

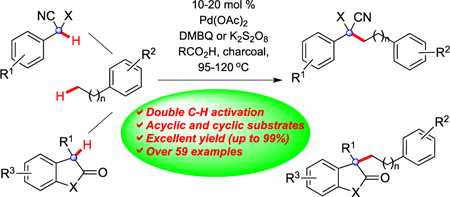
Abstract
The oxidative activation of alkyl C–H bonds vs arene C–H bonds with Pd(OAc)2 has been found to be generalizable to a number of nucleophilic substrates allowing the formation of a range of hindered quaternary centers. The substrates share a common mechanistic path wherein Pd(II) initiates an oxidative dimerization. The resultant dimer modifies the palladium catalyst to favor activation of alkyl C–H bonds in contrast to the trends typically observed via a concerted metalation deprotonation mechanism. Notably, insertion occurs at the terminus of the alkyl arene for hindered substrates. Two different oxidant systems were discovered that turn over the process. Parameters have been identified that predict, which substrates are productive in this reaction.
Keywords: cross dehydrogenative coupling, oxidative coupling, palladium, chemoselective activation, quaternary
Graphical Abstract
Introduction
The use of aromatic hydrocarbons as precursors to the construction of more complex structures has many intrinsic advantages. Aromatic hydrocarbons are inexpensive building blocks readily available from petroleum. Their direct functionalization to desired targets avoids the introduction of functional groups such as halides that require the use of hazardous materials or generate undesired byproducts. The aromatic hydrocarbons are also more stable than the corresponding functionalized equivalents such as benzylic halides. For example, naphthylic halide analogs are not stable and are not readily amenable to SN2 displacement.
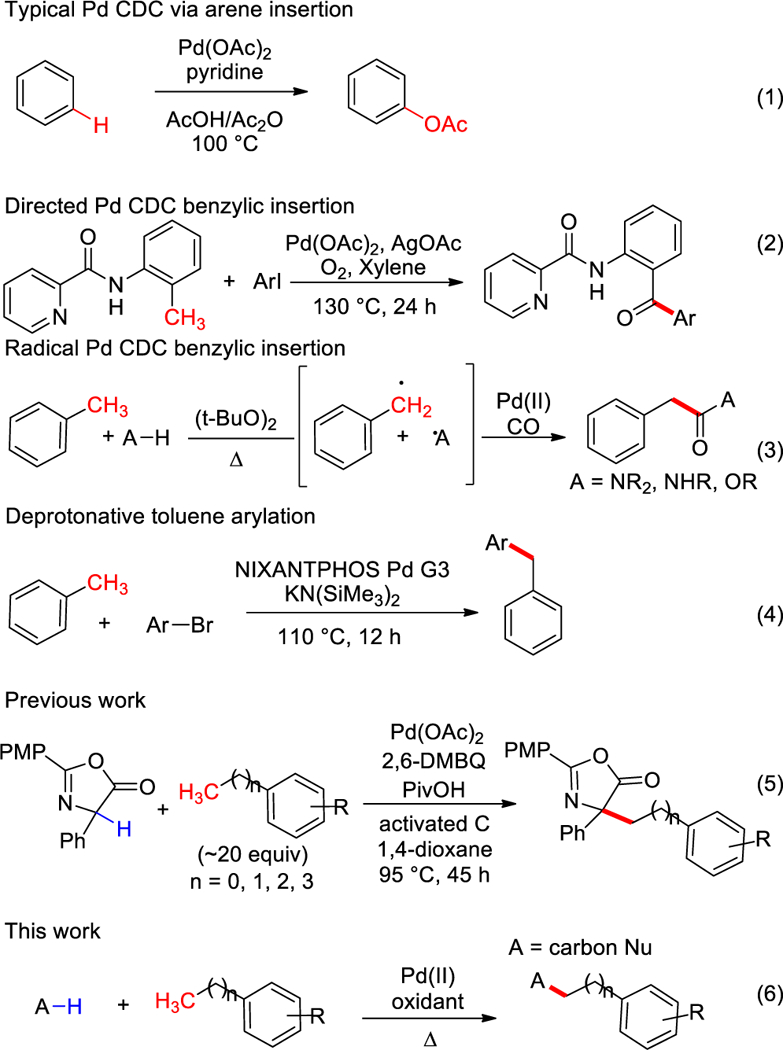
For these reasons, much research has focused on the effective use of aromatic hydrocarbons as substrates and metal catalyzed C–H activation chemistry has played a key role.1 The use of palladium, in particular, has proven highly effective especially in the activation of sp2 C–H bonds of arenes (e.g. Scheme 1, eq 1),2 even allowing C–H activation of two reaction partners where the only byproduct is dihydrogen.3 On the other hand, the activation of the sp3 C–H bonds of aromatic hydrocarbons by palladium occurs much less readily to the point where toluene derivatives can undergo selective sp2 C–H functionalization in the presence of one or more benzylic centers.4 Strategies to achieve benzylic activation have relied on directing groups (Scheme 1, eq 2),5 generation of benzylic radicals (Scheme 1, eq 3)6, or the lower acidity of benzylic sites (Scheme 1, eq 4).7
Scheme 1.
Pd-Catalyzed Activation of Arenes and Toluenes
Recently, we have reported an alternative strategy to activation of aromatic hydrocarbons wherein dehydrogenative coupling forms a hindered bond (Scheme 1, eq 5).8 One unique feature of this process is the catalytic activation of benzylic C–H bonds under oxidizing conditions with palladium that typically result in sp2 C–H activation. Another unusual feature is that coupling occurs at the terminus of the alkyl arene, presumably via a migration of the palladium center. In this article, we disclose our discovery of a range of acyclic and cyclic substrates that can participate in this process to generate hindered bonds selectively (eq 6). From the studies undertaken, guidelines are now available to predict which carbon nucleophiles will be effective.
Results and Discussion
Our analysis of the reaction from eq 5 revealed the intermediacy of a dimer from the azlactone, which readily forms under a range of oxidative conditions.9 Notably, others have found reactions that also proceed through dimers similar to those that we reported.10 Reasoning that other substrates which form similar dimeric intermediates might be subject to a similar reaction profile (eq 7), the literature was surveyed for compounds that readily undergo oxidative dimerization. A surprisingly large number of compounds with similar behavior was identified including malonates,11 malononitrile,12 cyanoacetate,13 oxindoles,14 isooxindoles,15 benzofurans,16 fluorenes,17 and hydroxycoumarins.18
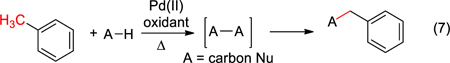
Acyclic Substrates: Malononitriles.
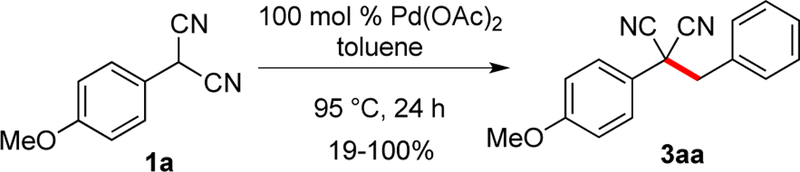
A primary concern was whether the reaction was restricted to cyclic nucleophiles. Thus, the initial priority was to explore acyclic substrates capable of oxidative dimerization. Malononitriles are also well-known to undergo oxidative dimerization.12 To probe the fundamental reactivity, trials were first conducted with stoichiometric Pd(OAc)2 using dinitrile 1a along with toluene (Scheme 2).
Scheme 2.
Variable Yields in Dinitrile Toluene Coupling
Good reactivity was observed, but the outcome was strongly affected by the batch of palladium used. Older batches of palladium were highly effective in the stoichiometric conversion of dinitrile 1a to 3aa with toluene (Scheme 2), while new batches were not. Different sources of Pd(OAc)2 are known to give much different results due to different composition arising from the preparation, such as the red-brown [Pd3(OAc)5NO2]. [Pd3(OAc)6] is a dark purple solid whereas [Pd(OAc)2]n polymer is a light grey-purple powder.19
Proceeding with older batches of Pd(OAc)2, the para-methoxyphenyl and para-methylphenyl malononitriles were found to be effective partners in the coupling of different toluenes (Scheme 3). Further, the same stoichiometric oxidant discovered for the azlactones, dimethylbenzoquinone (DMBQ), was found to be effective in turning over the process with catalytic amounts of Pd(OAc)2. In most cases, similar yields were obtained for the catalytic vs stoichiometric palladium conditions. Amongst the xylene isomers, para-xylene (27–36%) was less effective than meta-xylene (59–65%) or ortho-xylene (32–50%). In all cases, reactions were conducted until all starting material was consumed and the mass balance is decomposition with no specific byproducts being isolable. Notably, higher yields were obtained under catalytic conditions for para-methoxytoluene. We speculate that stochiometric Pd(OAc)2 can cause direct oxidation of the para-methoxytoluene, which was mitigated with lower amounts of Pd(OAc)2 in the presence of the mild oxidant DMBQ.
Scheme 3.
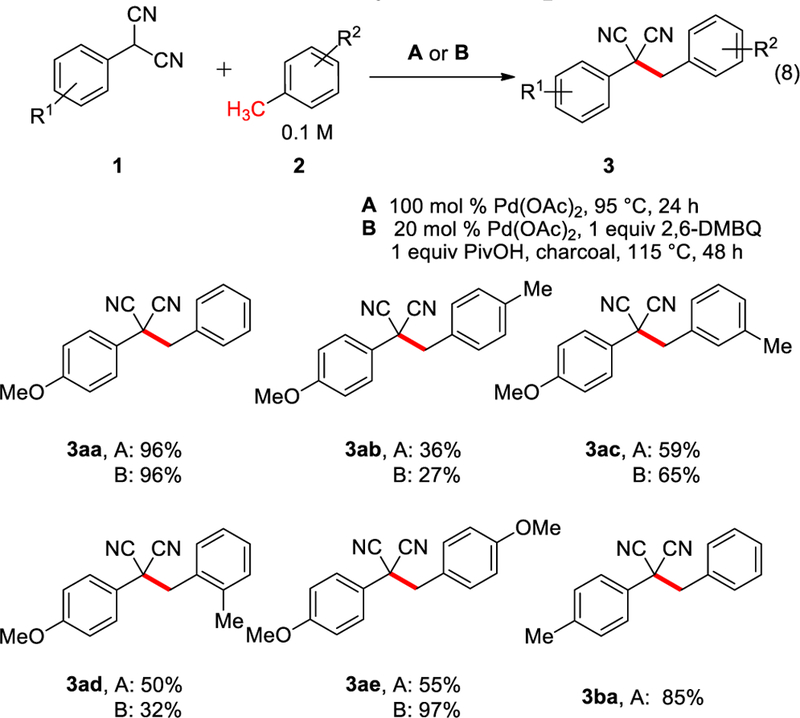
Catalytic Palladium Oxidative Coupling of Malononitriles with Methylarenes (eq 8)
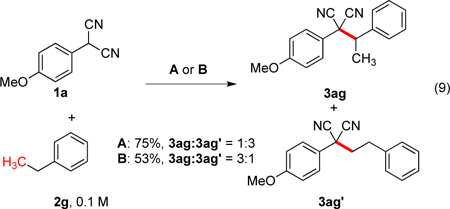
For ethylbenzene, a divergence in behavior was seen relative to our prior report with azlactone (eq 5)8 where exclusive functionalization of the terminal CH3 position occurred. With the malononitrile, a mixture of the terminal (methyl) vs internal (benzylic) products was observed (eq 9). The terminal product is proposed to arise from initial benzylic insertion followed by β-hydride elimination and migratory insertion.20 We hypothesize that the driving force for rearrangement to the terminal position is largely controlled by formation of the less hindered alkyl palladium species. However, these results indicate that the nucleophile also plays a role, where sterically small nucleophiles can react at either the α- or β-positions. Under stochiometric conditions, there is a preference for the latter 3ag’, whereas the former 3ag predominates under catalytic conditions. Under catalytic conditions, the higher effective concentration of the nucleophile relative to the palladium species may cause greater capture of the benzylic intermediate prior to rearrangement.
Acyclic Substrates: α-Cyanoacetates.
α-Cyanoacetates are also well known to undergo such dimerizations, although somewhat more slowly.11 To probe the fundamental reactivity, trials were first conducted with stoichiometric Pd(OAc)2 using cyanoacetate 4a along with 20 equivalents of alkylarene in dioxane (eq 10, Scheme 4). At 95 °C, the reaction with toluene proceeded well (81% yield). Again, para-xylene (38%) was less effective than meta-xylene (69%) or ortho-xylene (61%). Substrates with electron-donating groups (para-methoxytoluene) proceeded with moderate conversion (42%), but those with electron-withdrawing groups, such as para-trifluorotoluene or para-cyanotoluene gave no product. 2-Methylnaphthalene proceeded well (67%), whereas the more hindered 1-methylnaphthalene did not work (<10%). In contrast to the malononitriles, ethylbenzene gave the product arising exclusively from functionalization of the terminal position of the alkyl (5ag)
Scheme 4.
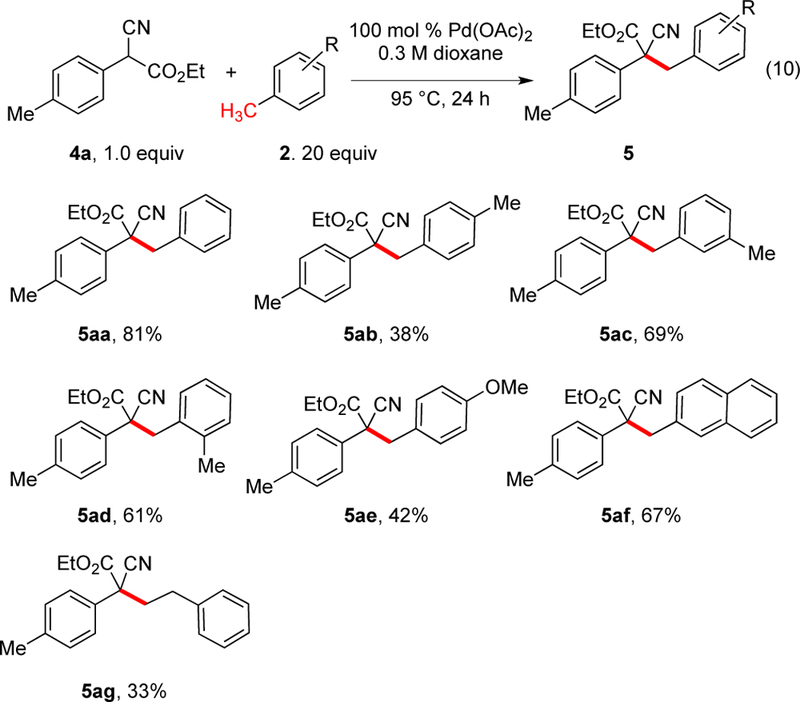
Stoichiometric Palladium Oxidative Coupling of Cyanoacetates with Alkylarenes (eq 10)
Initial trials using the same DMBQ oxidant as described above for the malononitriles led to poor yields. On the whole, the α-cyanoacetates were less reactive than malononitriles. As such, high throughput screening was undertaken to identify an oxidant that would permit turnover of the process. With varying amounts of Pd(OAc)2, several oxidants were examined (Scheme 5) and K2S2O8 was found to be superior. Having observed that carboxylic additives improved the yields with the azlactone substrate,8 presumably by ligating the palladium to prevent precipitation during the reduction, both acetic acid and pivalic acid additives were examined. Further, several solvents (DMF, NMP, 1,4-dioxane, 2-butanol, diglyme, i-PrOAc) were screened to minimize the amount of alkylarene employed (see SI). Overall acetic acid in dioxane performed best. Further examination of temperature (95, 110, 120 °C) revealed that higher temperatures (120 °C) were well tolerated causing little decomposition and higher yields (see SI).
Scheme 5.
Optimization of Oxidant for Cyanoacetates with Alkylarenes.a,b
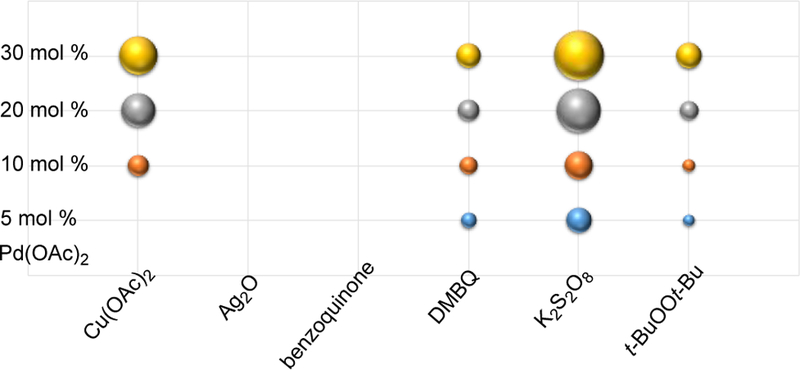
aSubstrate 4a, Pd(OAc)2, 0.1 M toluene, 95 °C, 15 h. bSphere size correlates with conversion vs an internal standard (see SI for quantitative values).
Applying the optimized conditions (eq 11) to a range of toluene substrates (top of Scheme 6) provided results as good as for the stoichiometric transformation with the exception of para-methoxytoluene, which likely undergoes an oxidation side reaction with the K2S2O8. Yet again, para-xylene (48%) was less effective than meta-xylene (68%) or ortho-xylene (62%). Further, the catalytic conditions performed well for several different cyanoacetates (bottom of Scheme 6) including those bearing electron-withdrawing groups (5ba) or electron-donating groups (5da).
Scheme 6.

Catalytic Palladium Oxidative Coupling of Cyanoacetates with Alkylarenes (eq 11)
Cyclic Substrates: Oxindoles.
Moving away from acyclic substrates, oxindoles are another class of compounds that readily dimerize.14 In addition, oxindoles bearing two substituents at the 3-position are found in natural products and pharmaceutical targets (Scheme 7).21 Notably, 3-aryl-3-benzyl motif that maps onto this method has been utilized by Hoffmann-LaRoche.21c Functionalization of oxindoles has been the focus of much work, including oxidative methods.22
Scheme 7.
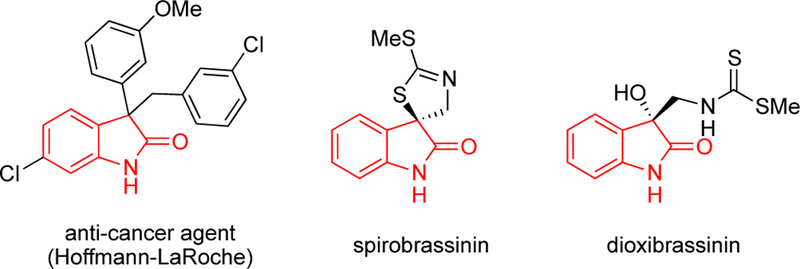
Bioactive Oxindole Structures
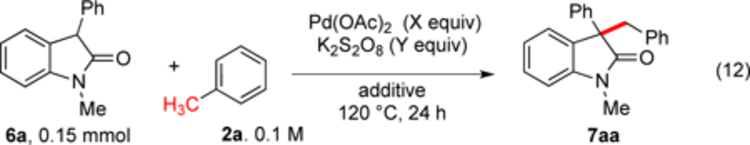
Very good initial results were obtained in the coupling of 3-phenyloxindole (6a) with toluene (eq 13) with stoichiometric Pd(OAc)2 (see SI). A survey of oxidants revealed K2S2O8 as highly effective while minimizing byproduct formation (see SI). In this case, using dioxane as a solvent was not as effective (see SI), so reactions were conducted in 0.1 M alkylarene. Further optimization (Table 1) revealed that the palladium catalyst loading could be lower when a pivalic acid additive and activated charcoal were employed (entry 6).
Table 1.
Optimization of 3-Phenyloxindole Toluene Coupling (eq 12)
 | ||||
|---|---|---|---|---|
| entry | X | Y | additive (equiv) | yield (%)b |
| 1 | 0.3 | 2 | 98 (97)c | |
| 2 | 0.3 | 1 | 76 | |
| 3 | 0.2 | 2 | 64 | |
| 4 | 0.2 | 2 | PivOH (1) | 72 |
| 5 | 0.3 | 2 | AcOH (1) | 80 |
| 6 | 0.2 | 2 | PivOH (1) | 96d |
| 7 | 0.3 | 2 | AcOH (1) | 77d |
| 8 | 0.2 | 2 | K2CO3 (1) | 51 |
| 9 | 0.2 | 2 | NaOAc (1) | 44 |
Reaction conditions: 6a (0.15 mmol), 2a (0.1 M), Pd(OAc)2 (X mol %), K2S2O8 (Y equiv) at 120 oC for 24 h under argon.
Isolated yield.
Run twice
Activated charcoal (10x weight of Pd) was added.
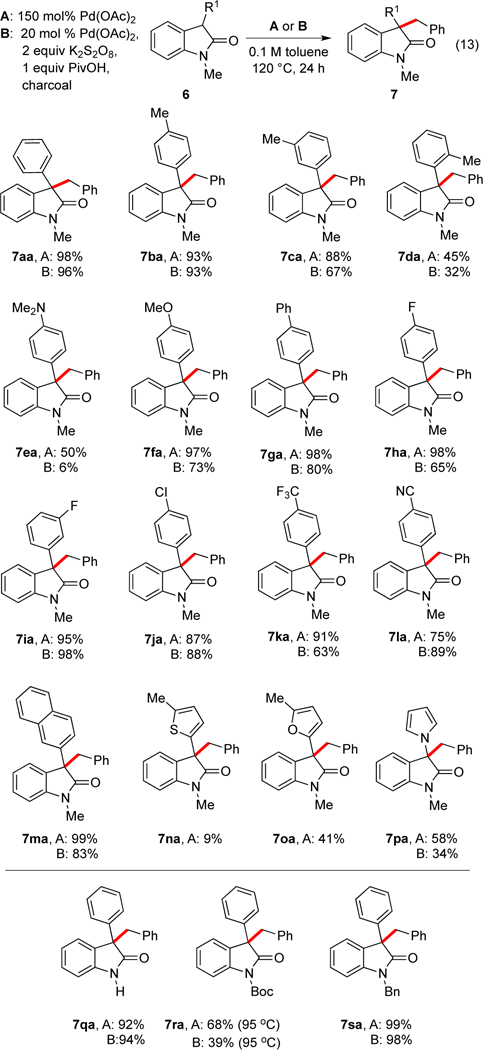
Additional 3-substituted oxindoles were synthesized following literature protocols23 and were examined in the coupling with toluene (eq 13, Scheme 8). Different 3-aryl groups with both electron-donating and electron-withdrawing substituents were well tolerated leading to high yields under both stoichiometric and catalytic conditions (7aa-7ma). In comparison generation of the oxindole cation initiates a Friedel-Crafts reaction with arenes; such a pathway cannot incorporate toluene at the benzylic carbon. Notably, this approach to 7ba, 7fa and similar structures is orthogonal to the oxidative coupling method reported by Li and workers24 wherein selective coupling of the oxindole occurs via a Friedel-Crafts reaction resulting in functionalization of the arene C–H of toluene rather than the benzylic C–H. The 2-naphthyl substrate formed product (7ma) in high yield whereas the hindered 1-naphthyl substrate reacted poorly.
Scheme 8.
Palladium Oxidative Coupling of Oxindoles with Toluene (eq 13)
Heterocyclic groups could also be employed at the 3-position including a case with a nitrogen substituent (7na-7pa). The oxindole could also be employed directly without protection of the nitrogen (7qa) or with other protecting groups (7ra, 7sa); however, the Boc group was somewhat unstable at the high reaction temperatures.
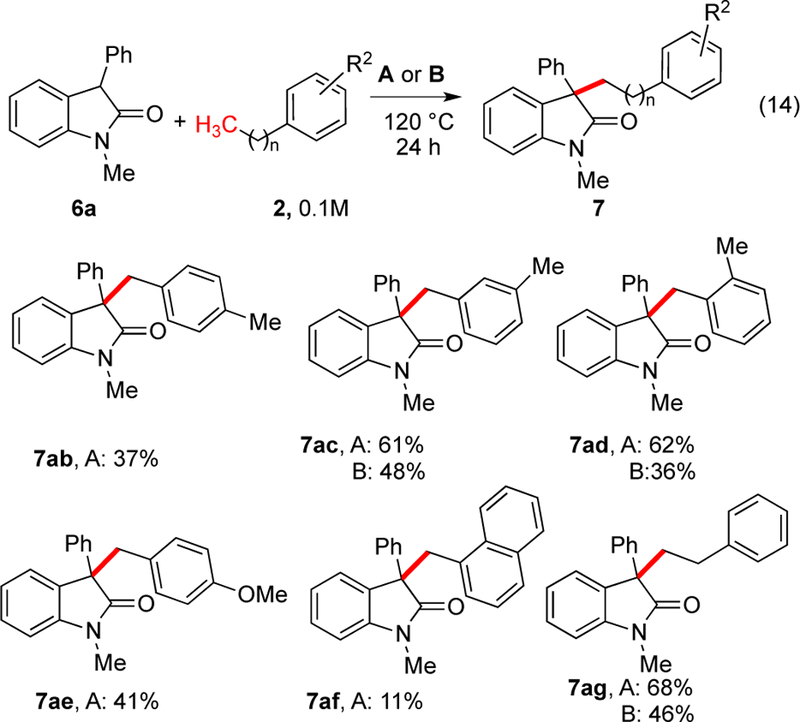
Similar trends were observed again with different arylalkanes with improved outcomes for ethylbenzene (eq 14, Scheme 9). Varying toluene, poor reactivity was again observed for substrates with electron-withdrawing groups such as para-methylbenzonitrile and para-chlorotoluene. The reaction was also unsuccessful for 2-methylfuran. To probe the β-hydride elimination and rearrangement, isobutylbenzene was examined. As expected, the addition of steric hindrance abrogated reactivity.
Scheme 9.
Palladium Oxidative Coupling of Parent Oxindole with Alkylarenes (eq 14)
Cyclic Substrates: Benzofuranones.
Benzofuranones are isoelectronic with oxindoles and also exhibit oxidative dimerization behavior.16 They are found in a number of natural products and pharmaceutical targets (Scheme 10).25
Scheme 10.
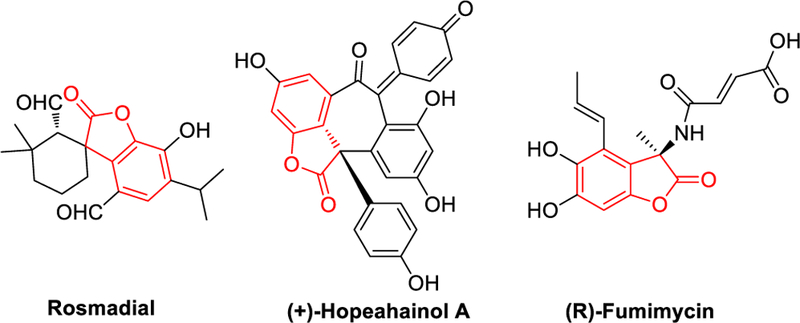
Bioactive Benzofuranone Structures
Using the same conditions developed for the oxindoles, toluene coupled very well with the parent 3-phenylbenzofuranone to generate 9aa under stoichiometric or catalytic conditions in 94% and 91% yield, respectively. Substrates varying the core ring system substituents (top Scheme 11) or the 3-aryl group (middle Scheme 11) were generated26 and were found to couple with toluene very well. Electron-donating (9fa, 9ja-9la) and electron-withdrawing groups (9ga, 9ha) could be employed. Notably, chloro groups could be employed providing an opportunity for further derivatization (9ha). Lower catalyst loadings could also be employed, but required longer reaction or resulted in lower yields (for 9ba with 5 mol % Pd(OAc)2, 59% and 75% product were obtained at reaction times of 24 h and 48 h, respectively).
Scheme 11.
Palladium Oxdiative Coupling of Benzofuranones with Alkylarenes (eq 15)a
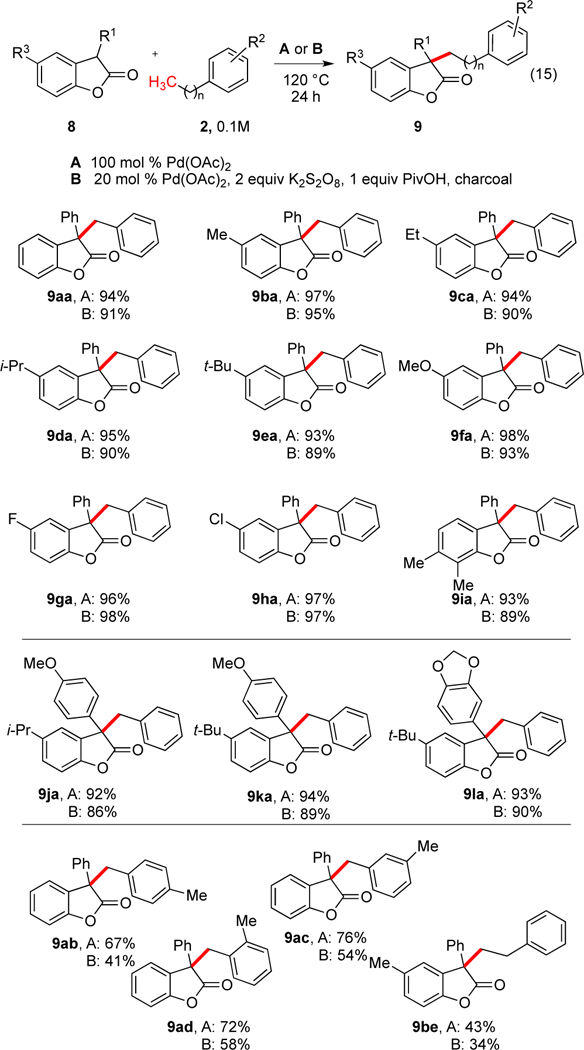
aFor 9ba, using conditions B with 5 mol % Pd(OAc)2, 59% and 75% product were obtained at reaction times of 24 h and 48 h, respectively.
A similar scope was seen with different alkylarenes as had been observed in the cases above (bottom, Scheme 11).
Substrate Properties Correlated with Reactivity.
In our prior report, the dimer of the azlactone (eq 5) was found to form under reaction conditions. When isolated and resubjected to the reaction conditions, this dimer also converted to product. The corresponding dimers were observed in the conversion of substrates 1, 4, 6, and 8 although very little of the dimer built up during the conversion of oxindoles 6 or benzofuranones 8. The dimer (10) of α-cyanoacetate 4a was readily formed with oxidants and was independently synthesized (Scheme 12) with Cu(TMEDA)ClOH. Resubjection of dimer 10 to the reaction conditions with Pd(OAc)2 and toluene again afforded the product 5aa (Scheme 12).
Scheme 12.
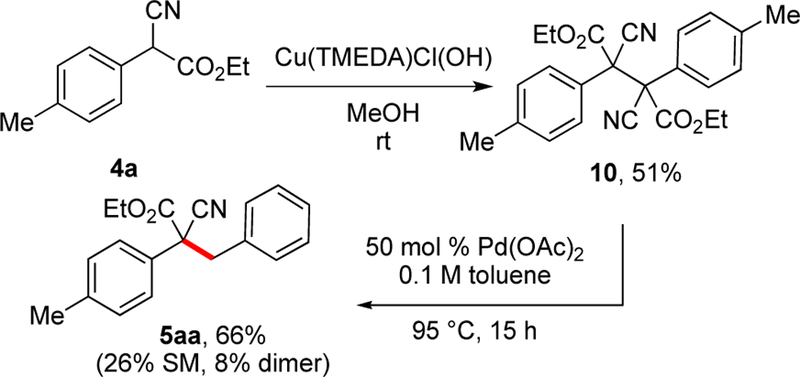
Dimer Intermediate from Cyanoacetate Substrate
Heating the dimer with toluene alone did not form the product. Similarly, subjection of substrate under the catalytic conditions without Pd(OAc)2 did not form the product. For example, when oxindole 6a was treated with 2 equiv K2S2O8, 1 equiv PivOH, charcoal, in 0.1 M toluene for 24 h at 120 °C, the outcome was 73% unreacted 6a, 25% dimer, and 0% product. Thus, it appears that Pd(OAc)2 plays a role in the C–H activation event of the toluene as well as in dimerization of the nucleophilic substrate.
Calculations of the bond dissociation energy (BDE) values for a range of potential nucleophilic substrates was undertaken (Table 2). If the ability to undergo dimerization via nonionic processes is the most important factor in determining reactivity, then the substrate C–H bond dissociation energy should correlate with reactivity in the overall process. A survey of all the substrates in Table 2, revealed that only entries 3–11, possessing C–H bond dissociation energies in the range of 61.0–71.6 kcal/mol, were effective in the transformation. Substrates with similarly weak C–H bonds (entries 1–2, and to a lesser extent, entries 12, 14), were found to be unreactive. All of these compounds have weaker bond dissociation energies than toluene (90 kcal/mol),27 which accounts for why dimer alone (see above) is unable to activate toluene.
Table 2.
Bond Dissociation Energies of A-H and A-A
| Entry | A–H | monomer BDEa (kcal/mol) | dimer BDEa,b (kcal/mol) | |
|---|---|---|---|---|
| 1 |  |
11 | 71.9 | 0.8 |
| 2 | 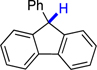 |
12 | 71.0 | 1.4 |
| 3 | 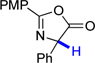 |
13 | 60.8 | 2.4 |
| 4 | 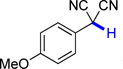 |
1a | 65.1 | 5.3 |
| 5 | 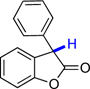 |
8a | 67.9 | 7.4 |
| 6 | 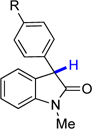 |
6f, R = OMe | 68.1 | 8.7 |
| 7 | 6b, R = Me | 69.0 | 10.0 | |
| 8 | 6a, R = H | 69.5 | 10.5 | |
| 9 | 6l, R = CN | 69.6 | 10.8 | |
| 10 | 6k, R = CF3 | 69.8 | 11.4 | |
| 11 | 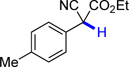 |
4a | 70.7 | 13.0 |
| 12 | 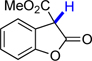 |
14 | 71.7 | 22.6 |
| 13 | 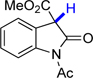 |
15 | 72.1 | 22.7 |
| 14 | 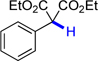 |
16 | 79.1 | 24.1 |
| 15 | 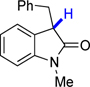 |
6u | 74.7 | 26.3 |
| 16 | 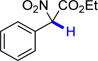 |
17 | 81.9 | 27.9 |
| 17 | 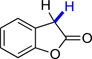 |
8m | 78.2 | 38.6 |
Computed at uB3LYP/6–31G*.
Meso diastereomer.
To further probe the importance of the initial oxidation to the dimer, the dimer of 6u was independently synthesized28 and resubjected to the reaction conditions in a similar manner as outlined in Scheme 8. Similar to monomer 6u, the dimer of 6u did not convert to the product in the presence of Pd(OAc)2 (Scheme 13) indicating that dimerization is necessary, but not sufficient in order for the process to occur.
Scheme 13.
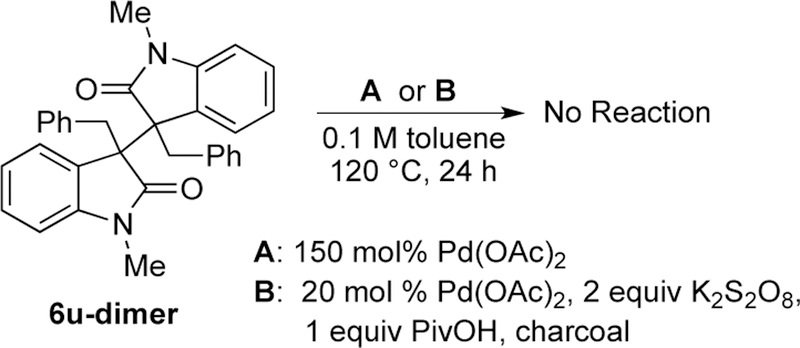
3-Benzyloxindole Dimer with Pd(OAc)2
The dimer BDE values were also computed and correlated more strongly with reactivity (Table 2). Substrates with dimers BDE values either below 2 kcal/mol or above 15 kcal/mol did not form product. That compounds with stronger dimer BDE values did not generate product, even when the dimer was preformed (see Scheme 13) indicates that the oxidizing power of the dimer is not the most important factor. Furthermore, the relative reactivity of the substrates in entries 3–11 followed the order of the dimer BDE values as judged by TLC monitoring.
Competition experiments between substrates were performed to probe the role of BDE values (Scheme 14). Reaction from the monomer and dimer gave different outcomes, consistent with two separate steps contributing to the overall rate: dimerization by palladium and reaction of the dimer with a palladium alkyl. For all pairs, the substrates with the stronger BDE values were less reactive.
Scheme 14.
Competition Experiments
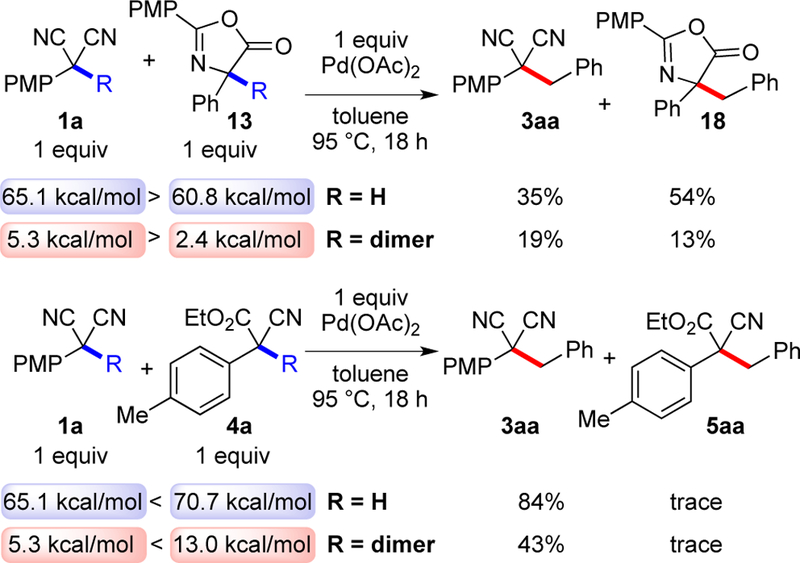
aYields from 1H NMR using 4,4’-di-t-Bubiphenyl as an internal standard.
All told, it appears that the dimer must be able to fragment readily, hence the poor results with compounds with higher dimer BDE (Table 2, entries 12–17). The poor results with dimers that cleave very readily (entries 1–2) are consistent with two scenarios. The dimer may play a partial role in assisting the palladium activation of toluene, in which case a modest oxidizing power is needed which the dimers of 11 and 12 lack. Alternately, the captodative radical from homolysis of the dimer must engage the palladium alkyl intermediate and cannot do so if it is too stable, as manifested in very low dimer BDE values.
Conclusions
The discovery of a range of nucleophilic substrates that can engage in the oxidative activation of alkyl C-H bonds vs arene C–H bonds with Pd(OAc)2 has been described. This process uses inexpensive alkylarene precursors derived from petroleum and allows the formation of a range of hindered quaternary centers. All of the substrates react by an initial oxidative dimerization initiated by the Pd(OAc)2 and/or the terminal oxidant. A number of terminal oxidants are compatible with the process with DMBQ or K2S2O8 being the most effective to date. The resultant dimer modifies the palladium catalyst to favor activation of alkyl C-H bonds in contrast to the trends typically observed via a concerted metalation deprotonation mechanism. Notably, insertion occurs at the terminus of the alkyl arene for hindered substrates via β-hydride elimination/migratory insertion, a pathway that is not typically seen in oxidative palladium chemistry. The bond dissociation energies of the dimeric intermediates have been identified that predict which substrates are productive in this reaction. These guidelines allow rationale selection of substrates to employ in this method and further mechanistic studies are underway to understand the discrete steps of this unusual and complex mechanism.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to the NSF (CHE1464778, CHE1764298) and the NIH (GM-08765) for financial support of this research. Partial instrumentation support was provided by the NIH and NSF (1S10RR023444, 1S10RR022442, CHE-0840438, CHE-0848460, 1S10OD011980) and computational support by was provided by XSEDE (TG-CHE120052). G.H., P.N., U.N.K., and A.V. thank the Chinese Scholarship Council, the University of Guanajuato and CONACyT, Dr. Reddy’s Laboratories (Dr. Rakeshwar Bandichhor), and the S. N. Bose Scholarship Program of India, respectively, for financial support. Drs. Rakesh Kohli and Charles W. Ross III are acknowledged for obtaining accurate mass data.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures for all experiments and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) For reviews on metal-catalyzed C–H activation, see: Activation C-H. In Topics in Current Chemistry Vol. 292; Yu J-Q; Shi Z, Eds. Springer: Berlin, 2010. [PubMed] [Google Scholar]; (b) Roudesly F; Oble J; Poli G Metal-catalyzed CH activation/functionalization: The fundamentals J. Mol. Cat. A.: Chemical 2017, 426, 275–296. [Google Scholar]; (c) Chen X; Engle KM; Wang DH; Yu J-Q Palladium(II)-Catalyzed C–H Activation/C–C Cross-Coupling Reactions: Versatility and Practicality. Angew. Chem. Int. Ed 2009, 48, 5094–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Arockiam PB; Bruneau C; Dixneuf PH; Ruthenium(II)-Catalyzed C–H Bond Activation and Functionalization. Chem. Rev 2012, 112, 5879–5918. [DOI] [PubMed] [Google Scholar]; (e) Pototschnig G; Maulide N; Schnȕrch M Direct Functionalization of C–H Bonds by Iron, Nickel, and Cobalt Catalysis. Chem. Eur. J 2017, 23, 1–28. [DOI] [PubMed] [Google Scholar]; (f) Usman M; Ren Z-H; Wang Y-Y; Guan Z-H Recent Developments in Cobalt Catalyzed Carbon–Carbon and Carbon-Heteroatom Bond Formation via C–H Bond Functionalization. Synthesis 2017, 49, 1419–1443. [Google Scholar]
- 2.Cook AK; Sanford MS Mechanism of the Palladium-Catalyzed Arene C–H Acetoxylation: A Comparison of Catalysts and Ligand Effects. J. Am. Chem. Soc 2015, 137, 3109–3118. [DOI] [PubMed] [Google Scholar]
- 3.(a) For reviews on CDC reaction, see: Lyons TM; Sanford MS Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wencel-Delord J; Dröge T; Liu F; Glorius F Towards Mild Metal-catalyzed C–H Bond Activation. Chem. Soc. Rev 2011, 40, 4740–4761. [DOI] [PubMed] [Google Scholar]; (c) Liu C; Zhang H; Shi W; Lei AW Bond Formations between Two Nucleophiles: Transition Metal Catalyzed Oxidative Cross-Coupling Reactions. Chem. Rev 2011, 111, 1780–1824. [DOI] [PubMed] [Google Scholar]; (d) Kozlowski MC Oxidative Coupling in Complexity Building Transforms. Acc. Chem. Res 2017, 50, 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Li B-J; Tian S-L; Fang Z; Shi Z-J. Multiple C−H Activations To Construct Biologically Active Molecules in a Process Completely Free of Organohalogen and Organometallic Components. Angew. Chem. Int. Ed 2008, 47, 1115 –1118. [DOI] [PubMed] [Google Scholar]; (b) Potavathri S; Pereira KC; Gorelsky SI; Pike A; LeBris AP; DeBoef B Regioselective Oxidative Arylation of Indoles Bearing N-Alkyl Protecting Groups: Dual C−H Functionalization via a Concerted Metalation−Deprotonation Mechanism. J. Am. Chem. Soc 2010, 132, 14676–14681. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Balcells D; Clot E; Eisenstein. C−H Bond Activation in Transition Metal Species from a Computational Perspective. Chem. Rev 2010, 110, 749–823. [DOI] [PubMed] [Google Scholar]; (d) Wang D-Y; Guo SH; Pan G-F; Zhu X-Q; Gao Y-R; Wang Y-Q Direct Dehydrogenative Arylation of Benzaldehydes with Arenes Using Transient Directing Groups. Org. Lett 2018, 20, 1794–1797. [DOI] [PubMed] [Google Scholar]
- 5.(a) Guin S; Rout SK; Banerjee A; Nandi S; Patel BK Four Tandem C−H Activations: A Sequential C−C and C−O Bond Making via a Pd-Catalyzed Cross Dehydrogenative Coupling (CDC) Approach. Org. Lett 2012, 14, 5294–5297. [DOI] [PubMed] [Google Scholar]; (b) Kianmehr E; Faghih Nasser; Khan KM Palladium-Catalyzed Regioselective Benzylation−Annulation of Pyridine N−Oxides with Toluene Derivatives via Multiple C−H Bond Activations: Benzylation versus Arylation. Org. Lett 2015, 17, 414–417. [DOI] [PubMed] [Google Scholar]
- 6.(a) Xie P; Xie YJ; Qian B; Zhou H; Xia C; Huang HM Palladium-Catalyzed Oxidative Carbonylation of Benzylic C−H Bonds via Nondirected C(sp3)−H Activation. J. Am. Chem. Soc 2012, 134, 9902–9905. [DOI] [PubMed] [Google Scholar]; (b) Xie P; Xia C; Huang HM Palladium-Catalyzed Oxidative Aminocarbonylation: A New Entry to Amides via C−H Activation. Org. Lett 2013, 15, 3370–3373. [DOI] [PubMed] [Google Scholar]; (c) Liu H; Laurenczy G; Yan N; Dyson PJ Amide Bond Formation via C(sp3)–H Bond Functionalization and CO Insertion. Chem. Commun 2014, 50, 341–343. [DOI] [PubMed] [Google Scholar]; (d) Tanaka T; Hashiguchi K; Tanaka T; Yazaki R; Ohshima T Chemoselective Catalytic Dehydrogenative Cross-Coupling of 2-Acylimidazoles: Mechanistic Investigations and Synthetic Scope. ACS Catal 2018, 8, 8430–8440. [Google Scholar]
- 7.Sha S-C; Tcyrulnikov S; Li M; Hu B; Fu Y; Kozlowski MC; Walsh PJ Cation-π Interactions in the Benzylic Arylation of Toluenes with Bi-metallic Catalysts. J. Am. Chem. Soc 2018, 140, 12415–12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curto JM; Kozlowski MC Chemoselective Activation of sp3 vs sp2 C−H Bonds with Pd(II). J. Am. Chem. Soc 2015, 137, 18–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Dixit VM; Bhat V; Trozzolo AM; George MV Sensitized photooxygenations of Δ2-Oxazolin-5-ones and Related Studies. J. Org. Chem 1979, 44, 4169–4173. [Google Scholar]; (b) Kato H; Tani K; Kurumisawa H; Tamura Y Photooxidation of Some Mesoionic and Related Systems. Chem. Lett 1980, 9, 717–720. [Google Scholar]; (c) Arlandini E; Clerici F; Erba E; Rossi LM Reaction of 5-Amino-4,5-dihydro-4-methylene-1,2,3-triazoles with 2,4-Diaryl-5(4H)-oxazolon-4-yl Radicals. Chem. Ber 1990, 123, 217–220. [Google Scholar]; (d) Rodriguez H; Marquez A; Chuaqui CA; Gomez B Oxidation of Mesoionic Oxazolones by Oxygen. Tetrahedron 1991, 47, 5681–5688. [Google Scholar]
- 10.(a) Andersen KK; Gloster DF; Bray DD; Shoja M; Kjær A Synthesis of Symmetrical 2,2′,4,4′-tetrasubstituted[4,4′-bioxazole]- 5,5′(4H,4′H)-diones and Their Reactions with Some Nucleophiles. J. Heterocycl. Chem 1998, 35, 317–324. [Google Scholar]; (b) Tanaka T; Tanaka T; Tsuji T; Yazaki R; Ohshima T Strategy for Catalytic Chemoselective Cross-Enolate Coupling Reaction via a Transient Homocoupling Dimer. Org. Lett 2018, 20, 3541–3544. [DOI] [PubMed] [Google Scholar]
- 11.(a) De Jongh HAP; De Jongh CRHI; Sinnige HJM; De Klein WJ; Huysmans WGB; Mijs WJ; Van Den Hoek WJ; Smidt J Oxidative carbon-carbon coupling. II. Effect of ring substituents on the oxidative carbon-carbon coupling of arylmalonic esters, arylmalodinitriles, and arylcyanoacetic esters J. Org. Chem 1972, 37, 1960–1966. [Google Scholar]; (b) De Jongh HAP; De Jongh CRHI; Huysmans WGB; Sinnige HJM; De Klein WJ Mijs WJ; Jaspers H Radical Initiation of Vinyl Polymerization By α, α, α’, α’-Tetrasubstituted Dibenzyls. Makromolekulare Chemie 1972, 157, 279–298. [Google Scholar]
- 12.(a) Hartzler HD Polycyano Radicals. J. Org. Chem 1966, 31, 2654–2658. [Google Scholar]; (b) Huang XL; Dannenberg JJ Theoretical Studies of Radical Recombination Reactions. 4. An AM1/CI Study of Reactions of Benzylic and Allylic Radicals. An Intrinsic Barrier to Bond Formation. J. Org. Chem 1991, 56, 6367–6371. [Google Scholar]
- 13.(a) De Jongh HAP; De Jongh CRHI; Mijs WJ J. Org. Chem Oxidative carbon-carbon coupling. I. Oxidative coupling of alpha-substituted benzylcyanides 1971, 36, 3160–3168. [Google Scholar]; (b) Kozlowski MC; Divirgilio ES; Malolanarasimhan K; Mulrooney CA Oxidation of Chiral α-phenylacetate Derivatives: Formation of Dimers with Contiguous Quaternary Stereocenters versus Tertiary Alcohols. Tetrahedron: Asymmetry 2005, 16, 3599–3605. [Google Scholar]
- 14.(a) Klasek A; Lycka A; Rouchal M; Rudolf O; Ruzicka A Reduction of 3-Aminoquinoline-2,4(1H,3H)-diones and Deamination of the Reaction Products. Helvetica Chimica Acta 2014, 97, 595–612. [Google Scholar]; (b) Wu H-R; Huang H-Y; Ren C-L; Liu L; Wang D; Li C-J FeIII-Catalyzed Cross-Dehydrogenative Arylation (CDA) between Oxindoles and Arenes under an Air Atmosphere. Chem.-Eur. J 2015, 21, 16744–16748. [DOI] [PubMed] [Google Scholar]; (c) Ghosh S; Chaudhuri S; Bisai A Oxidative Dimerization of 2-Oxindoles Promoted by KOtBu-I2: Total Synthesis of (±)-Folicanthine. Org. Lett 2015, 17, 1373–1376. [DOI] [PubMed] [Google Scholar]; (d) Bleith T; Deng Q-H; Wadepohl H; Gade LH Radical Changes in Lewis Acid Catalysis: Matching Metal and Substrate. Angew. Chem., Int. Ed 2016, 55, 7852–7856. [DOI] [PubMed] [Google Scholar]; (e) Wu H-R; Cheng L; Kong D-L; Huang H-Y; Gu C-L; Liu L; Wang D; Li C-J FeCl3-Mediated Radical Tandem Reactions of 3-Benzyl-2-oxindoles with Styrene Derivatives for the Stereoselective Synthesis of Spirocyclohexene Oxindoles. Org. Lett 2016, 18, 1382–1385. [DOI] [PubMed] [Google Scholar]; (f) Uraguchi D; Torii M; Ooi T Acridinium Betaine as a Single-Electron-Transfer Catalyst: Design and Application to Dimerization of Oxindoles. ACS Catal 2017, 7, 2765–2769. [Google Scholar]
- 15.Forrester AR; Ingram AS; Thomson RH Persulphate Oxidations. Part VII. Oxidation of o-benzyl- and o-benzoyl-benzamides. J. Chem. Soc., Perkin Trans. 1, 1972, 0, 2853–2857.
- 16.(a) Scaiano JC; Martin A; Yap GPA; Ingold KU A Carbon-Centered Radical Unreactive Toward Oxygen: Unusual Radical Stabilization by a Lactone Ring. Org. Lett 2000, 2, 899–901. [DOI] [PubMed] [Google Scholar]; (b) Frenette M; MacLean PD; Barclay LRC; Scaiano JC Radically Different Antioxidants: Thermally Generated Carbon-Centered Radicals as Chain-Breaking Antioxidants. J. Am. Chem. Soc 2006, 128, 16432–16433. [DOI] [PubMed] [Google Scholar]; (c) Dhotare BB; Kumar M; Nayak SK Catalytic Oxidation of 3-Arylbenzofuran-2(3H)-ones with PCC-H5IO6: Syntheses of 3-Aryl-3-hydroxy/3-amido-3-arylbenzofuran-2(3H)-ones. J. Org. Chem 2018, 83, 10089–10096. [DOI] [PubMed] [Google Scholar]
- 17.(a) Kitagawa T; Miyabo A; Fujii H; Okazaki T; Mori T; Matsudou M; Sugie T; Takeuchi K Self-Initiated Autoxidation of a Sterically Crowded Cycloheptatriene Derivative via Norcaradienyloxyl Radicals. J. Org. Chem 1997, 62, 888–892. [Google Scholar]; (b) Frenette M; Aliaga C; Sanchis EF; Scaiano JC Bond Dissociation Energies for Radical Dimers Derived from Highly Stabilized Carbon-Centered Radicals. Org. Lett 2004, 6, 2579–2582. [DOI] [PubMed] [Google Scholar]; (c) AI-Afyouni MH; Huang TA; Hung-Low F; Bradley CA Synthesis of Bifluorenes via Cobalt Halide Radical Coupling. Tetrahedron Lett 2011, 52, 3261–3265. [Google Scholar]
- 18.Li J-S; Fu D-M; Xue Y; Li Z-W; Li D-L; Da Y-D; Yang F; Zhang L; Lu C-H; Li G One-step Synthesis of Furocoumarins via Oxidative Annulation of 4-Hydroxycoumarins with DDQ. Tetrahedron 2015, 71, 2748–2752. [Google Scholar]
- 19.Carole WA; Colacot TJ Understanding Palladium Acetate from a User Perspective. Chem. Eur. J 2016, 22, 7686–7695. [DOI] [PubMed] [Google Scholar]
- 20.(a) For examples of Pd migration via β-hdyride elimination/ migration insertion, see: Sommer H; Juliá-Hernández F. Martin R; Marek I Walking Metals for Remote Functionalization. ACS Central Science 2018, 4, 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Le Bras J; Muzart J Palladium-Catalyzed Domino Dehydrogenation/Heck-Type Reactions of Carbonyl Compounds Adv. Synth. Catal 2018, 360, 2411–2428. [Google Scholar]; (c) Franzoni I; Mazet C Recent trends in Pd-catalyzed remote functionalization of carbonyl compounds Org. Biomol. Chem 2014, 12, 233–241. [DOI] [PubMed] [Google Scholar]
- 21.(a) Takasugi M; Monde K; Katsui N; Shirata A Spirobrassinin, a Novel Sulfur-containing Phytoalexin from the Daikon Rhaphanus sativus L. var. hortensis (Cruciferae). Chem. Lett 1987, 16, 1631–1632. [Google Scholar]; (b) Pedras MSC; Okanga FI; Zaharia IL Khan AQ Phytoalexins from Crucifers: Synthesis, Biosynthesis, And Biotransformation. Phytochemistry 2000, 53, 161–176. [DOI] [PubMed] [Google Scholar]; (c) Katayev D; Kundig EP Catalytic Enantioselective Synthesis of a 3-Aryl-3-benzyloxindole (=3-Aryl-3-benzyl-1,3-dihydro-2H-indol-2-one) Exhibiting Antitumor Activity. Helv. Chim. Acta 2012, 95, 2287–2295. [Google Scholar]
- 22.(a) Selected references: Huang H-Y; Wu H-R; Wei F; Wang D; Liu L Iodine-Catalyzed Direct Olefination of 2-Oxindoles and Alkenes via Cross-Dehydrogenative Coupling (CDC) in Air. Org. Lett 2015, 17, 3702–3705. [DOI] [PubMed] [Google Scholar]; (b) Hu R-B; Wang C-H; Ren W; Zhong L; Yang S-D Direct Allylic C−H Bond Activation To Synthesize [Pd(η3-cin)(IPr)Cl] Complex: Application in the Allylation of Oxindoles. ACS Catal 2017, 7, 7400–7404. [Google Scholar]; (c) Gao S; Liu H; Yang C; Fu Z; Yao H; Lin A. Accessing 1,3-Dienes via Palladium-Catalyzed Allylic Alkylation of Pronucleophiles with Skipped Enynes. Org. Lett 2017, 19, 4710–4713. [DOI] [PubMed] [Google Scholar]; (d) Klare HFT; Goldber AFG; Duquette DC; Stoltz BM Oxidative Fragmentations and Skeletal Rearrangements of Oxindole Derivatives. Org. Lett 2017, 19, 988–991. [DOI] [PubMed] [Google Scholar]
- 23.(a) Susanti D; Ng LLR; Chan PWH Silica Gel-Mediated Hydroamination/Semipinacol Rearrangement of 2-Alkylaminophenylprop-1-yn-3-ols: Synthesis of 2-Oxindoles from Alkynes and 1-(2-Aminophenyl) Ketones. Adv. Synth. Catal 2014, 356, 353–358. [Google Scholar]; (b) Li Y; Wang K; Ping Y; Wang Y; Kong W Nickel-Catalyzed Domino Heck Cyclization/Suzuki Coupling for the Synthesis of 3,3-Disubstituted Oxindoles. Org. Lett 2018, 20, 921–924. [DOI] [PubMed] [Google Scholar]; (c) Panyam PKR; Ugale B; Gandhi T Palladium(II)/N-Heterocyclic Carbene Catalyzed One-Pot Sequential α-Arylation/Alkylation: Access to 3,3-Disubstituted Oxindoles. J. Org. Chem 2018, 83, 7622–7632. [DOI] [PubMed] [Google Scholar]
- 24.Wu H-R; Huang H-Y; Ren C-L; Liu L; Wang D; Li C-J FeIII-Catalyzed Cross-Dehydrogenative Arylation (CDA) between Oxindoles and Arenes under an Air Atmosphere. Chem. Eur. J 2015, 21, 16744–16748. [DOI] [PubMed] [Google Scholar]
- 25.(a) Nicolaou KC; Snyder SA; Huang X; Simonsen KB; Koumbis AE; Bigot A Studies toward Diazonamide A: Initial Synthetic Forays Directed toward the Originally Proposed Structure. J. Am. Chem. Soc 2004, 126, 10162–10173. [DOI] [PubMed] [Google Scholar]; (b) Piacente S; Montoro P; Oleszek W; Pizza C Yucca schidigera Bark: Phenolic Constituents and Antioxidant Activity. J. Nat. Prod 2004, 67, 882–885. [DOI] [PubMed] [Google Scholar]; (c) Gu Q; Wang R-R; Zhang X-M; Wang Y-H; Zheng Y-T; Zhou J; Chen J-J A new benzofuranone and anti-HIV constituents from the stems of Rhus chinensis Planta. Med 2007, 73, 1–4. [DOI] [PubMed] [Google Scholar]; (d) Nicolaou KC; Wu TR; Kang Q; Chen DTK; Total Synthesis of Hopeahainol A and Hopeanol. Angew. Chem., Int. Ed 2009, 48, 3440–3443. [DOI] [PubMed] [Google Scholar]
- 26.(a) Yang M; Jiang X; Shi W-J; Zhu Q-L; Shi Z-J Direct Lactonization of 2-Arylacetic Acids through Pd(II)-Catalyzed C–H Activation/C–O Formation. Org. Lett 2013, 15, 690–693. [DOI] [PubMed] [Google Scholar]; (b) Cheng X-F; Li Y; Su Y-M; Yin F; Wang J-Y; Sheng J; Vora HU; Wang X-S; Yu J-Q Pd(II)-Catalyzed Enantioselective C–H Activation/C–O Bond Formation: Synthesis of Chiral Benzofuranones. J. Am. Chem. Soc 2013, 135, 1236–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cho BS; Chung YK Palladium-catalyzed Bisarylation of 3-Alkylbenzofurans to 3-Arylalkyl-2-Arylbenzofurans On Water: Tandem C(sp3)–H And C(sp2)–H Activation Reactions Of 3-Alkylbenzofurans. Chem. Commun 2015, 51, 14543–14546. [DOI] [PubMed] [Google Scholar]
- 27.Blanksby SJ; Ellison GB Bond Dissociation Energies of Organic Molecules Acc. Chem. Res 2003, 36, 255–263. [DOI] [PubMed] [Google Scholar]
- 28. Lee HJ; Lee S; Lim JW; Kim JN An Expedient Synthesis of Oxindole Dimers by Direct Oxidative Dimerization of Oxindoles. Bull. Korean Chem. Soc 2013, 34, 2446–2450. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.