

Phospholipid-dependent regulation of NF-κB contributes to mesenchymal-to-epithelial transition in pluripotency acquisition.
Abstract
Metabolic reprogramming has emerged as a key regulator of cell fate decisions. Roles of glucose and amino acid metabolism have been extensively documented, whereas lipid metabolism in pluripotency remains largely unexplored. Using a high-coverage lipidomics approach, we reveal dynamic changes in phospholipids occurring during reprogramming and show that the CDP-ethanolamine (CDP-Etn) pathway for phosphatidylethanolamine (PE) synthesis is required at the early stage of reprogramming. Mechanistically, the CDP-Etn pathway inhibits NF-κB signaling and mesenchymal genes in a Pebp1-dependent manner, without affecting autophagy, resulting in accelerated mesenchymal-to-epithelial transition (MET) and enhanced reprogramming. Furthermore, PE binding to Pebp1 enhances the interaction of Pebp1 with IKKα/β and reduces the phosphorylation of IKKα/β. The CDP-Etn-Pebp1 axis is associated with EMT/MET in hepatocyte differentiation, indicating that Etn/PE is a broad-spectrum MET/EMT-regulating metabolite. Collectively, our study reveals an unforeseen connection between phospholipids, cell migration, and pluripotency and highlights the importance of phospholipids in cell fate transitions.
INTRODUCTION
Somatic cells can be reprogrammed into induced pluripotent stem cells (iPSCs) by ectopic expression of defined factors (1). This process is a multistep process driven by both intracellular signaling and extracellular cues and associated with comprehensive and multilayered remodeling in cell morphology, epigenetics, metabolism, and organelles (2, 3). PSCs, which have unique capacities to self-renew and differentiate into almost any type of cell, have a unique metabolic profile that differs from somatic cells (4). PSCs rely mainly on glycolysis (3–5), while somatic cells depend on oxidative phosphorylation (OXPHOS) (3–5). A metabolic switch from OXPHOS to glycolysis occurs early during the reprogramming of somatic cells to pluripotency, whereas metabolism shifts from glycolysis to OXPHOS upon differentiation (5). These switches are critical for the respective acquisition and loss of pluripotency. PSCs also largely depend on one-carbon metabolism, which not only provides substrate for nucleic acid synthesis but also produces methyl donors for histone and DNA methylation (6, 7).
PSCs and somatic cells also display specific lipid profiles (8–10). PSCs are characterized by highly unsaturated lipids, whose modulation influences pluripotency maintenance (9). In addition, total fatty acid synthesis and oxidation are required for somatic cell reprogramming (11–13). However, due to the extraordinary variety and complexity of metabolic pathways, little has been reported on the distinct lipids in pluripotency regulation (14, 15). The rule and role of distinct lipids and their interconnection with multilayered intracellular and extracellular mechanisms contributing to reprogramming remain unknown. Phospholipids are the most abundant lipids in most mammalian cells, not only serving as structural components of all cellular membrane but also participating in many physiological and pathological processes, including signal transduction, epigenetic regulation, and tumorigenesis (16–18). Phospholipids are critical for normal growth and development. The elimination of many phospholipid biosynthesis genes, including Chka, Pcyt1, or Pcyt2, causes embryonic lethality (19). However, whether and how phospholipids influence pluripotency acquisition/escape and maintenance remains unclear.
Here, using a high-coverage lipidomics approach, we show that phosphatidylethanolamine (PE) increases at the early stage of somatic cell reprogramming, and its synthesis is required for reprogramming. Mechanistically, PE inhibits nuclear factor κB (NF-κB) signaling by enhancing the interaction of PE-binding protein 1 (Pebp1) with IκB kinase α (IKKα) and IKKβ, thus accelerating mesenchymal-to-epithelial transition (MET) and pluripotency acquisition. In addition, the expression of PEBP1 and genes involved in PE synthesis is closely correlated with the epithelial-to-mesenchymal transition (EMT)/MET process in hepatocyte differentiation, suggesting that Etn/PE is a broad-spectrum MET/EMT-regulating metabolite.
RESULTS
Lipidomics analysis reveals dynamic changes of phospholipids during reprogramming
The composition of phospholipids varies markedly across different types of cells and has been implicated in disease pathogenesis, early embryo development, and cell fate decisions (10, 16, 18). To determine whether phospholipid metabolism is associated with pluripotency acquisition, we conducted nontargeted lipidomics to examine the lipid profiles of mouse embryonic fibroblasts (MEFs), mouse embryonic stem cells (mESCs), and MEFs undergoing reprogramming in a serum-free, chemically defined medium (iCD1) (20, 21). A total of 758 lipid molecules were identified and quantified in all samples with six biological replicates. Although most lipids were more abundant in mESCs than in MEFs, not all of them increased gradually during the reprogramming process (Fig. 1A). We observed marked phospholipid remodeling during the whole process of reprogramming. Most PE and lyso-PE (LPE) species increased markedly during the first 4 days of reprogramming and then decreased during the latter days, while most phosphatidycholine (PC), diacylglycerol (DG), and triacylglycerol (TG) increased at the late phase (Fig. 1A and table S1). Other lipid species, including lyso-PC (LPC), phosphatidylserine (PS), phosphatidylinositol, and phosphatidylglycerol, are less in mESCs than in MEFs, and they decreased gradually during reprogramming (Fig. 1A and table S1). These results reveal the dynamic changes of phospholipids during reprogramming, implying the key roles of phospholipid remodeling in reprogramming.
Fig. 1. The CDP-Etn pathway is required for efficient somatic cell reprogramming.
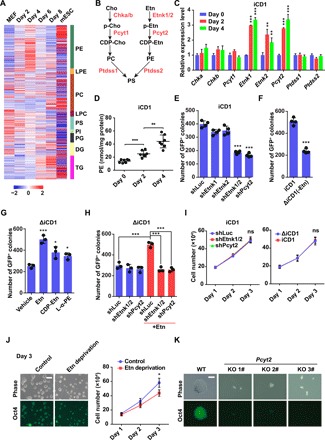
(A) Lipid profiles of MEFs, mESCs, and MEFs undergoing Sox2, Klf4, and Oct4 (SKO) reprogramming on days 2, 4, 6, and 8. n = 6. Lipid species identified are listed in table S1. (B) Schematic of key phospholipid synthesis pathways. (C) qRT-PCR analysis of expression of genes encoding rate-limiting enzymes in phospholipid synthesis pathways on days 0, 2, and 4 in MEFs transduced with SKO in iCD1 medium. Data are represented as mean ± SD (n = 3). **P < 0.01 and ***P < 0.001. (D) Cellular PE levels were measured in MEFs transduced with SKO in iCD1 medium on days 0, 2, and 4. Data are represented as mean ± SD (n = 7). **P < 0.01 and ***P < 0.001. (E) Knockdown of Etnk1 and Etnk2 (shEtnk1/2) or Pcyt2 (shPcyt2) impaired reprogramming efficiency. The numbers of Oct4-GFP+ colonies were counted on day 6. shRNA against luciferase (shLuc) was used as control. Data are represented as mean ± SD (n = 4). ***P < 0.001. (F) The numbers of Oct4-GFP+ colonies were counted on day 6 in MEFs transduced with SKO in iCD1 medium or iCD1 medium without Etn (ΔiCD1). Data are represented as mean ± SD (n = 4). ***P < 0.001. (G) The numbers of Oct4-GFP+ colonies were counted on day 6 in MEFs transduced with SKO in ΔiCD1 medium supplemented with vehicle, Etn, CDP-Etn, or L-α-PE. Data are represented as mean ± SD (n = 3). *P < 0.05 and ***P < 0.001. (H) Etn had no effect on reprogramming when Etnk1/2 or Pcyt2 was silenced by shRNAs. The numbers of Oct4-GFP+ colonies were counted on day 6. shLuc was used as control. Data are represented as mean ± SD (n = 3). ***P < 0.001. (I) Suppression of the CDP-Etn pathway by shRNA against Etnk1/2 or Pcyt2 or Etn deprivation had no effect on the proliferation of MEFs undergoing reprogramming. Data are represented as mean ± SD (n = 3). ns, not significant. (J) Etn deprivation inhibited ESC growth. Representative phase-contrast and Oct4-GFP images (left) and growth curves (right) of ESCs in complete medium (control) or medium deprived of Etn. Scale bar, 250 μm. Data are represented as mean ± SD (n = 3). *P < 0.05. (K) Knockout (KO) of Pcyt2 significantly impaired ESC growth. Representative phase-contrast and Oct4-GFP images of wild-type (WT) and Pcyt2 knockout ESCs. Scale bar, 100 μm.
The CDP-Etn pathway is required for efficient somatic cell reprogramming
The dynamic changes of the three most abundant phospholipids—PC, PE, and PS—during reprogramming prompted us to further investigate their roles in reprogramming. We performed quantitative reverse transcription polymerase chain reaction (qRT-PCR) on genes encoding the rate-limiting enzymes for PC, PE, and PS syntheses during somatic cell reprogramming (Fig. 1B) (19, 20). Among the genes examined, only genes involved in the cytidine diphosphate ethanolamine (CDP-Etn) pathway for PE synthesis—Etnk1, Etnk2, and Pcyt2—significantly increased (Fig. 1C). We further showed that cellular PE levels increased gradually during the early stage of reprogramming by an enzymatic assay (Fig. 1D), suggesting a role for PE in somatic cell reprogramming.
To determine whether PE synthesis is required for reprogramming, we individually knocked down Etnk1, Etnk2, and Pcyt2 using two different short hairpin RNAs (shRNAs) against each gene (fig. S1A). We found that, of the three genes, only Pcyt2 silencing impaired reprogramming efficiency (Fig. 1E and fig. S1B). Because Etnk1 and Etnk2 are complementary in catalyzing the phosphorylation of Etn (19), we then simultaneously knocked down Etnk1 and Etnk2 (hereafter, Etnk1/2) in reprogramming. As expected, simultaneous knockdown of Etnk1/2 resulted in a remarkable reduction in PE levels and reprogramming efficiency similar to Pcyt2 silencing (Fig. 1E and fig. S1, C and E). These results indicated that the CDP-Etn pathway for PE synthesis is required for efficient reprogramming.
To further clarify the effects of the CDP-Etn pathway on reprogramming, we prepared reprogramming medium lacking Etn (ΔiCD1), a precursor of PE, which successfully reduced cellular PE level (fig. S1D). We observed a significant decrease of reprogramming efficiency in ΔiCD1 compared with control, evidenced by green fluorescent protein–positive (GFP+) colonies and endogenous pluripotency gene expression (Fig. 1F and fig. S1, F and G), indicating that Etn is essential for efficient reprogramming. The resulting ΔiCD1-iPSC colonies expressed pluripotency markers, showed normal karyotype, and were able to generate chimeric mice capable of germline transmission similar to that of iCD1-iPSC colonies (fig. S2, A to C). Moreover, supplementation of Etn could restore reprogramming efficiency in ΔiCD1 medium in a dose-dependent manner (Fig. 1G and fig. S1H). We further showed that Etn promoted the reprogramming of mouse tail-tip fibroblasts (TTFs), suggesting that the effect of Etn on reprogramming may not be unique in MEFs (fig. S2D). To explore the effect of the timing of Etn supplementation, we performed experiments by adding Etn at different time points and intervals during reprogramming. Addition of Etn from day 0 to day 4 of reprogramming was sufficient to achieve optimal reprogramming efficiency, indicating that Etn exerts its effect during the early phase (fig. S1I).
To test whether Etn enhances reprogramming through the CDP-Etn pathway, we supplemented Etn in MEFs undergoing reprogramming while silencing Etnk1/2 or Pcyt2. Supplementation of Etn had little effect on reprogramming when the CDP-Etn pathway was blocked by silencing of Etnk1/2 or Pcyt2 (Fig. 1H and fig. S1J), suggesting that Etn promotes reprogramming via the CDP-Etn pathway. Moreover, addition of intermediates of the CDP-Etn pathway, CDP-Etn or L-α-PE, in ΔiCD1 medium also enhanced reprogramming efficiency (Fig. 1G).
Phospholipids are essential for cell growth (19, 22); however, we found that specific suppression of the CDP-Etn pathway by Etn deprivation or shRNAs against Etnk1/2 or Pcyt2 had no effect on the proliferation in MEFs undergoing reprogramming (Fig. 1I). We further tested the effects of Etn on mESCs, where we observed a mild proliferation reduction in culture medium lacking Etn (Fig. 1J). This discrepancy between the effects of Etn deprivation on mESCs and MEFs could be simply due to the different metabolic requirement of cell proliferation (22). We then generated Pcyt2 knockout mESC clones by CRISPR-Cas9–mediated gene editing (fig. S2E) and confirmed the indel mutations (fig. S2F). Knockout of Pcyt2 in mESCs led to a notable reduction of proliferation but had little effect on pluripotency, evidenced by the Oct4-GFP expression (Fig. 1K). It can thus be concluded that the CDP-Etn pathway does not regulate proliferation in MEFs undergoing reprogramming, although it is critical for the growth of mESC.
The CDP-Etn pathway accelerates MET during reprogramming
To better understand how the CDP-Etn pathway facilitates reprogramming, we blocked the CDP-Etn pathway in MEFs by Etn deprivation or shRNAs against Etnk1/2 or Pcyt2 and performed RNA sequencing (RNA-seq) at the early phase of reprogramming (Fig. 2A). Considering the 752 genes in the intersection regulated by Etn deprivation, shEtnk1/2, and shPcyt2, we found overrepresentation of genes involved in cell adhesion (Fig. 2, B and C), suggesting that the CDP-Etn pathway might affect MET during reprogramming. Specifically, RNA-seq showed that inhibition of the CDP-Etn pathway by silencing or Etn deprivation led to a decrease in the expression of epithelial genes and an increase in mesenchymal genes (Fig. 2D). We then examined the expression of several MET-related genes using qRT-PCR, which confirmed the RNA-seq data (Fig. 2E). Consistent results were also obtained at the protein level by Western blot analysis of the epithelial marker Cdh1 (Fig. 2F). To further show that the CDP-Etn pathway regulates MET, we assessed cell migration ability using the scratch assay. In line with our above results, suppression of the CDP-Etn pathway by silencing or Etn deprivation promoted cell migration during reprogramming (Fig. 2, G and H). Immunofluorescence and flow cytometric analysis also indicated that deprivation of Etn reduced surface Cdh1 levels and the percentage of Cdh1-positive cells during reprogramming (Fig. 2I). In addition, Etn could not enhance reprogramming in the presence of transforming growth factor–β1 (TGF-β1), TGF-β2, or TGF-β3, all of which could inhibit EMT and impair reprogramming (Fig. 2J) (23, 24). Together, these results suggested that the CDP-Etn pathway accelerated MET during reprogramming.
Fig. 2. The CDP-Etn pathway accelerates MET during reprogramming.
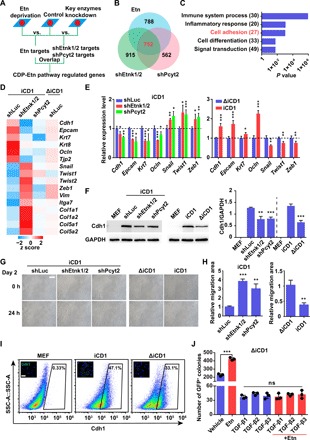
(A) Experimental design of RNA-seq for profiling of genes regulated by the CDP-Etn pathway during reprogramming. shLuc was used as control. (B) Venn diagram showing the substantial overlap of Etn-, shEtnk1/2-, and shPcyt2-regulated genes on day 2 of SKO reprogramming. (C) GO analysis of genes regulated by the CDP-Etn pathway on day 2 of SKO reprogramming. (D) Heatmap showing the expression of selected epithelial genes (Cdh1, Epcam, Krt7, Krt8, Ocln, and Tjp2) and mesenchymal genes (Snail, Twist1, Twist2, Zeb1, Vim, Itga7, Col1a1, Col1a2, Col5a1, and Col5a2) on day 2 of SKO reprogramming. (E) qRT-PCR analysis of expression of selected epithelial and mesenchymal genes on day 2 in MEFs transduced with SKO and shLuc, shEtnk1/2, or shPcyt2 in iCD1 medium (left) and in MEFs transduced with SKO in ΔiCD1 or iCD1 medium (right). Data are represented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001. (F) Western blot analysis (left) and corresponding quantification (right) of Cdh1 expression on day 2 in MEFs transduced with SKO and shLuc, shEtnk1/2, or shPcyt2 in iCD1 medium or in MEFs transduced with SKO in ΔiCD1 or iCD1 medium. Data are represented as mean ± SD (n = 3). **P < 0.01 and ***P < 0.001. (G and H) Representative images (G) and quantification (H) of scratch assays on day 2 in MEFs transduced with SKO and shLuc, shEtnk1/2, or shPcyt2 in iCD1 medium (left) or in MEFs transduced with SKO in ΔiCD1 or iCD1 medium (right). Scale bar, 100 μm. Data are represented as mean ± SEM (n = 4). **P < 0.01 and ***P < 0.001. (I) Flow cytometry analysis of Cdh1 in MEFs and MEFs transduced with SKO in ΔiCD1 or iCD1 medium. Representative images of immunofluorescence staining of Cdh1 (green) are shown. Scale bar, 100 μm. (J) Etn had no effect on reprogramming in the presence of TGF-β1, TGF-β2, or TGF-β3. The numbers of Oct4-GFP+ colonies were counted on day 6. Data are represented as mean ± SD (n = 3). ***P < 0.001.
The CDP-Etn pathway acceleration of MET depends on Pebp1
Lipids play signaling roles in a variety of physiological processes, including autophagy, where their functions arise from interactions with proteins (25–27). PE functions as a lipid anchor for microtubule-associated protein light chain 3 (LC3) (25, 27), which is essential for autophagy. Because activation of autophagy is an important early step in reprogramming (28, 29), we monitored autophagic flux upon suppression of the CDP-Etn pathway in reprogramming. Similarly, we observed that the LC3 lipidation increased in the absence and presence of bafilomycin A1 (Baf A1), indicating a robust induction of autophagy in our reprogramming system (fig. S3A). However, suppression of the CDP-Etn pathway had no effect on autophagy during reprogramming (fig. S3A). This result is probably due to minor residual phospholipids in medium and/or inadequate knockdown of PE-generating enzymes. In addition to autophagy, PE also regulates diverse kinase signaling pathways by interacting with Pebp1 (also known as Raf kinase inhibitory protein) (26, 30). Therefore, we sought to determine whether the CDP-Etn pathway exerts its function via Pebp1 during reprogramming. We found not only that Pebp1 expression increases over the course of reprogramming (Fig. 3A) but also that knockdown of Pebp1 inhibits reprogramming (Fig. 3B and fig. S3, B and C), strongly indicating a role for Pebp1 in reprogramming.
Fig. 3. The CDP-Etn pathway acceleration of MET depends on Pebp1.
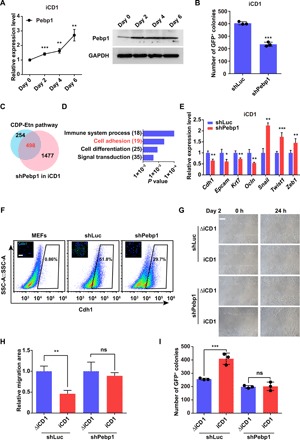
(A) qRT-PCR analysis (left) and Western blot analysis (right) of expression of Pebp1 on days 0, 2, 4, and 6 in MEFs transduced with SKO in iCD1 medium. Data are represented as mean ± SD (n = 3). **P < 0.01 and ***P < 0.001. (B) Knockdown of Pebp1 (shPebp1) impaired reprogramming efficiency. The numbers of Oct4-GFP+ colonies were counted on day 6. Data are represented as mean ± SD (n = 3). ***P < 0.001. (C) Venn diagram showing the overlap of genes regulated by the CDP-Etn pathway and Pebp1 on day 2 of SKO reprogramming. (D) GO analysis of genes regulated by both the CDP-Etn pathway and Pebp1 on day 2 of SKO reprogramming. (E) qRT-PCR analysis of expression of selected epithelial and mesenchymal genes on day 2 in MEFs transduced with SKO and shLuc or shPebp1 in iCD1 medium. Data are represented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001. (F) Flow cytometry analysis of Cdh1 in MEFs and MEFs transduced with SKO and shLuc or shPebp1 in iCD1 medium. Representative images of immunofluorescence staining of Cdh1 (green) are shown. Scale bar, 100 μm. (G and H) No significant differences in cell migration were observed between reprogramming in ΔiCD1 or iCD1 medium when Pebp1 was knocked down. Representative images (G) and quantification (H) of scratch assays were analyzed on day 2. Scale bar, 100 μm. Data are represented as mean ± SEM (n = 4). **P < 0.01. (I) No significant differences in reprogramming efficiency were observed between reprogramming in ΔiCD1 or iCD1 medium when Pebp1 was knocked down. The numbers of Oct4-GFP+ colonies were counted on day 6. Data are represented as mean ± SD (n = 3). ***P < 0.001.
We then asked whether Pebp1 was involved in the MET during reprogramming. We performed RNA-seq in MEFs undergoing reprogramming with or without Pebp1 silencing. About two-thirds of genes regulated by the CDP-Etn pathway were regulated by Pebp1 in reprogramming (Fig. 3C). Gene Ontology (GO) analysis revealed that these genes regulated by Pebp1 were also involved in the regulation of cell adhesion (Fig. 3D). In addition, Pebp1 silencing led to a decrease in the expression of epithelial genes and an increase in mesenchymal genes (Fig. 3E and fig. S3D). Likewise, immunofluorescence and flow cytometric analysis indicated a robust reduction in surface Cdh1 levels when Pebp1 was silenced (Fig. 3F). These data suggest that, during reprogramming, Pebp1 regulates MET in a very similar manner as the CDP-Etn pathway. Furthermore, we also performed scratch assays to assess the effect of Pebp1 silencing on the migration of MEFs undergoing reprogramming with or without Etn. Deprivation of Etn promoted migration in control-silenced cells but had no significant effect on Pebp1-silenced cells (Fig. 3, G and H). Consistent with these results, Etn deprivation did not have a noticeable effect on reprogramming when Pebp1 was silenced (Fig. 3I and fig. S3E). Together, these findings suggested that the CDP-Etn pathway accelerated MET in a Pebp1-dependent manner.
The CDP-Etn-Pebp1 axis modulates NF-κB signaling to inhibit mesenchymal genes
Pebp1 acts as a regulator of diverse kinase signaling pathways, several of which are implicated in somatic cell reprogramming such as the NF-κB and extracellular signal–regulated kinase (ERK) pathways (30–32). To determine whether the CDP-Etn pathway or Pebp1 modulates NF-κB and ERK signaling in reprogramming, we examined the nuclear translocation of NF-κB p65 and the levels and phosphorylation status of ERK. Inhibition of the CDP-Etn pathway by Etn deprivation or shRNAs against Etnk1/2 or Pcyt2 led to increased translocation of NF-κB p65 to the nucleus but had no effect on ERK activation (Fig. 4A and fig. S4A). Similar results were obtained from Pebp1 silencing experiments, suggesting that the CDP-Etn pathway and Pebp1 inhibit the activation of NF-κB p65, but not ERK, in reprogramming. Furthermore, upon Pebp1 silencing, Etn deprivation had no effect on nuclear translocation of NF-κB p65 or ERK activation (Fig. 4A and fig. S4, A to C), indicating that the CDP-Etn pathway inhibits NF-κB in a Pebp1-dependent manner. Pebp1 can interact with several kinases upstream of the NF-κB signaling (33), and phospholipids are involved in the association of Pebp1 with kinases (26), prompting us to ask whether phospholipid PE modulates the interaction between Pebp1 and kinases of the NF-κB signaling. To answer this question, we performed coimmunoprecipitation in MEFs undergoing reprogramming in the absence or presence of Etn and showed that Pebp1 interacts with IKKα and IKKβ, kinases upstream of the NF-κB signaling (Fig. 4B) (33). Furthermore, Etn could significantly increase the binding of Pebp1 to IKKα and IKKβ without any noticeable changes in the expression and distribution of Pebp1 (Fig. 4, B and C, and fig. S4, D to F). We also showed that Etn reduced the phosphorylation of IKKα/β in a Pebp1-dependent manner (Fig. 4C). PE binding to Pebp1 has been shown to disrupt the association of Pebp1 with certain kinases, including Raf-1 and MAPK kinase 1 (MEK1), whereas our results showed that PE enhanced the interaction between Pebp1 and kinases upstream of NF-κB. Thus, it is possible that Pebp1 interacts with IKKα and IKKβ by a mechanism that is distinct from Raf-1 and MEK1. Together, these results indicated that phospholipid PE increases the binding of Pebp1 to kinases upstream of NF-κB signaling, IKKα and IKKβ, to decrease the phosphorylation of IKKα/β and inhibit NF-κB in reprogramming.
Fig. 4. The CDP-Etn-Pebp1 axis modulates NF-κB signaling to inhibit mesenchymal genes.
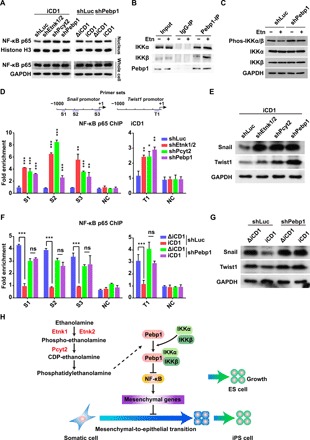
(A) Western blot analysis of NF-κB p65 in the nuclear fraction and whole cell on day 2 in MEFs transduced with SKO and shLuc, shEtnk1/2, shPcyt2, or shPebp1 in iCD1 medium (left) or in MEFs transduced with SKO and shLuc or shPebp1 in ΔiCD1 or iCD1 medium (right). (B) Coimmunoprecipitation analysis of the interaction between Pebp1 and IKKα or IKKβ on day 2 in MEFs undergoing reprogramming in the absence or presence of Etn. (C) Western blot analysis of phosphorylation of IKKα/β on day 2 in MEFs transduced with SKO and shLuc or shPebp1 in the absence or presence of Etn. (D) ChIP-qPCR analysis of NF-κB binding at the Snail (left) and Twist1 (right) promoters on day 2 in MEFs transduced with SKO and shLuc, shEtnk1/2, shPcyt2, or shPebp1 in iCD1 medium. Schematic of primer sets for ChIP-qPCR on the Snail and Twist1 promoters is shown above. Data are represented as mean ± SD (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001. (E) Western blot analysis of Snail and Twist1 expression on day 2 in MEFs transduced with SKO and shLuc, shEtnk1/2, shPcyt2, or shPebp1 in iCD1 medium. (F) ChIP-qPCR analysis of NF-κB binding at the Snail (left) and Twist1 (right) promoters on day 2 in MEFs transduced with SKO and shLuc or shPebp1 in ΔiCD1 or iCD1 medium. Data are represented as mean ± SD (n = 3). ***P < 0.001. (G) Western blot analysis of Snail and Twist1 expression on day 2 in MEFs transduced with SKO and shLuc or shPebp1 in ΔiCD1 or iCD1 medium. (H) Proposed model for the CDP-Etn pathway in pluripotency acquisition.
To further clarify the causal relationship between the CDP-Etn pathway and MET in reprogramming, we performed chromatin immunoprecipitation qPCR (ChIP-qPCR) to analyze the binding of NF-κB p65 on the promoter of Snail and Twist1, mesenchymal genes known to be regulated by NF-κB p65 (Fig. 4D). As predicted, knockdown of Etnk1/2, Pcyt2, or Pebp1 resulted in markedly increased binding of NF-κB p65 on these promoter regions and corresponding increases in the protein levels of Snail and Twist1 (Fig. 4, D and E). Moreover, Etn deprivation also enhanced the binding of NF-κB p65 on the promoters of Snail and Twist1 and increased Snail and Twist1 protein levels in control-silenced, but not Pebp1-silenced, cells (Fig. 4, F and G). As several mesenchymal genes, such as Snail and Twist1, are known as Cdh1 repressors (34), NF-κB might activate Snail and Twist1 directly, leading to repression of Cdh1. Together, these results suggest that the CDP-Etn-Pebp1 axis inhibits NF-κB signaling to suppress mesenchymal genes and thus accelerates MET during reprogramming (Fig. 4H).
MET and EMT are fundamental processes in development (35). The transition between mesenchymal and epithelial state is required for the differentiation of specialized cell types (35, 36), prompting us to test whether phospholipids are involved in EMT and MET during differentiation. Because differentiation of hepatocytes from human ESCs requires a sequential EMT-MET (36), we analyzed the expression of PEBP1 and genes involved in phospholipid synthesis in this context. Our results showed that the expression of PEBP1 and genes involved in PE synthesis decreased during EMT and increased during MET, whereas genes involved in PC and PS synthesis showed a lack of correlation with the EMT-MET process (fig. S4G). These observations suggest that our findings of connection between phospholipid PE and MET play a broad role in many biological processes involving MET or EMT.
DISCUSSION
Lipids have emerged as key regulators of cell fate decisions. However, little is known about their mechanisms of action. In this study, we revealed a previously unrecognized connection between phospholipids and cell fate decisions. We showed that the CDP-Etn pathway for PE synthesis is required at the early phase for efficient mouse somatic cell reprogramming. The CDP-Etn pathway inhibits NF-κB signaling and mesenchymal gene expression, leading to accelerated MET and enhanced reprogramming, thus revealing a possible mechanistic link of phospholipid metabolism to cell migration and cell fate.
We demonstrated the connection between phospholipids, MET, and pluripotency, further elucidating the multilayered mechanisms contributing to reprogramming. The mechanisms of somatic cell reprogramming have been revealed at many phenotypic levels: epigenetic, proteomic, metabolic, chromosomal, and morphological (2, 3, 23, 24, 28). Using a high-coverage lipidomics approach, we revealed dynamic changes of phospholipids occurring during reprogramming and elucidated the multilayered regulatory mechanism at metabolic, morphological, and organellar levels. First, our results indicate a regulatory link between metabolism and cell morphology. Somatic cell reprogramming requires a morphological change, from mesenchymal state to epithelial state, at the early phase of reprogramming (3, 23, 24). It has been commonly suggested that an interplay exists between cell morphology and metabolism because changes in morphology are always associated with metabolic remodeling (35, 37, 38). Yet, it has remained largely unknown how the two processes are intertwined, especially in pluripotency acquisition. Our observations thus suggest a model in which metabolic switch facilitates morphological changes during reprogramming. Second, our findings reveal a previously unknown role of phospholipid metabolism not via its typical autophagy-dependent manner. PC, PE, and PS are the three most abundant phospholipids in mammalian cells, serving as structural components of all cellular membrane systems (18, 19). Lipids are essential components of cellular membranes and are crucial for autophagy. Our results indicate that only the PE synthesis pathway, but not PC or PS, is activated at the early phase of reprogramming and is required for efficient reprogramming. We showed that the CDP-Etn pathway promoting pluripotency induction is not dependent on autophagy but on regulating the important signaling pathway of pluripotency. Last, our study identified Etn/PE as a previously unidentified extracellular lipid signaling metabolite contributing to pluripotency regulation. Phospholipids are bioactive signaling molecules able to act in many cellular processes, such as pluripotency maintenance. Lysophosphatidic acid (LPA) and sphingosine 1-phosphate (S1P) are the most widely studied, and their receptors are widely expressed in mouse and human ESCs (39). LPA has been reported to activate or cooperate with many signaling pathways, such as the Wnt signaling pathway and the JAK (Janus kinase)/STAT3 (signal transducer and activator of transcription 3) signaling pathway, leading to the expression of self-renewal genes (39). Both LPA and S1P stimulate ERK1/2 phosphorylation and are involved in ESC proliferation and differentiation (39). In addition, LPA activates ROCK signaling and cooperates with leukemia inhibitory factor (LIF) and bone morphogenetic protein 4 (BMP4) signaling to promote the conversion of primed mESCs to naïve state (40). Here, we identified phospholipid PE and its precursor Etn as previously unidentified extracellular lipid signaling metabolites, which inhibit NF-kB signaling to enhance somatic cell reprogramming and are necessary for mESC growth.
We tested our findings of phospholipids regulating MET during differentiation, implicating its broad importance in development. We showed that the expression of PEBP1 and genes involved in PE synthesis is closely associated with the EMT/MET process in differentiation, suggesting that Etn/PE is a broad-spectrum MET/EMT-regulating metabolite. Phospholipids are critical for normal growth and development, and the deficiency in many phospholipid biosynthesis genes leads to early embryonic lethality (19). Along with previous studies, we show that inhibition of the CDP-Etn pathway for phospholipid PE synthesis impairs mESC growth. However, our study now reveals that inhibition of the CDP-Etn pathway does not affect pluripotency genes. We also show that phospholipid PE regulates MET during the acquisition of pluripotency. As MET is critical for embryonic development, these data may suggest important mechanisms of phospholipid deficiency–induced early embryonic lethality.
In conclusion, we report a dynamic remodeling of phospholipids during pluripotency acquisition. The early increase in PE via the CDP-Etn pathway facilitates MET and is required for efficient reprogramming, indicating a link between metabolism and morphology. Mechanistically, PE binding to Pebp1 enhances the interaction of Pebp1 to IKKα and IKKβ, leading to inhibition of NF-κB and mesenchymal genes, thus facilitating MET and reprogramming. This Etn/PE-Pebp1 axis may also play a role in hepatocyte differentiation. Further investigation is needed to clarify whether phospholipids play a broad role in biological processes involving EMT/EMT.
MATERIALS AND METHODS
Cell culture
OG2 MEFs were derived from embryonic day 13.5 embryos carrying the Oct4-GFP transgenic allele. MEFs and Plat-E cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/high glucose (HyClone) supplemented with 10% fetal bovine serum (FBS) (Gibco), GlutaMAX (Gibco, 100×), and nonessential amino acids (NEAA) (Gibco, 100×). OG2 mESCs were cultured on 0.1% gelatin-coated plates in DMEM/high glucose supplemented with N2 (Gibco, 200×), B27 (Gibco, 100×), sodium pyruvate (Gibco, 100×), GlutaMAX (Gibco, 100×), NEAA (Gibco, 100×), 0.1 mM 2-mercaptoethanol (Gibco), LIF (1000 U/ml), 3 μM CHIR99021 (Selleck), and 1 μM PD0325901 (Selleck). TGF-β1, TGF-β2, and TGF-β3 (R&D Systems) were used at 2 ng/ml. Baf A1 (Gene Operation) was used at 100 nM and treated for 4 hours before sample collection. The cells were obtained with approval from the ethics committee of the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences (GIBH). All the animals were handled according to approved Institutional Animal Care and Use Committee protocols of the GIBH.
iPSC generation and characterization
Retroviral vectors expressing mouse Oct4, Sox2, and Klf4 were transfected in Plat-E cells using polyethylenimine (Polysciences) to produce retrovirus. A total of 2 × 104 OG2 MEFs or 2 × 104 OG2 TTFs within two passages were seeded in 12-well plates and infected twice with retroviral supernatants generated with Plat-E cells. After infection, the culture medium was changed to the chemically defined medium iCD1 or ΔiCD1 [iCD1 deprived of ethanolamine (Sigma)]. Oct4-GFP–positive colonies were counted on day 6 (MEFs) or day 14 (TTFs) after infection. iPSC colonies were picked up and characterized as described (20). Karyotype was analyzed by fixing with methanol–acetic acid (3:1) for 30 min and stained with 5% Giemsa (Sigma). Chimeras were generated by injecting iPSCs into blastocysts from ICR mice, followed by implantation into pseudopregnant ICR mice. F2 mice were then bred from chimeric and ICR mice for germline transmission as previously described (20).
Plasmids
The pMX retroviral vector expressing Oct4, Sox2, or Klf4 and the pX330 vector were purchased from Addgene. shRNA inserts were cloned into pRetroSuper retroviral vectors. shRNA target sequences were listed in table S2.
Lipid extraction
Lipids were extracted as described previously (21). Briefly, 1 × 107 cells were scraped and quenched in 400 μl of methanol/deionized water (3:1, v/v). After quenching, 1 ml of methyl tert-butyl ether was added and incubated for 1 hour at room temperature. Samples were then homogenized and incubated with 200 μl of deionized water. The samples were then centrifuged, and the upper organic phase containing the lipids was extracted into a clean tube. Lipid extracts were dried stored at −80°C until further analysis. Dried lipid films were resuspended in 120 μl of acetonitrile (ACN), isopropanol (IPA), and water (60:30:5, v/v/v) for mass spectrometry (MS) analysis. The internal standard mixture containing LPC 12:0 at 0.3 μg/ml, PC 38:0 at 0.375 μg/ml, PE 30:0 at 0.28 μg/ml, PE 34:0 at 0.3 μg/ml, sphingomyelin 12:0 at 0.24 μg/ml, ceramide 17:0 at 0.3 μg/ml, free fatty acids 16:0-d3 at 0.187 μg/ml, and TG 45:0 at 0.225 μg/ml was added to each kind of sample matrix before lipid extraction.
Nontargeted lipidomics analysis
An ACQUITY UPLC system (Waters, Milford, MA, USA) coupled via an electrospray ion source with a Q Exactive HF MS system (Thermo Fisher Scientific, Rockford, IL, USA) was used for nontargeted lipid profiling in both full-scan and data-dependent MS/MS (ddMS2) modes to obtain lipid information including retention time (tR), accurate masses, and/or MS/MS fragments. Data analysis was performed by Dalian Chem Data Solution Information Technology Co. Ltd.
A reversed-phase BEH C8 column (2.1 mm × 100 mm, 1.7 μm, Waters, Milford, MA, USA) was used for the chromatographic separation of lipids. Mobile phases A and B were ACN/H20 (60:40, v/v) and IPA/ACN (90:10, v/v), respectively, both containing 10 mM ammonium acetate (AmAc). The flow rate was 0.26 ml/min. The column temperature was 55°C. The elution gradient started with 32% B and was kept for 1.5 min, then linearly increased to 85% B at 15.5 min and to 97% B at 15.6 min, and held for 2.4 min. The gradient was back to 32% B at 18.1 min and kept for 1.9 min to equilibrate the column. The temperature of the sample manager was set to 10°C.
The mass spectrometer was operated with a capillary voltage of 3.5 kV in positive mode and 3.0 kV in negative mode. The capillary temperature was set to 300°C. Sheath gas flow rate and aux gas flow rate were set to 45 and 10 (in arbitrary units). Aux gas heater temperature was 350°C. The S-lens retention factor (RF) level was 50.0. The resolutions of 70,000 and 17,500 were set for full-scan MS and ddMS2 in both modes. The automatic gain control target and maximum ion injection time were 3 × 106 ion capacity and 100 ms in full-scan MS settings, while their values were 1 × 105 ion capacity and 50 ms in ddMS2 settings. The TopN (N, the number of top most abundant ions for fragmentation) was set to 8. The normalized collision energy was set to 25, 35, and 45 eV, respectively. The scan range was set to m/z (mass/charge ratio) 100 to 1500.
The identification of lipids was achieved by using a commercial database of metabolites called OSI/SMMS2 and a strategy in a previous report (21). To the molecules with successful matching of MS2, all information of tR, MS1, and MS2 was used for more accurate characterization of lipids. After this, LipidSearch (version 4.0) software was further used to validate the identification with comparison of details of MS2 fragments, including the length of the structural chain and the position of the double bond. The thresholds of parameters of tR, MS1, and MS2 were set to 0.25 min and 0.01 and 0.025 Da in the LipidSearch platform, respectively. To the molecules with no matching of MS2, the information of tR and MS1 was used to annotate the class of lipids, including the length of chain and the position of the double bond.
Quantitative reverse transcription polymerase chain reaction
Total RNA was extracted with TRIzol (Invitrogen), and 5 μg of RNA was used to generate complementary DNA. Transcriptional levels of genes were determined using Premix Ex Taq (Takara) and analyzed with a CFX-96 Real-Time system (Bio-Rad). The primers used in this study are listed in table S3.
Cellular PE measurement
Cells (1 × 106) were lysed in 1 ml of homogenization solution containing 5% Triton X-100, heated to 80°C, and cooled down to room temperature twice to solubilize all lipids. Cellular PE was measured with the PE Assay Kit (BioVision, K499) following the manufacturer’s instruction. The fluorescence signal was measured at excitation/emission = 535/587 nm.
Generation of Pcyt2 knockout mESCs
For generating CRISPR-Cas9–mediated Pcyt2 knockout in mESCs, guide RNAs (gRNAs) for Pcyt2 were designed on the CRISPR design website (crispr.mit.edu) and cloned into the pX330 vector. For targeting, 1 × 106 mESCs were electroporated with 3 μg of pX300 plasmid containing gRNA for Pcyt2 and then seeded in gelatin-coated plates. After 48 hours, ESC colonies were picked and verified by genomic PCR. The gRNA sequences are shown in fig. S2E.
RNA sequencing
Total RNA was extracted with TRIzol (Invitrogen). Libraries were prepared using an Illumina TruSeq RNA Sample Prep kit following the manufacturer’s instruction. The experiments were performed at the Annoroad Gene Technology Co. Ltd. (Beijing, China). Data were analyzed with RSEM software. The GO analysis was performed using DAVID database (https://david.ncifcrf.gov). The P values represent the modified Fisher’s exact corrected EASE score.
Western blot
Whole-cell extracts were obtained with radioimmunoprecipitation assay (RIPA) buffer (Beyotime). Nuclear protein extracts were prepared with the kit (Beyotime) according to the manufacturer’s instructions. Proteins were analyzed with SDS–polyacrylamide gel electrophoresis (PAGE) and then transferred to polyvinylidene difluoride (PVDF) membrane (Millipore). After incubation with antibodies, the membrane was exposed to X film. Relative band density was analyzed using ImageJ. The following primary antibodies were used: anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Bioworld, AP2063, 1:5000), anti-Cdh1 (Cell Signaling, 3195, 1:1000), anti–NF-κB p65 (Cell Signaling, 8242, 1:1000), anti–histone H3 (Abcam, ab1791, 1:5000), anti–phospho-IKKα/β (Cell Signaling, 2697, 1:1000), anti-Snail (Cell Signaling, 3879, 1:1000), anti-Twist1 (Proteintech, 25465-1-AP, 1:500), anti-ERK (Cell Signaling, 9102, 1:1000), anti–phospho-ERK (Cell Signaling, 9101, 1:1000), anti-LC3B (Cell Signaling, 2775, 1:1000), and anti-Pebp1 (Invitrogen, 36-0700, 1:1000). The secondary antibody was goat anti-rabbit (KangChen, KC-RB-035, 1:5000).
Cell migration assay
Scratch assay was performed to determine the migration ability of MEFs undergoing reprogramming. Briefly, MEFs in a confluent monolayer were scratched with pipette tips to create a cell-free scratch into which cells at the edges of the wound can migrate. Relative migration area was calculated by subtracting the total wound area at 24 hours from the total wound area at 0 hour after wounding and then normalized to control.
Immunofluorescence
Cells on glass dishes were fixed with 4% paraformaldehyde for 30 min, permeabilized in 0.1% Triton X-100 for 30 min, and then blocked in 10% FBS (Gibco) for 1 hour at room temperature. Cells were incubated with primary antibodies for 3 hours and then with secondary antibodies for 1 hour at room temperature. Next, the cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (0.1 μg/ml) (Sigma) and analyzed using a Zeiss LSM 710 confocal microscope. The following primary antibodies were used: anti-Cdh1 (Cell Signaling, 3195, 1:200), anti-Rex1 (Santa Cruz Biotechnology, sc-50668, 1:200), anti- SSEA1 (R&D Systems, MAB2155, 1:200), anti-Pebp1 (Invitrogen, 36-0700, 1:200), and anti–NF-κB p65 (Cell Signaling, 8242, 1:200). The secondary antibody was goat anti-rabbit DyLight 488 (Abcam, ab96883, 1:1000) or goat anti-rabbit DyLight 594 (Abcam, ab96885, 1:1000).
Flow cytometry
Cells were fixed with 4% paraformaldehyde for 30 min and blocked in 0.1% bovine serum albumin (BSA) for 30 min at room temperature. Cells were stained with anti-Cdh1 (Cell Signaling, 3195, 1:200) and then secondary antibody goat anti-rabbit DyLight 488 (Abcam, ab96883, 1:200). Next, cells were analyzed with a BD Accuri C6 Plus flow cytometer.
Immunoprecipitation
Cells were lysed in cell lysis buffer [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 1% NP-40, and protease inhibitor cocktail] for 30 min. Antibodies anti-Pebp1 (Invitrogen, 3E12D7, 1:200) and normal mouse immunoglobulin G (Merck Millipore, 12-371, 1:200) were linked to Dynabeads with protein A and G (Invitrogen) for 4 hours at 4°C in lysis buffer. Lysates were incubated with the washed antibody-linked Dynabeads overnight at 4°C. After immunoprecipitation, the beads were washed with lysis buffer five times and boiled in SDS buffer for 10 min. The eluents were analyzed by Western blot. The following primary antibodies were used: anti-Pebp1 (Invitrogen, 36-0700, 1:1000), anti-IKKα (Cell Signaling, 2682, 1:1000), and anti-IKKβ (Cell Signaling, 2370, 1:1000). The secondary antibody was goat anti-rabbit (KangChen, KC-RB-035, 1:5000).
Chromatin immunoprecipitation qPCR
Cells were cross-linked with 1% formaldehyde and then quenched with 125 mM glycine. Fixed cells were lysed in nuclei extraction buffer [50 mM Hepes KOH (pH 7.5), 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, and protease inhibitor cocktail]. Pellets were resuspended in protein extraction buffer [10 mM tris-HCl (pH 8.0), 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, and protease inhibitor cocktail] and sheared by sonication. Sheared chromatin was diluted 10 times with ChIP IP buffer [0.01% SDS, 1% Triton X-100, 2 mM EDTA, 50 mM tris-HCl (pH 8.0), 150 mM NaCl, and protease inhibitor cocktail]. Antibodies were linked to Dynabeads with protein A and G (Invitrogen) for 4 hours at 4°C in PBST (phosphate-buffered saline supplemented with 0.01% Tween 20). Diluted chromatin was incubated with antibodies overnight at 4°C. After immunoprecipitation, beads were washed with low-salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl (pH 8.0), and 150 mM NaCl], high-salt wash buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-HCl (pH 8.0), and 500 mM NaCl], LiCl wash buffer [0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, and 10 mM tris-HCl (pH 8.1)], and TE buffer [10 mM tris-HCl and 1 mM EDTA (pH 8.0)]. DNA was extracted with Chelex-100 and used for analysis. The antibody used for ChIP was anti–NF-κB p65 (Cell Signaling, 8242, 1:200). ChIP-qPCR primers are listed in table S4.
Statistical analysis
Data are presented as mean ± SD or mean ± SEM. Sample number (n) indicates the number of independent biological samples in each experiment. Statistical comparisons were performed using unpaired two-tailed Student’s t tests or one-way analysis of variance. P < 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 6.
Supplementary Material
Acknowledgments
We thank Jiekai Chen for providing iCD1 medium. We also thank all the members in the laboratory of X.L. Funding: This work was financially supported by the National Key Research and Development Program of China (2018YFA0107100), Strategic Priority Research Program of the Chinese Academy of Sciences (XDA16030505), National Key Research and Development Program of China (2017YFC1001602, 2017YFA0106300, 2017YFA0102900, and 2016YFA0100300), National Natural Science Foundation projects of China (U1601227, 31622037, 31631163001, 81570520, 31701281, 31701106, 31601176, 31601088, 31801168, 31900614, 31970709, and 81901275), Key Research Program of Frontier Sciences, CAS (QYZDB-SSW-SMC001), CAS STS Program, Guangzhou Health Care and Cooperative Innovation Major Project (201704020218 and 201604020009), Guangdong Province Science and Technology Program (2015TX01R047, 2017B020230005, 2017A020215056, 2017B030314056, 2018A030313825, and 2018GZR110103002), Guangzhou Science and Technology Program (201707010178 and 201807010067), Yangtze River Scholar Bonus Schemes (to X.L.), and CAS Youth Innovation Promotion Association (to K.C.). Author contributions: X.L. conceived, designed, and supervised the project. Y.W. and K.C. designed and performed the experiments and analyzed data. G.X. participated in plasmid construction. L.L. participated in iPSC generation and ChIP experiments. B.M. and Z.H. participated in qRT-PCR analysis. L.D. provided human hepatocyte differentiation samples. X.L., Y.W., and K.C. wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The shEtnk1, shEtnk2, and shPcyt2 plasmids can be provided by X.L. pending scientific review and a completed material transfer agreement. Requests for the shEtnk1, shEtnk2, and shPcyt2 plasmids should be submitted to X.L. RNA-seq data are deposited under accession number GSE116080 in the Gene Expression Omnibus database.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax7525/DC1
Fig. S1. The CDP-Etn pathway is required for somatic cell reprogramming.
Fig. S2. Characterization of iPSCs generated from iCD1 or ΔiCD1 and generation of Pcyt2 knockout mESCs.
Fig. S3. The CDP-Etn pathway acceleration of MET depends on Pebp1.
Fig. S4. The CDP-Etn-Pebp1 axis modulates NF-κB signaling to inhibit mesenchymal genes.
Table S1. Lipid species identified MEFs, mESCs, and MEFs undergoing SKO reprogramming on days 2, 4, 6, and 8.
Table S2. shRNA target sequences.
Table S3. Primers for qRT-PCR.
Table S4. Primers for ChIP-qPCR.
REFERENCES AND NOTES
- 1.Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S., Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Buganim Y., Faddah D. A., Jaenisch R., Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 14, 427–439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folmes C. D., Nelson T. J., Martinez-Fernandez A., Arrell D. K., Lindor J. Z., Dzeja P. P., Ikeda Y., Perez-Terzic C., Terzic A., Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab. 14, 264–271 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Teslaa T., Teitell M. A., Pluripotent stem cell energy metabolism: An update. EMBO J. 34, 138–153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moussaieff A., Rouleau M., Kitsberg D., Cohen M., Levy G., Barasch D., Nemirovski A., Shen-Orr S., Laevsky I., Amit M., Bomze D., Elena-Herrmann B., Scherf T., Nissim-Rafinia M., Kempa S., Itskovitz-Eldor J., Meshorer E., Aberdam D., Nahmias Y., Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 21, 392–402 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Wang J., Alexander P., Wu L., Hammer R., Cleaver O., McKnight S. L., Dependence of mouse embryonic stem cells on threonine catabolism. Science 325, 435–439 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shyh-Chang N., Locasale J. W., Lyssiotis C. A., Zheng Y., Teo R. Y., Ratanasirintrawoot S., Zhang J., Onder T., Unternaehrer J. J., Zhu H., Asara J. M., Daley G. Q., Cantley L. C., Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339, 222–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Panopoulos A. D., Yanes O., Ruiz S., Kida Y. S., Diep D., Tautenhahn R., Herrerias A., Batchelder E. M., Plongthongkum N., Lutz M., Berggren W. T., Zhang K., Evans R. M., Siuzdak G., Izpisua Belmonte J. C., The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 22, 168–177 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yanes O., Clark J., Wong D. M., Patti G. J., Sanchez-Ruiz A., Benton H. P., Trauger S. A., Desponts C., Ding S., Siuzdak G., Metabolic oxidation regulates embryonic stem cell differentiation. Nat. Chem. Biol. 6, 411–417 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meissen J. K., Yuen B. T., Kind T., Riggs J. W., Barupal D. K., Knoepfler P. S., Fiehn O., Induced pluripotent stem cells show metabolomic differences to embryonic stem cells in polyunsaturated phosphatidylcholines and primary metabolism. PLOS ONE 7, e46770 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Y., Chen K., Liu X., Huang L., Zhao D., Li L., Gao M., Pei D., Wang C., Liu X., Srebp-1 interacts with c-Myc to enhance somatic cell reprogramming. Stem Cells 34, 83–92 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Lin Z., Liu F., Shi P., Song A., Huang Z., Zou D., Chen Q., Li J., Gao X., Fatty acid oxidation promotes reprogramming by enhancing oxidative phosphorylation and inhibiting protein kinase C. Stem Cell Res. Ther. 9, 47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L., Zhang T., Wang L., Cai Y., Zhong X., He X., Hu L., Tian S., Wu M., Hui L., Zhang H., Gao P., Fatty acid synthesis is critical for stem cell pluripotency via promoting mitochondrial fission. EMBO J. 36, 1330–1347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rinschen M. M., Ivanisevic J., Giera M., Siuzdak G., Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 20, 353–367 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guijas C., Montenegro-Burke J. R., Warth B., Spilker M. E., Siuzdak G., Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat. Biotechnol. 36, 316–320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang B., Rong X., Palladino E. N. D., Wang J., Fogelman A. M., Martin M. G., Alrefai W. A., Ford D. A., Tontonoz P., Phospholipid remodeling and cholesterol availability regulate intestinal stemness and tumorigenesis. Cell Stem Cell 22, 206–220.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ye C., Sutter B. M., Wang Y., Kuang Z., Tu B. P., A metabolic function for phospholipid and histone methylation. Mol. Cell 66, 180–193.e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vance J. E., Tasseva G., Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta 1831, 543–554 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Vance D. E., Vance J. E., Physiological consequences of disruption of mammalian phospholipid biosynthetic genes. J. Lipid Res. 50, S132–S137 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J., Liu J., Chen Y., Yang J., Chen J., Liu H., Zhao X., Mo K., Song H., Guo L., Chu S., Wang D., Ding K., Pei D., Rational optimization of reprogramming culture conditions for the generation of induced pluripotent stem cells with ultra-high efficiency and fast kinetics. Cell Res. 21, 884–894 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xuan Q., Hu C., Yu D., Wang L., Zhou Y., Zhao X., Li Q., Hou X., Xu G., Development of a high coverage pseudotargeted lipidomics method based on ultra-high performance liquid chromatography-mass spectrometry. Anal. Chem. 90, 7608–7616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Locasale J. W., Cantley L. C., Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 14, 443–451 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samavarchi-Tehrani P., Golipour A., David L., Sung H. K., Beyer T. A., Datti A., Woltjen K., Nagy A., Wrana J. L., Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7, 64–77 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Li R., Liang J., Ni S., Zhou T., Qing X., Li H., He W., Chen J., Li F., Zhuang Q., Qin B., Xu J., Li W., Yang J., Gan Y., Qin D., Feng S., Song H., Yang D., Zhang B., Zeng L., Lai L., Esteban M. A., Pei D., A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Rockenfeller P., Koska M., Pietrocola F., Minois N., Knittelfelder O., Sica V., Franz J., Carmona-Gutierrez D., Kroemer G., Madeo F., Phosphatidylethanolamine positively regulates autophagy and longevity. Cell Death Differ. 22, 499–508 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao J., O’Donnell V. B., Balzar S., St. Croix C. M., Trudeau J. B., Wenzel S. E., 15-Lipoxygenase 1 interacts with phosphatidylethanolamine-binding protein to regulate MAPK signaling in human airway epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 108, 14246–14251 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Girardi J. P., Pereira L., Bakovic M., De novo synthesis of phospholipids is coupled with autophagosome formation. Med. Hypotheses 77, 1083–1087 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Wang S., Xia P., Ye B., Huang G., Liu J., Fan Z., Transient activation of autophagy via Sox2-mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell 13, 617–625 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Wu Y., Li Y., Zhang H., Huang Y., Zhao P., Tang Y., Qiu X., Ying Y., Li W., Ni S., Zhang M., Liu L., Xu Y., Zhuang Q., Luo Z., Benda C., Song H., Liu B., Lai L., Liu X., Tse H.-F., Bao X., Chan W.-Y., A. Esteban M., Qin B., Pei D., Autophagy and mTORC1 regulate the stochastic phase of somatic cell reprogramming. Nat. Cell Biol. 17, 715–725 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Al-Mulla F., Bitar M. S., Taqi Z., Yeung K. C., RKIP: Much more than Raf kinase inhibitory protein. J. Cell. Physiol. 228, 1688–1702 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Prieto J., León M., Ponsoda X., Sendra R., Bort R., Ferrer-Lorente R., Raya A., López-García C., Torres J., Early ERK1/2 activation promotes DRP1-dependent mitochondrial fission necessary for cell reprogramming. Nat. Commun. 7, 11124 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torres J., Watt F. M., Nanog maintains pluripotency of mouse embryonic stem cells by inhibiting NFκB and cooperating with Stat3. Nat. Cell Biol. 10, 194–201 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Yeung K. C., Rose D. W., Dhillon A. S., Yaros D., Gustafsson M., Chatterjee D., McFerran B., Wyche J., Kolch W., Sedivy J. M., Raf kinase inhibitor protein interacts with NF-κB-inducing kinase and TAK1 and inhibits NF-κB activation. Mol. Cell. Biol. 21, 7207–7217 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cano A., Perez-Moreno M. A., Rodrigo I., Locascio A., Blanco M. J., del Barrio M. G., Portillo F., Nieto M. A., The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2, 76–83 (2000). [DOI] [PubMed] [Google Scholar]
- 35.Pei D., Shu X., Gassama-Diagne A., Thiery J. P., Mesenchymal-epithelial transition in development and reprogramming. Nat. Cell Biol. 21, 44–53 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Li Q., Hutchins A. P., Chen Y., Li S., Shan Y., Liao B., Zheng D., Shi X., Li Y., Chan W.-Y., Pan G., Wei S., Shu X., Pei D., A sequential EMT-MET mechanism drives the differentiation of human embryonic stem cells towards hepatocytes. Nat. Commun. 8, 15166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tatapudy S., Aloisio F., Barber D., Nystul T., Cell fate decisions: Emerging roles for metabolic signals and cell morphology. EMBO Rep. 18, 2105–2118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shimizu Y., Satou M., Hayashi K., Nakamura Y., Fujimaki M., Horibata Y., Ando H., Watanabe T., Shiobara T., Chibana K., Takemasa A., Sugimoto H., Anzai N., Ishii Y., Matrix-assisted laser desorption/ionization imaging mass spectrometry reveals changes of phospholipid distribution in induced pluripotent stem cell colony differentiation. Anal. Bioanal. Chem. 409, 1007–1016 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Pitson S. M., Pebay A., Regulation of stem cell pluripotency and neural differentiation by lysophospholipids. Neurosignals 17, 242–254 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Qin H., Hejna M., Liu Y., Percharde M., Wossidlo M., Blouin L., Durruthy-Durruthy J., Wong P., Qi Z., Yu J., Qi L. S., Sebastiano V., Song J. S., Ramalho-Santos M., YAP induces human naive pluripotency. Cell Rep. 14, 2301–2312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax7525/DC1
Fig. S1. The CDP-Etn pathway is required for somatic cell reprogramming.
Fig. S2. Characterization of iPSCs generated from iCD1 or ΔiCD1 and generation of Pcyt2 knockout mESCs.
Fig. S3. The CDP-Etn pathway acceleration of MET depends on Pebp1.
Fig. S4. The CDP-Etn-Pebp1 axis modulates NF-κB signaling to inhibit mesenchymal genes.
Table S1. Lipid species identified MEFs, mESCs, and MEFs undergoing SKO reprogramming on days 2, 4, 6, and 8.
Table S2. shRNA target sequences.
Table S3. Primers for qRT-PCR.
Table S4. Primers for ChIP-qPCR.