

HEDGES, a tunable nonviral DNA-based gene therapy platform, durably expresses human therapeutic proteins in immunocompetent mice.
Abstract
Recombinant adeno-associated virus (AAV) vectors are transforming therapies for rare human monogenic deficiency diseases. However, adaptive immune responses to AAV and its limited DNA insert capacity, restrict their therapeutic potential. HEDGES (high-level extended duration gene expression system), a nonviral DNA- and liposome-based gene delivery platform, overcomes these limitations in immunocompetent mice. Specifically, one systemic HEDGES injection durably produces therapeutic levels of transgene-encoded human proteins, including FDA–approved cytokines and monoclonal antibodies, without detectable integration into genomic DNA. HEDGES also controls protein production duration from <3 weeks to >1.5 years, does not induce anti-vector immune responses, is reexpressed for prolonged periods following reinjection, and produces only transient minimal toxicity. HEDGES can produce extended therapeutic levels of multiple transgene-encoded therapeutic human proteins from DNA inserts >1.5-fold larger than AAV-based therapeutics, thus creating combinatorial interventions to effectively treat common polygenic diseases driven by multigenic abnormalities.
INTRODUCTION
In contrast to viral vector-based gene delivery approaches, nonviral DNA-based approaches have very large DNA insert capacity (1, 2) and the ability to efficiently reexpress reinjected genes in immunocompetent hosts (3, 4). Nonviral, systemic DNA-based approaches typically use cationic polymers or lipids to mediate delivery, either complexed to DNA (polyplex or lipoplex, respectively) (1, 2) or administered sequentially (5–7). Unfortunately, such approaches often produce subtherapeutic levels (as well as transient duration) of transgene-encoded protein production (2) and can elicit unacceptable host toxicity (8, 9). To date, these three critical limitations have precluded the successful translation of nonviral, DNA-based approaches as human therapeutics. To overcome these limitations, we developed HEDGES (high-level extended duration gene expression system), a nonviral DNA- and liposome-based in vivo gene delivery and expression platform.
RESULTS
Durable therapeutic expression of hG-CSF following HEDGES delivery
Human granulocyte-colony stimulating factor (hG-CSF) or Neupogen is a U.S. Food and Drug Administration (FDA)–approved cytokine widely used to minimize chemotherapy-induced neutropenia (10). We first assessed the duration of therapeutic serum hG-CSF levels produced in immunocompetent, outbred CD-1 mice following the intravenous cationic liposome-based injection of hG-CSF cDNA expression vectors. Previously, one systemic, lipoplex-based injection of an hG-CSF cDNA vector produced detectable hG-CSF serum levels for <3 days in mice (4). In the present study, we systematically tested the effects of different combinations of DNA vector, lipid carrier, and/or vector:carrier coformulation modifications to identify those combinations that increased systemic expression of HEDGES-delivered cDNAs and/or reduced its toxicity. The HEDGES gene delivery platform that produced maximal gene expression and minimal toxicity included the following: (i) sequential cationic liposome-DNA injection (liposomes are intravenously coinjected first, followed 2 minutes later by the intravenous injection of plasmid DNA alone); (ii) use of DNA lacking CpG dinucleotide sequences (3, 11); and (iii) coinjecting cationic plus neutral liposomes, each stably incorporating dexamethasone palmitate in the lipid bilayer. Selective modifications of DNA vector regulatory elements and/or expression cassettes, as well as of liposome mean diameter and lamellar structure, further improved HEDGES transfection efficiency (the level of the transgene protein product produced over time), as well as the ability to control the duration of transgene protein production after a single injection (by selectively modifying aspects of either DNA vector design and/or cationic liposome formulation).
The duration of hG-CSF serum protein expression after one intravenous HEDGES injection into outbred immunocompetent CD-1 mice is shown in Fig. 1. Outbred CD-1 mice, which have greater diversity of major histocompatibility complex (MHC) complex alleles than genetically inbred animals, were used to more realistically mimic human studies in which MHC polymorphisms would be expected to have a considerable impact on undesirable immunogenicity occurring as part of gene therapy. We injected either a single expression cassette vector, in which the human elongation factor-1α (EF-1α) promoter drove a single hG-CSF cDNA (EF-1pro/hG-CSF) or a dual-cassette vector in which the EF-1α promoter element was used to separately drive expression of an hG-CSF and a luciferase cDNA (EF-1pro/Luc–EF-1pro/hG-CSF) (see table S1). The dual-cassette EF-1pro/Luc–EF-1pro/hG-CSF vector produced therapeutic serum hG-CSF levels (>100 pg/ml) for <15 days. Absolute neutrophil counts (ANCs) were elevated fivefold over controls at day 8 but dropped to less than twofold by day 15. In contrast, the single-cassette EF-1α/hG-CSF vector produced therapeutic hG-CSF serum levels for at least 57 days in mice. In addition, the ANC remained greater than 20-fold above normal throughout this period. As previously achieved in healthy rats using an adeno-associated virus (AAV)–hG-CSF vector (12), HEDGES treatment of healthy mice produced serum hG-CSF levels >100 pg/ml as well as increased ANCs >15-fold from baseline. A similar fold change in ANC in neutropenic patients reduced opportunistic infections (13, 14). However, hG-CSF serum concentrations required to increase ANC >15-fold in human patients have been reported to be significantly higher [in the 4 to 5 ng/ml range (15)].
Fig. 1. One intravenous HEDGES hG-CSF cDNA injection produces prolonged hG-CSF protein production and bioactivity in mice.

(A) Serum hG-CSF levels are shown from groups of mice (three mice per group) treated with liposomes followed by either an EF1/hG-CSF or an EF1/Luc–EF1/hG-CSF expression vector or LRS (control) at indicated times. Differences between EF1/hG-CSF vector and EF1/Luc–EF1/hG-CSF vector are significant (P < 0.05) by t test at days 1, 22, and 29. hG-CSF levels in sera from untreated control mice, as well as from mock-treated control mice (mice receiving HEDGES-mAb cDNAs), were undetectable in this hG-CSF ELISA. (B) Neutrophil counts are displayed from groups of mice treated as in (A). Differences are significant (P < 0.01) at days 8, 15, and 22. (C) Serum hG-CSF levels from groups of mice (five per group) injected with the following DNA/liposome combinations: mCMVenh:hCMVpro/hG-CSF and MLV, mCMVenh:EF1pro/hG-CSF and SUV, and hCMVenh:hCMVpro/hG-CSF and MLV. Differences between MLV and SUV groups subsequently injected with EF1/hG-CSF are significant at days 154, 161, 182, and 197. (D) Hematoxylin and eosin–stained spleen (top) and bone marrow (bottom) tissue sections harvested from control mice or mice injected 582 days earlier with SUV liposomes and EF1/hG-CSF. Scale bars, 25 μm. (E) hG-CSF serum levels from groups of mice (n = 3) injected 24 hours earlier with EF1/hG-CSF plasmid DNA or PCR-generated (linear, closed-end linear, or circularized) EF1/hG-CSF DNA. (F) hG-CSF serum levels at indicated time points from groups of mice (n = 3) injected with EF1/hG-CSF either as plasmid DNA or as PCR-generated circularized DNA. A third group, initially injected with circularized PCR DNA underwent reinjection 35 days after the initial injection. Reinjected PCR and plasmid groups are significantly different at day 106. (A, B, C, E and F) Graphs represent mean ± SEM. One representative result from two to five independent experiments is shown. Statistical significance was tested by the two-tailed Student’s t test or ANOVA where appropriate.
We then selectively modified either the cationic liposome formulation or DNA vector enhancer-promoter elements in an attempt to further control the duration of hG-CSF serum protein production by the single-cassette HEDGES EF-1pro/hG-CSF cDNA vector. Figure 1C shows that the use of different enhancer-promoter combinations as well as of different cationic liposome formulations can each independently control the duration of hG-CSF protein production. Therapeutic serum hG-CSF levels produced by one intravenous injection of a HEDGES-based murine cytomegalovirus (CMV) enhancer and EF-1α promoter cDNA construct (mCMVenh:EF-1pro/hG-CSF cDNA vector) (table S1) 2 minutes after intravenous injection of DOTAP (1,2-dioleoyl-3-trimethylammonium-propane chloride) in the form of small unilamellar vesicles (SUVs), with a mean diameter of 74 nm as measured by laser light scattering (lls), persisted for >575 days. In contrast, hG-CSF serum levels in mice receiving the same mCMVenh:EF-1pro/hG-CSF vector following injection of larger DOTAP multilamellar vesicles (MLVs), with a mean diameter of 339 nm as measured by lls, while initially as high as in the SUV-injected mice, became undetectable by day 200 following injection. The significant differences in hG-CSF protein production over time produced by injecting DOTAP MLV versus SUV liposomes are intriguing. This difference cannot be reflective of a longer duration of the SUV themselves, since all liposomes will be eliminated within a time much shorter than the period of hG-CSF protein production. Furthermore, the shorter duration of hG-CSF protein production associated with MLV does not appear to be caused by its transfecting fewer cells, since expression for MLV is initially higher than for SUV. Therefore, it seems unlikely that the shorter duration is, in essence, caused by a statistical decay in expression over time. Instead, these differences may relate to the effects that particle size has on the vascular areas transfected and that expression in some cell types or certain areas of the vasculature may be of longer duration than others.
In addition, while the human CMV enhancer/human CMV immediate early promoter (hCMVenh:hCMVpro) and mCMV enhancer/EF1 promoter (mCMVenh:EF1pro) combinations preinjected with MLV in mice (table S1) each produced similar initial hG-CSF serum levels for the next 200 days, the hCMVenh:hCMV proinjected group’s hG-CSF levels remained therapeutic for at least 60 days longer. Together, our results show that a single intravenous HEDGES injection can substantially prolong therapeutic hG-CSF serum levels compared to those produced by intravenous injection of conventional lipoplex (4). Our results also demonstrate that selectively modifying the DNA vector enhancer-promoter, the liposome formulation and size, or the number of expression cassettes per DNA vector can each control the duration of nonvirally delivered, cDNA-encoded protein production. By combining selected DNA vector, carrier, and/or coformulation modifications, the HEDGES platform was able to produce therapeutic hG-CSF levels for periods ranging from <21 to >575 days (Fig. 1, A to C). Since recombinant hG-CSF protein is typically administered to patients as a 10-day course (10), the ability to control the duration of expression of HEDGES-delivered genes without requiring the use of gene switches should potentially confer significant advantages.
One HEDGES SUV-based EF-1-hG-CSF cDNA injection also increased tissue neutrophil progenitor cell infiltration into murine spleen and bone marrow for extended periods, documenting that the cDNA-encoded hG-CSF protein remained bioactive for ≥582 days after injection. Figure 1D shows replacement of normal erythroid elements in mouse spleen as well as in bone marrow with nearly solid sheets of immature polymorphonuclear leukocytes at day 582 after injection. As expected from prolonged expression of a human protein in immunocompetent mice, mouse anti-human G-CSF antibodies appeared in EF-1pro/hG-CSF cDNA-injected mice by day 22 following injection (fig. S1), although their ongoing presence did not appear to significantly limit hG-CSF–mediated induction of neutrophilia over time.
Previously, lipoplex-based vectors injected intravenously were shown primarily to transfect vascular endothelial cells (VECs), as well as monocyte/macrophages, as revealed by both light and electron microscopy 24 hours after injection (16). Consistent with these findings, we found that HEDGES delivery of a human p53 cDNA resulted in VEC expression of hTP53, a nonsecreted protein, 24 hours later (fig. S2). In addition, despite producing therapeutic hG-CSF serum protein levels as well as bioactivity for >582 days after a single HEDGES hG-CSF cDNA injection (Fig. 1, C and D), integration of the HEDGES DNA vector into genomic DNA of treated mice was undetectable in any of four tissues assayed (lung, liver, spleen, and thymus) (fig. S3). However, assaying a wider range of tissues would be required to more completely exclude this possibility. The lung is the organ most efficiently transfected by sequential injection of cationic liposomes followed by plasmid DNA (6). To more precisely assess the specific cell types HEDGES transfects, as well as the percentage of cells transfected, we measured Escherichia coli β-galactosidase (β-GAL) transgene expression by fluorescent flow cytometric analysis, 24 hours after intravenous, HEDGES-based injection of the β-GAL cDNA. As shown in fig. S4, approximately 25% of lung VECs were transfected. Similar to HEDGES-based delivery of the human p53 cDNA (fig. S2), VECs in the lung were the primary cell type transfected by HEDGES-based delivery of the β-GAL cDNA. As normal lung VECs are largely nondividing over time (17), these results are consistent with the hypothesis that HEDGES DNA vectors are both retained and expressed as episomes for prolonged periods within nondividing VECs.
We then compared the effects on hG-CSF serum levels produced following one intravenous HEDGES injection of an hG-CSF cDNA vector delivered as plasmid DNA or as linear-, closed-end linear-, or circularized polymerase chain reaction (PCR)–generated DNA in CD-1 mice. Only the circularized, PCR DNA-generated DNA conformation produced significantly higher hG-CSF serum levels than those produced by plasmid DNA (Fig. 1E). Lipoplex formulations have been shown to be reexpressed efficiently following reinjection in immunocompetent mice (3, 4). Similarly, we found that a single reinjection of the EF1pro/hG-CSF cDNA as circularized, PCR-generated DNA further increased hG-CSF levels for >100 days compared to non-reinjected mice (Fig. 1F). Thus, a single reinjection of circularized PCR-DNA can further elevate ongoing serum levels of HEDGES-produced therapeutic proteins.
Safety and performance of the HEDGES delivery platform
Intravenous lipoplex gene therapy in mice has demonstrated significant host toxicity. Specifically, intravenous lipoplex-based therapy increases alanine and aspartate aminotransferase (ALT and AST) levels to 40-fold within the first 48 hours after injection into mice (9), induces cytokine responses that significantly limit the duration of transgene expression (18), and can produce 100% mortality within 24 hours at high DNA doses (100 μg per mouse) (8). Prior work has shown that sequential intravenous injection of cationic liposomes followed by intravenous plasmid DNA could simultaneously reduce inflammation (7) that may promote hepatotoxicity as well as increase transgene expression (6). Similarly, the use of CpG-free DNA for nonviral gene delivery has also resulted in less innate immune stimulation, which was associated with decreased toxicity and increased gene expression after intravenous or aerosol administration (3, 11). Here, we combined these approaches with HEDGES. The sequential intravenous injection of DOTAP followed by ≥100 μg of CpG dinucleotide-free DNA increased ALT and AST levels only fourfold above background levels 24 hours after injection, which returned to background levels by 48 hours (Fig. 2A). Despite using DNA doses that produced 100% mortality within 48 hours when injected as lipoplex (8), no mortality was observed in HEDGES-injected mice receiving up to a 120-μg DNA dose (Fig. 2A).
Fig. 2. Evolving HEDGES platform incrementally lowers toxicity while increasing expression.
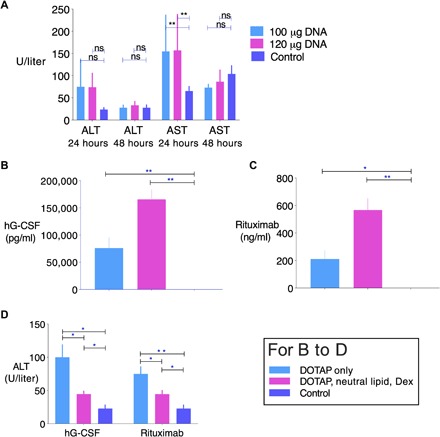
(A) Alanine transaminase (ALT) and aspartate transaminase (AST) levels at 24 and 48 hours from mice injected with LRS (control) or 1000 nmol of DOTAP liposomes, followed by 100 or 120 μg of an EF1pro/hG-CSF DNA vector. Means ± SEM are displayed. **P < 0.01 compared to control by ANOVA. ns, not significant. No mortality was observed in HEDGES-injected mice receiving up to a 120-μg dose of DNA. (B) hG-CSF and (C) rituximab serum levels 24 hours after HEDGES injection. DOTAP only received 1120 nmol DOTAP liposomes; DOTAP, neutral lipid, and Dex received 1120 nmol DOTAP liposomes incorporating 2.5 mol % Dex-Palm, 1000 nmol DMPC liposomes incorporating 5 mol % Dex-Palm, and intraperitoneal water-soluble dexamethasone (40 mg/kg) 2 hours before intravenous injections. Mice receiving liposomes also received 88 μg of DNA vector encoding hG-CSF or rituximab. (D) Serum ALT levels from groups of mice in (B) and (C) 24 hours after injection. *P < 0.05 and **P < 0.01 by the two-tailed unpaired Student’s t test. One representative result from two to three independent experiments is shown.
Production of prolonged therapeutic monoclonal antibody (mAb) serum levels following systemic administration of nonviral DNA- plus lipid-based approaches has not previously been reported (19). In contrast, HEDGES produced extended serum levels of three different mAbs. Specifically, HEDGES produced extended therapeutic serum levels of an anti-human CD20 mAb, rituximab, which is approved to treat human B lymphocyte–derived malignancies and autoimmune diseases (20). We modified our original HEDGES dual expression cassette DNA vector to express the CpG-depleted anti-human CD20 heavy and light immunoglobulin (IgG) chain cDNAs for expression codon-optimized for mice. To minimize or obviate proinflammatory mediators that contribute to lipoplex-mediated toxicity as well as inhibit transgene expression, we intraperitoneally administered one dose of aqueous soluble dexamethasone before injecting HEDGES. In addition, we stably inserted dexamethasone palmitate into both liposome bilayers of a coinjected cationic and neutral liposome mixture, an approach that specifically targets dexamethasone to the cells that internalize the liposomes to which they are coupled. This approach has been used to effectively treat human patients with lethal macrophage activation syndrome resistant to conventional corticosteroid therapy (21). One intravenous injection of this dexamethasone-modified HEDGES anti-human CD20 mAb platform produced bioactive levels that persisted for >275 days (Fig. 3A). Sera harvested from these mice for at least 288 days postinjection lysed CD20-positive Raji human B cell lymphoma target cells as effectively as recombinant rituximab protein (Fig. 3B). In contrast to postinjection hG-CSF levels (Fig. 1A), anti-human CD20 mAb serum levels did not drop significantly from day 1 to day 8 after injection. This likely reflected the significantly longer protein serum half-life of rituximab of approximately 10 days (22) [due, at least in part, to Fc receptor recycling of the mAb from the tissues into the blood (23)], compared to hG-CSF’s serum half-life of less than 5 hours (24).
Fig. 3. One intravenous HEDGES injection of multicassette DNA vectors encoding cognate mAb heavy- and light-chain cDNAs produce prolonged bioactive mAb serum proteins.
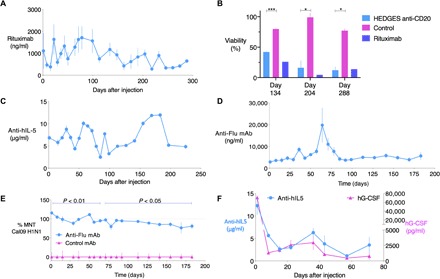
On day 0, groups of CD-1 mice (n = 3) were given dexamethasone intraperitoneally 2 hours before intravenous coinjection of 1120 nmol DOTAP + Dex and 1000 nmol DMPC + Dex liposomes and 2 minutes later intravenously, and then 88 μg of mAb heavy- and light-chain cDNA vector intravenously. Control mice received LRS. (A) Mean ± SEM rituximab serum levels at indicated time points were measured by ELISA. mAb levels in sera from untreated control mice, as well as from mock-treated control mice (mice receiving HEDGES encoding unrelated mAb cDNAs), were undetectable in the rituximab, 5J8 mAb, and mepolizumab ELISAs, respectively. (B) Mean ± SEM viability of CD20+ Raji lymphoma target cells is shown following incubation with human plasma and sera from anti-CD20 or control HEDGES-treated mice isolated at indicated time points. Target Raji cells incubated with recombinant rituximab (10 mg/ml) is also presented for comparison. ***P < 0.001 and *P < 0.05 by ANOVA method. (C) Mean ± SEM anti–IL-5/mepolizumab serum levels are shown over time by ELISA. 5J8 anti-H1 IAV mAb serum levels were measured by ELISA (D) and tested in parallel for capacity to neutralize Cal09 H1N1 IAV in a microneutralization assay (MNT) at indicated time points (E). Serum from HEDGES-rituximab–treated mice (control) served as controls at all time points in ELISA and MNT. (F) Mepolizumab and hG-CSF serum levels over time following intravenous HEDGES injection of an 8.8-kb triple-cassette cDNA vector encoding hG-CSF and mepolizumab heavy- and light-chain cDNAs. Graphs represent mean ± SEM. One representative result from two to three independent experiments is shown.
While HEDGES-based injection of the rituximab heavy- and light-chain cDNAs produced ongoing serum levels of rituximab, these levels varied significantly over time (Fig. 3B). HEDGES injection of cDNA for rituximab, a humanized mAb, into fully immunocompetent, outbred CD-1 mice, created the potential to elicit a mouse anti-human protein antibody immune response. We examined this possibility by assessing whether HEDGES-rituximab cDNA-injected mice produced mouse anti-rituximab antibodies over time, as well as whether the presence of such antibodies altered serum rituximab levels. As shown in fig. S5, only a minority of the HEDGES rituximab cDNA-injected mice per group developed mouse anti-rituximab antibodies, an observation consistent with the use of outbred mice. This variability in antitransgene immune response between individual mice likely reflects the genetic diversity in the MHC locus of the outbred strain used for these experiments (25). In each mouse that developed mouse anti-rituximab antibodies, serum rituximab levels then fell toward background levels. In contrast, rituximab serum levels were maintained in the mice that did not develop these antibodies. This immunological heterogeneity between individual outbred mice conferred significant variability in serum rituximab levels produced over time within individual mouse groups. In addition, it has previously been shown that altering the expression of endogenous, transfection-controlling cellular genes can produce significant alterations in the expression levels of transgenes transfected by lipoplex-based methods (26). Thus, it is possible that physiologic variations in the expression of endogenous, transfection-controlling genes in mice may modulate ongoing transgene expression levels in transfected VECs over time.
We also observed that pretreatment with dexamethasone combined with the HEDGES platform using dexamethasone-modified liposomes significantly increased hG-CSF (Fig. 2B) as well as anti-human CD20 mAb (Fig. 2C) serum levels following injection of either the hG-CSF or anti-human CD20 mAb heavy and light IgG chain cDNAs, respectively. Dexamethasone addition also significantly further reduced ALT levels compared to the initial, dexamethasone-free HEDGES platform (Fig. 2, A and D).
mAB expression with HEDGES delivery
To determine whether HEDGES could be generally used to achieve prolonged cDNA-based mAb expression, we then evaluated this platform’s production of mepolizumab, a humanized anti–human interleukin-5 (hIL-5) mAb approved to treat severe asthma, as well as of 5J8, a human mAb directed against H1 hemagglutinin and capable of neutralizing the 1918 and 2009 H1N1 pandemic influenza A virus (IAV) strains (27, 28). One HEDGES injection of the anti–hIL-5 mAb DNA vector encoding the heavy and light IgG chain cDNAs into CD-1 mice produced serum mAb levels in the 5 μg/ml range within 24 hours of injection, which persisted for >220 days (Fig. 3C). Similarly, one HEDGES-based injection of a DNA vector encoding the heavy and light IgG chain cDNAs of 5J8 into CD-1 mice also produced serum 5J8 mAb levels in the 5 μg/ml range for at least 180 days (Fig. 3D). The 5J8 mAb transgene-encoded protein product was functionally active, as sera from HEDGES 5J8-treated CD-1 mice neutralized the H1N1 2009 (Cal/09) IAV pandemic strain. This neutralization activity was present by 24 hours after injection and persisted for at least 180 days (Fig. 3E).
Last, as proof of concept for using HEDGES to provide therapy involving multiple gene products, we tested its ability to efficiently coexpress three different genes from a single DNA vector. Figure 3F shows that a single intravenous HEDGES-based injection of a triple expression cassette plasmid DNA vector containing the cDNAs for hG-CSF and the anti–hIL-5 mAb heavy and light IgG chains simultaneously produced therapeutic serum levels of hG-CSF and anti–hIL-5 mAb for at least 70 days. Thus, one HEDGES-based injection of an 8.8-kb DNA vector containing a 7.2-kb DNA insert produced therapeutic levels of three different cDNA-encoded therapeutic proteins (one complete mAb and one cytokine) for an extended period.
DISCUSSION
Ideally, DNA-based in vivo gene transfer and expression platforms should be able to efficiently produce one or more cDNA-encoded therapeutic proteins for controlled periods, ranging from durable to short term after one injection, without requiring the use of gene on/off or suicide switches. They should also be able to serially reexpress these cDNA-encoded proteins upon reinjection into fully immunocompetent hosts. They should do so without eliciting significant host toxicity, antivector-specific adaptive immune responses, or detectable DNA vector integration into host genomic DNA.
Results presented here show that HEDGES, a systemic, nonviral DNA-based injection platform, can achieve all these goals in outbred, immunocompetent mice. We have used HEDGES to express a range of different human protein therapies in fully immunocompetent, outbred mice to maximize their translational relevance for human studies. Our demonstration that HEDGES can produce bioactive levels of human protein therapies for prolonged periods in immunocompetent, outbred mice is previously unknown as nonviral, DNA-based mAb delivery studies have routinely used inbred mice, rendered immunodeficient either genetically (29) or by preinjection of anti-mouse T cell–depleting antibodies (30). The use of outbred rather than genetically inbred animals should more realistically recapitulate the more variable intragroup results characteristic of human studies than inbred mouse strains (31). A salient feature of HEDGES is its ability to produce biologically active levels of single or multiple transgene-encoded proteins for extended periods of time, accompanied by only minimal, transient postinjection toxicity. In addition, despite persistent transgene expression following a single treatment, integration of the HEDGES DNA vector into mouse genomic DNA was undetectable in each of the four tissues examined here, indicating that the vector DNA was likely maintained episomally in the nucleus of long-lived, nondividing cells, primarily VECs. Furthermore, the duration of hG-CSF DNA-encoded protein production was controllable across a broad temporal range by selectively modifying the DNA vector, cationic carrier system, and/or their coformulation, without using ligand-induced suicide or on/off switches. HEDGES cDNA-encoded proteins were efficiently reexpressed following reinjection into immunocompetent mice. In contrast, injection of recombinant AAV vectors elicits anti-AAV adaptive immune responses that limits or prevents transgene reexpression following AAV re-injection into immunocompetent hosts (32). Similarly, HEDGES’ ability to efficiently coexpress three different therapeutic proteins from a >7-kb DNA insert contrasts with AAV’s more limited DNA insert capacity of <4.8 kb (33), and therefore its more limited ability to coexpress multiple different cDNAs.
When compared to liposome-DNA (lipoplex)–based approaches, HEDGES is significantly less toxic [100 μg per mouse lipoplex DNA doses are associated with 100% mortality within 24 hours (8), whereas HEDGES DNA doses >100 μg per mouse produced no mortality (Fig. 2B)]. HEDGES also produces substantially longer duration of injected transgene expression (Fig. 1A) than lipoplex. Specifically, one systemic HEDGES-based injection of the hG-CSF cDNA produced >100 pg/ml serum hG-CSF levels for at least the next 582 days (Fig. 1C), whereas one systemic lipoplex-based injection of the hG-CSF cDNA produced detectable serum hG-CSF levels for <3 days (4). Systemic mRNA-based nucleic acid delivery approaches can produce very high peak levels of mRNA-expressed proteins in a variety of different cell types. However, mRNA’s short half-life generally produces encoded proteins only for relatively short periods after injection (34, 35). The combination of short-term protein production plus the potential for significant host toxicity following repeated reinjection of mRNA-carrier complexes currently limits their utility for longer-term protein production (34, 35). To further improve HEDGES efficacy and safety profile, efforts to modify it to efficiently express larger DNA inserts (>10 kb) in vivo as well as to further reduce HEDGES host toxicity to background level are ongoing.
HEDGES’ ability to produce serum mAb levels that provide effective virus neutralization within 24 hours of injection that persisted for >180 days suggests that it may provide rapid-onset, long-lasting immunity to highly pathogenic viruses, even in immunocompromised hosts who are refractory to conventional vaccination approaches. Thus, HEDGES-based therapy may be applicable to a wide range of clinical situations in which the expression of one or more different therapeutic and/or protective genes for controlled periods would be beneficial. HEDGES will continue to evolve as a modular platform that may be adapted for treating a spectrum of genetic diseases caused by the aberrant expression of either one or more genes, as well as for conferring prolonged protection against a range of infectious diseases.
MATERIALS AND METHODS
In vivo protocols and mice
Female outbred Hsd:ICR (CD-1®) mice (7 to 10 weeks old, from Envigo or Charles River Laboratories) were used for all studies. All mouse studies were conducted according to protocols approved by the Institutional Animal Care and Use Committee at the California Pacific Medical Center Research Institute.
Plasmid construction
The CpG-free gBlock fragments (Integrated DNA Technologies) containing R6K, Kanr, and multiple cloning sites (Dra III, Eco RI, and Nhe I) were assembled using the Gibson Assembly Method (NEB) as a 1.2-kb base vector. To accommodate multiple rounds of cloning into the base vector, the expression cassette was constructed in pUC19 by placing Eco RI–Nhe I and Xba I restriction sites at the 5′ and 3′ ends, respectively, flanking the expression cassette. The open reading frame (ORF) was inserted at Bst EII–Bgl II, and the enhancer/promoter was located between the Nhe I and Bst EII sites. The polyadenylation site was between the Bgl II and Xba I sites. The Eco RI–Xba I expression cassette fragment was then inserted into the CpG-free base vector at the Eco RI–Nhe I sites. The ORFs of hG-CSF/CSF3 (GenBank X03438.1) and soLux (11) were CpG free. The ORFs of rituximab (U.S. patent US5736137), anti-Flu mAb (GenBank JF791169.1 and X57821.1), and anti–hIL-5 mAb (U.S. patent US20030059429) were constructed as CpG free and codon-optimized using GeneArt (Thermo Scientific) software.
DNA vector production
Plasmid was produced using the GeneJet Endo-free Maxiprep Kit (Thermo Fisher), according to the manufacturer’s instructions. Plasmid was eluted with lactated Ringer’s solution (LRS) and filter-sterilized with a 0.2-μm sterile syringe polyethersulfone membrane (Millipore).
Liposome production
DOTAP and DMPC (14:0 PC 1,2-dimyristoyl-sn-glycero-3-phosphocholine) lipids were obtained from Avanti Polar Lipids (SKU 890890C and 850345C). Dexamethasone 21-palmitate (Dex-Palm) was obtained from Toronto Research Chemicals (D298830). MLV stocks were prepared by suspending DOTAP or DMPC, with or without Dex-Palm, in 5% dextrose in water. Cationic SUV stocks were prepared by suspending DOTAP, with or without Dex-Palm, in 5% dextrose in water and sonicating in a water bath sonicator (Avanti Polar Lipids).
HEDGES injections
For intravenous injections, mice were injected in the lateral tail vein with liposomes suspended in LRS. Approximately 2 minutes later, they were injected in the lateral tail vein with DNA vectors suspended in LRS. For intraperitoneal injections of glucocorticoids, mice were injected with water-soluble dexamethasone (SLBW5982; Sigma-Aldrich) suspended in LRS 2 hours before the sequential intravenous injections of liposomes and DNA vectors. For cationic and neutral liposome intravenous coinjection, DOTAP cationic liposomes and DMPC neutral liposomes were combined.
Blood collection
Mice were anesthetized by isoflurane inhalation and bled via submandibular vein puncture. Blood was collected into serum separator tubes or lithium heparin-coated tubes (365967 or 365985; Becton Dickinson).
Tissue harvest
Mice were euthanized via CO2 inhalation, and blood was cleared by perfusion of phosphate-buffered saline. For flow cytometry, lungs were harvested and minced, and then frozen in CELLBANKER 2 freezing medium. For histopathology, organs were harvested and fixed in 10% neutral buffered formalin before transfer into 70% ethanol for staining and analysis at the University of Washington Histology and Imaging Core.
Complete blood count and serum chemistries
Whole blood collected from mice and plasma or sera isolated from blood were analyzed at the Comparative Pathology Laboratory at University of California, Davis for complete blood counts (ANC, absolute lymphocyte count, and platelets) and chemistries (ALT and AST).
ELISA analysis of plasma and serum
Plasma levels of hG-CSF were analyzed using the hG-CSF Quantikine enzyme-linked immunosorbent assay (ELISA) kit (SCS50; R&D Systems). Serum rituximab levels were analyzed using the rituximab ELISA kit (IG-AB106; Eagle Biosciences). For anti–hG-CSF, 5J8, and anti–IL-5 mAb ELISAs, immunoassay plates were coated with antigen (BioLegend 578606 and Sino Biologicals 11085-V08H and 15673-HNCE, respectively) and blocked with 3% bovine serum albumin. After binding of expressed serum proteins, detection was carried out with enzyme-linked anti-human IgG and 3,3′,5,5′-tetramethylbenzidine substrate (AP113P; Millipore). For the anti-rituximab ELISA, the coat protein was rituximab from InvivoGen, and the detection antibody was goat anti-mouse IgG H+L horseradish peroxidase from Thermo Fisher.
Histopathology
Tissue samples were paraffin-embedded, sectioned, and stained with hematoxylin and eosin. Slides for immunohistochemistry were processed on a Leica Bond Max Automated Immunostainer. Antigen retrieval on slides was performed using EDTA solution, and 10% normal goat serum was used as blocking agent. Antibodies were used against CD31 (SZ31; Dianova) and p53 (2527; Cell Signaling). Additional blocking for endogenous peroxidase was performed with Leica peroxide block (DS9800). Slides were visualized using Leica Bond Mixed Refine detection. Tissues were hematoxylin counterstained, dehydrated with 100% ethanol, cleared in xylene, and mounted with resin mounting medium.
Cell viability assay
Raji cells (1 × 105 cells per well of a 96-well plate) were incubated for 1 hour at room temperature with either recombinant rituximab, sera from mice injected with HEDGES vector encoding rituximab and lipid, or control mouse sera (mice injected with hG-CSF and lipid). Twenty microliters of pooled normal human plasma (from Innovative Research) was then added to all wells, and the plates were incubated for another 12 hours at 37°C. Samples were run in duplicate. Cell viability was measured using the Promega CellTiter-Glo reagent according to the manufacturer’s instruction (G7570).
IAV microneutralization
Prediluted sera (1:40), heat-treated for complement inactivation, from mice receiving HEDGES treatment for heavy and light IgG chain genes of 5J8 (anti-H1) or rituximab (anti-hCD20, a negative control) were tested in triplicate for the capacity to neutralize 100× TCID50 (median tissue culture infectious dose) Cal09 H1N1 IAV (strain X-179a) in a standard in vitro microneutralization assay (36). Data were normalized by setting control serum to 0% and uninfected wells to 100%.
Genomic integration analysis
Integration analysis by nonrestricted linear amplification-mediated PCR was performed as previously described (37).
Statistics
P values for ELISA assays, viability assay, and toxicity markers were calculated by two-sided, unpaired Student’s t test or analysis of variance (ANOVA) using GraphPad Prism software.
Supplementary Material
Acknowledgments
Funding: We acknowledge that funding for this research was provided solely by DNARx LLC. Author contributions: C.H., A.L.Y., S.A.C., T.D.H., D.L., D.B.L., and R.D. designed the study; C.H., A.L.Y., S.A.C., L.S., M.Ma., M.My., R.J.I., R.T., S.J.U., and Y.L. performed experiments; C.H., A.L.Y., S.A.C., and R.T. collected and analyzed data; M.K.-S., D.B.L., T.D.H., D.L., and R.D. constructed and wrote the manuscript; L.S., M.K.-S., D.L., T.D.H., and D.B.L. gave expert advice. Competing interests: We wish to disclose a potential conflict of interest on the part of authors C.H., A.L.Y., S.A.C., L.S., M.Ma., R.T., M.My., Y.L., M.K.-S., T.D.H., D.L., D.B.L., and R.D., each of whom has some financial ownership of DNARx LLC, the entity that funded this work. The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Materials described in this paper can be provided by DNARx pending scientific review and a completed material transfer agreement. Please send communications to the corresponding author to initiate material transfer requests.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax0217/DC1
Fig. S1. HEDGES hG-CSF cDNA injection elicits the production of endogenous mouse anti–hG-CSF antibodies.
Fig. S2. Intravenous HEDGES injection of the human TP53 cDNA is expressed predominantly in mouse VECs.
Fig. S3. Intravenous injection of HEDGES vector DNA does not detectably integrate into genomic DNA of injected mice.
Fig. S4. HEDGES treatment with a β-GAL reporter-containing plasmid results in expression of reporter in CD31+CD45− lung cells in mice.
Fig. S5. Development of mouse anti-rituximab antibodies suppresses rituximab levels in HEDGES-treated mice.
Table S1. HEDGES expression plasmids.
REFERENCES AND NOTES
- 1.Kamimura K., Suda T., Zhang G., Liu D., Advances in gene delivery systems. Pharmaceut Med 25, 293–306 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Templeton N. S., Cationic liposome-mediated gene delivery in vivo. Biosci. Rep. 22, 283–295 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Yew N. S., Zhao H., Wu I. H., Song A., Tousignant J. D., Przybylska M., Cheng S. H., Reduced inflammatory response to plasmid DNA vectors by elimination and inhibition of immunostimulatory cpg motifs. Mol. Ther. 1, 255–262 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Tu G., Kirchmaier A. L., Liggitt D., Liu Y., Liu S., Yu W. H., Heath T. D., Thor A., Debs R. J., Non-replicating epstein-barr virus-based plasmids extend gene expression and can improve gene therapy in vivo. J. Biol. Chem. 275, 30408–30416 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Liu Y., Liggitt D., Zhong W., Tu G., Gaensler K., Debs R., Cationic liposome-mediated intravenous gene delivery. J. Biol. Chem. 270, 24864–24870 (1995). [DOI] [PubMed] [Google Scholar]
- 6.Song Y. K., Liu F., Liu D., Enhanced gene expression in mouse lung by prolonging the retention time of intravenously injected plasmid DNA. Gene Ther. 5, 1531–1537 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Tan Y., Liu F., Li Z., Li S., Huang L., Sequential injection of cationic liposome and plasmid DNA effectively transfects the lung with minimal inflammatory toxicity. Mol. Ther. 3, 673–682 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Firozi P., Zhang W., Chen L., Quiocho F. A., Worley K. C., Templeton N. S., Identification and removal of colanic acid from plasmid DNA preparations: Implications for gene therapy. Gene Ther. 17, 1484–1499 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tousignant J. D., Gates A. L., Ingram L. A., Johnson C. L., Nietupski J. B., Cheng S. H., Eastman S. J., Scheule R. K., Comprehensive analysis of the acute toxicities induced by systemic administration of cationic lipid: Plasmid DNA complexes in mice. Hum. Gene Ther. 11, 2493–2513 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Lyman G. H., Dale D. C., Culakova E., Poniewierski M. S., Wolff D. A., Kuderer N. M., Huang M., Crawford J., The impact of the granulocyte colony-stimulating factor on chemotherapy dose intensity and cancer survival: A systematic review and meta-analysis of randomized controlled trials. Ann. Oncol. 24, 2475–2484 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hyde S. C., Pringle I. A., Abdullah S., Lawton A. E., Davies L. A., Varathalingam A., Nunez-Alonso G., Green A. M., Bazzani R. P., Sumner-Jones S. G., Chan M., Li H., Yew N. S., Cheng S. H., Boyd A. C., Davies J. C., Griesenbach U., Porteous D. J., Sheppard D. N., Munkonge F. M., Alton E. W., Gill D. R., Cpg-free plasmids confer reduced inflammation and sustained pulmonary gene expression. Nat. Biotechnol. 26, 549–551 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Bonham L., Palmer T., Miller A. D., Prolonged expression of therapeutic levels of human granulocyte colony-stimulating factor in rats following gene transfer to skeletal muscle. Hum. Gene Ther. 7, 1423–1429 (1996). [DOI] [PubMed] [Google Scholar]
- 13.Dale D. C., Bonilla M. A., Davis M. W., Nakanishi A. M., Hammond W. P., Kurtzberg J., Wang W., Jakubowski A., Winton E., Lalezari P., Robinson W., Glaspy J. A., Emerson S., Gabrilove J., Vincent M., Boxer L. A., A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood 81, 2496–2502 (1993). [PMC free article] [PubMed] [Google Scholar]
- 14.Migliaccio A. R., Migliaccio G., Dale D. C., Hammond W. P., Hematopoietic progenitors in cyclic neutropenia: Effect of granulocyte colony-stimulating factor in vivo. Blood 75, 1951–1959 (1990). [PubMed] [Google Scholar]
- 15.Wang B., Ludden T. M., Cheung E. N., Schwab G. G., Roskos L. K., Population pharmacokinetic-pharmacodynamic modeling of filgrastim (r-methug-csf) in healthy volunteers. J. Pharmacokinet. Pharmacodyn. 28, 321–342 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Liu Y., Mounkes L. C., Liggitt H. D., Brown C. S., Solodin I., Heath T. D., Debs R. J., Factors influencing the efficiency of cationic liposome-mediated intravenous gene delivery. Nat. Biotechnol. 15, 167–173 (1997). [DOI] [PubMed] [Google Scholar]
- 17.Schlereth K., Weichenhan D., Bauer T., Heumann T., Giannakouri E., Lipka D., Jaeger S., Schlesner M., Aloy P., Eils R., Plass C., Augustin H. G., The transcriptomic and epigenetic map of vascular quiescence in the continuous lung endothelium. eLife 7, e34423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sellins K., Fradkin L., Liggitt D., Dow S., Type I interferons potently suppress gene expression following gene delivery using liposome(−)DNA complexes. Mol. Ther. 12, 451–459 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Hollevoet K., Declerck P. J., State of play and clinical prospects of antibody gene transfer. J. Transl. Med. 15, 131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feugier P., A review of rituximab, the first anti-CD20 monoclonal antibody used in the treatment of B non-Hodgkin’s lymphomas. Future Oncol. 11, 1327–1342 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Nakagishi Y., Shimizu M., Kasai K., Miyoshi M., Yachie A., Successful therapy of macrophage activation syndrome with dexamethasone palmitate. Mod. Rheumatol. 26, 617–620 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Kagan L., Zhao J., Mager D. E., Interspecies pharmacokinetic modeling of subcutaneous absorption of rituximab in mice and rats. Pharm. Res. 31, 3265–3273 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Awwad S., Angkawinitwong U., Overview of antibody drug delivery. Pharmaceutics 10, E83 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kearns C. M., Wang W. C., Stute N., Ihle J. N., Evans W. E., Disposition of recombinant human granulocyte colony-stimulating factor in children with severe chronic neutropenia. J. Pediatr. 123, 471–479 (1993). [DOI] [PubMed] [Google Scholar]
- 25.Rai D., Pham N. L., Harty J. T., Badovinac V. P., Tracking the total cd8 t cell response to infection reveals substantial discordance in magnitude and kinetics between inbred and outbred hosts. J. Immunol. 183, 7672–7681 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Handumrongkul C., Zhong W., Debs R. J., Distinct sets of cellular genes control the expression of transfected, nuclear-localized genes. Mol. Ther. 5, 186–194 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Deeks E. D., Mepolizumab: A review in eosinophilic asthma. BioDrugs 30, 361–370 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Krause J. C., Tsibane T., Tumpey T. M., Huffman C. J., Basler C. F., Crowe J. E. Jr., A broadly neutralizing human monoclonal antibody that recognizes a conserved, novel epitope on the globular head of the influenza h1n1 virus hemagglutinin. J. Virol. 85, 10905–10908 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muthumani K., Block P., Flingai S., Muruganantham N., Chaaithanya I. K., Tingey C., Wise M., Reuschel E. L., Chung C., Muthumani A., Sarangan G., Srikanth P., Khan A. S., Vijayachari P., Sardesai N. Y., Kim J. J., Ugen K. E., Weiner D. B., Rapid and long-term immunity elicited by DNA-encoded antibody prophylaxis and DNA vaccination against chikungunya virus. J Infect Dis 214, 369–378 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esquivel R. N., Patel A., Kudchodkar S. B., Park D. H., Stettler K., Beltramello M., Allen J. W., Mendoza J., Ramos S., Choi H., Borole P., Asija K., Bah M., Shaheen S., Chen J., Yan J., Durham A. C., Smith T. R. F., Broderick K., Guibinga G., Muthumani K., Corti D., Humeau L., Weiner D. B., In vivo delivery of a DNA-encoded monoclonal antibody protects non-human primates against zika virus. Mol. Ther. 27, 974–985 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aldinger K. A., Sokoloff G., Rosenberg D. M., Palmer A. A., Millen K. J., Genetic variation and population substructure in outbred cd-1 mice: Implications for genome-wide association studies. PLOS ONE 4, e4729 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vandamme C., Adjali O., Mingozzi F., Unraveling the complex story of immune responses to aav vectors trial after trial. Hum. Gene Ther. 28, 1061–1074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salganik M., Hirsch M. L., Samulski R. J., Adeno-associated virus as a mammalian DNA vector. Microbiol Spectr 3, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laina A., Gatsiou A., Georgiopoulos G., Stamatelopoulos K., Stellos K., RNA therapeutics in cardiovascular precision medicine. Front. Physiol. 9, 953 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu M. A., A comparison of plasmid DNA and mrna as vaccine technologies. Vaccines (Basel) 7, E37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harmon M. W., Rota P. A., Walls H. H., Kendal A. P., Antibody response in humans to influenza virus type b host-cell-derived variants after vaccination with standard (egg-derived) vaccine or natural infection. J. Clin. Microbiol. 26, 333–337 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paruzynski A., Arens A., Gabriel R., Bartholomae C. C., Scholz S., Wang W., Wolf S., Glimm H., Schmidt M., von Kalle C., Genome-wide high-throughput integrome analyses by nrlam-pcr and next-generation sequencing. Nat. Protoc. 5, 1379–1395 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax0217/DC1
Fig. S1. HEDGES hG-CSF cDNA injection elicits the production of endogenous mouse anti–hG-CSF antibodies.
Fig. S2. Intravenous HEDGES injection of the human TP53 cDNA is expressed predominantly in mouse VECs.
Fig. S3. Intravenous injection of HEDGES vector DNA does not detectably integrate into genomic DNA of injected mice.
Fig. S4. HEDGES treatment with a β-GAL reporter-containing plasmid results in expression of reporter in CD31+CD45− lung cells in mice.
Fig. S5. Development of mouse anti-rituximab antibodies suppresses rituximab levels in HEDGES-treated mice.
Table S1. HEDGES expression plasmids.