (Figure 4B) and IFNγ (Th1) and TNFα (Th1) demonstrated a trend of decrease though not significant (Figure S8).
Figure 4.

MSC-derived exosomes have immunosuppressive effects and induce Tregs in vitro. (A) Human PBMCs were labeled with CFSE and activated for 4 d with Dyna beads, in the absence or presence of exosomes produced by native or IFNγ-stimulated MSC. (B) Addition of the MSC-derived exosomes to the PBMCs culture enhances the secretion of IDO, while reduces the secretion of IL-17AF, IL-22, IL-12p70, and IL-6 compared to activated PBMCs (unpaired t tests were used to determine the p values; n.s. is nonsignificant, *p < 0.05, **p < 0.01, ***p < 0.001). (C-F) FOXP3-eGFP mice splenocytes were stimulated with anti-CD3 + IL-2 with or without TGFβ and were further cultured with indicated concentrations of IFNγ-Exo. Representative FACS plots of CD4+ gate (C) and its quantification (D) or CD8+ gate (E) and its quantification (F). Unpaired t tests were used to determine p values (n = 3 per concentration; *p < 0.05). Statistical analyses were conducted between 0 μg and 20 μg IFNγ-Exo.
To investigate if MSC exosomes induce Tregs in vitro, murine splenocytes from the FOXP3-eGFP “Treg reporter” mice were stimulated with anti-CD3 and IL-2 with or without TGFβ (a critical factor for driving the expression of FOXP3 in the precursors and ultimately generating conventional Tregs)44,45 and were further cultured in the presence of different concentrations of IFNγ-Exo, Native-Exo, or nonconditioned medium control. Tregs are marked by expression of FOXP3 and high levels of IL-2 receptor alpha chain (IL-2Rα, or CD25). In the presence of TGFβ, IFNγ-Exo (n = 3 for each concentration) enhanced the frequency of CD4+CD25+FOXP3+ Tregs in a dose-dependent manner; nil (18.1% ± 4.9%), 0.2 μg/well (19.6% ± 4.7%), 2 μg/well; (22.3% ± 5.4%), and 20 μg/well (24.4% ± 2.8%) (Figure 4C, D) as well as CD8+CD25+FOXP3+ Tregs: nil (4.9% ± 0.5%), 0.2 μg/well (5.2% ± 0.9%), 2 μg/well (7.4% ± 2.1%), and 20 μg/well (7.4% ± 0.2%) (Figure 4E,F). Finally, we confirmed that nonconditioned medium control failed to induce Tregs in splenocyte cultures (Figure S10), which indicates tha tour exosome depletion protocol is sufficient to remove biologically functional exosomes from FBS. Interestingly, in the absence of TGFβ, both Native-Exo and IFNγ-Exo (n = 3 for each concentration) enhanced the frequency of CD4+CD25+FOXP3+ and CD8+CD25+FOXP3+ Tregs in a dose-dependent manner. The fact that exosomes can induce Tregs in the absence of TGFβ suggests that they might either provide TGFβ on their surface46,47 and/or provide additional signals directly on lymphocytes or indirectly on accessory spleen cells (e.g., B cells, dendritic cells (DCs), splenic macrophages) that cumulatively induce or enhance Treg production. Native-Exo also enhanced Treg induction in vitro but generally to a lesser extent than IFNγ-Exo (Figure S11). Note that IFNγ-Exo enhanced Treg induction in splenocyte cultures, but not in cultures of purified CD4+ and CD8+ T cells activated with plate-bound anti-CD3 and soluble cytokines (data not shown), suggesting that IFNγ-Exo may target an accessory cell such as antigen-presenting cells (APCs) rather than T cells directly, which is in agreement with recent studies.48–50
Collectively, these data support the hypothesis that MSC-derived exosomes can ameliorate EAE by, at least partly, suppressing pathological T cell subset activation and by inducing Tregs.
MSC Exosome Traffic into Spinal Cord in Vivo.
To further reveal mechanisms underlying exosome-mediated effects in neurologic functions in EAE, we next investigated the fate and biodistribution of administered exosomes. In this set of experiments, IFNγ-Exo and Native-Exo were labeled with DiR, a lipophilic dye that is commonly used for in vivo and ex vivo imaging.51 DiR itself was also included as a negative control. EAE and healthy mice were administered the same amount of DiR-exosomes as we did for previous treatment studies. For each experimental group, the mice were sacrificed at 3 and 24 h after the exosome injection, and freshly dissected tissues were analyzed immediately for the fluorescence signal using a IVIS imaging system. Exosomes were mostly found in the liver and spleen of healthy and EAE mice (Figure 5A,C), consistent with previous reports.52,53 No major difference was observed in the biodistribution profile of IFNγ-Exo in the liver, lung, spleen, kidney, and brain between WT and EAE animals. Similar results were observed for Native-Exo (Figure S12). It is also worthwhile to note that little signal was observed in the lungs of exosome-treated animals, suggesting that exosomes, unlike MSCs, can bypass the small lung vasculature bed owing to their small size. Importantly, dye-labeled IFNγ-Exo was observed in the spinal cords of EAE, but not in healthy animals, at 3 h following administration (Figure 5B,D), further suggesting the involvement of IFNγ-Exo in the lesions. However, IFNγ-Exo signal was not detectable in spinal cord tissues at 24 h post-injection (Figure 5B,D). That exosomes cannot be detected after a short period of time in vivo suggests that their long-lasting efficacy even with a single injection might be through the proposed “hit and run” mechanism,54 probably through immunomodulatory mechanisms, particularly through the induction of Tregs for immune tolerance.55 A similar biodistribution trend (i.e., localization in the spleen, liver, and spinal cord) was observed for Native-Exo (data not shown).
Figure 5.

Biodistribution of exosomes in vivo. (A) Representative IVIS images of different organs harvested at 3 and 24 h following i.v. infusion of DiR-labeled IFNγ-Exo into healthy and EAE mice. Control groups received DiR dye alone (EAE-DiR, healthy-DiR) to determine the background. (B) Representative IVIS images of spinal cords 3 and 24 h post-injection of DiR-labeled IFNγ-Exo. (C) Quantification of the fluorescent signal in different organs including the liver, spleen, kidney, brain, and lung at 3 and 24 h following infusion of DiR-labeled IFNγ-Exo (n = 3; n.s. is nonsignificant, **p < 0.01). (D) Quantification of the fluorescent signal in spinal cords at 3 and 24 h post-injection of DiR-labeled IFNγ-Exo. (n = 3; n.s. is nonsignificant, *p < 0.05; **p < 0.01). Unpaired t tests were used to determine p values.
RNA Inactivation Partially Impairs the Ability of Exosomes To Induce Tregs.
Exosomes comprise a complex repertoire of surface and intravesicular markers, including proteins and nucleic acids (particularly RNA), that might work in concert to enhance therapeutic effects compared to separated factors,7,56 but also complicate identification of their active ingredients, contributing to the exosome MOA. Identifying the main molecular targets is important since this will help us to develop therapeutic approaches that can further enhance efficacy by overexpressing selected factor(s). Exosomes allow the exchange of genetic information between cells, such as mRNA, long noncoding RNA (lncRNA), and microRNA.57,58 Therefore, the RNA cargo of exosomes is believed to be a key mediator of exosome functions.59,60 We obtain approximately 20 ng total RNA per 150 μg exosome preparation based on Nanodrop and Bioanalyzer analyses. We decided to first take a global approach by eliminating all RNAs to assess whether RNA plays any role in exosome’s function.
It is well-known that UV-light inactivates RNA functions.61,62 We first exposed IFNγ-Exo and Native-Exo to UV-light (254 nm) for various time points (220 s, 540 s, and 1800 s) to identify an optimal time point that sufficiently inactivates RNAs but without cross-linking or degrading the protein components. RT-qPCR was then performed for β-actin (IFNγ-Exo) and GAPDH (Native-Exo). β-actin and GAPDH were chosen because they are the most abundant RNAs in IFNγ-Exo and Native-Exo fractions, respectively, based on our deep sequencing data (Table S1). Inactivation of RNAs was confirmed by RT-qPCR at 220 s (Figure S13A) and longer treatments (540 s and 1800 s) (data not shown). It is also known that UV exposure could damage proteins.63,64 Therefore, we studied the effect of UV exposure on potential protein cross-linking and degradation of IFNγ-Exo using Blue BANDit protein gel staining. The protein bands for samples with 220 s of UV illumination were found to be relatively similar to the control, suggesting that this UV exposure regimen had minimal effect on protein cross-linking and fragmentation (Figure S13B). In the sample with 1 h of UV illumination, however, high molecular weight moieties appeared at the top of the lane, as observed in another report.65 Therefore, we chose to use 220 s of UV exposure to inactivate RNA for our experiments.
We then performed our in vitro assay measuring exosome potentiation of Treg induction under different conditions (with or without TGFβ) with UV-treated exosomes. In the absence of TGFβ, UV-treated IFNγ-Exo (n = 3, 67.2 ± 18.7%; p < 0.05) and Native-Exo (n = 3, 65.3 ± 15.3%; p < 0.05) both show a significant reduction in CD4+CD25+FOXP3+ percentage compared to their untreated controls (Figure 6A,B). In the presence of TGFβ, a similar reduction in CD4+ Treg induction was observed for IFNγ-Exo (n = 3, 45.7 ± 9.9%; p < 0.01) and Native-Exo (n = 3, 50.7 ± 4.4%; p < 0.01) compared to their untreated controls. These trends are generally applied to CD8+CD25+FOXP3+ Tregs that have been only recently characterized in the literature.66 In the absence of TGFβ, UV-treated IFNγ-Exo (n = 3, 86.3 ± 8.9%) and UV-treated Native-Exo (n = 3, 76.6 ± 23.1%) both show a trend of reduction in CD8+CD25+FOXP3+ percentage compared to their untreated controls (n = 3), although not significant (Figure 6C,D). Similarly, in the presence of TGFβ, UV-treated IFNγ-Exo (n = 3, 65.6 ± 2.5%; p < 0.01) and Native-Exo (n = 3, 60.6 ± 16.1%; p < 0.05) both show a noticeable reduction in CD8+CD25+FOXP3+ percentage compared to untreated controls (n = 3). These results suggest that the exosomal Treg induction capability, at least in part, is mediated by exosomal RNAs and implicates an important role for RNA in exosome functions.
Figure 6.

Treg induction of exosomes is partly impaired by UV treatment. Murine splenocytes from Foxp3-eGFP mice were activated with anti-CD3 + IL-2 with or without TGFβ and were further cultured in the presence or absence of IFNγ-Exo, UV IFNγ-Exo, Native-Exo, and UV Native-Exo. (A) Representative flow cytometry plots and (B) quantification of percentage reduction of CD4+ gate for exosomes with or without UV treatment. (C) Representative flow cytometry plots and (D) quantification of CD8+ gate for exosomes with or without UV treatment. Twenty μg exosomes was used in this study. Paired t tests were used to determine p values (n = 3 for each group; n.s. is nonsignificant, *p < 0.05; **p < 0.01).
Deep RNA Sequencing Reveals Differential Enrichment of MSC Exosomes in Anti-Inflammatory RNAs.
Thus, far we observed that the RNA is partly responsible for exosome’s efficacy. To further delineate exosome-mediated recovery mechanism in EAE mice, we decided to perform deep RNA sequencing to get insight into the types of RNAs, including both mRNAs and small noncoding RNAs, which are enriched in exosomes and might be responsible for their therapeutic effects. Clinical recovery was observed in both IFNγ-Exo and Native-Exo with IFNγ-Exo showing more potency. Therefore, we decided to compare IFNγ-Exo and Native-Exo RNA components to understand the differences or possible enrichment of particular RNA, which might explain the increased potency in the IFNγ-Exo group.
Normalized counts of long RNA sequencing demonstrate that both Native-Exo and IFNγ-Exo have less protein-coding transcripts compared to their MSC counterparts (Figure 7A,B,D and Figure S14A,B). Interestingly, there are a large number of noncoding RNAs detected in Native-Exo and IFNγ-Exo and significantly enriched in the IFNγ-Exo group (Figure 7A) compared to MSC groups (Figure 7A and Figure S14). This enrichment of noncoding RNAs in exosome fraction is particularly important since noncoding RNAs have regulatory roles and one transcript can affect multiple pathways at the same time. These noncoding RNAs include miRNA, tRNA, lincRNA, and antisense RNA, etc. (Figure 7C, E).
Figure 7.

Deep RNA whole transcriptome analysis of native MSC, IFNγ-MSC, and their exosomes. (A) Protein coding (mRNA) transcript and noncoding transcripts of MSCs and their exosomes. (B) Whole RNA sequence summary of Native-Exo. (C) Noncoding RNA sequence summary (percentage of different types of noncoding RNAs) of Native-Exo. (D) Whole RNA sequence summary of IFNγ-Exo. (E) Noncoding RNA sequence summary (percentage of different types of noncoding RNAs) of IFNγ-Exo.
In our study, we found that a number of mRNAs are loaded by cells into exosomes which can potentially subsequently be delivered and translated into proteins in recipient cells. In particular, we found in our RNaseq analysis that IFNγ-Exo is highly enriched in several mRNAs with anti-inflammatory properties compared to Native-Exo (Table 1). Of particular importance, for example, is IDO that has been widely demonstrated as a key player in MSC-mediated immunomodulation67,68 and in the down modulation of neuroinflammation in EAE.69,70 Specifically, for any 1 million RNAs in our exosome preparation, there are 0 IDO transcripts in Native-Exo and approximately ~48 IDO transcripts in the IFNγ-Exo. We further demonstrated using RT-PCR that IDO mRNAs are present in full-length (Figure S15). Together with our observation of increased IDO protein level in PBMC co-culture with IFNγ-Exo, this evidence further supports that the observed MSC-derived exosome’s suppressive effect on PBMC proliferation could potentially be mediated, at least in part, by the transferred exosome IDO mRNA cargo to recipient PBMCs which are then translated and secreted into the medium.
Table 1.
List of Anti-Inflammatory mRNAs Enriched in IFNγ-Exo
| RNA | Function |
|---|---|
| Indoleamine 2,3- dioxygenase 1 | Anti-inflammatory |
| Thymosin beta 10 pseudogene 1 | Potent anti-inflammatory, Inhibition of cell migration |
| CD74 molecule | Interact with Macrophage migration inhibitory factor |
| Major histocompatibility complex, class 11, DR alpha | Antigen presentation |
| Interferon-induced protein with tetratricopeptide repeats 2 | Inhibitor of Cell migration |
| Guanylate binding protein 5 | Inhibit proliferation of endothelial cells |
| Interferon stimulated exonuclease gene 20kDa | Host defense against RNA viruses |
| Interleukin IS binding protein | Inhibitor of Pro-inflammatory IL-18 |
| Glia maturation factor, gamma | Stimulation of neural regeneration, and inhibition of proliferation of tumor cells |
| Retinoic acid receptor responder (tazarotene induced) 3 | Anti-angiogenic effect |
| Interferon-induced protein with tetratricopeptide repeats 3 | Inhibitor of Cell migration |
| Apolipoprotein L | Apoptosis |
| Lectin, galactoside-binding, soluble, 9 | Anti-inflammatory |
| Tripartite motif containing 14 | Antiviral activity |
| Chemokine (C-X-C motif) ligand 10 | Antitumor activity, Inhibition of angiogenesis |
| Major histocompatibility complex, class II, DR beta 1 | Antigen presentation |
| Bone marrow stromal cell antigen 2 | Antiviral activity |
Investigating each of the individual noncoding RNA classes and molecules is beyond the scope of this work. Instead, we took an initial attempt with a focus on miRNA to examine the main canonical pathways. Using ingenuity pathway analysis database of gene ontology (GO), we narrowed down the top three canonical pathways affected by miRNAs enriched in IFNγ-Exo (Table S2). Of particular relevance is the phosphoinositide 3-kinase (pI3K/AKT) signaling pathway—our analysis demonstrating that 19 out of 123 genes in this pathway are affected—that has been linked to and investigated as a drug target for autoimmune disease71 including CNS inflammation.72 Furthermore, we noticed several individual miRNAs existed in IFNγ-Exo including miR-146b which is involved in anti-inflammatory processes.73,74 These findings warrant future investigation toward the molecular mechanism of miRNA in IFNγ-Exo’s efficacy. Furthermore, a previous study demonstrated that microparticles secreted from MSCs were preferentially enriched in pre-microRNAs,73 while the miRNAs discussed above in our study are in their mature form. Therefore, it will be interesting in future work to further characterize exosomal miRNAs found in different stages of their biogenesis and to determine the role of RNA-induced silencing complex in exosomal miRNA-mediated therapeutic function in recipient cells.75
Proteomic Analysis Reveals Differential Enrichment of MSC Exosomes in Anti-Inflammatory and Neuroprotective Proteins.
We demonstrated previously that even after UV irradiation, exosomes still are able to induce Tregs to some extent. This could be due to the protein content of exosomes. In order to further investigate the potential roles of exosomal proteins in clinical recovery, proteomics analysis of exosomes and their corresponding MSC was performed. Comparison of MSC with exosomes shows that both carry their unique proteins (Figure 8A). In particular, peptide composition comparison of Native-Exo with IFNγ-Exo revealed that 310 peptides as shared peptides and 228 and 104 peptides are unique for Native-Exo and IFNγ-Exo, respectively (Figure 8B). As IFNγ-Exo-treated groups displayed more robust efficacy, compared to Native-Exo, to identify and narrow down potential therapeutic proteins, we decided to focus on IFNγ-Exo. Our proteomics analysis identified multiple proteins enriched in IFNγ-Exo with anti-inflammatory or neuroprotective properties (Table 2) including macrophage inhibitory cytokine 1 (MIC-1), galectin-1 (Gal-1), heat shock protein 70 (HSP70), and latent-transforming growth factor β-binding protein (LTBP) that are anti-inflammatory and immunomodulatory and/or neuroprotective.76–80 Western blotting for two representative protein markers HSP70 and Gal-1 was performed to confirm that they were indeed present and enriched in IFNγ-Exo compared to Native-Exo (Figure 8C). Moreover, our IFNγ activated MSCs overexpress immunomodulatory MHCII81 and PD-L1,42 and future work will examine if they get transferred to the surface of exosomes. Indeed, these protein markers including especially Gal-1 and PD-L1 have been implicated in EV’s immunosuppressive effects.47,67,82–85
Figure 8.

Proteomics of MSCs and exosomes. (A) Comparison of protein composition of Native and IFNγ MSC and their correspondent exosomes. (B) Comparison of protein composition of Native-Exo and IFNγ-Exo. (C) HSP70 and Gal-1 are enriched in IFNγ-Exo compared to Native-Exo.
Table 2.
Peptides Found in IFNγ-Exo Samples
| Neuronal Protection | Anti-inflammatory | ||
|---|---|---|---|
| Peptide Name | Function(s) | Peptide Name | Function(s) |
| Laminin subunit beta-2 |
1. Axon extension involved in regeneration 2. Axon guidance 3. Astrocyte development |
Extracellular sulfatase Sulf-1 | Negative regulation of cell migration |
| Aggrecan | Central nervous system development | Macrophage inhibitory cytokine 1; Growth differentiation factor-15 | Treg induction-Negative regulation of macrophages |
| Testican-1 |
1. Central nervous system neuron differentiation 2. Nervous system development 3. Neurogenesis 4. Neuron migration |
Slit homolog 2 protein |
1. Negative regulation of cel! migration 2. Negative regulation of leukocyte, neutrophil, and monocyte chemotaxis 3. Negative regulation of mononuclear cell migration |
| Versican core protein | Central nervous system development | Gremlin-1 | Negative regulation of monocyte chemotaxis |
| Periostin |
1. Neuron projection extension 2. Negative regulation of cell-matrix adhesion |
Thy-Tmembrane glycoprotein |
1. Negative regulation of cell migration 2. Negative regulation of T cell receptor signaling pathway |
| Glypican-1 |
1. Axon guidance 2. Myelin assembly 3. Schwann cell differentiation |
Annexin A4 |
1. Negative regulation of interleukin-8 secretion 2. Negative regulation of NF-kappaB transcription factor activity |
| Hyaluronan and proteoglycan link protein 3 | Central nervous system development | Endoglin |
1. Negative regulation of cell migration 2. Promotes transforming growth factor beta-mediated Smad 1/5/8 signaling |
| Hyaluronan and proteoglycan link protein 1 | Central nervous system development | Latent-transforming growth factor P-binding protein |
1. TGFβ stability 2. Treg Induction |
| Regucalcin | Negative regulation of apoptotic process | 5’-nueleotidase (CD73) | Converting AMP to adenosine |
| Galectin-1 | Inhibition of soluble and cellular mediators of the inflammatory response | Galectin-1 | Inhibition of soluble and cellular mediators of the inflammatory response |
| Cofilin-1 | Negative regulation of apoptotic process | ||
| HSP70 | Anti apoptotic | ||
| Myosin-14 | Neuronal action potential | ||
CONCLUSION
This study facilitates the understanding of potential mechanisms by which MSCs exert therapeutic function. In the context of cell therapy, the majority of i.v.-infused MSCs get entrapped in filter organs without significantly homing to sites of injury, and yet they frequently exhibit therapeutic activity in numerous animal models.2 The mechanisms to this puzzling phenomenon remain unclear. Our unpublished data showed that lung entrapped MSCs under physical and mechanical stresses could shed small vesicles into the circulation that could reach distant organs. Recent evidence suggests infused MSCs and their apoptotic products can be phagocytosed, which led to the generation of third-party phagocytes that ultimately mediate the observed immunomodulatory effects.4–6,86 The fact that MSC-derived exosomes can recapitulate MSC’s efficacy in our EAE model and that their effects on T cells are mediated at least partly by third-party cells (e.g., macrophage) implies that transplanted MSCs could mediate their efficacy in vivo through secreted EVs, likely by first interfacing with APCs and phagocytes.7,13,49,74,85,87
Our data demonstrate that exosomes derived from IFNγ activated MSCs (1) suppress PBMC cell proliferation (likely through IDO), reduce pro-inflammatory cytokines, and enhance induction of Tregs in vitro and (2) are distributed to the inflamed, but not healthy, spinal cords, reduce neuroinflammation and demyelination, and improve functional outcomes in chronic EAE murine model. This study asserts that stem cell-derived exosomes can represent a viable approach to treat autoimmune and neurodegenerative disorders (as proposed in Figure 9), which remains a major unmet clinical need. There are several potential advantages for the exploitation of exosomes as therapeutics and as vehicles for therapeutic delivery, compared to cells and conventional drug delivery systems (e.g., liposomes and nanoparticles): (1) The smaller size of exosomes allows reduced entrapment in small capillaries following systemic infusion and potentially improves the therapeutic delivery to the diseased sites, (2) exosomes are expected to have a significantly longer shelf life, lower side effects, and other risks compared to cells because of their acellular status, (3) exosomes possess a complex mixture of factors targeting different therapeutic pathways and synergize to enhance therapeutic function compared to using individual factors, and (4) the natural cell origin of exosomes enhances our ability to genetically modify the cell of origin to produce exosomes with overexpressed agents for improved efficacy, biocompatibility, and reduced immunogenicity.
Figure 9.

Overview of proposed mechanisms of MSC exosome-mediated recovery in EAE. Upon injection, exosomes could bypass the blood spinal cord barrier either through gaps between endothelial cells (paracellular route) or by first being endocytosed and then exocytosed by endothelial cells (transcellular route). Possessing a complex mixture of RNA and proteins targeting different pathways enable exosomes to exert their therapeutic efficacy probably by downregulating inflammation and inducing tolerance by increasing the number of Tregs. Over time, resolving inflammation in spinal cord is associated with decreased demyelination, resulting in recovery.
We demonstrated that MSC exosomes showed potential to induce a tolerogenic immune response and therefore a sustained clinical recovery. Moreover, further MOA investigation on the reduction of Th17 cells and their associated pro-inflammatory cytokines can lead to development of exosome therapeutics that target disease pathways such as Th17 cytokines for patients who are refractory to the current treatment.35 We demonstrated MSC-derived exosomes host a mixture of proteins and RNAs, some of which are implicated in anti-inflammatory, antigen presenting, or neuronal protection pathways. Future work will investigate individual markers (e.g., IDO) and dissect the cellular and molecular mechanisms to identify a panel of molecular markers that are responsible for exosome’s therapeutic effect. This effort will allow us to (1) optimize exosome manufacturing and preparation processes to standardize potential batch-to-batch variations and develop quality control and release assays for clinical applications88 and (2) engineer exosomes to overexpress selected candidates in the future to improve their efficacy.7,89 Future work will also optimize treatment timing, especially at the onset of the disease, which might be more effective in attenuating inflammatory responses during the induction phase. It will also be interesting to further characterize how doses and numbers of injections affect outcomes, especially considering that increased doses in the MSC field have resulted in mixed outcomes,90 presumably due to the host immune responses (e.g., antibodies) that were raised to attenuate the therapy.91 Intravenous infusion was chosen as an administration route in this study due to its minimal invasiveness and convenience, however, other routes such as local injection of exosomes15 to inflamed spinal cords would be an interesting approach to explore in the future work to optimize their therapeutic outcome.
MATERIALS AND METHODS
Cells.
Human bone marrow derived MSCs were obtained from Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine and was the source of MSC used in this study unless stated otherwise. Human umbilical cord tissue-derived MSCs (UC-MSCs) were isolated from discarded umbilical cords and prepared following an established protocol.92 UC-MSCs were used only for the data presented in Figure S6 and Figure S7, where we investigated MSC source variability on efficacy and early intervention. These cells were fully characterized. They are adherent on tissue culture plastic, express markers CD73, CD105, and CD90, do not express hematopoietic markers CD34 and CD45, and can differentiate into osteoblasts and adipocytes in vitro. αMEM supplemented with 4 mM l-glutamine and 15% fetal bovine serum (FBS) was used to culture MSCs. Passages 2–3 were used for all the experiments. To activate the MSCs, 10 ng/mL of IFNγ (PeproTech Inc., USA) was used for 48 h. As IFNγ has been reported to ameliorate EAE,93 ELISA assay LEGEND MAX (cat. no. 430107, Biolegend) was performed to confirm there was no significant carryover of IFNγ from the culture medium to the IFNγ-Exo preparation (Figure S16). For exosome collection, 80% confluent cells were cultured with complete media supplemented with 15% exosome-depleted FBS for 3 days. To deplete exosomes, FBS was centrifuged at 120,000 × g for 18 h, which was demonstrated to be sufficient to remove approximately 95% of FBS extracellular vesicles.94 However, we caution that different protocols may contribute to mixed FBS exosome depletion efficiency.95 It is always important to run a nonconditioned medium control to ensure FBS exosomes are sufficiently depleted.
Isolation of Exosomes.
Conditioned media from cultures of MSC cells grown as described above, with or without IFNγ treatment, were centrifuged at 300 × g for 10 min to remove any cells or large cellular fragments. Supernatants were then collected and transferred to ultracentrifuge tubes (Polyallomer Quick-Seal centrifuge tubes 25 × 89 mm, Beckman Coulter). Samples were centrifuged using an ultracentrifuge (Optima L-90 K or Optima XE- 90 Ultracentrifuge, Beckman Coulter) for 20 min at 16,500 × g (Type Ti 45, Beckman Coulter), to remove microvesicles. Supernatants were carefully collected and centrifuged for 2.5 h with a Type 45 Ti rotor at 120,000 × g at 4 °C. The exosome pellet was reconstituted in PBS and stored at −80 °C. For RNA sequencing and proteomics experiments, exosomes prepared from four different MSC batches were pooled to help reduce any potential impact of batch-to-batch variations on their molecular markers.
Protein Quantification.
Prepared exosomes in PBS were mixed with 1 × RIPA buffer (Cell signaling technologies, USA) and underwent three consecutive sonications (5 min each with vortexing in between). Protein contents were analyzed using a BCA protein assay kit (Thermo Scientific Pierce, Rockford, IL, USA). Twenty-five μL of sample (or 25 μL BSA standard) was transferred to a 96-well plate, to which 200 mL of working reagent (50:1 ratio of assay reagents A and B) was added. The plate was incubated at 37 °C for 30 min and then analyzed using a SpectraMax 384 Plus spectrophotometer with the SoftMax Pro software (Molecular Devices, 1311 Orleans Drive, Sunnyvale, CA, USA).
Western Blotting.
Prepared MSC exosomes following sonication, as described above ,with volumes corresponding to 25 μg protein were analyzed using a gradient precast polyacrylamide gel (Mini-PROTEAN; Bio-Rad laboratories, Hercules, CA, USA). Samples were then transferred onto a nitrocellulose membrane which was blocked using 5% blotting grade Blocker Non-Fat Dry Milk (Bio-Rad Laboratories) in Tris-buffer saline (TBS) for 2 h. The membrane was subsequently incubated with primary antibodies against Calnexin (clone H-70, Santa Cruz Biotechnology, Santa Cruz, CA, USA), Galectin-1/LGALS1 (clone D608T) Rabbit mAb, HSP70 (clone D69) RabbitpAb, TSG101 (1:1000, clone 4A10, Abcam, Cambridge, UK), or CD81 (clone H-121, Santa Cruz Biotechnology) in 0.25% blotting grade Blocker Non-Fat Dry Milk in TBS-Tween (TBST) overnight at 4 °C. The membrane was then washed with TBST for 10 min, three times. Secondary antibodies (for calnexin and CD81:1:10,000; ECL antirabbit IgG horseradish peroxidase-linked F(ab′)2 fragment (donkey, antirabbit); for TSG101, HSP70, and galectin-1:1:2000; ECL antimouse IgG horseradish peroxidase-linked F(ab′)2 fragment (sheep, antimouse); (GE Healthcare, Buckinghamshire, UK) diluted in 0.25% blotting grade Blocker Non-Fat Dry Milk in TBST were incubated with the membrane for 1.5 h. The membranes were finally analyzed with ECL Prime Western Blotting Detection (GE Healthcare) and a VersaDoc 4000 MP (Bio-Rad Laboratories).
Flow Cytometry.
MSC exosomes were labeled with anti-CD63-modified magnetic beads (15 μg of exosome per 7 μL of beads, Exosome Isolation CD63, lot OK527, Life Technologies AS, Oslo, Norway) overnight with gentle agitation. The beads were washed with 1% exosome-depleted FBS in PBS and then incubated with human IgG (Sigma-Aldrich) for 15 min at 4 °C. Following another washing step, the beads were incubated with PE-labeled antibodies against CD9, CD63, and CD81 or isotype control (BD Bioscience, Erembodegem, Belgium) for 40 min with gentle agitation at room temperature (RT). After another washing step, the samples were analyzed using a FACSAria (BD Bioscience), and data were processed using FlowJo Software (Tri Star, Ashland, OR, USA). For flow cytometry experiments on T cells isolated from various organs, lymph nodes, spleen, and spinal cords of mice were removed and kept in ice cold RPMI supplemented with 10% heat inactivated FBS and 1% antibiotics. Cells were then collected by homogenization using a cell strainer, which were then centrifuged at 500 × g for 5 min, prior to RBC lysis with ACK buffer. Lymphocytes were then collected using Percoll media density gradient centrifugation and subsequently stained for T cell subsets mentioned in the manuscript using the following antibodies: BV605 IL17 (cat. no.: 506927), PE-IFNγ (cat. no.: 505807), BV785 CD4 (cat. no.: 317441), and BV510 CD25 (cat. no.: 302639), all purchased from Biolegend.
Electron Microscopy.
MSC exosomes were analyzed using electron microscopy following our published protocol.96 For immunogold labeling for electron microscopy, we followed a previously published protocol.97
Nanoparticle Tracking analysis (NTA).
NTA was performed using the Nanosight system (Malvern instruments, USA). Exosomes suspended in PBS (approximately 2 μg/μL) were first diluted 100-fold prior to analysis. Exosomes were analyzed based on light scattering using an optical microscope aligned perpendicularly to the beam axis. A 60 s video was recorded and subsequently analyzed using NTA software.
RNA Isolation for Deep RNA Sequencing.
MSC exosomes (~800 μg) were prepared for RNA sequencing following our previously established protocol.98
Whole Transcriptome (WT) Sequencing.
For sequencing RNA prepared as mentioned above, a library for small and long RNA was created. Illumina HiSeq 2500 was then used to perform the sequencing of both the WT and small-RNA to a read depth of approximately 50 million for the long RNA and 10 million for the small RNA following our previous protocol.96 Briefly, the raw sequence image.bcl files obtained from the Illumina HiSeq 2500 were converted to fastq format. We also checked to make sure the quality scores did not deteriorate too much at the read ends. The fastqs were kept untrimmed for the long RNA but were trimmed to remove the adapters for the small-RNA. The fastqs were aligned to the reference human genome (hg19, Ensembl version 75) using Bowtie for the small-RNA and STAR v2.3.0 for the long-RNA. The STAR/Bowtie aligned.sam files were converted to .bam files, which were then sorted by coordinates using SAMtools v0.1.19.
For WT sequencing, the read counts and FPKMs for genes were obtained using the hg19 gene reference from Ensembl 75. The read counts were generated using htseq-count (intersection nonempty mode), and FPKMs were generated using Cufflinks v2.2.1. A total library normalization was conducted by scaling the sample libraries to the total number of reads mapping to the reference human genome. TPMs for each gene i was calculated by TPMi = FPKMi/∑FPKMi. See each of the figures for more details on the analysis.
For small-RNA sequencing, the samples were processed using sRNAbench, which permits mapping the reads to several RNA libraries using Bowtie. The reads were first mapped to the miRNA database (miRBase v21) and were then competitively and simultaneously mapped to the other RNA libraries, that is, rRNA (rRNA from NCBI), Mt_tRNA (Mitochondrial tRNA from Ensembl 75), tRNA (tRNA from UCSC http://gtrnadb.ucsc.edu/Hsapi19/hg19-tRNAs.fa), piRNA (piRBase v1.0), snoRNA (small nucleolar RNA from Ensembl 75), snRNA (small nuclear RNA from Ensembl 75), Vault RNA (Ensembl 75), and Y RNA (Ensembl 75).
Log2-fold changes were calculated for the comparisons between cells treated with IFNγ and control cells (Cells_IFNγ vs Cells_C) and exosomes from cells treated with IFNγ vs exosomes from control cells (Exo_IFNγ vs Exo_C) for both the WT sequencing and miRNA reads. TPMS was used for the WT sequencing runs, whereas read counts normalized for total library size were used for the miRNA comparisons. For the WT sequencing, the top 350 genes with the highest absolute log2-fold changes were put into ingenuity pathway analysis (IPA) to see which pathways were affected by the IFNγ treatment. Also, for the small RNA sequencing runs, the target mRNA was found by miRNA target explorer from IPA. The list was whittled down to include only those mRNA targets for the miRNA, which were experimentally observed.
RT-PCR Experiment of IDO-1 mRNA.
Total RNA from 250 μg of MSC-derived exosomes was extracted using QIAGEN RNeasy kit (cat. no.: 74104) using manufacturer’s instructions. cDNA was reverse transcribed using a BioRad iScript cDNA synthesis kit (cat. no.: 1708891). Forward primer (tggcacacgctatggaaaac) and reverse primer (accttccttcaaaagggatttct) specific to IDO1 were used to amplify IDO1 gene from the cDNA product. Primers without cDNA were run as negative controls. The PCR was conducted at an annealing temperature of 65 °C for 35 cycles.
Proteomic Analysis.
MSCs and their exosomes samples (~0.2 mg per sample) were digested with trypsin after reduction and alkylation with (methylmethanethiosulfonate. Peptides were analyzed on an Orbitrap Fusion Tribrid MS interfaced with a nanoLC system with a C18 column and an acetonitrile gradient. Raw spectra were analyzed with MaxQuant (Cox & Mann 2008), and LFQ intensities were compared to determine protein spectra with 2-fold change in intensity. GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were then performed on proteins with a 2-fold change in LFQ intensity between samples using the Database for Annotation, Visualization and Integrated Discovery. The top 20 GO and KEGG categories with a Benjamani adjusted p-value <0.05 were plotted.
EAE Induction and Cell/Exosome Transplantation.
The 6–8-week-old female C57BL/6J mice were purchased from Charles River laboratories Inc. (San Diego, USA). Animals were housed in a pathogen-free facility and cared per the Animal Ethical Committee guidelines of University of California, Irvine. Mice were immunized with complete Freund adjuvant (IFA; Difco, Detroit, MI) containing 4 mg/mL Mycobacterium tuberculosis (strain H37Ra; Difco) and 200 μg MOG35–55 (AnaSpec, CA, USA).
To induce EAE, female C57BL/6J mice were immunized with complete Freund’s adjuvant (CFA), MOG35–55, and pertussis toxin (Sigma-Aldrich) (injected 400 ng on days 0 and 2), and approximately 15–20 days later, mice displayed the peak of the disease showing complete paralysis of the tail and hind limbs with flattened posture. Native and IFNγ-MSC (1 million) and their respective exosomes (150 μg) were injected i.v. at the peak of the disease. Each mouse was graded blind every other day and assigned a clinical score ranging from 0 to 4: No obvious changes in motor function compared to non-immunized mice (score 0), tip of tail is limp (score 0.5), limp tail (score 1.0), limp tail and hind leg inhibition (score 1.5), limp tail and weakness of hind legs (score 2.0), limp tail and dragging of hind legs (score 2.5), limp tail and complete paralysis of hind legs (score 3.0), limp tail and complete paralysis of hind legs and partial front leg paralysis (score 3.5), and mouse is minimally moving around the cage and is minimally alert (score 4.0). Mice were monitored for at least 40 days after immunization. The cumulative disease score was determined by summing the daily neurologic scores for each mouse for the study period. For “Foxp3-eGFP” mice, the animals were obtained from the Cahalan Laboratory at UC Irvine, which were purchased from the Jackson Laboratories (stock number 006772). During disease establishment, we found that these mice were more prone to EAE pathology. Thus, we used half of a dose of pertoxin and MOG33–35 for this group. The treatment procedure was same as described for the C57BL/6J mice. To assess the effect of earlier intervention and number of injections, two doses of MSC-derived IFNγ-Exo at days 12 and 14, respectively, post-immunization, were transplanted and assessed as described above.
Histology and Immunofluorescence Staining.
At the time of sacrifice, mice were transcardially perfused with 4% paraformaldehyde. Spinal cords were removed and fixed in the same fixative for 24 h. They were washed using PBS and embedded in OCT in cryomolds. Sections were cut at 8-μm thickness using a cryostat. The sections were desiccated for 1 h. Samples were doused with 10% normal goat serum or 0.5% triton in PBS for 1 h at RT, dried, doused with a primary antibody using CD4 (1:200 dilution; BD Pharmingen) and IBA-1 (1:400 dilution; Wako), and placed in a 4 °C room overnight. The samples were then washed 3 times with PBS for 5 min, and incubated with the secondary antibodies for 1 h at RT. The following secondary antibodies were used: Alexa Fluor 488 goat antirabbit IgG and Alexa Fluor 594 goat antihamster IgG (Invitrogen). Following washing using PBS 3 times for 5 min, the samples were mounted with DAPI. The slides were finally analyzed using Nikon (Nikon, Tokyo, Japan, http://www.nikon.com) microscope.
The luxol fast blue (LFB) stain determined myelin differentiation. Spinal cord sections from mice that received PBS or exosomes were desiccated for 1 h and then went through a series of washing steps with ethanol, lithium carbonate, Harris hematoxylin, acid alcohol, ammonium hydroxide, and Eosin Y. For LFB, 3 different animals were examined with at least 10 sections per animal. Quantification of myelinated and unmyelinated axons was performed using multiple sections from at least three different spinal cord regions from three animals. All spinal cord slides were analyzed using Nikon (Nikon, Tokyo, Japan).
For hematoxylin and eosin staining, spinal cord slides from PBS or exosome-treated mice were hydrated in deionized water (DI) and left in hematoxylin for 5 min. The samples went through a series of washes and left in Eosin Y for 1 min and 20 s. The samples went through another series of ethanol washes and analyzed using Nikon.
PBMC Isolation, Activation, and Proliferation Assay.
PBMCs were isolated from buffy coats of blood samples obtained from healthy, anonymous donors (UCI Institute for Clinical and Transitional Science) using density gradient centrifugation (Ficoll-Pague plus, GE Healthcare). Twenty-five μg of MSC exosomes was incubated with 1 × 105 CFSE solution (Molecular Probes, Eugene, OR) labeled PBMCs. To activate T-cell proliferation, Dynabeads Human T-Activator CD3/CD28 for T Cell Expansion and Activation was used with a 1:1 ratio of PBMCs:Dynabeads. T-cell proliferation was analyzed after 4 days using flow cytometry (FACSAria, BD), and data were analyzed using the FlowJo.
Cytokine Measurements.
In PBMC co-culture, ELISA assays for prostaglandin E2 (pGE2, Abcam cat. no.: ab133021), tumor necrosis factor alpha-inducible protein 6 (TSG-6, Sigma-Aldrich, cat. no.: RAB1092–1KT), and TGFβ1 (Invitrogen, cat. no.: 88-8350-22) were performed per the manufacturer’s protocols. Additionally, latent TGFβ1 was activated by acidification of the cell culture supernatants, based on manufacturer instructions. Ineffective acidification led to negative absorbance values in several wells in TGFβ1 ELISA assay which were eliminated from the presented data. For the Luminex assay, 50 μL of PBMC culture supernatants were collected and either frozen at −80 °C or immediately analyzed using a human custom ProcartaPlex (11plex, ThermoFisher Scientific, Vienna, Austria) with Luminex MAGPIX. Results were then reported as mean fluorescence intensity.
To measure inflammatory cytokines in spinal cords, the supernatant after cell isolations of spinal cords was harvested. Supernatant solutions (100 μL) were then immediately kept in −80 °C and thawed immediately before performing the Luminex assay. Thawed samples were centrifuged at 10,000 × g for 5 min to extract cells and debris from the solution. We then performed a Luminex assay using Th1/Th2/Th9/Th17/Th22/Treg Cytokine 17-Plex Mouse ProcartaPlex Panel (ThermoFisher Scientific, Vienna, Austria).
Treg Induction Assay.
The 8–12-week-old “Foxp3-eGFP” mice were sacrificed using CO2 asphyxiation followed by cervical dislocation. The spleen was removed and homogenized using a cell strainer. Cells were collected by centrifugation at 500 × g for 5 min, followed by RBC lysis using ACK buffer. Splenocyte suspensions were prepared and stimulated with anti-CD3 clone 145–2c11 (1 μg/mL) and IL-2 (25 ng/mL) without or with TGFβ (Biolegend, USA). The splenocytes were further cultured with the indicated concentrations of exosomes from native or IFNγ-stimulated MSC or nonconditioned medium controls. After 4 d, cultures were harvested and stained for flow cytometry analysis of CD4+CD25+FOXP3+ T cells and CD8+CD25+FOXP3+ T cells.
Biodistribution Experiments.
Male wild-type and EAE induced C57BL/6 mice were injected i.v. with Native-Exo and IFNγ-Exo labeled with DiR lipophilic dye (PerkinElmer). Briefly, 4 μL of DiR dye was added to 1 mL of Diluent C (Fluorescent Cell Linker Kit for General Cell Membrane Labeling according to the manufacturer’s protocol; Sigma-Aldrich cat. no.: CGLDIL-6 × 10 ML) before incubated with exosomes or the control for 5 min. Two mL of 1% BSA was then added. The samples were then filtered using 300 kDa Vivaspin filters (Sartorius Stedim Biotech GmbH, Goettingen, Germany) by centrifugation at 4000 × g. The exosome samples were washed 3 times with 5 mL of PBS. The same amount of dye was used as the dye control in all the experiments. EAE and wild-type mice were imaged at 3 and 24 h using IVIS. This imager uses a highly sensitive CCD camera to image the high wavelength fluorescence. Mice were also sacrificed at 3 and 24 h, and organs were imaged at both time points. Mice were sedated with isoflurane and live imaged using 1–2 min exposure and ICG filter at 745 nm excitation for 3 and 24 h mouse groups. After each live imaging, the mice organs were isolated and imaged using 1–2 s exposure (ICG filter at 745 nm excitation). Fluorescence for each mice and organ image was quantified using Live Imaging Software in IVIS.
Blue BANDit Staining To Detect Protein Damage Caused by UV.
The Blue BADIT protein assay was conducted to explore the effect of UV illumination on proteins of exosomes. Retrieved exosomes (2 μg/μL) were divided into three identical sub-batches. The first sub-batch was maintained without UV illumination as a control. The second and third sub-batches were exposed to 120 mJ/cm2 UV illumination (Spectrolinker XL-1000, NY, USA) for 220 s and 1 h, respectively. Then, exosome proteins were retrieved using RIPA (as described earlier). Next, 10 μL of each sample was added to 10 μL of Laemmli 2X sample buffer (BIORAD, USA) and loaded into Mini-PROTEAN TGX 10% precast gel (BIORAD, USA). Five μL of PageRuler Plus protein ladder was also loaded into the gel. Following electrophoresis, the gel was washed 3 times for 5 min in 200 mL of deionized water with gentle agitation. Water was discarded, and at least 20 mL of Blue BANDit protein stain was added, ensuring the gel is completely submerged. The gel was covered with aluminum foil and stained for 1 h with gentle agitation. Next, the gel was rinsed with several changes of at least 200 mL of deionized water overnight with gentle agitation. Stained proteins began to appear as blue bands on a clear background.
Supplementary Material
ACKNOWLEDGMENTS
We thank L. Liu for help on UC-MSC isolation, M. Lodoen for discussion on cytokine analyses, L. BenMohamed and R. Srivastava for discussions on spinal cord cytokine analyses, and J. Zimak for discussion on PCR assay. We also thank the Cahalan lab at UCI for providing eGFP-FOXP3 mice for this study. This work was supported by the National Institute of Health (1DP2CA195763–01). M.R. was supported by a National Institute of Neurological Disorders and Stroke (NINDS/NIH) Training Grant (award no. NS082174). J.J. acknowledges a Otto. W. Shaler scholarship, and A.I.S. was supported by a postdoctoral fellowship from the Fondation ARC pour la recherche sur le cancer (SAE20150602901).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.9b01004.
Figures S1–S16 and Table S1 and S2, which depict additional results (PDF)
The authors declare the following competing financial interest(s): W.Z. is a co-founder of Velox Biosystems Inc., Baylx Inc., and Amberstone Biosciences Inc. J.L. has equity in Codiak BioSciences and holds the rights to multiple extracellular vesicle diagnostics and therapeutics patents.
REFERENCES
- (1).Singer NG; Caplan AI Mesenchymal Stem Cells: Mechanisms of Inflammation. Annu. Rev. Pathol.: Mech. Dis 2011, 6, 457–478. [DOI] [PubMed] [Google Scholar]
- (2).Galipeau J; Sensébé L, Mesenchymal Stromal Cells: Clinical Challenges and Therapeutic Opportunities. Cell Stem Cell 2018, 22, 824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fischer UM; Harting MT; Jimenez F; Monzon-Posadas WO; Xue H; Savitz SI; Laine GA; Cox CS Jr. Pulmonary Passage Is A Major Obstacle for Intravenous Stem Cell Delivery: The Pulmonary First-Pass Effect. Stem Cells Dev. 2009, 18, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).de Witte SFH; Luk F; Sierra Parraga JM; Gargesha M; Merino A; Korevaar SS; Shankar AS; O’Flynn L; Elliman SJ; Roy D; Betjes MGH; Newsome PN; Baan CC; Hoogduijn MJ Immunomodulation by Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC by Monocytic Cells. Stem Cells 2018, 36, 602–615. [DOI] [PubMed] [Google Scholar]
- (5).Galleu A; Riffo-Vasquez Y; Trento C; Lomas C; Dolcetti L; Cheung TS; von Bonin M; Barbieri L; Halai K; Ward S; Weng L; Chakraverty R; Lombardi G; Watt FM; Orchard K; Marks DI; Apperley J; Bornhauser M; Walczak H; Bennett C; et al. Apoptosis in Mesenchymal Stromal Cells Induces in vivo Recipient-Mediated Immunomodulation. Sci. Transl. Med 2017, 9, No. eaam7828. [DOI] [PubMed] [Google Scholar]
- (6).Luk F; de Witte SFH; Korevaar SS; Roemeling-van Rhijn M; Franquesa M; Strini T; van den Engel S; Gargesha M; Roy D; Dor FJMF; Horwitz EM; de Bruin RWF; Betjes MGH; Baan CC; Hoogduijn MJ Inactivated Mesenchymal Stem Cells Maintain Immunomodulatory Capacity. Stem Cells Dev. 2016, 25, 1342–1354. [DOI] [PubMed] [Google Scholar]
- (7).Riazifar M; Pone EJ; Lotvall J; Zhao W Stem Cell Extracellular Vesicles: Extended Messages of Regeneration. Annu. Rev. Pharmacol. Toxicol 2017, 57, 125–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Thery C; Ostrowski M; Segura E Membrane Vesicles as Conveyors of Immune Responses. Nat. Rev. Immunol 2009, 9, 581–593. [DOI] [PubMed] [Google Scholar]
- (9).Marino J; Babiker-Mohamed MH; Crosby-Bertorini P; Paster JT; LeGuern C; Germana S; Abdi R; Uehara M; Kim JI; Markmann JF; Tocco G; Benichou G Donor Exosomes Rather Than Passenger Leukocytes Initiate Alloreactive T Cell Responses After Transplantation. Sci. Immunol 2016, 1, aaf8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Viaud S; Ploix S; Lapierre V; Théry C; Commere P-H; Tramalloni D; Gorrichon K; Virault-Rocroy P; Tursz T; Lantz O; Zitvogel L; Chaput N Updated Technology to Produce Highly Immunogenic Dendritic Cell-derived Exosomes of Clinical Grade: A Critical Role of Interferon-γ. J. Immunother 2011, 34, 65–75. [DOI] [PubMed] [Google Scholar]
- (11).Pusic AD; Pusic KM; Clayton BL; Kraig RP IFNgamma-Stimulated Dendritic Cell Exosomes as A Potential Therapeutic for Remyelination. J. Neuroimmunol 2014, 266, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Shigemoto-Kuroda T; Oh JY; Kim D.-k.; Jeong HJ; Park SY; Lee HJ; Park JW; Kim TW; An SY; Prockop DJ; Lee RH MSC-derived Extracellular Vesicles Attenuate Immune Responses in Two Autoimmune Murine Models: Type 1 Diabetes and Uveoretinitis. Stem Cell Rep. 2017, 8, 1214–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lankford KL; Arroyo EJ; Nazimek K; Bryniarski K; Askenase PW; Kocsis JD Intravenously Delivered Mesenchymal Stem Cell-Derived Exosomes Target M2-type Macrophages in The Injured Spinal Cord. PLoS One 2018, 13, No. e0190358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Laso-García F; Ramos-Cejudo J; Carrillo-Salinas FJ; Otero-Ortega L; Feliú A; Gómez-de Frutos M; Mecha M; Díez-Tejedor E; Guaza C; Gutiérrez-Fernández M Therapeutic Potential of Extracellular Vesicles Derived From Human Mesenchymal Stem Cells in A Model of Progressive Multiple Sclerosis. PLoS One 2018, 13, No. e0202590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Bai L; Shao H; Wang H; Zhang Z; Su C; Dong L; Yu B; Chen X; Li X; Zhang X Effects of Mesenchymal Stem Cell-Derived Exosomes on Experimental Autoimmune Uveitis. Sci. Rep 2017, 7, 4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fujii S; Miura Y; Fujishiro A; Shindo T; Shimazu Y; Hirai H; Tahara H; Takaori-Kondo A; Ichinohe T; Maekawa T Graft-Versus-Host Disease Amelioration by Human Bone Marrow Mesenchymal Stromal/Stem Cell-Derived Extracellular Vesicles Is Associated with Peripheral Preservation of Naive T Cell Populations. Stem Cells 2018, 36, 434–445. [DOI] [PubMed] [Google Scholar]
- (17).Silva AKA; Perretta S; Perrod G; Pidial L; Lindner V; Carn F; Lemieux S; Alloyeau D; Boucenna I; Menasché P; Dallemagne B; Gazeau F; Wilhelm C; Cellier C; Clément O; Rahmi G Thermoresponsive Gel Embedded with Adipose Stem-Cell-Derived Extracellular Vesicles Promotes Esophageal Fistula Healing in a Thermo-Actuated Delivery Strategy. ACS Nano 2018, 12, 9800–9814. [DOI] [PubMed] [Google Scholar]
- (18).Lai RC; Yeo RWY; Tan KH; Lim SK Exosomes for Drug Delivery — A Novel Application for The Mesenchymal Stem Cell. Biotechnol. Adv 2013, 31, 543–551. [DOI] [PubMed] [Google Scholar]
- (19).Dendrou CA; Fugger L; Friese MA Immunopathology of Multiple Sclerosis. Nat. Rev. Immunol 2015, 15, 545–558. [DOI] [PubMed] [Google Scholar]
- (20).Ransohoff RM; Hafler DA; Lucchinetti CF Multiple Sclerosis-A Quiet Revolution. Nat. Rev. Neurol 2015, 11, 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Hogancamp WE; Rodriguez M; Weinshenker BG The Epidemiology of Multiple Sclerosis. Mayo Clin. Proc 1997, 72, 871–878. [DOI] [PubMed] [Google Scholar]
- (22).Constantinescu CS; Farooqi N; O’Brien K; Gran B Experimental Autoimmune Encephalomyelitis (EAE) as A Model for Multiple Sclerosis (MS). Br. J. Pharmacol 2011, 164, 1079–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Duijvestein M; Wildenberg ME; Welling MM; Hennink S; Molendijk I; van Zuylen VL; Bosse T; Vos ACW; de Jonge-Muller ESM; Roelofs H; van der Weerd L; Verspaget HW; Fibbe WE; te Velde AA; van den Brink GR; Hommes DW Pretreatment with Interferon-γ Enhances the Therapeutic Activity of Mesenchymal Stromal Cells in Animal Models of Colitis. Stem Cells 2011, 29, 1549–1558. [DOI] [PubMed] [Google Scholar]
- (24).Polchert D; Sobinsky J; Douglas GW; Kidd M; Moadsiri A; Reina E; Genrich K; Mehrotra S; Setty S; Smith B; Bartholomew A IFN-γ Activation of Mesenchymal Stem Cells for Treatment and Prevention of Graft Versus Host Disease. Eur. J. Immunol 2008, 38, 1745–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sheng H; Wang Y; Jin Y; Zhang Q; Zhang Y; Wang L; Shen B; Yin S; Liu W; Cui L; Li N A Critical Role of IFNγ in Priming MSC-Mediated Suppression of T Cell Proliferation Through Up-Regulation of B7-H1. Cell Res. 2008, 18, 846–857. [DOI] [PubMed] [Google Scholar]
- (26).Stadelmann C; Wegner C; Bruck W Inflammation, Demyelination, and Degeneration - Recent Insights From MS Pathology. Biochim. Biophys. Acta, Mol. Basis Dis 2011, 1812, 275–282. [DOI] [PubMed] [Google Scholar]
- (27).Naegele M; Martin R The Good and The Bad of Neuroinflammation in Multiple Sclerosis. Handb. Clin. Neurol 2014, 122, 59–87. [DOI] [PubMed] [Google Scholar]
- (28).Vogel DY; Vereyken EJ; Glim JE; Heijnen PD; Moeton M; van der Valk P; Amor S; Teunissen CE; van Horssen J; Dijkstra CD Macrophages in Inflammatory Multiple Sclerosis Lesions Have An Intermediate Activation Status. J. Neuroinflammation 2013, 10, 809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Leong SK; Ling EA Amoeboid and Ramified Microglia: Their Interrelationship and Response to Brain Injury. Glia 1992, 6, 39–47. [DOI] [PubMed] [Google Scholar]
- (30).Ayoub AE; Salm AK Increased Morphological Diversity of Microglia in The Activated Hypothalamic Supraoptic Nucleus. J. Neurosci 2003, 23, 7759–7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zozulya AL; Wiendl H The Role of Regulatory T Cells in Multiple Sclerosis. Nat. Clin. Pract. Neurol 2008, 4, 384–398. [DOI] [PubMed] [Google Scholar]
- (32).Carbone F; De Rosa V; Carrieri PB; Montella S; Bruzzese D; Porcellini A; Procaccini C; La Cava A; Matarese G Regulatory T Cell Proliferative Potential Is Impaired in Human Autoimmune Disease. Nat. Med 2014, 20, 69–74. [DOI] [PubMed] [Google Scholar]
- (33).Fletcher JM; Lalor SJ; Sweeney CM; Tubridy N; Mills KH T Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Clin. Exp. Immunol 2010, 162, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Matheu MP; Othy S; Greenberg ML; Dong TX; Schuijs M; Deswarte K; Hammad H; Lambrecht BN; Parker I; Cahalan MD Imaging regulatory T Cell Dynamics and CTLA4-Mediated Suppression of T Cell Priming. Nat. Commun 2015, 6, 6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wagner CA; Goverman JM Novel Insights and Therapeutics in Multiple Sclerosis. F1000Research 2015, 4, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Johnson MC; Pierson ER; Spieker AJ; Nielsen AS; Posso S; Kita M; Buckner JH; Goverman JM Distinct T Cell Signatures Define Subsets of Patients with Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm 2016, 3, e278–e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Murphy ÁC; Lalor SJ; Lynch MA; Mills KHG Infiltration of Th1 and Th17 Cells and Activation of Microglia in The CNS During The Course of Experimental Autoimmune Encephalomyelitis. Brain, Behav., Immun 2010, 24, 641–651. [DOI] [PubMed] [Google Scholar]
- (38).Caravagna C; Jaouën A; Desplat-Jégo S; Fenrich KK; Bergot E; Luche H; Grenot P; Rougon G; Malissen M; Debarbieux F Diversity of Innate Immune Cell Subsets across Spatial and Temporal Scales in An EAE Mouse Model. Sci. Rep 2018, 8, 5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Thakker P; Leach MW; Kuang W; Benoit SE; Leonard JP; Marusic S IL-23 Is Critical in the Induction but Not in the Effector Phase of Experimental Autoimmune Encephalomyelitis. J. Immunol 2007, 178, 2589. [DOI] [PubMed] [Google Scholar]
- (40).Dennis G Jr.; Holweg CTJ; Kummerfeld SK; Choy DF; Setiadi AF; Hackney JA; Haverty PM; Gilbert H; Lin WY; Diehl L; Fischer S; Song A; Musselman D; Klearman M; Gabay C; Kavanaugh A; Endres J; Fox DA; Martin F; Townsend MJ Synovial Phenotypes in Rheumatoid Arthritis Correlate with Response to Biologic Therapeutics. Arthritis Res. Ther 2014, 16, R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).François M; Romieu-Mourez R; Li M; Galipeau J Human MSC Suppression Correlates with Cytokine Induction of Indoleamine 2,3-Dioxygenase and Bystander M2Macrophage Differentiation. Mol. Ther 2012, 20, 187–195. [DOI] [PubMed] [Google Scholar]
- (42).Tipnis S; Viswanathan C; Majumdar AS Immunosuppressive Properties of Human Umbilical Cord-Derived Mesenchymal Stem Cells: Role of B7-H1 and IDO. Immunol. Cell Biol. 2010, 88, 795–806. [DOI] [PubMed] [Google Scholar]
- (43).Meisel R; Zibert A; Laryea M; Göbel U; Däubener W; Dilloo D Human Bone Marrow Stromal Cells Inhibit Allogeneic T-Cell Responses by Indoleamine 2,3-Dioxygenase-Mediated Tryptophan Degradation. Blood 2004, 103, 4619. [DOI] [PubMed] [Google Scholar]
- (44).Fu S; Zhang N; Yopp AC; Chen D; Mao M; Chen D; Zhang H; Ding Y; Bromberg JS TGF-β Induces Foxp3 + T-Regulatory Cells from CD4 + CD25 − Precursors. Am. J. Transplant 2004, 4, 1614–1627. [DOI] [PubMed] [Google Scholar]
- (45).Sakaguchi S; Yamaguchi T; Nomura T; Ono M Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [DOI] [PubMed] [Google Scholar]
- (46).Yin Y; Shelke GV; Jang SC; Lasser C; Wennmalm S; Hoffmann HJ; Nilsson J; Li L; Gho YS; Lotvall J Regulation of Mesenchymal Stem Cell Function by TGFβ-1 On Mast Cell Extracellular Vesicles - Role of Endosomal Retention. 2017, bioRxiv.org e-Print archive. https://www.biorxiv.org/content/10.1101/172213v1.
- (47).Mokarizadeh A; Delirezh N; Morshedi A; Mosayebi G; Farshid A-A; Mardani K Microvesicles Derived From Mesenchymal Stem Cells: Potent Organelles for Induction of Tolerogenic Signaling. Immunol. Lett 2012, 147, 47–54. [DOI] [PubMed] [Google Scholar]
- (48).Zhang B; Yin Y; Lai RC; Tan SS; Choo AB; Lim SK Mesenchymal Stem Cells Secrete Immunologically Active Exosomes. Stem Cells Dev. 2014, 23, 1233–1244. [DOI] [PubMed] [Google Scholar]
- (49).Zhang B; Yeo RWY; Lai RC; Sim EWK; Chin KC; Lim SK Mesenchymal Stromal Cell Exosome-Enhanced Regulatory T-cell Production through An Antigen-Presenting Cell-Mediated Pathway. Cytotherapy 2018, 20, 687–696. [DOI] [PubMed] [Google Scholar]
- (50).English K; Ryan JM; Tobin L; Murphy MJ; Barry FP; Mahon BP Cell Contact, Prostaglandin E2 and Transforming Growth Factor Beta 1 Play Non-Redundant Roles in Human Mesenchymal Stem Cell Induction of CD4+CD25High forkhead box P3+ Regulatory T Cells. Clin. Exp. Immunol 2009, 156, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Merian J; Gravier J; Navarro F; Texier I Fluorescent Nanoprobes Dedicated to in vivo Imaging: From Preclinical Validations to Clinical Translation. Molecules 2012, 17, 5564–5591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Lai CP; Mardini O; Ericsson M; Prabhakar S; Maguire CA; Chen JW; Tannous BA; Breakefield XO Dynamic Biodistribution of Extracellular Vesicles in vivo Using A Multimodal Imaging Reporter. ACS Nano 2014, 8, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wiklander OP; Nordin JZ; O’Loughlin A; Gustafsson Y; Corso G; Mager I; Vader P; Lee Y; Sork H; Seow Y; Heldring N; Alvarez-Erviti L; Smith CI; Le Blanc K; Macchiarini P; Jungebluth P; Wood MJ; Andaloussi SE Extracellular Vesicle in vivo Biodistribution Is Determined by Cell Source, Route of Administration and Targeting. J. Extracell. Vesicles 2015, 4, 26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Von Bahr L; Batsis I; Moll G; Hägg M; Szakos A; Sundberg B; Uzunel M; Ringden O; Le Blanc K Analysis of Tissues Following Mesenchymal Stromal Cell Therapy in Humans Indicates Limited Long-Term Engraftment and No Ectopic Tissue Formation. Stem Cells 2012, 30, 1575–1578. [DOI] [PubMed] [Google Scholar]
- (55).Rosenblum MD; Way SS; Abbas AK Regulatory T Cell Memory. Nat. Rev. Immunol 2016, 16, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Colombo M; Raposo G; Thery C Biogenesis, Secretion, and Intercellular Interactions of Exosomes and Other Extracellular Vesicles. Annu. Rev. Cell Dev. Biol 2014, 30, 255–289. [DOI] [PubMed] [Google Scholar]
- (57).Ekstrom K; Valadi H; Sjostrand M; Malmhall C; Bossios A; Eldh M; Lotvall J Characterization of mRNA and microRNA in Human Mast Cell-Derived Exosomes and Their Transfer to Other Mast Cells and Blood CD34 Progenitor Cells. J. Extracell. Vesicles 2012, 1, 18389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kogure T; Yan IK; Lin WL; Patel T Extracellular Vesicle-Mediated Transfer of A Novel Long Noncoding RNA TUC339: A Mechanism of Intercellular Signaling in Human Hepatocellular Cancer. Genes Cancer 2013, 4, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Valadi H; Ekstrom K; Bossios A; Sjostrand M; Lee JJ; Lotvall JO Exosome-Mediated Transfer of mRNAs and microRNAs Is A Novel Mechanism of Genetic Exchange Between Cells. Nat. Cell Biol. 2007, 9, 654–659. [DOI] [PubMed] [Google Scholar]
- (60).Eldh M; Ekstrom K; Valadi H; Sjostrand M; Olsson B; Jernas M; Lotvall J Exosomes Communicate Protective Messages During Oxidative Stress; Possible Role of Exosomal Shuttle RNA. PLoS One 2010, 5, No. e15353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Wurtmann EJ; Wolin SL RNA under Attack: Cellular Handling of RNA Damage. Crit. Rev. Biochem. Mol. Biol 2009, 44, 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Greenberg JR Ultraviolet Light-Induced Crosslinking of mRNA to Proteins. Nucleic Acids Res. 1979, 6, 715–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Rule Wigginton K; Menin L; Montoya JP; Kohn T Oxidation of Virus Proteins During UV(254) and Singlet Oxygen Mediated Inactivation. Environ. Sci. Technol 2010, 44, 5437–5443. [DOI] [PubMed] [Google Scholar]
- (64).Kochevar IE UV-Induced Protein Alterations and Lipid Oxidation in Erythrocyte Membranes. Photochem. Photobiol 1990, 52, 795–800. [DOI] [PubMed] [Google Scholar]
- (65).Li Y; Xia W; Liu Y; Remmer HA; Voorhees J; Fisher GJ Solar Ultraviolet Irradiation Induces Decorin Degradation in Human Skin Likely via Neutrophil Elastase. PLoS One 2013, 8, No. e72563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Churlaud G; Pitoiset F; Jebbawi F; Lorenzon R; Bellier B; Rosenzwajg M; Klatzmann D Human and Mouse CD8+CD25+FOXP3+ Regulatory T Cells at Steady State and During Interleukin-2 Therapy. Front. Immunol 2015, 6, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chen W; Huang Y; Han J; Yu L; Li Y; Lu Z; Li H; Liu Z; Shi C; Duan F; Xiao Y Immunomodulatory Effects of Mesenchymal Stromal Cells-Derived Exosome. Immunol. Res 2016, 64, 831–840. [DOI] [PubMed] [Google Scholar]
- (68).Murphy MB; Moncivais K; Caplan AI Mesenchymal Stem Cells: Environmentally Responsive Therapeutics for Regenerative Medicine. Exp. Mol. Med 2013, 45, No. e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Sakurai K; Zou JP; Tschetter JR; Ward JM; Shearer GM Effect of Indoleamine 2,3-dioxygenase On Induction of Experimental Autoimmune Encephalomyelitis. J. Neuroimmunol 2002, 129, 186–196. [DOI] [PubMed] [Google Scholar]
- (70).Mbongue JC; Nicholas DA; Torrez TW; Kim NS; Firek AF; Langridge WH The Role of Indoleamine 2, 3-Dioxygenase in Immune Suppression and Autoimmunity. Vaccines (Basel, Switz.) 2015, 3, 703–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Patel RK; Mohan C PI3K/AKT Signaling and Systemic Autoimmunity. Immunol. Res 2005, 31, 47–55. [DOI] [PubMed] [Google Scholar]
- (72).Li H; Park D; Abdul-Muneer PM; Xu B; Wang H; Xing B; Wu D; Li S PI3Kgamma Inhibition Alleviates Symptoms and Increases Axon Number in Experimental Autoimmune Encephalomyelitis Mice. Neuroscience 2013, 253, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Chen TS; Lai RC; Lee MM; Choo ABH; Lee CN; Lim SK Mesenchymal Stem Cell Secretes Microparticles Enriched in Pre-microRNAs. Nucleic Acids Res. 2010, 38, 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Song Y; Dou H; Li X; Zhao X; Li Y; Liu D; Ji J; Liu F; Ding L; Ni Y; Hou Y Exosomal miR-146a Contributes to The Enhanced Therapeutic Efficacy of Interleukin-1β-Primed Mesenchymal Stem Cells Against Sepsis. Stem Cells 2017, 35, 1208–1221. [DOI] [PubMed] [Google Scholar]
- (75).Rupaimoole R; Slack FJ MicroRNA Therapeutics: Towards a New Era for The Management of Cancer and Other Diseases. Nat. Rev. Drug Discovery 2017, 16, 203. [DOI] [PubMed] [Google Scholar]
- (76).Bootcov MR; Bauskin AR; Valenzuela SM; Moore AG; Bansal M; He XY; Zhang HP; Donnellan M; Mahler S; Pryor K; Walsh BJ; Nicholson RC; Fairlie WD; Por SB; Robbins JM; Breit SN MIC-1, A Novel Macrophage Inhibitory Cytokine, Is A Divergent Member of The TGF-β Superfamily. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 11514–11519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Rabinovich GA; Sotomayor CE; Riera CM; Bianco I; Correa SG Evidence of A Role for Galectin-1 in Acute Inflammation. Eur. J. Immunol 2000, 30, 1331–1339. [DOI] [PubMed] [Google Scholar]
- (78).Nonaka M; Fukuda M Galectin-1 for Neuroprotection? Immunity 2012, 37, 187–189. [DOI] [PubMed] [Google Scholar]
- (79).Mansilla MJ; Montalban X; Espejo C Heat Shock Protein 70: Roles in Multiple Sclerosis. Mol. Med. (Manhasset, NY, U. S.) 2012, 18, 1018–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Saharinen J; Hyytiainen M; Taipale J; Keski-Oja J Latent Transforming Growth Factor-Beta Binding Proteins (LTBPs)–Structural Extracellular Matrix Proteins for Targeting TGF-Beta Action. Cytokine Growth Factor Rev. 1999, 10, 99–117. [DOI] [PubMed] [Google Scholar]
- (81).Johnson DB; Nixon MJ; Wang Y; Wang DY; Castellanos E; Estrada MV; Ericsson-Gonzalez PI; Cote CH; Salgado R; Sanchez V; Dean PT; Opalenik SR; Schreeder DM; Rimm DL; Kim JY; Bordeaux J; Loi S; Horn L; Sanders ME; Ferrell PB Jr.; et al. Tumor-specific MHC-II Expression Drives A Unique Pattern of Resistance to Immunotherapy Via LAG-3/FCRL6 Engagement. JCI Insight 2018, 3, 120360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Del Fattore A; Luciano R; Pascucci L; Goffredo BM; Giorda E; Scapaticci M; Fierabracci A; Muraca M Immunoregulatory Effects of Mesenchymal Stem Cell-Derived Extracellular Vesicles on T Lymphocytes. Cell Transplant 2015, 24, 2615–2627. [DOI] [PubMed] [Google Scholar]
- (83).Chen G; Huang AC; Zhang W; Zhang G; Wu M; Xu W; Yu Z; Yang J; Wang B; Sun H; Xia H; Man Q; Zhong W; Antelo LF; Wu B; Xiong X; Liu X; Guan L; Li T; Liu S; et al. Exosomal PD-L1 Contributes to Immunosuppression and Is Associated with Anti-PD-1 Response. Nature 2018, 560, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Kilpinen L; Impola U; Sankkila L; Ritamo I; Aatonen M; Kilpinen S; Tuimala J; Valmu L; Levijoki J; Finckenberg P; Siljander P; Kankuri E; Mervaala E; Laitinen S Extracellular Membrane Vesicles From Umbilical Cord Blood-Derived MSC Protect Against Ischemic Acute Kidney Injury, A Feature That Is Lost After Inflammatory Conditioning. J. Extracell. Vesicles 2013, 2, 21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Goncalves F. d. C.; Luk F; Korevaar SS; Bouzid R; Paz AH; López-Iglesias C; Baan CC; Merino A; Hoogduijn MJ Membrane Particles Generated From Mesenchymal Stromal Cells Modulate Immune Responses by Selective Targeting of Pro-inflammatory Monocytes. Sci. Rep 2017, 7, 12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Braza F; Dirou S; Forest V; Sauzeau V; Hassoun D; Chesné J; Cheminant-Muller M-A; Sagan C; Magnan A; Lemarchand P Mesenchymal Stem Cells Induce Suppressive Macrophages through Phagocytosis in a Mouse Model of Asthma. Stem Cells 2016, 34, 1836–1845. [DOI] [PubMed] [Google Scholar]
- (87).Domenis R; Cifu A; Quaglia S; Pistis C; Moretti M; Vicario A; Parodi PC; Fabris M; Niazi KR; Soon-Shiong P; Curcio F Pro Inflammatory Stimuli Enhance The Immunosuppressive Functions of Adipose Mesenchymal Stem Cells-Derived Exosomes. Sci. Rep 2018, 8, 13325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Reiner AT; Witwer KW; van Balkom BWM; de Beer J; Brodie C; Corteling RL; Gabrielsson S; Gimona M; Ibrahim AG; de Kleijn D; Lai CP; Lötvall J; del Portillo HA; Reischl IG; Riazifar M; Salomon C; Tahara H; Toh WS; Wauben MHM; Yang VK; et al. Concise Review: Developing Best-Practice Models for the Therapeutic Use of Extracellular Vesicles. Stem Cells Transl. Med 2017, 6, 1730–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Armstrong JPK; Holme MN; Stevens MM Re-Engineering Extracellular Vesicles as Smart Nanoscale Therapeutics. ACS Nano 2017, 11, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Bouffi C; Bony C; Courties G; Jorgensen C; Noël D IL-6-Dependent PGE2 Secretion by Mesenchymal Stem Cells Inhibits Local Inflammation in Experimental Arthritis. PLoS One 2010, 5, No. e14247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Griffin MD; Ritter T; Mahon BP Immunological Aspects of Allogeneic Mesenchymal Stem Cell Therapies. Hum. Gene Ther. 2010, 21, 1641–1655. [DOI] [PubMed] [Google Scholar]
- (92).Lu LL; Liu YJ; Yang SG; Zhao QJ; Wang X; Gong W; Han ZB; Xu ZS; Lu YX; Liu D; Chen ZZ; Han ZC Isolation and Characterization of Human Umbilical Cord Mesenchymal Stem Cells with Hematopoiesis-Supportive Function and Other Potentials. Haematologica 2006, 91, 1017. [PubMed] [Google Scholar]
- (93).Voorthuis JAC; Uitdehaag BMJ; De Groot CJA; Goede PH; Meide PHVD; Dijkstra CD Suppression of Experimental Allergic Encephalomyelitis by Intraventricular Administration of Interferon-Gamma in Lewis Rats. Clin. Exp. Immunol 1990, 81, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Shelke GV; Lasser C; Gho YS; Lotvall J Importance of Exosome Depletion Protocols to Eliminate Functional and RNA-Containing Extracellular Vesicles from Fetal Bovine Serum. J. Extracell. Vesicles 2014, 3, 24783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Liao Z; Muth DC; Eitan E; Travers M; Learman LN; Lehrmann E; Witwer KW Serum Extracellular Vesicle Depletion Processes Affect Release and Infectivity of HIV-1 in culture. Sci. Rep 2017, 7, 2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Lasser C; Shelke GV; Yeri A; Kim D; Crescitelli R; Raimondo S; Sjostrand M; Gho YS; Jensen KVK; Lotvall J Two Distinct Extracellular RNA Signatures Released by A Single Cell Type Identified by Microaara and Next-Generation Sequencing. RNA Biol. 2017, 14, 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Pospichalova V; Svoboda J; Dave Z; Kotrbova A; Kaiser K; Klemova D; Ilkovics L; Hampl A; Crha I; Jandakova E; Minar L; Weinberger V; Bryja V Simplified Protocol for Flow Cytometry Analyses of Fluorescently Labeled Exosomes and Microvesicles Using Dedicated Flow Cytometer. J. Extracell. Vesicles 2015, 4, 25530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Cvjetkovic A; Lotvall J; Lasser C The Influence of Rotor Type and Centrifugation Time On The Yield and Purity of Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.