

Abstract
PURPOSE
The primary objective of the Children’s Oncology Group study ANBL0531 (ClinicalTrials.gov identifier: NCT00499616) was to reduce therapy for subsets of patients with intermediate-risk neuroblastoma using a biology- and response-based algorithm to assign treatment duration while maintaining a 3-year overall survival (OS) of 95% or more for the entire cohort.
PATIENTS AND METHODS
Children younger than age 12 years with intermediate-risk stage 2A/2B or stage 3 tumors with favorable histology; infants younger than age 365 days with stage 3, 4 or 4S disease; and toddlers from 365 to younger than 547 days with favorable histology, hyperdiploid stage 4, or unfavorable histology stage 3 tumors were eligible. Patients with MYCN-amplified tumors were excluded. Patients were assigned to initially receive two (group 2), four (group 3), or eight (group 4) cycles of chemotherapy with or without surgery on the basis of prognostic markers, including allelic status of chromosomes 1p and 11q; ultimate duration of therapy was determined by overall response.
RESULTS
Between 2007 and 2011, 404 evaluable patients were enrolled. Compared with legacy Children’s Oncology Group studies, subsets of patients had a reduction in treatment. The 3-year event-free survival and OS rates were 83.2% (95% CI, 79.4% to 87.0%) and 94.9% (95% CI, 92.7% to 97.2%), respectively. Infants with stage 4 tumors with favorable biology (n = 61) had superior 3-year event-free survival compared with patients with one or more unfavorable biologic features (n = 47; 86.9% [95% CI, 78.3% to 95.4%] v 66.8% [95% CI, 53.1% to 80.6%]; P = .02), with a trend toward OS advantage (95.0% [95% CI, 89.5% to 100%] v 86.7% [95% CI, 76.6% to 96.7%], respectively; P = .08). OS for patients with localized disease was 100%.
CONCLUSION
Excellent survival was achieved with this treatment algorithm, with reduction of therapy for subsets of patients. More-effective treatment strategies still are needed for infants with unfavorable biology stage 4 disease.
INTRODUCTION
Neuroblastoma is characterized by a broad array of clinical behavior,1,2 and biomarkers are used to classify risk and stratify treatment.3 Previous multi-institutional and cooperative group risk-based clinical trials have led to substantial improvement in outcomes for children with neuroblastoma,4 with increasingly intensive multimodality approaches for high-risk patients5-8 and successive reductions in therapy for patients with low- and intermediate-risk disease.9-11 The overall objective of Children’s Oncology Group (COG) ANBL0531 (ClinicalTrials.gov identifier: NCT00499616) was to further reduce chemotherapy exposure and surgical morbidity for subsets of intermediate-risk patients while maintaining 90% or greater 3-year overall survival (OS) within each clinical and biologic biomarker-defined treatment stratum (group) and 95% or greater 3-year OS for the entire cohort. The study also tested the efficacy of a standard retrieval approach for patients who developed progressive, nonmetastatic disease.
PATIENTS AND METHODS
Study Design and Oversight
This study was a prospective, single-arm, phase III clinical trial with a comparison benchmark selected on the basis of a historical cohort. Chemotherapy treatment assignment was stratified by the degree of surgical resection for patients with stage 2A/2B tumors, the presence of symptoms for patients with stage 4S disease, the allelic status of chromosomes 1p36 and 11q23,12 age at diagnosis, International Neuroblastoma Staging System (INSS) stage, histology, MYCN status, and ploidy.
Patients and Tumor Assessments
Patients were staged according to the INSS,13 and response was assessed using a protocol-specific modification of the International Neuroblastoma Response Criteria.13 Primary tumor response was defined by tumor volume as follows: partial response (PR; 50% to 90% reduction), very good PR (VGPR; greater than 90% reduction), or complete response. Tumors were classified as favorable or unfavorable biology on the basis of ploidy for infants younger than 365 days and toddlers age 365 to younger than 547 days with stage 4 disease, histology (favorable histology [FH], unfavorable histology [UH]) according to the International Neuroblastoma Pathology Classification system,14 and the allelic status of chromosome 1p36 and 11q23. Patients with MYCN-amplified tumors were excluded. The study cohort included patients 12 years of age or younger with stage 2A/2B tumors with less than 50% resection or biopsy only, infants younger than 365 days with stage 3 or 4 neuroblastoma, patients 365 days to 12 years of age with FH stage 3 tumors, toddlers age 365 days to younger than 547 days with UH stage 3 disease, toddlers age 365 to younger than 547 days with stage 4 biologically favorable tumors (FH, hyperdiploid, any 1p/11q status), and infants younger than 365 days with stage 4S neuroblastoma who were either symptomatic or had biologically unfavorable (UH, diploid) tumors. The cohort of patients with stage 4S disease was reported separately.15 Written informed consent was obtained from parents or guardians after the study was approved by the National Cancer Institute Pediatric Central institutional review board and the local institutional review board.
MYCN copy number was determined by fluorescence in situ hybridization,16 DNA index by flow cytometry,17 and allelic status of 1p36 and 11q23 by polymerase chain reaction–based analysis of microsatellite repeat loci, all at the COG reference laboratory. Histology was centrally reviewed and classified according to the International Neuroblastoma Pathology Classification system.14
Initial Treatment Assignment
Eligible patients were assigned to initially receive two (group 2), four (group 3), or eight (group 4) cycles of chemotherapy on the basis of age at diagnosis, INSS stage, histology, and genetic features of the tumor. If data were missing for any tumor biomarker, the result was classified as unfavorable. Table 1 lists the clinical and biologic characteristics of the patients initially assigned to groups 2, 3, and 4 and shows the comparison of treatment assignments in ANBL0531 versus the legacy COG clinical trials. Group 1 patients were not eligible for this trial.
TABLE 1.
Intermediate-Risk Neuroblastoma Initial Treatment Assignment

The chemotherapy regimens in ANBL0531 and A3961 (ClinicalTrials.gov identifier: NCT00003093) were the same10 (Appendix Table A1, online only). Second-look surgery of the primary tumor was scheduled after the assigned number of chemotherapy cycles, although not required for group 2 and 3 patients who achieved PR or better of the primary tumor with chemotherapy alone. The treatment end point for group 3 patients with stage 4 or 4S disease was resolution of nonliver/nonskin metastases and PR or better of the primary tumor. Group 2 and 3 patients who did not achieve the treatment end point with the assigned regimen received up to eight cycles of front-line chemotherapy, and up to six additional cycles of cyclophosphamide and topotecan (CPM + TOPO; Appendix Table A1). The treatment end point for all group 4 patients was VGPR or better of the primary tumor and resolution of nonliver/nonskin sites for patients with metastatic disease. Six cycles of isotretinoin were administered to group 4 toddlers who achieved a VGPR or better after eight cycles of chemotherapy with or without surgery (Appendix Table A1). Group 4 patients who did not achieve VGPR were treated with CPM plus TOPO.
For tumors with intraspinal extension and cord compression, the criteria to stop therapy included the achievement of both the group-defined treatment end point and sufficient shrinkage of the intraspinal tumor to minimize the risk of ongoing or impending neurologic impairment. Laminectomy for surgical debulking was not recommended as primary therapy.
Retrieval Therapy for Progressive, Nonmetastatic Disease
Up to six cycles of CPM plus TOPO (Appendix Table A1) were used as salvage therapy for progressive, nonmetastatic disease within 3 years of study enrollment. The treatment end point was VGPR. Patients with recurrent or progressive metastatic disease at any time were removed from protocol therapy.
Toxicity Monitoring
All patients underwent evaluations of hematologic, hepatic, and renal function every 3 weeks. Cardiac function was evaluated at diagnosis, after cycle 4, and at end of therapy. Auditory function was assessed at diagnosis and end of therapy. Toxic effects were defined according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0), and all grade 3 or greater toxicities were documented.
Statistical Analysis
The primary end point was OS time calculated from the date of diagnosis to the occurrence of death as a result of any cause. Event-free survival (EFS) time was calculated from the date of diagnosis to the first occurrence of an event (relapse, progression, death as a result of any cause, or secondary cancer). Patients who did not die (OS) or experience an event (EFS) were censored on the date of last contact. The analyses of EFS and OS were performed as intention to treat. Survival estimates on the basis of the Kaplan-Meier method18 were reported at 3 years, with SEs per Peto et al.19 Interim monitoring for sufficiently high 3-year EFS was conducted within each group as an early surrogate for OS. In addition, patients age 365 days to younger than 547 days were monitored for early relapse/progression as an indication that reduction in therapy was unsuccessful. Complete statistical considerations are found in the study protocol (Data Supplement).
The primary analysis applied herein differs from the original protocol plan; it was not possible to implement the planned analysis of a comparison of ANBL0531 with an analogous historical cohort because of missing risk factor data, including 1p and 11q status. For the primary objective, the sample size provides sufficient precision for placement of a 95% CI (n = 404; SE, 1.13%; interval width, 4.43%) on the 3-year OS.20 If the upper limit of the 95% CI was above the benchmark (95% overall, 90% within each group), we would conclude that the observed 3-year OS was not statistically significantly lower than the benchmark. Two-sided log-rank tests were used to compare survival curves by subgroups.
RESULTS
Patient Characteristics, Group Assignment, and Treatment
Patients (N = 464) were enrolled between October 8, 2007, and June 30, 2011. Eighteen were deemed ineligible, and 42 were excluded because they were subsequently classified as high risk (Fig 1). The study cohort comprised the remaining 404 evaluable patients. Initial treatment assignments and patient characteristics are listed in Tables 1 and 2. Compared with the legacy COG trials,9,10 therapy reduction for group 2 and 3 patients was prescribed by assignment to fewer chemotherapy cycles and/or by the targeted PR treatment end point for the primary site rather than VGPR. Initial treatment assignment also was reduced significantly for group 4 toddlers with stage 3 or 4 disease (n = 10) previously treated with high-risk regimens.5 Treatment assignment for infants with stage 3 or 4 disease with either diploid or UH tumors was unchanged from A3961. However, for group 4 infants with hyperdiploid, FH stage 4 disease with loss of heterozygosity (LOH) at 1p36/11q23 or missing allelic status data (n = 32), initial treatment assignment was increased.
FIG 1.

CONSORT diagram of the 464 patients enrolled in the Children’s Oncology Group study ANBL0531 between October 8, 2007, and June 30, 2011, and nonrandomized treatment assignment of the 404 eligible and evaluable patients. INPC, International Neuroblastoma Pathology Classification.
TABLE 2.
Characteristics of Patients and Outcomes

Treatment received according to initial group assignment is listed in Table 3. In total, 97 group 2 patients (55.4%), 31 group 3 patients (22.0%), and 16 group 4 patients (18.2%) received fewer cycles of chemotherapy compared with legacy COG studies. Among the 83 patients with stage 2A/2B or 3 disease who received additional cycles of chemotherapy, eight had ganglioneuroblastoma; seven had neuroblastoma, differentiating subtype; and two had ganglioneuroma, maturing subtype. Forty-two of these 83 patients had paraspinal/intraspinal tumors.
TABLE 3.
Assigned and Actual Number of Chemotherapy Courses Received

Surgery and Radiotherapy
Surgical data were available for 391 patients (96.8%). At diagnosis, 33 patients underwent more than 90% tumor resection, 21 underwent between 50% and 90% resection, and 337 had a biopsy only or less than 50% resection. The degree of resection at diagnosis was unknown in eight patients, and five infants with stage 4S disease did not undergo a diagnostic procedure. Second-look surgery was performed after chemotherapy in 38 patients; 28 had more than 90% resection, eight had between 50% and 90% resection, and two underwent a less than 50% resection.
No significant difference in OS was observed according to the degree of resection at diagnosis (more than 90% resection v between 50% and 90% resection, biopsy only, or less than 50% resection; P = .74). Surgery-related complications were reported in 38 patients (9.7%), including pulmonary complications (2.0%), intestine or liver resection (0.8%), and/or wound infection (0.5%). No vascular or nerve injuries were reported, and no patient underwent nephrectomy. Eight patients received radiotherapy, including five for progressive hepatomegaly15 and three with spinal cord compression.
Chemotherapy-Related Toxicity
Reversible myelosuppression was the most commonly observed grade 3 or greater toxicity (Appendix Table A2, online only). Nonhematologic organ toxicity was rare, and no patient reported hearing loss.
Up to six additional chemotherapy cycles of CPM plus TOPO were administered to 27 patients (Table 3; Appendix Table A3, online only): 20 because of inadequate response to initial therapy (Appendix Table A4, online only), and seven because of development of progressive, nonmetastatic disease. Toxicities associated with cycles 9 to 14 included neutropenia in 44.4% of patients and infections in 22.2%. Similar toxicities were observed in the seven patients who received up to six cycles of salvage therapy after developing progressive, nonmetastatic disease (Appendix Table A3).
Survival According to Initial Group Assignment and 1p36/11q23 Allelic Status
Three-year EFS and OS rates for the entire study cohort were 83.2% (95% CI, 79.4% to 87.0%) and 94.9% (95% CI, 92.7% to 97.2%), respectively (Fig 2A). The respective 3-year EFS rates for group 2 (n = 175), group 3 (n = 141), and group 4 (n = 88) patients were 87.2% (95% CI, 82.0% to 92.4%), 86.5% (95% CI, 80.8% to 92.2%), respectively, and 69.5% (95% CI, 59.6% to 79.5%; Fig 2B), and 3-year OS rates were 99.4% (95% CI, 98.3% to 100%), 94.3% (95% CI, 90.4% to 98.2%), and 87.1% (95% CI, 79.8% to 94.4%; Fig 2C), respectively. No statistically significant differences in EFS or OS were observed for patients with (n = 41) versus those without (n = 300) 1p36 LOH (3-year EFS rate: 87.8% [95% CI, 77.8% to 97.8%] v 83.7% [95% CI, 79.3% to 88.0%; P = .41]; 3-year OS rate: 95.1% [95% CI, 88.5% to 100%] v 95.5% [95% CI, 93.1% to 98.0%; P = .44]; Figs 2D and 2E). Inferior EFS was observed for patients with 11q23 LOH (n = 26) versus those without 11q23 LOH (n = 314; 68.4% [95% CI, 50.1% to 86.6%] v 85.4% [95% CI, 81.4% to 89.5%]) at 3 years (P = .02; Fig 2F), and a trend toward inferior OS was observed for patients with 11q23 LOH (88.0% [95% CI, 75.3% to 100%] v 96.1% [95% CI, 93.8% to 98.3%]) at 3 years (P = .09; Fig 2G). Twenty-seven patients (four in group 2, six in group 3, and 17 in group 4) received CPM plus TOPO. Eighteen remain event free, nine experienced relapse, and two died.
FIG 2.

(A) Event-free survival (EFS) and overall survival (OS) for evaluable, intermediate-risk patients (n = 404). (B) EFS for evaluable, intermediate-risk patients according to initial treatment group assignment (n = 404; P = .0012). (C) OS for evaluable, intermediate-risk patients according to initial treatment group assignment (n = 404; P < .001). (D) EFS for evaluable, intermediate-risk patients according to 1p loss of heterozygosity (LOH) status (n = 341; P = .41). (E) OS for evaluable, intermediate-risk patients according to 1p LOH status (n = 341; P = .44). (F) EFS for evaluable, intermediate-risk patients according to 11q LOH status (n = 340; P = .02). (G) OS for evaluable, intermediate-risk patients according to 11q LOH status (n = 340; P = .09).
Outcome by Stage
Three-year EFS and OS rates for patients with stage 2A/2B or 3 tumors (n = 222) were 87.6% (95% CI, 83.1% to 92.1%) and 100%, respectively. Patients with stage 4S neuroblastoma (n = 49) had 3-year EFS and OS rates of 79.5% (95% CI, 67.8% to 91.3%) and 81.4% (95% CI, 70.1% to 92.7%), respectively.15 Three-year EFS and OS rates for infants with stage 4 disease (n = 125) were 78.1% (95% CI, 70.7% to 85.5%) and 91.0% (95% CI, 85.8% to 96.1%; Fig 3A). EFS was statistically significantly better for infants with stage 4 disease with favorable biology (n = 61) compared with those with confirmed unfavorable biology (n = 47) tumors (3-year EFS rate: 86.9% [95% CI, 78.3% to 95.4%] v 66.8% [95% CI, 53.1% to 80.6%]; P = .02; Fig 3B), although OS was not significantly different (3-year OS rate: 95.0% [95% CI, 89.5% to 100%] v 86.7% [95% CI, 76.6% to 96.7%]; P = .08; Fig 3C). Among the 24 group 4 infants with stage 4 disease with confirmed diploid or UH tumors, with or without 1p36/11q23 LOH, the 3-year EFS and OS estimates were 63.9% (95% CI, 43.8% to 84.0%) and 77.3% (95% CI, 59.2% to 95.3%), respectively. For infants with stage 4 hyperdiploid, FH tumors assigned to group 4 because of 1p36/11q23 LOH or unknown allelic status (n = 32), 3-year EFS and OS estimates were 68.6% (95% CI, 52.2% to 85.1%) and 93.8% (95% CI, 85.2% to 100%), respectively. The EFS and OS estimates for the eight toddlers with stage 4 hyperdiploid, FH tumors were 62.5% (95% CI, 28.9% to 96.1%) and 100%, respectively. The two group 4 toddlers with stage 3 disease remain event free.
FIG 3.

(A) Event-free survival (EFS) and overall survival (OS) for evaluable infants with stage 4 disease younger than 365 days of age (n = 125). (B) EFS for evaluable infants with stage 4 disease younger than 365 days of age according to tumor biology (n = 108; P = .02). (C) OS for evaluable infants with stage 4 disease younger than 365 days of age according to tumor biology (n = 108; P = .08). Favorable biology constituted hyperdiploid tumors with favorable histology and normal 1p and 11q alleles. Unfavorable biology tumors had at least one unfavorable feature (diploidy, unfavorable histology, 1p loss of heterozygosity, or 11q loss of heterozygosity).
Disease Progression and Death
Seven patients with progressive, nonmetastatic disease were treated with the prescribed salvage regimen; six remain alive, three experienced relapse (two metastatic), and one died. With the exclusion of these seven patients, events occurred in 63 patients, including disease progression (n = 53 [24 metastatic, 28 locoregional progression, one unknown]), death as first event (n = 9), and secondary malignancy (one acute myeloid leukemia). Twenty-one patients died. Causes of death were complications of hepatomegaly in infants younger than 90 days of age (n = 5); relapsed metastatic disease (n = 7); and death as a result of other causes, including infection (n = 3), liver transplantation (n = 1), second malignancy (n = 1), and drowning (n = 1). The cause of death for three patients is unknown.
DISCUSSION
Treatment duration in ANBL0531 was assigned according to clinical features and a tumor biology- and response-based algorithm. In addition to established COG risk group criteria, treatment was stratified by allelic status of 1p36 and 11q23.12 Compared with the legacy study,10 an increase in treatment duration was prescribed for a subset of infants with stage 4 FH, hyperdiploid tumors with 1p36 and/or 11q23 LOH (or unknown allelic status), whereas treatment was reduced for specific subsets of patients with favorable biology tumors, including normal 1p36 and 11q23. Additional therapy reduction was achieved by using PR of the primary tumor as the end point of therapy for group 2 and 3 patients in lieu of VGPR.10 Only 90 patients (23%) required surgery to achieve PR or better of the primary site, which reflects the efficacy of the chemotherapy regimen. Subgroups of toddlers ages 365 to younger than 547 days with stage 3 and 4 disease,21-23 previously classified as high risk, received significantly reduced therapy on the basis of the favorable outcome observed in previous studies.21,22
Three-year EFS and OS rates of 88% (95% CI, 84% to 90%) and 96% (95% CI, 94% to 97%), respectively, were achieved in the A3961 study10 by assigning four cycles of chemotherapy plus surgery for patients with favorable biology tumors (hyperdiploid/FH) and eight cycles of chemotherapy plus surgery for those with unfavorable biology (diploid and/or UH). The International Society of Pediatric Oncology European Neuroblastoma Research Network studies similarly demonstrated excellent survival with reductions in treatment of infants with unresectable locoregional tumors or with disseminated disease that lacked MYCN amplification.24,25
Excellent outcome also was achieved in ANBL0531, with 3-year EFS and OS rates of 83.2% (95% CI, 79.4% to 87.0%) and 94.9% (95% CI, 92.7% to 97.2%), respectively. An analogous comparison with a historical cohort (primarily A396110), as originally planned, was not possible because the criteria used to classify patients as intermediate risk were modified in 200726 and the required risk factors were unknown in the historical data. In addition, the revised eligibility criteria for ANBL0531 did not align with A3961. Specifically, patients with stage 2A/2B disease with less than 50% resection were eligible for ANBL0531, whereas these patients were previously classified as low risk.9 In addition, infants with stage 4S disease with unknown biology were eligible for this study. Furthermore, two toddler cohorts (ages 365 to younger than 547 days) previously classified as high risk5 were eligible for ANBL0531. Although none of the 10 toddlers have died, additional studies with larger numbers of patients will be needed to assess further the efficacy of intermediate-risk therapy in this cohort.
Numerous studies have demonstrated an association between 1p36 or 11q23 LOH and inferior survival of children with neuroblastoma.27,28 More-recent studies that analyzed the whole genome have demonstrated that other segmental chromosomal abnormalities (SCAs) also are associated with poorer outcome.29,30 Previously, the presence of 1p36 or 11q23 LOH was shown to be independently associated with decreased progression-free survival in a multivariable model among low- and high-risk patients,12 and a significantly inferior EFS also was seen for patients enrolled in ANBL0531 whose tumors harbored 11q23 LOH. However, EFS was not significantly different for patients with tumors with versus without 1p36 LOH, which indicates that the prognostic value of specific SCAs differs in this cohort.
Using primary tumor PR as a treatment end point for favorable biology tumors was a successful strategy, with 100% survival observed in patients with localized disease. Treatment was not reduced for infants with hyperdiploid, FH, stage 4 disease with normal 1p36/11q23 compared with that in A3961,10 and excellent EFS and OS were observed. Future prospective studies will be needed to determine whether it is possible to achieve excellent outcome with reduced therapy for this cohort. For the 32 infants with hyperdiploid, FH, stage 4 tumors with LOH at 1p36 or 11q23 or missing LOH data, intensification of treatment in ANBL0531 compared with A396110 did not reduce the risk of relapse previously reported for infants with tumors that harbor an SCA genomic profile.31 The 3-year EFS rate for this cohort was 68.6% (95% CI, 52.2% to 85.1%) v 86.9% (95% CI, 78.3% to 95.4%) for infants with stage 4 disease with favorable biology (P = .04). OS was not significantly different, which indicates that many of the infants with 1p36/11q23 aberrations underwent successful salvage therapy. Of the 20 patients who received CPM plus TOPO because of an inadequate response to initial therapy, nine achieved VGPR or better. However, six of the 20 patients developed progressive disease or experienced relapse, and one died, which indicates that more-effective treatment is needed for patients who do not meet the defined treatment end point after eight cycles of chemotherapy (Appendix Table A4).
It is important to emphasize that intermediate-risk neuroblastoma is a heterogeneous collection of neuroblastoma phenotypes. Using this response- and biology-based algorithm that includes the allelic status of 1p36 and 11q23, excellent survival was achieved with refined treatment strategies for specific subsets of intermediate-risk patients. In extension of these results, the observation of favorable biology L2 tumors that lack SCAs in patients younger than 18 months of age is being evaluated in COG ANBL1232 (ClinicalTrials.gov identifier: NCT02176967). Additional genomic biomarkers also are being investigated in ANBL1232, in European clinical trials,32 and through the ongoing Therapeutically Applicable Research to Generate Effective Treatments genomic sequencing initiative.33 The incorporation of these biomarkers may enable more precise prognostication and treatment stratification, which will improve further the outcome for children with intermediate-risk neuroblastoma.
ACKNOWLEDGMENT
Additional authors and contributions: David Baker (Princess Margaret Hospital for Children, Perth, Western Australia, Australia), study design and protocol review; Sandra Gorges (University of California, Davis, Sacramento, CA; deceased), development of imaging guidelines for protocol, central radiology review for image-defined risk factors, and study objective; and Fredric A. Hoffer (University of Washington, Seattle, WA), central radiology review for image-defined risk factors and study objective.
Appendix
TABLE A1.
Chemotherapy Regimens Used

TABLE A2.
Episodes of Grade 3, 4, or 5 Toxicity During Cycles 1 to 8 of Treatment

TABLE A3.
Episodes of Grade 3, 4, or 5 During Retrieval Therapy Cycles 9 to 14
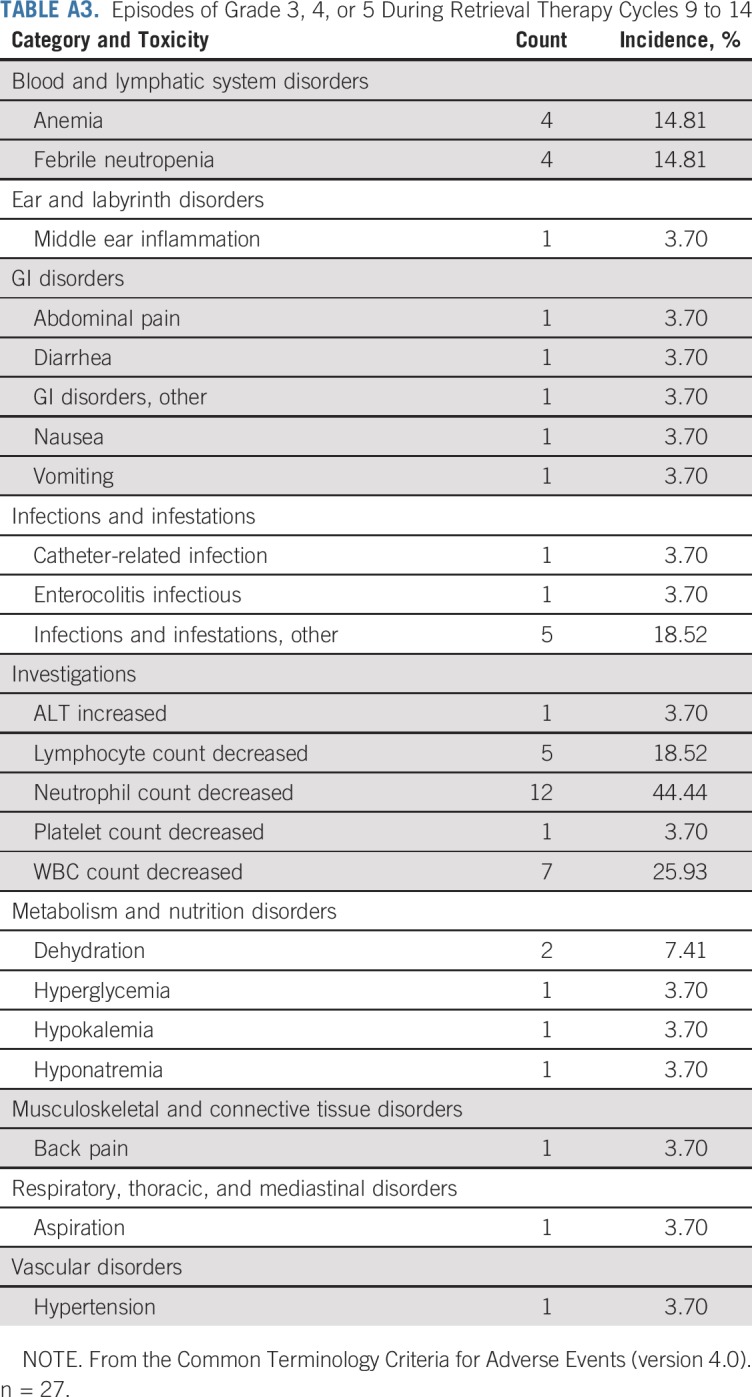
TABLE A4.
Outcome for Patients Receiving CPM Plus TOPO for Inadequate Initial Response to Chemotherapy

Footnotes
Presented at the 2014 Advances in Neuroblastoma Research Meeting, Cologne, Germany, May 13-16, 2014, and 2014 American Society of Clinical Oncology Annual Meeting, Chicago, IL, May 30-June 3, 2014.
Supported in part by National Cancer Institute grant U10 CA180899 (Children’s Oncology Group Statistics and 39 Data Center), National Clinical Trials Network Operations Center grant U10 CA180886, and the St Baldrick’s Foundation.
Clinical trial information: NCT00499616.
AUTHOR CONTRIBUTIONS
Conception and design: Clare J. Twist, Mary Lou Schmidt, Wendy B. London, Araz Marachelian, Margaret H. Collins, Natia Esiashvili, E. Stanton Adkins, Michael Handler, Howard Katzenstein, Edward Attiyeh, Michael D. Hogarty, Elizabeth Wagner, Katherine K. Matthay, Julie R. Park, John M. Maris, Susan L. Cohn
Administrative support: Wendy B. London
Provision of study material or patients: Araz Marachelian, Katherine K. Matthay, John M. Maris, Susan L. Cohn
Collection and assembly of data: Clare J. Twist, Mary Lou Schmidt, Arlene Naranjo, Wendy B. London, Araz Marachelian, Hiroyuki Shimada, Margaret H. Collins, Natia Esiashvili, E. Stanton Adkins, Edward Attiyeh, Julie Gastier-Foster, Julie R. Park, Susan L. Cohn
Data analysis and interpretation: Clare J. Twist, Mary Lou Schmidt, Arlene Naranjo, Wendy B. London, Sheena C. Tenney, Araz Marachelian, Natia Esiashvili, E. Stanton Adkins, Peter Mattei, Michael D. Hogarty, Julie Gastier-Foster, Katherine K. Matthay, Julie R. Park, John M. Maris, Susan L. Cohn
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Maintaining Outstanding Outcomes Using Response- and Biology-Based Therapy for Intermediate-Risk Neuroblastoma: A Report From the Children’s Oncology Group Study ANBL0531
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Arlene Naranjo
Consulting or Advisory Role: Novartis
Wendy B. London
Consulting or Advisory Role: ArQule, United Therapeutics
Travel, Accommodations, Expenses: ArQule
Araz Marachelian
Research Funding: Pfizer (Inst), United Therapeutics (Inst)
Margaret H. Collins
Honoraria: Allakos
Research Funding: Shire (Inst), Meritage (Inst), Regeneron Pharmaceuticals (Inst)
Travel, Accommodations, Expenses: Allakos, Regeneron Pharmaceuticals
Natia Esiashvili
Consulting or Advisory Role: Varian Medical Systems
Speakers’ Bureau: Varian Medical Systems
Travel, Accommodations, Expenses: Varian Medical Systems
Michael Handler
Travel, Accommodations, Expenses: Brainlab
Edward Attiyeh
Employment: Janssen Pharmaceuticals
Stock and Other Ownership Interests: Johnson & Johnson
Julie Gastier-Foster
Research Funding: Bristol-Myers Squibb (Inst), Incyte (Inst)
Katherine K. Matthay
Honoraria: EUSA Pharma, Progenics
Julie R. Park
Honoraria: Bristol-Myers Squibb
John M. Maris
Consulting or Advisory Role: Eli Lilly, Bayer AG
Susan L. Cohn
Stock and Other Ownership Interests: United Therapeutics (I), Varian Medical Systems (I), United Therapeutics, Vermillion, Resmed (I), Merck (I), Merck, Stryker (I), Stryker, Amgen (I), Pfizer (I), AbbVie, Amgen, Jazz Pharmaceuticals, Eli Lilly, Sanofi, Varex Imaging, Vermillion, Pfizer
Research Funding: United Therapeutics (Inst), Merck (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Irwin MS, Park JR. Neuroblastoma: Paradigm for precision medicine. Pediatr Clin North Am. 2015;62:225–256. doi: 10.1016/j.pcl.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Bagatell R, Cohn SL. Genetic discoveries and treatment advances in neuroblastoma. Curr Opin Pediatr. 2016;28:19–25. doi: 10.1097/MOP.0000000000000296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohn SL, Pearson AD, London WB, et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J Clin Oncol. 2009;27:289–297. doi: 10.1200/JCO.2008.16.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinto NR, Applebaum MA, Volchenboum SL, et al. Advances in risk classification and treatment strategies for neuroblastoma. J Clin Oncol. 2015;33:3008–3017. doi: 10.1200/JCO.2014.59.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kreissman SG, Seeger RC, Matthay KK, et al. Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): A randomised phase 3 trial. Lancet Oncol. 2013;14:999–1008. doi: 10.1016/S1470-2045(13)70309-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Park JR, Kreissman SG, London WB, et al: A phase III randomized clinical trial (RCT) of tandem myeloablative autologous stem cell transplant (ASCT) using peripheral blood stem cell (PBSC) as consolidation therapy for high-risk neuroblastoma (HR-NB): A Children’s Oncology Group (COG) study. J Clin Oncol 34, 2016 (suppl; abstr LBA3)
- 7.Ladenstein R, Pötschger U, Pearson ADJ, et al. Busulfan and melphalan versus carboplatin, etoposide, and melphalan as high-dose chemotherapy for high-risk neuroblastoma (HR-NBL1/SIOPEN): An international, randomised, multi-arm, open-label, phase 3 trial. Lancet Oncol. 2017;18:500–514. doi: 10.1016/S1470-2045(17)30070-0. [DOI] [PubMed] [Google Scholar]
- 8.Yu AL, Gilman AL, Ozkaynak MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strother DR, London WB, Schmidt ML, et al. Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: Results of Children’s Oncology Group study P9641. J Clin Oncol. 2012;30:1842–1848. doi: 10.1200/JCO.2011.37.9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker DL, Schmidt ML, Cohn SL, et al. Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med. 2010;363:1313–1323. doi: 10.1056/NEJMoa1001527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Bernardi B, Mosseri V, Rubie H, et al. Treatment of localised resectable neuroblastoma. Results of the LNESG1 study by the SIOP Europe Neuroblastoma Group. Br J Cancer. 2008;99:1027–1033. doi: 10.1038/sj.bjc.6604640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Attiyeh EF, London WB, Mossé YP, et al. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med. 2005;353:2243–2253. doi: 10.1056/NEJMoa052399. [DOI] [PubMed] [Google Scholar]
- 13.Brodeur GM, Pritchard J, Berthold F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11:1466–1477. doi: 10.1200/JCO.1993.11.8.1466. [DOI] [PubMed] [Google Scholar]
- 14.Shimada H, Ambros IM, Dehner LP, et al. The International Neuroblastoma Pathology Classification (the Shimada system) Cancer. 1999;86:364–372. [PubMed] [Google Scholar]
- 15.Twist CJ, Naranjo A, Schmidt ML, et al. Defining risk factors for chemotherapeutic intervention in infants with stage 4S neuroblastoma: A report from the Children’s Oncology Group Study ANBL0531. J Clin Oncol. 2019;37:115–124. doi: 10.1200/JCO.18.00419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathew P, Valentine MB, Bowman LC, et al. Detection of MYCN gene amplification in neuroblastoma by fluorescence in situ hybridization: A Pediatric Oncology Group study. Neoplasia. 2001;3:105–109. doi: 10.1038/sj.neo.7900146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Look AT, Hayes FA, Shuster JJ, et al. Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma: A Pediatric Oncology Group study. J Clin Oncol. 1991;9:581–591. doi: 10.1200/JCO.1991.9.4.581. [DOI] [PubMed] [Google Scholar]
- 18.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 19.Peto R, Pike MC, Armitage P, et al. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. II. Analysis and examples. Br J Cancer. 1977;35:1–39. doi: 10.1038/bjc.1977.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cantor AB. Projecting the standard error of the Kaplan-Meier estimator. Stat Med. 2001;20:2091–2097. doi: 10.1002/sim.856. [DOI] [PubMed] [Google Scholar]
- 21.George RE, London WB, Cohn SL, et al. Hyperdiploidy plus nonamplified MYCN confers a favorable prognosis in children 12 to 18 months old with disseminated neuroblastoma: A Pediatric Oncology Group study. J Clin Oncol. 2005;23:6466–6473. doi: 10.1200/JCO.2005.05.582. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt ML, Lal A, Seeger RC, et al. Favorable prognosis for patients 12 to 18 months of age with stage 4 nonamplified MYCN neuroblastoma: A Children’s Cancer Group Study. J Clin Oncol. 2005;23:6474–6480. doi: 10.1200/JCO.2005.05.183. [DOI] [PubMed] [Google Scholar]
- 23.London WB, Castleberry RP, Matthay KK, et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. J Clin Oncol. 2005;23:6459–6465. doi: 10.1200/JCO.2005.05.571. [DOI] [PubMed] [Google Scholar]
- 24.Rubie H, De Bernardi B, Gerrard M, et al. Excellent outcome with reduced treatment in infants with nonmetastatic and unresectable neuroblastoma without MYCN amplification: results of the prospective INES 99.1. J Clin Oncol. 2011;29:449–455. doi: 10.1200/JCO.2010.29.5196. [DOI] [PubMed] [Google Scholar]
- 25.De Bernardi B, Gerrard M, Boni L, et al. Excellent outcome with reduced treatment for infants with disseminated neuroblastoma without MYCN gene amplification. J Clin Oncol. 2009;27:1034–1040. doi: 10.1200/JCO.2008.17.5877. [DOI] [PubMed] [Google Scholar]
- 26.Maris JM, Hogarty MD, Bagatell R, et al. Neuroblastoma. Lancet. 2007;369:2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 27.Schleiermacher G, Delattre O, Peter M, et al. Clinical relevance of loss heterozygosity of the short arm of chromosome 1 in neuroblastoma: A single-institution study. Int J Cancer. 1996;69:73–78. doi: 10.1002/(SICI)1097-0215(19960422)69:2<73::AID-IJC1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 28.Maris JM, Weiss MJ, Guo C, et al. Loss of heterozygosity at 1p36 independently predicts for disease progression but not decreased overall survival probability in neuroblastoma patients: A Children’s Cancer Group study. J Clin Oncol. 2000;18:1888–1899. doi: 10.1200/JCO.2000.18.9.1888. [DOI] [PubMed] [Google Scholar]
- 29.Janoueix-Lerosey I, Schleiermacher G, Michels E, et al. Overall genomic pattern is a predictor of outcome in neuroblastoma. J Clin Oncol. 2009;27:1026–1033. doi: 10.1200/JCO.2008.16.0630. [DOI] [PubMed] [Google Scholar]
- 30.Schleiermacher G, Janoueix-Lerosey I, Ribeiro A, et al. Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol. 2010;28:3122–3130. doi: 10.1200/JCO.2009.26.7955. [DOI] [PubMed] [Google Scholar]
- 31.Schleiermacher G, Michon J, Ribeiro A, et al. Segmental chromosomal alterations lead to a higher risk of relapse in infants with MYCN-non-amplified localised unresectable/disseminated neuroblastoma (a SIOPEN collaborative study) Br J Cancer. 2011;105:1940–1948. doi: 10.1038/bjc.2011.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oberthuer A, Juraeva D, Hero B, et al. Revised risk estimation and treatment stratification of low- and intermediate-risk neuroblastoma patients by integrating clinical and molecular prognostic markers. Clin Cancer Res. 2015;21:1904–1915. doi: 10.1158/1078-0432.CCR-14-0817. [DOI] [PubMed] [Google Scholar]
- 33.Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]