

Extensive clonal T cell expansion is associated with response to immunotherapy.
Abstract
In both human and murine systems, we have developed an adoptive cellular therapy platform against medulloblastoma and glioblastoma that uses dendritic cells pulsed with a tumor RNA transcriptome to expand polyclonal tumor-reactive T cells against a plurality of antigens within heterogeneous brain tumors. We demonstrate that peripheral TCR Vβ repertoire analysis after adoptive cellular therapy reveals that effective response to adoptive cellular therapy is concordant with massive in vivo expansion and persistence of tumor-specific T cell clones within the peripheral blood. In preclinical models of medulloblastoma and glioblastoma, and in a patient with relapsed medulloblastoma receiving adoptive cellular therapy, an early and massive expansion of tumor-reactive lymphocytes, coupled with prolonged persistence in the peripheral blood, is observed during effective therapeutic response to immunotherapy treatment.
INTRODUCTION
Adoptive T cell therapies using tumor-infiltrating lymphocytes (TILs) and chimeric antigen receptor T cells have been demonstrably efficacious against several advanced cancers (1–8). The efficacy of these approaches requires selection of tumor-reactive T cells or ex vivo engineering of antigen-specific T cells using previously identified tumor-specific antigens. Despite these advances, the application of these approaches for brain tumors faces major limitations such as their antigenic heterogeneity and largely uncharacterized tumor-specific antigens. We have pioneered an adoptive cellular therapy (ACT) platform using total tumor RNA (ttRNA)–pulsed dendritic cells (DCs) to generate a polyclonal population of 80% CD8+ tumor-specific T cells with the capacity to target refractory intracranial brain tumors in preclinical models of glioblastoma, medulloblastoma, and brainstem glioma (9, 10). Tumor-reactive T cells for ACT are generated without prior knowledge of the relevant tumor rejection antigen(s) yet are highly tumor specific. To monitor the immunologic responses against uncharacterized and patient-specific antigens expressed within tumors, we identified tumor-specific T lymphocytes from the bulk of ex vivo activated T cells through evaluation of T cell receptor (TCR) Vβ repertoire expansion using flow cytometry–based spectratyping, functional analysis of T cell reactivity, and TCR DNA sequencing.
RESULTS AND DISCUSSION
Previous studies have demonstrated the feasibility of using TCR Vβ flow cytometric analysis in detection of lymphoproliferation in the peripheral blood of patients (11, 12). In this study, we first established the TCR Vβ family repertoire in TILs in two preclinical models of brain malignancies, orthotopic molecular subtype group 3 medulloblastoma (NSC) (13) and cortical high-grade glioma (KR158B) (14). Of the 15 families analyzed, we found no significant hyperexpansion of any one family relative to all other TCR Vβ families in untreated intracranial NSC or intracranial KR158B (fig. S1).
We next demonstrate that flow cytometric analysis of TCR Vβ expression can be used to both identify and enrich for the tumor-specific T cells from a bulk population of lymphocytes during ACT against NSC medulloblastoma and KR158B high-grade glioma. This is significant because tumor-associated antigens in brain cancers are largely uncharacterized and tumor-associated antigens in these preclinical models are completely uncharacterized. Despite this, we have identified and expanded the tumor-reactive T lymphocytes against multiple tumor models using ACT. To demonstrate this, C57BL/6 mice bearing orthotopic cerebellar NSC medulloblastoma were treated with ACT (Fig. 1A). Spleens were harvested from treated asymptomatic mice at 30, 50, and 120 days after ACT, and TCR sequencing was conducted on bulk splenocytes. Analysis revealed selective expansion of T cell clones within four TCR Vβ families, TCR Vβ 13-02+, 16-01+, 19-01+, and 24-01+, by 120 days after treatment, as compared to the ex vivo expanded T cells before intravenous infusion (Fig. 1B). Clonal analysis revealed that within the T cells that express TCR Vβ 13-02+, there is an approximate 10-fold clonal T cell expansion of productive frequency in treated mice over time, from 0.168% productive frequency in ex vivo activated T cells to 1.737% productive frequency in mice 120 days after ACT (Fig. 1C).
Fig. 1. Clonal T cell expansion in murine splenocytes after ACT against medulloblastoma.
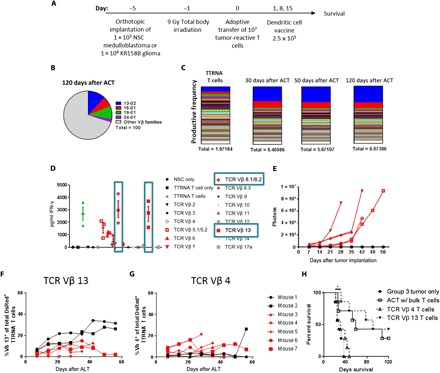
(A) Experimental layout of ACT in tumor-bearing mice. Intracranial tumor–bearing hosts received 9 gray (Gy) total body irradiation for host conditioning followed by adoptive transfer of tumor-reactive T cells and weekly DC vaccines. Hosts also receive hematopoietic stem cell transfer after total body irradiation to protect from bone marrow failure. (B) Splenocytes of treated animals were harvested at 30, 50, and 120 days for long-term survivors, were analyzed for TCR sequencing, and revealed selective expansion of four TCR Vβ families. (C) Intrafamily analysis revealed that TCR Vβ 13-02 family T cells experienced clonal expansion of one clone. Other families did experience clonal expansion. (D) Fifteen TCR Vβ families from bulk tumor-reactive T cells were isolated using FACS, and each family was cocultured against target NSC tumor cells. IFN-γ secretion was measured as an indication of the recognition of cognate tumor antigen. (E) NSC express a luciferase reporter, allowing for bioluminescent imaging of tumor growth that was conducted in mice treated with ACT. (F) Relative expansion of adoptively transferred T cells in the TCR Vβ 13 family in murine peripheral blood mononuclear cells (PBMCs) was measured in concurrence with tumor growth using flow cytometry. (G) Relative expansion of adoptively transferred T cells in the TCR Vβ 4 family in murine PBMCs was also measured as a control as this Vβ family did not demonstrate prior antitumor reactivity. (H) Cerebellar NSC medulloblastoma-bearing mice received ACT with either bulk ex vivo expanded tumor-reactive T cells or tumor-reactive T cells that only express TCR Vβ 13 or Vβ 4. Experiments (D) to (G) were conducted at least three times with 7 to 10 mice per experiment. (H) was conducted twice with n = 7 mice per group.
To determine whether this expansion of TCR Vβ 13+ T cells contributes to efficacy of ACT against this group 3 medulloblastoma, sterile fluorescence-activated cell sorting (FACS) was used to isolate 15 different TCR Vβ families from the bulk medulloblastoma-specific T cells. Ex vivo activated tumor-reactive T cells from each of the isolated TCR Vβ families were then cocultured against NSC medulloblastoma tumor cells. Supernatant interferon-γ (IFN-γ) was measured by enzyme-linked immunosorbent assay (ELISA) to indicate recognition of cognate tumor antigen. As expected, the bulk T cell population secreted IFN-γ (2681.67 ± 534.618 pg/ml) in response to tumor, while in the sorted populations, only T cells that express TCR Vβ 5.1/5.2+, 6+, 7+, 8.1/8.2+, or 13+ secreted statistically similar amounts of IFN-γ in response to tumor targets (P = 0.400, 0.100, 0.100, 0.700, and >0.999, respectively) (Fig. 1D). Other TCR Vβ families were unresponsive against NSC tumor cells, secreting little to no IFN-γ.
We next sought to determine whether the observed expansion of TCR Vβ 13+ T cells correlates with increased survival and efficacy against NSC medulloblastoma. NSC tumors were implanted into the cerebellums of mice, and tumor-bearing mice were treated with ACT using NSC-specific T cells generated from DsRed+ transgenic mice. Tumor growth was monitored over time using bioluminescent imaging (Fig. 1E). Peripheral blood was also sampled and measured for relative frequency of adoptively transferred ex vivo activated antitumor T cells that expressed TCR Vβ 13+ (Fig. 1F). Mice that were responsive to treatment and had long-term survival demonstrated increased relative frequency of TCR Vβ 13+ T cells over time. Other DsRed+ T cells from other TCR Vβ families including TCR Vβ 4+ did not demonstrate lymphocyte expansion (Fig. 1G). Next, we determined whether TCR Vβ 13+ T cells provide protective immunity against NSC medulloblastoma. NSC group 3 medulloblastoma was implanted into the cerebellum of mice, which were then treated with ACT using total bulk ex vivo expanded tumor-reactive T cells (9, 10). Other cohorts received only expanded T cells that express either TCR Vβ 13+, isolated by FACS, which has demonstrated antitumor function, or Vβ 4+, which has not had any prior antitumor reactivity. The group that received only T cells from the TCR Vβ 13+ family had an observed survival benefit equal to the ACT group that received the total bulk tumor-reactive T cells (P = 0.5369) (Fig. 1H). In a subsequent experiment, we adoptively transferred only TCR Vβ 6+, 7+, 8.1/8.2+, or 11+ T cells, but no survival benefit was observed over tumor-only controls (fig. S2). Although previous in vitro experiments show that TCR Vβ 8.1/8.2+ T cells demonstrate antitumor reactivity, adoptive transfer of these cells alone did not provide immunological protection against NSC tumors. This may be due to a lack of in vivo expansion of TCR Vβ 8.1/8.2+ T cells, although this mechanism of escape remains unclear. This further demonstrates that tumor-reactive T cells expressing TCR Vβ 13+ play a major role in the immunological rejection of orthotopic NSC medulloblastoma.
We then conducted experiments to seek out differences in relative frequencies of T cells in spleens of responders versus nonresponders to therapy. Responders were defined as mice that were asymptomatic and demonstrated absence of luminescence signal at day 90, while nonresponders were defined as mice that had become symptomatic and showed tumor growth by bioluminescence after ACT. In splenocytes of mice treated with ACT, responders demonstrated selective in vivo expansion of six TCR Vβ families Vβ 5.1/5.2+, 6+, 7+, 8.1/8.2+, 8.3+, and 13+ relative to nonresponders (Fig. 2A). Spleens of the responders were collected and were sorted via FACS for the adoptively transferred DsRed+ tumor–reactive T cells, and then further isolated by each TCR Vβ family. These T cells were then used as effector T lymphocytes in a functionality assay targeting tumor cells. DsRed+ T cells from TCR Vβ 5.1/5.2+, 6+, 8.1/8.2+, and 13+ families retained antitumor reactivity in vivo in long-term survivors (Fig. 2B). Splenic T cells were also harvested from nonresponders to therapy upon detection of intracranial tumor growth by bioluminescent imaging, and DsRed+ T cells from TCR Vβ 5.1/5.2+, 8.1/8.2+, 11+, and 13+ families were FACS-isolated and then used as effector T cells in a functionality assay targeting primary NSC tumor cells (Fig. 2C). TCR Vβ 8.1/8.2+ and 13+ retained their antitumor reactivity when targeted against the NSC cell line, demonstrating that the adoptively transferred T cells maintain their antitumor function upon tumor escape. This indicates that loss of T cell function is likely not responsible for escape from this adoptive immunotherapy platform.
Fig. 2. Selective expansion of the tumor-reactive TCR Vβ family in mice responsive to ACT.
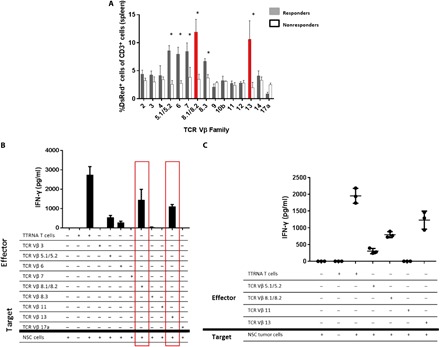
To generate antitumor T cells, total RNA is extracted from tumor cells and electroporated into syngeneic bone marrow–derived DCs. These cells are then cocultured with splenocytes from a previously immunized mouse with interleukin-2 for 5 to 7 days generating a polyclonal population of CD8+ T cells. After this ex vivo activation, 107 T cells are adoptively transferred into tumor-bearing mice followed by vaccination with 2.5 × 105 RNA-pulsed DCs. In the preclinical model of ACT, C57BL/6 mice receive orthotopic tumor followed by host conditioning with total body irradiation and hematopoietic stem cell transfer to protect from bone marrow failure. (A) Mice implanted with cerebellar NSC medulloblastoma were treated with ACT using DsRed+ tumor–reactive T cells. Spleens were harvested from all mice, and relative abundance of each TCR Vβ family was measured in both responders and nonresponders. Here, 25 mice are implanted with tumor and treated with ACT. The first five nonresponders that succumb to tumor are taken at humane end point and spleens were analyzed. The five responders are treated mice that demonstrate no evidence of tumor after 120 days. This experiment was repeated twice with the same results as shown. n = 5 to 7 mice per group. (B) Spleens of five asymptomatic long-term survivors were harvested at 90 days after ACT. DsRed+ T cells were isolated and separated by the TCR Vβ family. Each TCR Vβ family was cocultured in vitro against tumor cells, and IFN-γ secretion was measured. (C) Splenic T lymphocytes were harvested from nonresponders to therapy upon detection of tumor via bioluminescent imaging. DsRed+ T cells were FACS-isolated and sorted into TCR Vβ families and then used as effectors against the primary NSC cell line. IFN-γ was measured to determine antitumor reactivity.
These experiments were repeated in a KR158B-luc glioma (KR158B stably transfected with luciferase) (14) preclinical model of invasive high-grade glioma, which corroborated the finding that tumor-reactive T cells can be identified by isolation of hyper-expanded TCR Vβ families after ACT and tested ex vivo. We generated KR158B-reactive T cells ex vivo and used FACS to isolate T cells by the TCR Vβ family. Each family was tested for antitumor reactivity against KR158B tumor cells in a coculture assay, and IFN-γ was detectable from TCR Vβ 5.1/5.2+, 6+, and 8.1/8.2+ T cells (Fig. 3A). In mice treated with ACT against KR158B-luc glioma (using tumor-reactive T cells generated from DsRed+ mice to allow for longitudinal studies), responders demonstrated robust expansion of adoptively transferred TCR Vβ 6+ T cells in the spleen but no other Vβ families (Fig. 3B). The spleens of long-term responders were harvested 120 days after treatment, and DsRed+ T cells were FACS-sorted into TCR Vβ families. Each family was cocultured to target KR158B tumor cells, and IFN-γ was measured, revealing that T cells in the Vβ 6+ and 8.1/8.2+ families retained antitumor functionality in long-term survivors (Fig. 3C).
Fig. 3. Identification of tumor-specific T lymphocytes in a preclinical glioma model.
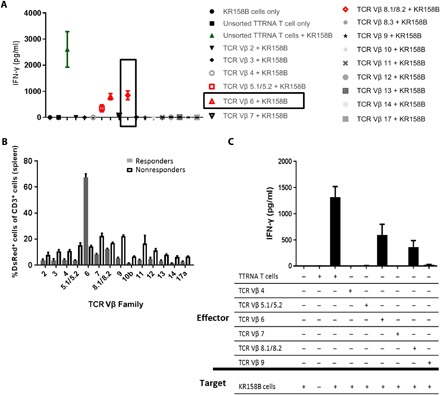
(A) Tumor-reactive T cells were generated in vitro and separated into 15 TCR Vβ families using sterile FACS isolation. T cells (4 × 105) per Vβ family were cocultured against 4 × 104 KR158B tumor target cells overnight, and supernatant IFN-γ was measured as an indication of the recognition of cognate tumor antigen. All conditions were conducted in triplicate, and the experiment was repeated an additional three times with the same results. (B) Fifteen mice received ACT using DsRed+ tumor–reactive T cells. Relative frequencies of TCR Vβ families within the adoptively transferred DsRed+ T cell population were compared between the first five nonresponders to therapy and five long-term survivors with no signs of tumor. (C) Spleens of the asymptomatic long-term survivors were also harvested for DsRed+ T cells, which were further separated by the TCR Vβ family using FACS. Each TCR Vβ family was cocultured in vitro against tumor cells as above, and IFN-γ secretion was measured.
To determine whether TCR Vβ 6+ glioma-specific T cells demonstrate relative expansion in the peripheral blood over time, ACT using 3 × 107 DsRed+ tumor–specific T cells were administered to mice bearing KR158B-luc glioma (KR158B stably transfected with luciferase), and tumor growth was followed after treatment (Fig. 4, A and B). Peripheral blood was also sampled, and relative frequency of DsRed+ KR158B-specific T cells was measured over time. DsRed+ tumor–specific T cells that expressed TCR Vβ 6+ experienced marked expansion in the peripheral blood in mice that responded to therapy but not in those that failed treatment (Fig. 4C). Other DsRed+ T cells from other TCR Vβ families including TCR Vβ 2+ and 8.1/8.2+ did not demonstrate lymphocyte expansion (fig. S3, A and B).
Fig. 4. TCR Vβ 6+ T cells are required for efficacy of ACT against glioma.
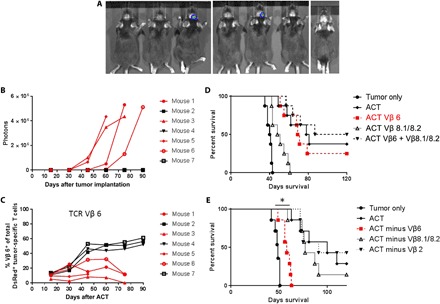
(A) Seven C57BL/6 mice received orthotopic KR158B tumor by implanting 104 tumor cells into the right caudate nucleus of the cortex. (B) This tumor line has a luciferase reporter that allowed for in vivo bioluminescent imaging of tumor growth of mice over time. Relative frequency of (C) TCR Vβ 6+. (D) Intracranial tumor–bearing mice received either ACT using bulk tumor-reactive T cell population, ACT using only TCR Vβ 6+ T cells, ACT using only TCR Vβ 8.1/8.2+ T cells, or ACT using only TCR Vβ 6+ and Vβ 8.1/8.2+ T cells. No significant differences in survival was found between bulk ACT and the group that received TCR Vβ 6+ T cells, n = 7 mice per group. (E) Intracranial tumor–bearing mice received either ACT, ACT with T cells depleted of Vβ 6+ T cells using FACS, ACT with T cells depleted of TCR Vβ 8.1/8.2+ T cells, or ACT with TCR Vβ 2+ T cells. We found a significant decrease in survival in the group where TCR Vβ 6+ cells were depleted as compared to bulk ACT, *P = 0.0003, n = 7 mice per group.
This strongly suggests that TCR Vβ 6+ T cells are the drivers of antitumor immunity against KR158B glioma. To test this hypothesis directly, we generated ex vivo expanded tumor-specific T cells and isolated either TCR Vβ 6+ or TCR Vβ 8.1/8.2+ cells using FACS. These isolated T cell populations were then used for ACT in KR158B tumor–bearing mice, and their survival was compared to a cohort receiving bulk T cells for ACT, which has been previously demonstrably efficacious (Fig. 4D). The prominent survival benefit of ACT was maintained in the group that received only TCR Vβ 6+ T cells (no significant difference in survival was found between the two groups, P = 0.5925) but not TCR Vβ 8.1/8.2+ T cells (tumor P = 0.0152). A group that received both T cell populations had no increase in survival benefit over the ACT control group (Fig. 4D). Conversely, depletion of TCR Vβ 6+ T cells from the tumor-specific cell population before adoptive transfer significantly decreased the survival benefit of ACT (P = 0.0003) (Fig. 4E). Mice that received ACT using tumor-reactive T cells depleted of either TCR Vβ 8.1/8.2+ or TCR Vβ 2+ T cells had no statistically significant difference in median survival compared to the group that received bulk T cells. These data together indicate that TCR Vβ6+ tumor-specific T cells are the functional drivers of antitumor immunity provided by ACT in this specific preclinical model of malignant glioma. These studies also demonstrated that in vivo evaluation of the hyperexpanded T cell Vβ repertoire in responding versus nonresponding cohorts could reveal tumor-specific T cells associated with clinical responsiveness.
In treated mice that eventually succumbed to disease, outgrown tumors were harvested, and the TCR Vβ repertoire was analyzed to determine relative expansion of adoptively transferred T cells. For these experiments, tumor-reactive T cells were generated using DsRed+ mice to enable longitudinal tracking of adoptively transferred cells. In NSC tumor-bearing mice that received ACT, we found no hyperexpansion of any T cell families derived from the adoptively transferred cells (fig. S4A). In KR158B-treated mice, we observed that TCR Vβ 7+ and 8.1/8.2+ DsRed+ T cells experienced a higher frequency of expansion relative to the other TCR Vβ families at the tumor site (fig. S4B). We FACS-isolated these DsRed+ TCR Vβ 6+, 7+, and 8.1/8.2+ T cells from the tumors of mice that failed therapy. When used as effectors in an in vitro functionality assay, we observed that these Vβ 8.1/8.2+ T cells secreted IFN-γ upon recognition of the in vitro passaged KR158B cell line, indicating that antitumor T cell function was not the cause of escape (fig. S4C). We also isolated other DsRed+ TCR Vβ families including Vβ 7+ T cells from the escaped tumors and found that these T cells did not demonstrate antitumor reactivity (fig. S4C). We then sought to increase survival against KR158B in tumor-bearing mice after treatment with ACT by using combinatorial anti-programmed cell death protein 1 (αPD-1) with ACT (Fig. 5A). Groups of mice that received ACT + αPD-1 experienced an average of 71.42% long-term survival (range between 60 and 85.7%), a larger proportion than those that received ACT alone (range between 42.8 and 57.1%) (this experiment was repeated a total of three times). As previously discussed above, KR158B-bearing mice that responded to ACT demonstrated an expansion of TCR Vβ 6+ T cells (Fig. 5B). In groups treated with ACT + αPD-1, we also observed that responders to therapy exhibited an expansion of TCR Vβ 6+ T cells (Fig. 5C). Although a greater proportion of mice survived in response to ACT + αPD-1, the exact mechanism by which this occurs is not yet clear.
Fig. 5. PD-1 immune checkpoint blockade increases expansion of tumor-specific T cells.
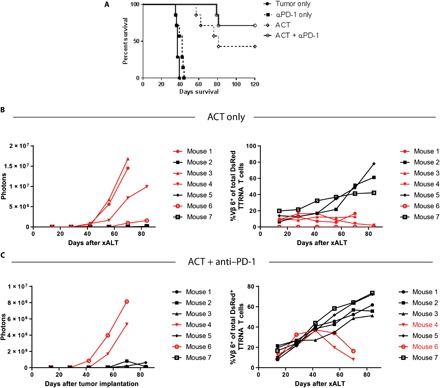
(A) Twenty-eight C57BL/6 mice received orthotopic KR158B glioma by implanting 104 tumor cells into the right caudate nucleus and then randomized into four groups. Mice received either no treatment, αPD-1 only, ACT only, or ACT + αPD-1 (n = 7 mice per group). There was no statistically significant increase in survival between ACT and ACT + αPD-1 groups, P = 0.1755. (B) KR158B tumor–bearing mice received ACT using DsRed+ tumor–reactive T cells alone, or (C) ACT + αPD-1. Tumor growth was followed weekly with bioluminescent in vivo imaging. At the same time points, peripheral blood was drawn to follow relative frequencies of DsRed+ TCR Vβ 6+ T cells over time in both groups, n = 7 mice per group.
In patients with durable responses after adoptive immunotherapy with autologous TIL therapy, sequencing of TCR Vβ demonstrated multiple T cell clonotypes in the adoptively transferred cells, but responses were associated with clonotypic expansion of only a select few over time, and these clones persisted in peripheral blood for one or more months after transfer (8, 15, 16). We have an ongoing phase 1/2 study evaluating the safety and feasibility of ACT using autologous ex vivo activated T cells in children with recurrent medulloblastoma or primitive neuroectodermal tumors (PNETs) that have failed definitive radiation therapy [Re-MATCH; FDA IND no. BB-14058; Principal Investigator (PI): D.M.]. Patients with recurrent medulloblastoma receive either nonmyeloablative salvage chemotherapy or induction chemotherapy followed by myeloablative chemotherapy and stem cell rescue followed by ACT consisting of ex vivo expanded autologous lymphocyte transfer and amplified tumor RNA–pulsed DC vaccines (DC + xALT therapy) and autologous peripheral blood stem cells (fig. S5). A patient who underwent induction and consolidation therapy followed by ACT demonstrated long-term progression-free survival (13 months) that exceeded any patient in our historical cohort of recurrent medulloblastoma patients treated with myeloablative chemotherapy and stem cell rescue alone (16). The longitudinal peripheral blood samples as well as ex vivo expanded T cell products were analyzed by TCR DNA sequencing to evaluate TCR Vβ clonal expansion (table S1). Patient peripheral blood mononuclear cells (PBMCs) were evaluated at pretreatment, in ex vivo expanded tumor-specific T cells, and then at 2, 4, 6, and 16 weeks after T cell infusion. Analysis demonstrated massive and selective expansion of tumor-reactive TCR Vβ clones in the peripheral blood up to 4 months (16 weeks) after treatment (Fig. 6A and table S2).
Fig. 6. Clonal T cell expansion in recurrent medulloblastoma patient PBMCs after ACT.
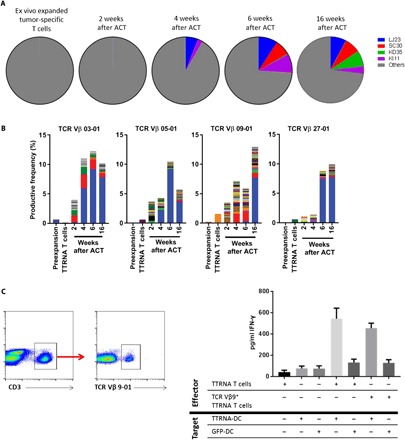
A patient with recurrent medulloblastoma was treated with ACT using autologous ex vivo activated T cells and experienced long-term survival with >2-year nonprogressing tumor. (A) TCR sequencing was conducted on the patient’s tumor-reactive T cells and on patient PBMCs taken at 2, 4, 6, and 16 weeks after ACT. All analysis and bioinformatics were conducted by Adaptive Biotechnologies (Seattle, WA). Productive frequencies of 59 TCR Vβ families were analyzed. Clonal analysis revealed hyperexpansion of five T cell clones, each expanding to greater than 5% productive frequency in PBMCs 16 weeks after treatment. Combined, these five clones make up 28.72% productive frequency of patient PBMCs. (B) Top four TCR Vβ families with the highest expression at 16 weeks after adoptive T cell transfer were plotted relative to the other 54 families at all time points (TCR Vβ 3, Vβ 5-01, Vβ 9-01, and Vβ 27-01). Clonal analysis of the 1000 clones within each family was conducted, revealing that the expanded families are largely composed of the expansion of a single clone. Each color on the graphs represents a single sequence. (C) TCR Vβ 9-01+ T cells from PBMCs were FACS-isolated and cocultured against tumor RNA–pulsed autologous DCs or GFP RNA–pulsed DCs in triplicate. IFN-γ secretion was measured.
Analysis of PBMCs at 16 weeks after treatment shows hyperexpansion of four TCR Vβ clones expressing unique sequences qAGGTRg.14426B03B02S07L14 (herein referred to as LJ23), lFNRGRn.12644B27S01B02S01L13 (herein referred to as SC30), sASGGPh.23441B09S01B01S05L13 (herein referred to as KD35), and sRISGGVq.526441B05S01B02S05L13 (herein referred to as KI11) (Fig. 6A). These four clones comprised 26.82% of all T cells found in PBMCs at 16 weeks after ACT. This patient’s lymphocyte counts normalized after therapy, demonstrating an extraordinary expansion of selective T cell clones after therapy and significant remodeling of the peripheral repertoire after therapy. Preexpansion PBMCs as well as ex vivo expanded T cells contain <1% productive frequency of these four clones combined, but all four clones demonstrated massive in vivo expansion after ACT (Fig. 6B). LJ23 is observed in patient PBMCs before ACT at a productive frequency of 0.000077% but expands to 7.7787833% at 16 weeks after ACT (>100,000-fold in vivo expansion). SC30 is observed in patient mononuclear cells (MNCs) before ACT at a productive frequency of 0.002525% and expands to 7.698551% at 16 weeks after treatment (>3000-fold in vivo expansion). KD35 is observed to be at a productive frequency of 0.001377% before treatment but expands to 7.708697% after ACT (>5500-fold in vivo expansion). Last, KI11 is observed at 0.002448% before ACT and expands to 3.619983% after treatment (>1400-fold in vivo expansion) (table S1).
Fifty-nine TCR Vβ families were expressed in this patient’s PBMCs, revealing that four TCR Vβ families (03+, 05-15+, 09-01+, and 27+) had productive frequencies greater than 5% at 16 weeks after treatment (Fig. 6B). Clonal analysis of each family revealed that each had relative expansion of one dominant clone as outlined in table S2. KD35 T cells, which are in the TCR Vβ 09-01+ family, demonstrated remarkable clonal expansion in patient PBMCs from pretreatment MNCs to bulk tumor-specific T cells, 2, 4, 6, and 16 weeks after T cell transfer (Fig. 6A and table S1). Clone KD35 experienced a remarkable expansion in PBMCs, composed of 7.708697% productive frequency of all T cells and 59.6% of the TCR Vβ 09-01+ family T cells (with no other clone within the 09-01+ reaching >0.5% as seen in Fig. 6B). Since clonal expansion of antigen-specific T cells was an indicator of antitumor reactivity in our preclinical studies, we next wanted to evaluate tumor reactivity of KD35 T cells against the primary tumor. We did not have prior knowledge of the peptide or antigen, which may drive KD35 T cell recognition but were armed with the data that the K35 clone makes up most of the TCR Vβ 9-01+ family T cells. Since monoclonal antibodies for TCR Vβ families are readily available, we isolated TCR Vβ9-01+ family T cells from cryopreserved PBMCs collected at 16 weeks after ACT. These cells were tested for antitumor function and used as effector cells against autologous DCs pulsed with ttRNA isolated from the original patient tumor tissue or control RNA [green fluorescent protein (GFP)]. The use of autologous DCs as surrogate targets allows for all ubiquitously expressed self-antigens to be expressed on the same cell type and control of specificity with the loaded RNA species. IFN-γ secretion was measured to indicate recognition of cognate tumor antigen. TCR Vβ 9+ T cells secreted IFN-γ (454 pg/ml), equal amounts to the bulk ex vivo expanded antitumor T cell population that secreted IFN-γ (543 pg/ml) (P = 0.2321) against tumor RNA–pulsed DCs. T cells targeted against autologous DCs pulsed with GFP RNA demonstrated selective specificity for tumor antigens (P = 0.002) (Fig. 6C). These results demonstrate the in vivo hyperexpansion of TCR Vβ 9-01+ tumor-reactive T cells and persistence of these tumor-reactive T cells for up to 4 months after treatment in the peripheral blood of a patient achieving long-term disease remission.
A recurrent medulloblastoma patient who underwent ACT but did not respond to therapy (<70 days to progression) was also examined for clonal T cell expansion using TCR Vβ sequencing with interesting results. PBMCs were collected at 1 day before ACT, and at 2, 4, and 6 weeks after ACT, and TCR DNA sequencing was conducted. At each time point, the relative productive frequency of the top 1000 clones was determined. At each time point, hyperexpanded TCR clones were found, but most importantly, at each time point, the hyperexpanded clones were different clones from the previous time points. In the previously discussed long-term responder to ACT, we observed the expansion of the same four clones over time. On the contrary, in the nonresponder to therapy, we did not find hyperexpansion of any single clones over time, and no longitudinal detection of hyperexpanded TCR clones was observed (fig. S6).
These data together strongly suggest that clonal expansion of TCR clones in the peripheral blood over time is a predictive biomarker of response to ACT for malignant brain tumors. Functional analysis of the TCR Vβ family for monitoring T cell expansion in peripheral blood of treated patients is now a useful modality that allows the prospective identification and biological characterization of viable tumor-reactive lymphocytes in patients undergoing ACT.
This study describes a method to identify tumor-reactive T cells in ACT against central nervous system malignancies by TCR Vβ spectratyping. We also demonstrate that despite the significant prolongation of life, not all treated hosts experience immunological rejection of their intracranial orthotopic tumors. This study demonstrates that the ex vivo activated tumor-specific T lymphocytes that adoptively transferred retain their antitumor function against the primary tumor, which they were initially activated to target. In these highly heterogeneous tumors, these multimodal immunotherapies are likely to have multiple mechanisms of immune escape. For example, in Fig. 5, the combination of αPD-1 with ACT increased the amount of overall long-term survivors, but a proportion of animals still succumbed to disease. Immunotherapy has been demonstrably promising against solid tumors, but it is imperative to determine mechanisms of failure to make more significant impacts against disease. Given our findings, potential actions of tumor escape could include antigen escape variants, which is highly likely in both high-grade gliomas and medulloblastomas, as these are highly heterogeneous tumors. Identification of these mechanisms is imperative to further progress the efficacy of adoptive immunotherapy.
MATERIALS AND METHODS
Mice
Female six- to eight-week-old C57BL/6 mice (stock 000664) and transgenic DsRed mice (stock 006051) were purchased from the Jackson Laboratory. The investigators adhered to the Guide for the Care and Use of Laboratory Animals as proposed by the committee on care of the Laboratory Animal Resources Commission on Life Sciences, National Research Council. The facilities at the University of Florida Animal Care Services are fully accredited by the American Association for Accreditation of Laboratory Animal Care, and all studies were approved by the University of Florida Institutional Animal Care and Use Committee.
Adoptive cellular therapy
As described in our previous publication by Flores et al. (9), tumor-bearing experiments were performed in a C57BL/6 background with two different cell lines implanted intracranially via stereotaxic frame (Stoelting, Wood Dale, IL). KR158B-luc tumors are intracranial murine astrocytomas (9, 14), and NSC tumors are intracranial murine medulloblastomas (13). Treatment of tumor-bearing mice began with 9-Gy x-ray myeloablation on day 5 after intracranial injection (X-RAD 320, Precision X-ray, North Branford, CT). On day 6 after intracranial injection, mice received 5 × 104 lineage-depleted hematopoietic stem cells (per the manufacturer’s protocol, Miltenyi Biotec, catalog no. 130090858) in addition to 107 autologous ex vivo expanded ttRNA T cells via a single intravenous injection in the tail vein. DC vaccine was injected intradermally posterior to the pinna weekly for 3 weeks with 2.5 × 105 cells beginning day 7 after intracranial injection.
RNA isolation
ttRNA isolation was performed with the RNAeasy Mini Kit from QIAGEN (QIAGEN, catalog no. 74104) per the manufacturer’s protocol for each respective tumor cell line.
Dendritic cells
DCs were generated from a 9-day differentiation and selection protocol as previously described (9). At day 8 of culture, DCs were electroporated as previously described with 25 μg of ttRNA. On day 9 of culture, DCs are available for priming mice, vaccinating tumor-bearing mice, or ex vivo expanding splenocytes to become tumor-reactive T cells.
Autologous T cells
Generation of autologous tumor-reactive T cells was performed as previously described (9). Naïve C57BL/6 mice are primed with 2.5 × 105 ttRNA-pulsed DCs, and 1 week later, primed splenocytes are expanded in a coculture with ttRNA DCs. Between days 5 and 7 of expansion, 107 T cells were isolated and injected intravenously into tumor-bearing mouse tail veins. In the case of TCR Vβ family–specific transplants, cells were FACS-sorted for the TCR Vβ family and transplanted to tumor-bearing hosts.
Flow cytometry
Flow cytometry was performed on FSC/SSC gating on the BD Biosciences FACS Calibur. Cells from transgenic DsRed mice were detected at FL2 and FACS-sorted using the BD Biosciences FACSAria II. Spectratyping TCR Vβ families was performed using a FACSAria II cell sorter with fluorescein isothiocyanate–conjugated antibodies from BD Biosciences anti-mouse TCR Vβ Screening Panel (BD Biosciences, catalog no. 557004). Cells were stained per protocol with 20 μl of antibody per 106 T cells.
TCR sequencing
Human PBMCs were isolated with a buffy coat preparation. Human TCR sequencing was performed by Adaptive Biotechnologies (Seattle, WA) through their immunoSEQ Platform. This assay uses a multiplex polymerase chain reaction system to amplify 87 base pairs spanning the rearranged CDR3β VDJ regions. Sequencing is completed using the Illumina platform (17). All data reported were acquired using their immunoSEQ Analyzer 2.0 TCR sequencing.
T cell functionality restimulation assay
In vitro experiments that analyzed IFN-γ release were performed with effector cells and targets in 96-well U-bottom plates. For the example of TCR-specific T cells and KR158B-luc cells, the coculture was performed with a 20:1 ratio of T cells/KR158B-luc cells. IFN-γ release was measured in picograms per milliliter by IFN-γ Platinum ELISA (eBiosciences, catalog no. BMS606) to determine degree of antitumor activity. ELISA was performed on harvested and frozen acellular media from the supernatants of the 96-well coculture plates after 1 day of culture.
Bioluminescent in vivo imaging
In vivo imaging of intracranial KR158B-luc tumors was performed using an IVIS Kinetic (PerkinElmer). Mice were imaged every 30 s until the peak photon reading is reached minutes after injection with 100 μl of luciferin substrate (Sigma-Aldrich). Bioluminescence for all animals was acquired with 1-s exposure.
TCR nomenclature
Nomenclature of TCRs was adapted from Reilly et al. (14). Briefly, V(D)J rearrangement sequences containing the V gene, CDR3, NDN, and J gene were obtained from Adaptive Biotechnology. The TCR nomenclature is made of five components: (i) the CDR3 amino acid identifier, (ii) the nucleotide sequence identifier, (iii) the TCR V region identifier, (iv) the TCR J region identifier, and (v) the CDR3 length identifier. The lowercase letters at the start and end of the amino acid sequence represent the last amino acids from the V and J segments, respectively, while the uppercase letters represent amino acids that are encoded by the NDN region. The series of numbers following the period represents the nucleotide identifier. Genetic codons are assigned nucleotide identification numbers from the standard codon table. The TCR V and J region identifiers follow the nucleotide sequence identifier, and these gene families were also sequenced by Adaptive Biotechnology. The final component, the CDR3 length identifier, represents the number of amino acids between the C-terminal–conserved cysteine of the V region and the phenylalanine of the J region.
Anti–PD-1
Administration of αPD-1 checkpoint inhibitor (Bio X Cell, catalog no. BP0146) began on the same day as adoptive T cell transfer and continued every 5 days for a total of four doses at 10 mg/kg.
Statistics
All experiments were analyzed in Prism 7 (GraphPad). The median survival for tumor-bearing animals is 25 to 42 days. Tumor-bearing survival experiments in this manuscript are no less than seven animals per group and analyzed with the Mantel-Cox log-rank test. An unpaired Mann-Whitney rank sum test or an unpaired Student’s t test was applied for two-group comparisons for in vivo or in vitro experiments, respectively. Significance is determined as P < 0.05. Animal studies were powered to include n = 5 randomized mice per group unless otherwise noted.
Supplementary Material
Acknowledgments
Funding: This research was supported by the National Cancer Institute R01 CA195563 (D.M.), the V Foundation for Cancer Research Translational Research Award (D.M.), the Hyundai Hope On Wheels Quantum Award (D.M.), the American Brain Tumor Association Research Collaboration Grant (C.F.), Alex’s Lemonade Stand Young Investigator Grant (C.F.), the Florida Center for Brain Tumor Research Grant (C.F.), the University of Florida Health Cancer Center Predoctoral Award (T.W.), and the Wells Foundation (D.M.). Author contributions: Conceptualization: D.M. and C.F. Data curation: C.F., T.W., B.D.D., G.M., J.Dr., R.A., C.M., J.G., D.S., and C.P. Formal analysis: C.F., T.W., B.D.D., O.Y., C.Y., and J.De. Funding acquisition: D.M., C.F., and T.W. Investigation: C.F., T.W., B.D.D., G.M., J.Dr., R.A., and J.G. Methodology: C.F. and D.M. Project administration: D.M. Resources: C.F. and D.M. Software: Adaptive Biosciences. Supervision: C.F., D.M., J.Kr., J.Ki., G.G., T.D., J.Ku., R.M., and S.G. Validation: C.F., B.D.D., T.W., G.M., J.Dr., R.A., and J.G. Visualization: C.F. and T.W. Writing original draft: C.F. Writing review and editing: C.F., T.W., D.M., and B.D.D. Competing interests: C.F. and D.M. are cofounders of iOncologi Inc., an immuno-oncology biotechnology company. C.F. and D.M. hold interests in iOncologi Inc., a biotechnology company focused on immuno-oncology. C.F. and D.M. have patents that have options to be licensed to iOncologi Inc. filed by the University of Florida (no. 2016303489, filed 29 July 2016; no. 2994241, filed 29 July 2016; no. 16833610.5, filed 29 July 2016; no. 2018-504814, filed 29 July 2016, published 16 August 2018; no. 10-2018-7005904, filed 29 July 2016; no. 18114083.9, filed 5 November 2018; no. PCT/US2016/044718, filed 29 July 2016, published 9 February 2017; no. 62/296,826, filed 18 February 2016; no. 62/296,849, filed 18 February 2016; no. 62/296,866, filed 18 February 2016; no. 62/199,916, filed 31 July 2015; no. 2017382271, filed 21 December 2017; no. 2017382271, filed 21 December 2017; no. 2017382271, filed 21 December 2017; no. 3046095, filed 21 December 2017; no. 2017800785846, filed 21 December 2017; no. 17885383.4, filed 21 December 2017; no. 2019-534399, filed 21 December 2017; no. PCT/US2017/067914, filed 21 December 2017, published 28 June 2018; no. 10-2019-7020802, filed 21 December 2017, no. 16/472,618, filed 21 June 2019; and no. 62/437,582, filed 21 December 2016). All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data including sequencing data may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaav9879/DC1
Fig. S1. TILs in untreated mice.
Fig. S2. Adoptive transfer of TCR Vβ6, 7, 8.1/8.2, or 11 does not provide survival benefit against NSC.
Fig. S3. Relative expansion of TCR Vβ families in peripheral blood after ACT.
Fig. S4. Tumor-reactive T cells retain function within tumor.
Fig. S5. ACT in recurrent medulloblastoma and PNET.
Fig. S6. Lack of single clonal expansion over time in nonresponder to ACT.
Table S1. Clonal expansion in patient PBMCs after ACT.
Table S2. Productive frequency of TCR Vβ families in patient MNCs before ACT, ex vivo expanded ttRNA T cells, and at follow-up after T cell infusion.
REFERENCES AND NOTES
- 1.Bishop D. C., Xu N., Tse B., O’Brien T. A., Gottlieb D. J., Dolnikov A., Micklethwaite K. P., PiggyBac-engineered T cells expressing CD19-specific CARs that lack IgG1 Fc spacers have potent activity against B-ALL xenografts. Mol. Ther. 26, 1883–1895 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.June C. H., O’Connor R. S., Kawalekar O. U., Ghassemi S., Milone M. C., CAR T cell immunotherapy for human cancer. Science 359, 1361–1365 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Bagley S. J., Desai A. S., Linette G. P., June C. H., O’Rourke D. M., CAR T cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro Oncol. 20, 1429–1438 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fraietta J. A., Lacey S. F., Orlando E. J., Pruteanu-Malinici I., Gohil M., Lundh S., Boesteanu A. C., Wang Y., O’Connor R. S., Hwang W. T., Pequignot E., Ambrose D. E., Zhang C., Wilcox N., Bedoya F., Dorfmeier C., Chen F., Tian L., Parakandi H., Gupta M., Young R. M., Johnson F. B., Kulikovskaya I., Liu L., Xu J., Kassim S. H., Davis M. M., Levine B. L., Frey N. V., Siegel D. L., Huang A. C., Wherry E. J., Bitter H., Brogdon J. L., Porter D. L., June C. H., Melenhorst J. J., Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 24, 563–571 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Restifo N. P., Dudley M. E., Rosenberg S. A., Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 12, 269–281 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg S. A., Restifo N. P., Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenberg S. A., Yang J. C., Sherry R. M., Kammula U. S., Hughes M. S., Phan G. Q., Citrin D. E., Restifo N. P., Robbins P. F., Wunderlich J. R., Morton K. E., Laurencot C. M., Steinberg S. M., White D. E., Dudley M. E., Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 17, 4550–4557 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zacharakis N., Chinnasamy H., Black M., Xu H., Lu Y. C., Zheng Z., Pasetto A., Langhan M., Shelton T., Prickett T., Gartner J., Jia L., Trebska-McGowan K., Somerville R. P., Robbins P. F., Rosenberg S. A., Goff S. L., Feldman S. A., Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 24, 724–730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flores C., Pham C., Snyder D., Yang S., Sanchez-Perez L., Sayour E., Cui X., Kemeny H., Friedman H., Bigner D. D., Sampson J., Mitchell D. A., Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology 4, e994374 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wildes T. J., Grippin A., Dyson K. A., Wummer B. M., Damiani D. J., Abraham R. S., Flores C. T., Mitchell D. A., Cross-talk between T cells and hematopoietic stem cells during adoptive cellular therapy for malignant glioma. Clin. Cancer Res. 24, 3955–3966 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsieh Y. C., Chang S. T., Huang W. T., Kuo S. Y., Chiang T. A., Chuang S. S., A comparative study of flow cytometric T cell receptor Vβ repertoire and T cell receptor gene rearrangement in the diagnosis of large granular lymphocytic lymphoproliferation. Int. J. Lab. Hematol. 35, 501–509 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Feng B., Jorgensen J. L., Hu Y., Medeiros L. J., Wang S. A., TCR-Vβ flow cytometric analysis of peripheral blood for assessing clonality and disease burden in patients with T cell large granular lymphocyte leukaemia. J. Clin. Pathol. 63, 141–146 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Pham C. D., Flores C., Yang C., Pinheiro E. M., Yearley J. H., Sayour E. J., Pei Y., Moore C., McLendon R. E., Huang J., Sampson J. H., Wechsler-Reya R., Mitchell D. A., Differential immune microenvironments and response to immune checkpoint blockade among molecular subtypes of murine medulloblastoma. Clin. Cancer Res. 22, 582–595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reilly K. M., Loisel D. A., Bronson R. T., McLaughlin M. E., Jacks T., Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat. Genet. 26, 109–113 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Zhou J., Dudley M. E., Rosenberg S. A., Robbins P. F., Persistence of multiple tumor-specific T-cell clones is associated with complete tumor regression in a melanoma patient receiving adoptive cell transfer therapy. J. Immunother. 28, 53–62 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gururangan S., Krauser J., Watral M. A., Driscoll T., Larrier N., Reardon D. A., Rich J. N., Quinn J. A., Vredenburgh J. J., Desjardins A., McLendon R. E., Fuchs H., Kurtzberg J., Friedman H. S., Efficacy of high-dose chemotherapy or standard salvage therapy in patients with recurrent medulloblastoma. Neuro Oncol. 10, 745–751 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robins H., Desmarais C., Matthis J., Livingston R., Andriesen J., Reijonen H., Carlson C., Nepom G., Yee C., Cerosaletti K., Ultra-sensitive detection of rare T cell clones. J. Immunol. Methods 375, 14–19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaav9879/DC1
Fig. S1. TILs in untreated mice.
Fig. S2. Adoptive transfer of TCR Vβ6, 7, 8.1/8.2, or 11 does not provide survival benefit against NSC.
Fig. S3. Relative expansion of TCR Vβ families in peripheral blood after ACT.
Fig. S4. Tumor-reactive T cells retain function within tumor.
Fig. S5. ACT in recurrent medulloblastoma and PNET.
Fig. S6. Lack of single clonal expansion over time in nonresponder to ACT.
Table S1. Clonal expansion in patient PBMCs after ACT.
Table S2. Productive frequency of TCR Vβ families in patient MNCs before ACT, ex vivo expanded ttRNA T cells, and at follow-up after T cell infusion.