

Abstract
The majority of patients with B-cell non-Hodgkin lymphoma (NHL) can be cured with standard chemoimmunotherapy. However, patients who fail first line therapy have dismal outcomes, particularly if they have disease that is resistant to salvage therapy, including chemoimmunotherapy, radiation and/or autologous stem cell transplantation. Indolent B-NHLs, such as follicular lymphoma (FL), although not generally considered curable may be treated over many years with good prognosis. However, a subset of B-NHLs can undergo histologic transformation into more aggressive subtypes with outcomes similar to aggressive B-NHLs. In recent years, T cells genetically modified with chimeric antigen receptors (CARs), have demonstrated a remarkable capacity to induce complete and durable clinical responses in patients with chemotherapy-refractory lymphomas. Indeed, two autologous CD19-directed CAR-modified T cell products have now been FDA-approved for the treatment of patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), primary mediastinal B-cell lymphoma (PMBCL) and transformed FL, while a plethora of other CAR-T cell targets are being explored in ongoing clinical trials. The purpose of this review is to summarize the clinical efficacy and unique toxicities of individually developed CAR-T cell products for the treatment of lymphomas, and their evolution from the laboratory bench to commercialization.
Keywords: cancer, CAR-T cell, chimeric antigen receptor, immunotherapy, lymphoma, oncology
Introduction
Non-Hodgkin lymphomas (NHL), the most common hematologic malignancy, are comprised of a heterogeneous group of lymphoid malignancies derived from both B cell and T cell progenitors or mature T cells, and rarely natural killer (NK) cells. Among the heterogeneous NHL subtypes, diffuse large B cell lymphoma (DLBCL) is by far the most common aggressive lymphoma, accounting for 30–35% of all NHLs diagnosed. The majority of patients can be cured with combination chemoimmunotherapy upfront or with salvage high dose chemotherapy and autologous hematopoietic stem cell transplantation (ASCT).1 However, 15–20% of patients will relapse or develop chemo-refractory disease 1–4 and these patients have a dismal prognosis, particularly if they are chemo-resistant to salvage therapy or relapse following ASCT.5–8 A recent retrospective meta-analysis of patients who achieved stable disease or progressive disease as the best response to chemotherapy, or who relapsed within 12 months of autologous transplant, showed an overall response rate (ORR) of only 26% to salvage therapy and median overall survival (OS) of 6.3 months.8 The outcomes for other aggressive NHL subtypes are equally poor after the failure of combination chemotherapy. This study further highlights the unmet need for novel curative therapeutic options for these patients as well as the identification of predictors of response.
The use of adoptive T cell transfer as a therapeutic modality to treat malignant neoplasms has evolved rapidly over the past decade. Genetically modified T cells expressing chimeric antigen receptors (CARs) targeting specific tumor-associated antigens have recently demonstrated impressive potency and durable responses in multiply treatment-refractory B cell malignancies, resulting in the approval of two CD19-specific commercial CAR-T cell products by the U.S. Food and Drug Administration (FDA). Herein, we review the clinical data on CAR-T cells for the treatment of NHL with an emphasis on the efficacy of the various products, challenges of use, ongoing clinical trials, and mechanisms of enhancing the clinical application of these products.
CAR-T design
Chimeric antigen receptors (CAR) are synthetic constructs that combine an extracellular antigen recognition domain derived from a monoclonal antibody specific for a tumor cell surface antigen with an intracellular T cell signaling domain, which results in T cell activation upon antigen binding.9,10 The intracellular signaling domain(s) were identified as an extremely important component of the CAR construct after “first generation” CARs, which only integrated a CD3ζ signaling domain, exhibited limited expansion and persistence despite the ability to respond to antigens and thus short-lived antitumor activity.11,12 The introduction of additional co-stimulatory domains such as CD28, 4–1BB, and OX40 in second and third generation constructs led to significantly improved expansion, persistence, and efficacy. 13–16 While the best co-stimulatory domain is still an area of active investigation, CD28 and 4–1BB have been the most widely used in CAR-T clinical trials with the CD28 CAR construct demonstrating higher peak expansion, and the 4–1BB based constructs showing longer persistence. 17 Which co-stimulatory domain will have the most impact on clinical outcomes remains to be seen.
Clinical outcomes of CD19.CAR T-cells
CD19 is a cell surface protein with expression restricted to both normal and malignant B cells, making it an attractive target for immunotherapy approaches. Many centers have tested the ability of adoptive transfer of CD19.CAR-T cells to eradicate CD19-positive malignancies. The first report of a clinical response in a patient with NHL using CD19 CAR-T cell therapy was published by Kochenderfer et al., in which they described a patient with multiply-relapsed follicular lymphoma (FL) who achieved a durable partial response after two courses of CAR-T cell therapy in combination with high dose IL-2.18 The authors later reported that four patients achieved complete remissions (two with DLBCL, NOS and two with primary mediastinal B-cell lymphoma with chemotherapy-refractory DLBCL), of whom three had durable responses.19 In a larger phase 1 study that included 22 patients, of whom 19 had DLBCL, the NCI group were able show an ORR of 68% with a complete remission (CR) rate of 47% among patients with DLBCL20 using a CD19-CD28 second generation CAR. The CRs were durable with 11 of 12 ongoing for durations of 7–24 months and 12-month progression-free survival (PFS) of 63% for all participants at the time of study publication. The group also demonstrated that administration of low dose cyclophosphamide plus fludarabine (Cy/Flu) prior to CAR-T cell infusion was adequate to deplete lymphocytes and increase serum levels of IL-15, which positively correlated with peak CAR-T cell expansion and a higher likelihood of obtaining a CR or PR. The toxicity profile in this study was acceptable, as supportive care alone was sufficient to manage the majority of patients who developed cytokine release syndrome (CRS) or neurological symptoms.
Promising results from initial early phase clinical trials lead to licensing studies followed by FDA approval of two commercial CAR-T cell products, tisagenlecleucel (Kymriah; tisa-cel)21 and axicabtagene ciloleucel (Yescarta; axi-cel)22, with a third product (lisocabtagene maraceucel; liso-cel) likely to request FDA licensing approval in the near future. Comparing the efficacy between different products directly is difficult given key differences in manufacturing and design of the clinical trials that led to licensure; but overall the clinical outcomes are similar amongst the three products with initial ORR reported between 60–80% and CR rates of 40–60% (Table 1).
Table 1:
Multi-center trials of CD19-CAR T cells for lymphoma
| Trial (Name/ID) | DX | N | Construct | LDC | Cell Dose | ORR | CR | DOR | OS | CRS ≥gr3 | CRES ≥gr3 | Notes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KITE; KTE-C19 (axicabtagene ciloleucel) | ||||||||||||
| ZUMA-1 52 Phase 1 |
DLCBL | 7 | CD3z/CD28 | Flu/Cy | 2×106/kg | 71% | 57% | - | - | 14% | 57% | 3 pts w/ ongoing CR at 12+ mo |
| ZUMA-1 23 Phase 2 |
DLBCL, tFL, PMBCL | 101 | CD3z/CD28 | Flu/Cy | 2×106/kg | 82% | 54% | 11.1 mo | 52% @ 18 mo | 13% | 28% | 40% CR @ 1 yr 3 deaths from SAE |
| NOVARTIS; CTL019 (tisagenlecleucel) | ||||||||||||
| JULIET 26 Phase 2 |
DLCBL | 99 | CD3z/4–1BB | Flu/Cy Benda |
0.1 – 6×108 | 53% | 39.5% | NR | 64.5% @ 6 mo | 23% | 12% | 30% CR @ 6 mo |
| JUNO; JCAR017 (lisocabtagene maraceucel) | ||||||||||||
| TRANSCEND 85 Phase 1 |
DLBCL; tFL (CORE cohort) |
49 | CD3z/4–1BB | Flu/Cy | 1×108 | 84% | 61% | 9.2 mo | 88% @ 6 mo | - | - | 52% CR @ 6 mo |
| TRANSCEND 85 Phase 1 |
All DLBCL subtypes (FULL cohort) |
68 | CD3z/4–1BB | Flu/Cy | 0.5 – 1×108 | 74% | 52% | 5.0 mo | 75% @ 6 mo | 1% | 14% | Higher 3 mo ORR/CR seen w/ DL2 |
Benda: bendamustine, CR: complete response, CRES: Cytokine-related Encephalopathy syndrome, CRS: cytokine release syndrome, cy: cyclophosphamide, DL: dose level, DOR: duration of response, Flu: fludarabine, f/u: follow-up, gr3: grade 3, LDC: lymphodepleting chemotherapy, mo: month(s), NR: not reached, ORR: overall response rate, OS: overall survival, PMBCL: primary mediastinal B-cell lymphoma, pts: patients, SAE: severe adverse event, tFL: transformed follicular lymphoma, yr: year.
The parent construct (formerly known as KTE-C19; now designated axicabtagene ciloleucel) tested in the early NIH trials was ultimately taken to phase I/II multi-center studies for commercial development sponsored by Kite Pharma. Results from the phase II portion of this pivotal clinical trial (ZUMA-1) demonstrated an ORR of 82% with 54% CRs.23 The time to response ranged from 0.6 to 6 months with a median time of 1 month. Very few relapses were seen in patients who remained in CR at 3–6 months post-infusion with a median duration of response of 11.1 months. Forty percent of patients remained in CR at 1 year and the overall survival (OS) at 18 months was 52%. Of those patients who did not achieve a CR at 1-month post-treatment, 11 of 35 and 12 of 25 patients with a PR and stable disease, respectively, went on to achieve a CR without additional therapies. Two-year follow-up data published in Lancet Oncology confirmed prior findings with an ORR of 83% and a CR rate of 58% at a median follow-up of 27.1 months.24 The median duration of response remained 11.1 months and median overall survival was not reached.
Tisagenlecleucel (formerly CTL019), a 4–1BB containing CAR, was developed at the University of Pennsylvania and tested in 28 r/r NHL patients in a case-series study. Sixty four percent of patients had a response, with 43% of patients with DLBCL and 71% of patients with FL (10 of 14) achieving a CR. Eight-six percent and 89% of patients with DLBCL and FL, respectively, maintained a sustained response at a median follow-up of 28.6 months.25 In the phase II multicenter registration trial (JULIET) that led to licensing approval, 40% of 99 patients achieved a CR with 30% remaining in CR at 6 months.26 Four patients (3 FL, 1 DLBCL) with a PR at 3 months achieved a CR by 6 months. The median duration of response and OS was not yet reached at the time of publication, and no patients who achieved a response proceeded to stem cell transplant.
Researchers at the Fred Hutchinson Cancer Research center treated 32 patients with NHL with a 1:1 ratio of CD4:CD8 CD19 CAR-T cells using a 4–1BB costimulatory domain. Their study demonstrated an ORR of 63% with a CR rate of 33% across all lymphoma subtypes.27 Comparable to the ZUMA-1 study, patients who received Cy/Flu prior to infusion had better CAR-T cell expansion and persistence, and a higher CR rate of 50% compared to patients who did not receive fludarabine-containing regimens. Due to two deaths at higher cell doses, subsequent patients were treated with lower doses and still demonstrated responses. Similar findings were noted in the phase I multicenter TRANSCEND NHL study testing JCAR017 (now referred to as liso-cel), which included patients with high risk DLBCL infused with a 1:1 ratio of CD4:CD8 CD19 CAR-T cells.28 In an updated analysis presented at the 2018 ASCO Annual Meeting, the 6-month ORR for this study was 49% with a CR rate of 46% for the pivotal core cohort. Of patients who achieved a CR at 3 months, 88% remained in CR at 6 months, while those who achieved only a PR had a median duration of response of 2.1 months. Liso-cel received FDA breakthrough therapy designation for NHL in December 2016, and a licensing application will likely be submitted in upcoming months if results remain positive. While the aforementioned studies were primarily based in the United States and Europe, several early phase studies conducted in China and recently presented in abstract form have also shown encouraging results.29–33
To date, the efficacy of CAR-T cells has been shown to be independent of classic prognostic markers such as cell of origin (ABC versus GBC), International Prognostic Index (IPI) score, prior number if lines of therapy, or by biologic factors such as intensity of CD19 expression. 23,26,34 However, peak CAR-T cell expansion has been significantly associated with response and development of neurologic toxicities, with an area under the curve (AUC) 5 times higher observed in responders versus non-responders for axi-cel.23 Continued long-term follow-up is necessary to determine the curative potential of CAR-T cells, as some patients who initially achieved only partial response (PR) later went on to develop CR as late as 1 year without intervening therapy, suggesting that responses may deepen over time.34 Nevertheless, in patients achieving only PR as the best response, the median duration of response was a dismal 2 months with the axi-cel and liso-cel products.
CAR-T cells for the treatment of T cell lymphomas
Unlike B cell lymphomas, T cell lymphomas are associated with an overall poor prognosis and have limited therapeutic options.35,36 Targeting T cell malignancies with CAR-T cells is more challenging due to shared antigen expression between normal, malignant, and therapeutic CAR-T cells, potentially leading to CAR-T cell fratricide (self-killing) and prolonged T cell aplasia.37–39 Unlike the manageable B cell aplasia caused by CD19 CAR-T cells, T-cell targeted CAR-Ts may cause profound immunodeficiency similar to that seen following allogeneic SCT leading to increased risk of severe infections. Furthermore, it can be challenging to harvest an adequate number of normal autologous T cells without contamination by malignant T cells. CD30 is a cell surface molecule overexpressed in peripheral T cell lymphomas (PTCLS), such as anaplastic large cell lymphoma (ALCL), but has limited expression in normal tissues and is only expressed on a subset of T cells. The CD30 antibody-drug conjugate brentuximab vedotin has proven efficacy for the treatment of CD30-positive PTCLs40–42 and for these reasons was the first antigen explored for targeting T-cell lymphomas with CARs. In a phase 1 study evaluating the use of CD30-directed CAR-T cells for relapsed/refractory EBV-, CD30+ lymphoid malignancies, one patient with ALK+ systemic ALCL achieved a complete response lasting 9 months.43
CD5 and CD7 have both been identified as potential targets with significant anti-tumor activity seen in preclinical models.39,44 There is currently one ongoing phase 1 clinical trial (NCT03081910) investigating the use of CD5 CAR-T cells for treatment of CD5 positive T cell malignancies. The results are eagerly awaited, as there is currently a huge unmet need for the treatment of these diseases for which ASCT is the only potentially curative option for patients who are able to achieve adequate remission to upfront therapies. A phase 1/2 study in China for CD7 positive leukemias and lymphomas utilizing anti-CD7 CAR in NKs cells with TCRζ, CD28, and 4–1BB signaling domains is actively recruiting but no results are yet available (NCT02742727). A phase 1 anti-CD7 CAR-T trial at our center (NCT03690011) aims to circumvent fratricide of infused T cells by knocking out CD7 by CRISPR-Cas in the T cells expressing the CD7.CAR.44 Given the potential for excessive toxicity due to pan-T cell aplasia, the CD5 and CD7 trials are designed as transplant-enabling studies. This strategy should prevent severe complications and provide an opportunity for cure until more data is available on the long-term durability of responses.
An alternative approach to selectively eradicate T cell malignancies is targeting the T cell receptor β-chain constant domains (TRBC1/TRBC2). While normal T cell populations are generally comprised of a mixture of cells expressing either TRCB1 or TRCB2, malignant clonal T cells often exclusively express only one receptor. Maciocia et al. were able to show proof of concept using an anti-TRBC1.CAR that was able to selectively kill both normal and malignant TRBC1 T cells while sparing TRBC2+ T cells, which maintained their normal immune function.45
Toxicities of CAR-T therapy
One of the most pressing challenges limiting widespread use of CAR-T therapy is the management of toxicities. Cytokine release syndrome (CRS), is a common yet potentially severe and life-threatening adverse event following CAR-T cell treatment, due to the release of massive inflammatory cytokines and markers, such as IL-6, INFγ, TNFα, IL-2, IL-10, GM-CSF, C-reactive protein (CRP) and ferritin, upon activation of CAR-T cells. Timing of symptom onset and symptom severity vary based on the product type and cell dose administered, disease burden, use of lymphodepleting chemotherapy, and the magnitude of immune activation. Symptoms can range from mild to severe, and include fever, fatigue, hypotension, capillary leak syndrome, coagulopathy, and multiorgan dysfunction. Mild cases can be treated with intensive supportive care, but severe cases require treatment with immunosuppressive therapy. Severe or life-threatening CRS occurs in up to 25% of patients (Table 1).15 The ZUMA-1 trial for axi-cel reported grade 3 or higher CRS and neurotoxicity events of 13% and 28%, respectively.23 In the JULIET trial, grade 3/4 CRS occurred in 23% of patients and severe neurotoxicity in only 12%.26 Impressively, the TRANSCEND trial reported the lowest rates of grade 3 or 4 CRS and neurotoxicity at 1% and 14%, respectively.
Tocilizumab, an IL-6 receptor antagonist, has been approved for treatment of severe or life-threatening CRS with one or two doses generally being sufficient to induce rapid reversal and improve symptoms within 7 days.27,46–48 Based on current available evidence, tocilizumab does not appear to affect the clinical efficacy of CAR-T cells. 16,49,50 Patients who do not respond to initial treatment with tocilizumab may be treated with corticosteroids, which has a broader effect on the immune system. Since corticosteroids can induce T cell suppression, their use is reserved for management of severe (grades III and higher) adverse events which can be life threatening.
Given the association of CRS with multiple cytokine elevations, it seems reasonable that these could be used as biomarkers of disease severity or response. However, while elevated cytokine levels have been associated with severe CRS, there are no extensively validated biomarkers predictive of severe CRS and cytokine assays are not widely available. Thus, real-time cytokine analysis is not currently used for clinical decision making in the treatment of CRS. To make cytokine assays more accessible to treating physicians, Faramand et al. presented outcomes on 20 patients with DLBCL treated with axi-cel who underwent a “point of care” cytokine assay designed to predict CRS and/or CRES. They were able to determine a correlation between elevated day 1 levels of IL-6 and angiopoietin 2/angiopoietin 1 ratio with severe CRS. Larger confirmatory studies are underway to apply cytokine testing in CAR-T recipients broadly. A potential surrogate marker that is widely available is serial monitoring of CRP and ferritin levels, which can be obtained real-time to help guide therapy.16 The overarching goal of CRS management is to maximize therapeutic benefit, while minimizing risk of life-threatening complications. Modifications to lymphodepletion regimens, CAR-T dosing, and management strategies for toxicities are being actively investigated in ongoing studies in an attempt to mitigate the toxicities of CAR-T cell therapy.
Neurotoxicity, also known as CAR-T cell Related Encephalopathy Syndrome (CRES)47, and now termed immune effector cell (IEC)—associated neurotoxicity syndrome (ICANS)51 is considered a separate entity from CRS although they may be associated, and is also a well-recognized adverse event. Patients may develop symptoms at the time of onset of CRS, following resolution of CRS, or in the absence of CRS, although patients who develop severe CRS also tend to exhibit some degree of neurotoxicity. Symptoms may manifest as encephalopathy, aphasia, ataxia, seizures, obtundation, and rarely with potentially fatal cerebral edema or hemorrhage.52 The majority of patients who develop neurotoxicity will present within the first 2 weeks following T cell infusion, with spontaneous resolution occurring within 7 to 14 days from onset.47 Severe toxicity has been associated with high cell doses and cytokine levels, primarily IL-2, GM-CSF, and ferritin.23 Tocilizumab has been used to treat neurotoxicity although the benefit is not as striking as that seen with its use in CRS — possibly due to the inability of tocilizumab to cross the blood brain barrier.46,53 Gust and colleagues recently identified central nervous system endothelial activation resulting in disruption of the blood brain barrier as the potential underlying mechanism of neurotoxicity associated with CAR-T cell therapy.53 While further studies are needed to better elucidate the mechanism of action and associated risk factors, their report provides potential for discovery of therapeutics specifically targeting endothelial activation as a means to prevent and/or treat ICANS. The ASBMT (now TCT) recently published a consensus grading scale for CRS and ICANS to provide a more accurate and standardized grading system that can be applied broadly across institutions and CAR-T products; this will allow more prompt recognition and treatment of symptoms and permit easier comparisons across trials.51
B cell aplasia is an expected “on target off tumor” toxicity of CD19 CAR-T therapy, as CD19 is a pan-B cell marker found on both normal and malignant B cells. However, most patients treated with CAR-T cells have very low levels of circulating B cells and hypogammaglobulinemia due to the heavy pretreatment with anti-CD20 immunotherapy. Long-term persistence of CD19 CAR-T cells may lead to prolonged severe B cell aplasia and increased infectious complications,54 but this can be managed with intravenous immunoglobulin (IVIG) supplementation. Recovery of polyclonal B cells has occurred in patients who achieved a response without relapse of lymphoma.15,19,20,24,25
Mechanisms of and potential solutions to overcome immune failure
Explicit comparison of the different trial results is impossible due to the wide variability in CAR-T cell constructs, manufacturing process, underlying disease, patient populations, and clinical trial design. Despite the exciting results seen in current studies, much more research is needed to determine specific CAR, disease, and patient-related factors that will provide the best chance for long-term remissions. One CAR construct may not be adequate when considering the complex nature of the diseases being targeted, and the therapy itself. In order to improve the treatment paradigm and make it more broadly available, several areas must be considered. For instance, selection of more appropriate targets (based on tissue distribution, antibody affinity, etc.) may enhance tumor killing and optimized CAR constructs may increase activation and killing while limiting toxicities. For example a recent report where a CD19 CAR was directed to the T-cell receptor alpha constant (TRAC) locus showed that such edited cells had greater activity than regular CD19 CAR transduced T cells in a murine acute lymphoblastic leukemia model.55
Loss of CD19 surface expression is a well described phenomenon of antigen escape in relapses after CD19 CAR-T cell therapy. 49,56–58 Several early phase clinical trials have shown safety and modest efficacy in targeting other antigens in lymphoma such as CD30,43 kappa light chains,59 and CD20.12,60–62 raising the possibility of constructing CARs targeting multiple antigen epitopes or administration of multiple CARs (simultaneously or sequentially) with different targets. For lymphoid malignancies, there are currently several trials underway targeting dual antigens with use of combined CARs (Table 2).
Table 2.
Ongoing non-CD19 or dual targeted CAR-T Trials for NHL in the US
| NCT | Phase | Target | Disease | Location |
|---|---|---|---|---|
| NCT03081910 | 1 | CD5 | R/R lymphoma or leukemia | BCM |
| NCT03105336 | 2 | CD19 | R/R Indolent B-NHL | Multi-center |
| NCT03019055 | 1 | CD19/20 | R/R B-NHL | MCW |
| NCT03233854 | 1 | CD19/22 | R/R B-NHL or ALL | Stanford |
| NCT03448393 | 1 | CD19/22 | R/R B-NHL or ALL | NCI |
| NCT03330691 | 1/2 | CD19/22 | R/R lymphoma | SCH |
| NCT02153580 | 1 | CD19/EGFR | R/R B-NHL | COH |
| NCT03277729 | 1/2 | CD20 | R/R B-NHL | FHCRC |
| NCT03244306 | 1 | CD22/EGFR | R/R lymphoma or leukemia | SCH |
| NCT03049449 | 1 | CD30 | R/R lymphoma | NCI |
| NCT02917083 | 1 | CD30 | R/R lymphoma | BCM |
| NCT02663297 | 1 | CD30 | Lymphoma s/p autoSCT | UNC |
| NCT02690545 | 1/2 | CD30 | R/R lymphoma | UNC |
| NCT03602157 | 1 | CD30/CCR4 | R/R lymphoma | UNC |
ALL: acute lymphoblastic leukemia; BCM: Baylor College of Medicine; B-NHL: B-cell non-Hodgkin lymphoma; COH: City of Hope; FHCRC: Fred Hutchinson Cancer Research Center; MCW: Medical College of Wisconsin; NCI: National Cancer Institute; SCH: Seattle Children’s Hospital; UNC: University of North Carolina.
Like endogenous T cells, CAR-T cells are also susceptible to immune inhibition via checkpoint blockade present in the tumor microenvironment. Upregulation of PDL-1, CTLA-4, and LAG3 expression were observed following treatment with axicabtagene ciloleucel (axi-cel), and, more importantly, greater than one-third of patients who failed to respond or relapsed following treatment with axi-cel had high levels of PD-L1 expression on tumor cells.63 Two other studies have also shown increased PD-1 expression on CD19 CAR-T cells following infusion in lymphoma patients.19,64 Thus, combining CAR-T cells with immune checkpoint inhibitors might enhance their clinical efficacy.
Preclinical testing of CAR-T and checkpoint inhibitor therapy in murine models has demonstrated augmented activity, 65,66 and there have also been reports of clinical responses to PD-1 blockade in a small number of patients refractory to CD19 CAR-T cell therapy67 leading to multiple clinical trials investigating combination strategies (NCT02706405, NCT03310619, and NCT03287817). Results from ZUMA-6 trial testing axi-cel in combination with atezolizumab for refractory DLBCL showed a reasonable safety profile with a 90% ORR in 9 of 10 evaluable patients (60% CR).68 A phase 2 trial is opening soon. Other studies are exploring the use of PD-1 blockade incorporated into CAR-T cells (e.g. anti-PD-1 secreting CAR-T cells; NCT03208556, NCT03298828) based on data from xenograft mouse models showing enhanced antitumor activity and prolonged overall survival. 69
Allogeneic CAR-T cell therapy
While autologous PBMCs have been the primary source for CAR-T manufacturing, allogenic (donor-derived) CD19 CAR-T cell generation and infusion for relapse after ASCT also is feasible and safe. 70–73 All of the studies using donor-derived CAR-T cells demonstrated objective clinical responses without inducing significant graft‐versus‐host disease (GVHD), suggesting that an “off‐the‐shelf” CAR T cell bank could be a feasible solution to reduce the time to treatment and cost (Figure 1). Use of gene-editing technologies such as transcription activator-like effector nuclease (TALEN), CRISPR or zinc-finger nucleases to knock out endogenous T cell receptor function has been shown to be a feasible strategy to create off-the-shelf allogeneic CAR-T cell products 74,75.
Figure 1.
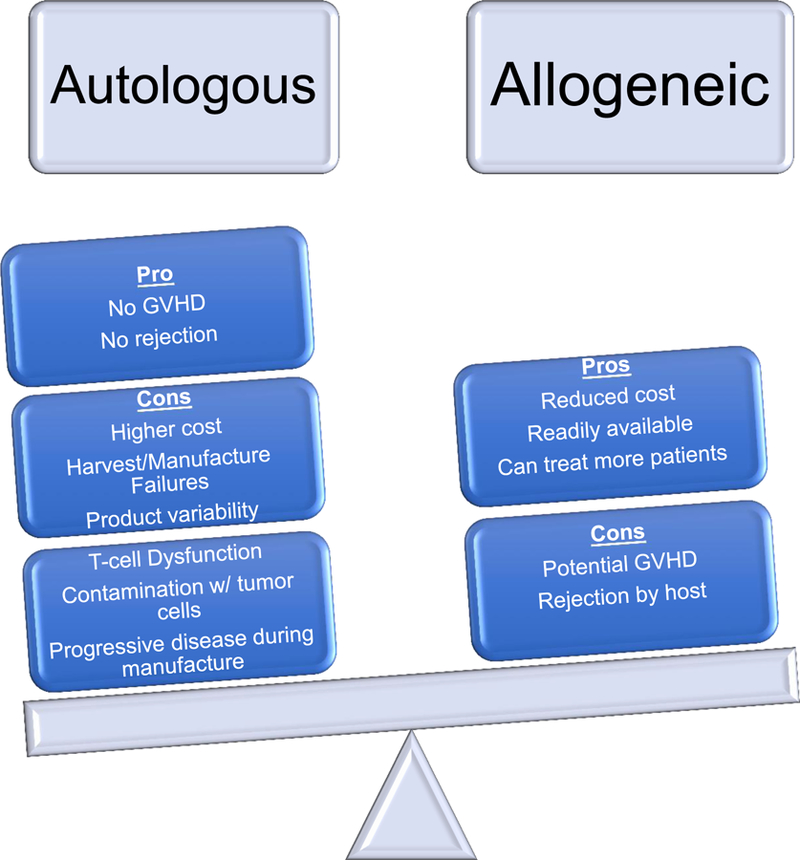
Potential Limitations & Benefits of Autologous versus Allogeneic CAR-T cells
Another approach is to use non-alloreactive allogeneic cells such as virus-specific T cells (VSTs), NK, NK-T, or gamma delta T cells to target tumor-associated proteins.76,77 Allogeneic NK cells offer promising potential as an off-the shelf product as they cause minimal GVHD78,79 and have a relatively short life span, which could presumably limit the on-target off-tumor effects seen with prolonged persistence of CAR-T cells. Despite the safety and early efficacy seen with use of NK-CARs in phase 1 studies 77,80–83, there are some limitations to the use of NK cells. While the short life span maybe beneficial for reducing severe toxicity, poor persistence could negatively impact anti-tumor efficacy. Several studies incorporating cytokine support have shown prolonged NK survival and improved tumor control.77,84 Thus, more research is needed to address unanswered questions and better elucidate the clinical efficacy of NK-CAR cells and the potential role they may play in CAR-T therapy.
Conclusions
CAR-T cells have shown great promise in the treatment of relapsed/refractory B cell malignancies, when few other options are available. However, there continues to be room for improvement in the efficacy and safety profile of CAR-T therapy, which will require collaboration from the immunotherapy/cell therapy fields to make this a potentially front-line therapy. Many clinical trials are currently underway in an effort to build upon the successes seen in prior studies. CAR constructs continue to be optimized, and new data is emerging on how to best combine CAR-T cells with other immunotherapies or targeted therapies to further improve efficacy. Additionally, more studies must be performed to determine the optimal timing of CAR-T cell therapy, as current studies have only treated patients with relapsed or refractory disease. Indeed as a next step, both commercially available CAR-T cell products for DLBCL are being tested in comparison with autologous HSCT in upcoming or ongoing phase III clinical trials (BELINDA and ZUMA-7).
Given that very few patients in the ZUMA-1 or JULIET trials proceeded to SCT following CAR-T induced remissions, the jury is still out regarding the question of consolidation with SCT following CAR-T cell induced remissions as the long-term follow-up is still ongoing.
If indeed we are able to achieve the ultimate goal of maximizing “on-target” response while limiting severe “off-tumor” toxicity, CAR-T cells could be paradigm changing and potentially eliminate the need for more toxic therapies such as ASCT, as well as have a significant impact on cost of care. Lastly, the incorporation of genetic, molecular, and response biomarkers may also help better guide future clinical trials and treatment decisions to improve outcomes.
Acknowledgments
This work was supported by the National Institutes of Health National Cancer Institute (grant 3P50CA126752 – LH and HEH), Leukemia and Lymphoma Society Specialized Center of Research award (PL and HEH), American Society of Hematology Scholar Award (PL) and the American Society for Blood and Marrow Transplantation young investigator award (PL).
Footnotes
Conflict of Interest Statement
HEH is a co-founder of ViraCyte and Marker Therapeutics, has received research support from Cell Medica and Tessa Therapeutics and served on advisory boards for Cytosen, Novartis and Gilead Sciences.
Ethics Statement
The authors confirm that the ethical policies of the journal, as noted on the journal’s author guidelines page, have been adhered to. No ethical approval was required as this is a review article with no original research data.
References:
- 1.Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood 2015;125(1):22–32. [DOI] [PubMed] [Google Scholar]
- 2.Hitz F, Connors JM, Gascoyne RD, et al. Outcome of patients with primary refractory diffuse large B cell lymphoma after R-CHOP treatment. Ann Hematol 2015;94(11):1839–1843. [DOI] [PubMed] [Google Scholar]
- 3.Feugier P, Van Hoof A, Sebban C, et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d’Etude des Lymphomes de l’Adulte. J Clin Oncol 2005;23(18):4117–4126. [DOI] [PubMed] [Google Scholar]
- 4.Camus V, Tilly H. Managing early failures with R-CHOP in patients with diffuse large B-cell lymphoma. Expert Rev Hematol 2017;10(12):1047–1055. [DOI] [PubMed] [Google Scholar]
- 5.Nagle SJ, Woo K, Schuster SJ, et al. Outcomes of patients with relapsed/refractory diffuse large B-cell lymphoma with progression of lymphoma after autologous stem cell transplantation in the rituximab era. Am J Hematol 2013;88(10):890–894. [DOI] [PubMed] [Google Scholar]
- 6.Seshadri T, Stakiw J, Pintilie M, Keating A, Crump M, Kuruvilla J. Utility of subsequent conventional dose chemotherapy in relapsed/refractory transplant-eligible patients with diffuse large B-cell lymphoma failing platinum-based salvage chemotherapy. Hematology 2008;13(5):261–266. [DOI] [PubMed] [Google Scholar]
- 7.Telio D, Fernandes K, Ma C, et al. Salvage chemotherapy and autologous stem cell transplant in primary refractory diffuse large B-cell lymphoma: outcomes and prognostic factors. Leuk Lymphoma 2012;53(5):836–841. [DOI] [PubMed] [Google Scholar]
- 8.Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood 2017;130(16):1800–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature 2017;545(7655):423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer discovery 2013;3(4):388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2010;16(9):1245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Till BG, Jensen MC, Wang J, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 2008;112(6):2261–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest 2011;121(5):1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milone MC, Fish JD, Carpenito C, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009;17(8):1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kochenderfer JN, Dudley ME, Feldman SA, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012;119(12):2709–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Science translational medicine 2014;6(224):224ra225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov 2015;14(7):499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kochenderfer JN, Wilson WH, Janik JE, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010;116(20):4099–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kochenderfer JN, Dudley ME, Kassim SH, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 2015;33(6):540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kochenderfer JN, Somerville RPT, Lu T, et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J Clin Oncol 2017;35(16):1803–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yescarta [package insert] In. Santa Monica, CA: Kite Pharma; 2017. [Google Scholar]
- 22.Kymriah [package insert] In. East Hanover, NJ: Novartis Pharmaceuticals; 2018. [Google Scholar]
- 23.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med 2017;377(26):2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Locke FL, Ghobadi A, Jacobson CA, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. The Lancet Oncology 2019;20(1):31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuster SJ, Svoboda J, Chong EA, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med 2017;377(26):2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schuster SJ, Bishop MR, Tam CS. Primary Analysis of Juliet: A Global, Pivotal, Phase 2 Trial of CTL019 in Adult Patients with Relapsed or Refractory Diffuse Large B-Cell Lymphoma. Blood 2017;130(Suppl 1):577. [Google Scholar]
- 27.Turtle CJ, Hanafi LA, Berger C, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Science translational medicine 2016;8(355):355ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abramson JS, Gordon LI, Palomba M, et al. Updated safety and long term clinical outcomes in TRANSCEND NHL 001, pivotal trial of lisocabtagene maraleucel (JCAR017) in R/R aggressive NHL. J Clin Oncol 2018;36(suppl; abstr 7505). [Google Scholar]
- 29.Huang L, Wang N, Cao Y, et al. CAR22/19 Cocktail Therapy for Patients with Refractory/Relpased B-cell Malignancies. Blood 2018;132 (Suppl 1):1408. [Google Scholar]
- 30.Chang L, Li Y, Tu S, et al. Phase I//II Trial of Multi-Target Chimeric Antigen Receptor Modeified T Cells (4SCAR2.0) Against Relapsed or Refractory Lymphomas. Blood 2018;132 (Suppl 1):225. [Google Scholar]
- 31.Ying Z, Wang W, Wang X, et al. CD19 CAR T Cell Product Exhibits High Remission Rate in Adult Relapsed and/or Refracrtory B-Cell Non-Hodgkin Lymphoma. Blood 2018;132 (Suppl). [Google Scholar]
- 32.Ying Z, Long L, Liu H, et al. ET190L1-Artemis T Cell Therapy Results in Durable Disease Remissions with No Cytokine Release Syndrome or Nuerotoxicity in Patients with Relapsed and Refrectory B-Cell Lymphoma. Blood 2018;132 (Suppl 1) [Google Scholar]
- 33.Wang D, Xiao Y, Li C, et al. Anti-CD30 Chimeric Antigen Receptor T Cell Therapy for CD30+ Relapsed/Refractory Hodgkin Lymphoma and Anaplastic Large Cell Lymphoma Patients. Blood 2018;132 (Suppl 1). [Google Scholar]
- 34.Locke FL, Ghobadi A, Jacobson CA, et al. Durability of response in ZUMA-1, the pivotal phase 2 study of axicabtagene ciloleucel (Axi-cel) in patients (Pts) with refractory large B-cell lymphoma. J Clin Oncol 2018;36(suppl; abstr 3003). [Google Scholar]
- 35.Swerdlow SH, World Health Organization, International Agency for Research on Cancer. WHO classification of tumours of haematopoietic and lymphoid tissues Revised 4th edition. ed. Lyon: International Agency for Research on Cancer; 2017. [Google Scholar]
- 36.Foss FM, Zinzani PL, Vose JM, Gascoyne RD, Rosen ST, Tobinai K. Peripheral T-cell lymphoma. Blood 2011;117(25):6756–6767. [DOI] [PubMed] [Google Scholar]
- 37.Gomes-Silva D, Atilla E, Atilla PA, et al. CD7 CAR T Cells for the Therapy of Acute Myeloid Leukemia. Mol Ther 2019;27(1):272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mamonkin M, Mukherjee M, Srinivasan M, et al. Reversible Transgene Expression Reduces Fratricide and Permits 4–1BB Costimulation of CAR T Cells Directed to T-cell Malignancies. Cancer Immunol Res 2018;6(1):47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood 2015;126(8):983–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pro B, Advani R, Brice P, et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: results of a phase II study. J Clin Oncol 2012;30(18):2190–2196. [DOI] [PubMed] [Google Scholar]
- 41.Horwitz SM, Advani RH, Bartlett NL, et al. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood 2014;123(20):3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fanale MA, Horwitz SM, Forero-Torres A, et al. Brentuximab vedotin in the front-line treatment of patients with CD30+ peripheral T-cell lymphomas: results of a phase I study. J Clin Oncol 2014;32(28):3137–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramos CA, Ballard B, Zhang H, et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J Clin Invest 2017;127(9):3462–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gomes-Silva D, Srinivasan M, Sharma S, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 2017;130(3):285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maciocia PM, Wawrzyniecka PA, Philip B, et al. Targeting the T cell receptor beta-chain constant region for immunotherapy of T cell malignancies. Nat Med 2017. [DOI] [PubMed]
- 46.Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014;124(2):188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol 2018;15(1):47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Le RQ, Li L, Yuan W, et al. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist 2018;23(8):943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371(16):1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Science translational medicine 2015;7(303):303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee DW, Santomasso BD, Locke FL, et al. ASBMT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 2018.
- 52.Locke FL, Neelapu SS, Bartlett NL, et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol Ther 2017;25(1):285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gust J, Hay KA, Hanafi LA, et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer discovery 2017;7(12):1404–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hill JA, Li D, Hay KA, et al. Infectious complications of CD19-targeted chimeric antigen receptor-modified T-cell immunotherapy. Blood 2018;131(1):121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017;543(7643):113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sotillo E, Barrett DM, Black KL, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer discovery 2015;5(12):1282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 2017;129(25):3322–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shalabi H, Kraft IL, Wang HW, et al. Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematologica 2018;103(5):e215–e218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramos CA, Savoldo B, Torrano V, et al. Clinical responses with T lymphocytes targeting malignancy-associated kappa light chains. J Clin Invest 2016;126(7):2588–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang Y, Zhang WY, Han QW, et al. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin Immunol 2014;155(2):160–175. [DOI] [PubMed] [Google Scholar]
- 61.Zhang WY, Wang Y, Guo YL, et al. Treatment of CD20-directed Chimeric Antigen Receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an early phase IIa trial report. Signal Transduct Target Ther 2016;1:16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Till BG, Jensen MC, Wang J, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4–1BB domains: pilot clinical trial results. Blood 2012;119(17):3940–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galon J, Rossi J, Turcan S, et al. Characterization of anti-CD19 chimeric antigen receptor (CAR) T cell-mediated tumor microenvironment immune gene profile in a multicenter trial (ZUMA-1) with axicabtagene ciloleucel (axi-cel, KTE-C19). Journal of Clinical Oncology 2017;35(15_suppl):3025. [Google Scholar]
- 64.Brudno JN, Somerville RP, Shi V, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J Clin Oncol 2016;34(10):1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.John LB, Kershaw MH, Darcy PK. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology 2013;2(10):e26286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cherkassky L, Morello A, Villena-Vargas J, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest 2016;126(8):3130–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chong EA, Melenhorst JJ, Lacey SF, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 2017;129(8):1039–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jacobson CA, Locke FL, Miklos DB, et al. End of Phase 1 Results from Zuma-6: Axicabtagene Ciloleucel (Axi-Cel) in Combination with Atezolizumab for thr Treatment of Patients with Refractory Diffuse Large B Cell Lymphoma. Blood 2018;132 (Suppl 1). [Google Scholar]
- 69.Li S, Siriwon N, Zhang X, et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin Cancer Res 2017;23(22):6982–6992. [DOI] [PubMed] [Google Scholar]
- 70.Cruz CR, Micklethwaite KP, Savoldo B, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood 2013;122(17):2965–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kochenderfer JN, Dudley ME, Carpenter RO, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 2013;122(25):4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kebriaei P, Singh H, Huls MH, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest 2016;126(9):3363–3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith M, Zakrzewski J, James S, Sadelain M. Posttransplant chimeric antigen receptor therapy. Blood 2018;131(10):1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Poirot L, Philip B, Schiffer-Mannioui C, et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res 2015;75(18):3853–3864. [DOI] [PubMed] [Google Scholar]
- 75.Qasim W, Zhan H, Samarasinghe S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science translational medicine 2017;9(374). [DOI] [PubMed] [Google Scholar]
- 76.Tzannou I, Papadopoulou A, Naik S, et al. Off-the-Shelf Virus-Specific T Cells to Treat BK Virus, Human Herpesvirus 6, Cytomegalovirus, Epstein-Barr Virus, and Adenovirus Infections After Allogeneic Hematopoietic Stem-Cell Transplantation. J Clin Oncol 2017;35(31):3547–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu E, Tong Y, Dotti G, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018;32(2):520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010;115(21):4293–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Curti A, Ruggeri L, D’Addio A, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood 2011;118(12):3273–3279. [DOI] [PubMed] [Google Scholar]
- 80.Tang X, Yang L, Li Z, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res 2018;8(6):1083–1089. [PMC free article] [PubMed] [Google Scholar]
- 81.Tonn T, Schwabe D, Klingemann HG, et al. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013;15(12):1563–1570. [DOI] [PubMed] [Google Scholar]
- 82.Oelsner S, Friede ME, Zhang C, et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017;19(2):235–249. [DOI] [PubMed] [Google Scholar]
- 83.Boyiadzis M, Agha M, Redner RL, et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy 2017;19(10):1225–1232. [DOI] [PubMed] [Google Scholar]
- 84.Fujisaki H, Kakuda H, Shimasaki N, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res 2009;69(9):4010–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Abramson JS, Palomba ML, Gordan L, et al. High Durable CR Rates in Relapsed/Refractory (R/R) Aggressive B-NHL Treated with the CD19-Directed CAR T Cell Product JCAR017 (TRANSCEND NHL 001): Defined Composition Allows for Dose-Finding and Definition of Pivotal Cohort. Abstract presented at Blood; 2017.