

Abstract
The NuRD chromatin remodeling and deacetylation complex, which includes MTA1, MBD3, CHD4 and HDAC1 among other components, is of importance for development and cancer progression. The oncogenic MUC1-C protein activates EZH2 and BMI1 in the epigenetic reprogramming of triple-negative breast cancer (TNBC). However, there is no known link between MUC1-C and chromatin remodeling complexes. Here we showed that MUC1-C binds directly to the MYC HLH-LZ domain and identified a previously unrecognized MUC1-C→MYC pathway that regulates the NuRD complex. MUC1-C/MYC complexes selectively activated the MTA1 and MBD3 genes and post-transcriptionally induced CHD4 expression in basal-but not luminal-type BC cells. In turn, MUC1-C formed complexes with these NuRD components on the ESR1 promoter. Downregulating MUC1-C decreased MTA1/MBD3/CHD4/HDAC1 occupancy and increased H3K27 acetylation on the ESR1 promoter with induction of ESR1 expression and downstream estrogen response pathways. Targeting MUC1-C and these NuRD components also induced expression of FOXA1, GATA3 and other markers associated with the luminal phenotype. These findings support a model in which MUC1-C activates the NuRD complex to drive dedifferentiation and reprogramming of TNBC cells.
Keywords: MUC1-C, MYC, NuRD, TNBC, dedifferentiation
Introduction
The mucin 1 (MUC1) C-terminal subunit (MUC1-C) is an oncogenic transmembrane protein that is overexpressed in triple-negative breast carcinomas (TNBCs) (1,2). MUC1-C interacts with receptor tyrosine kinases, such as EGFR, at the cell membrane and transduces prosurvival signals to the nucleus (2). In this capacity, MUC1-C directly interacts with transcription factors (TFs) and contributes to the regulation of their target genes (2–4). For instance, MUC1-C associates with β-catenin/TCF4 and activates the WNT pathway CCND1 and MYC target genes (5–7). MUC1-C also activates the IKK→NF-κB p65 pathway, binds to NF-κB p65 and promotes expression of the downstream BCL-XL and ZEB1 genes (8,9). MUC1-C activates gene expression in part by increasing TF occupancy on target genes and by recruiting the p300 acetyltransferase (5,6). Other studies have shown that MUC1-C induces DNA methyltransferase 1 (DNMT1) and DNMT3b, and thereby DNA methylation and repression of the CDH1 tumor suppressor gene (TSG) (3,4). MUC1-C also induces expression of the BMI1, RING1 and RING2 components of the Polycomb Repressive Complex 1 (PRC1) and contributes to BMI1-mediated repression of the CDKN2 TSG (3,4). These observations were extended by the demonstration that MUC1-C induces the EZH2, SUZ12 and EED subunits of PRC2 and binds directly to EZH2 (3,4). In turn, MUC1-C increases H3K27 trimethylation on the BRCA1 TSG and downregulates BRCA1 expression (3,4). In concert with involvement in epigenetic repression of TSGs, MUC1-C has been linked to plasticity as characterized by induction of the epithelial-mesenchymal transition (EMT), the cancer stem cell (CSC) state and drug resistance (3,4). These findings suggested that MUC1-C may play a role in other epigenetic pathways of gene repression and activation associated with hallmarks of the cancer cell. Of importance for the present work, there is no available evidence for involvement of MUC1-C in the regulation of ATP-dependent chromatin-remodeling complexes (10).
The nucleosome remodeling and histone deacetylase (NuRD) complex regulates chromatin assembly and reorganization, and is of importance for metazoan development and cancer progression (10). The highly conserved NuRD complex consists of different subunits with enzymatic and non-enzymatic functions that broadly occupy active promoters and enhancers in different cell types (11,12). The core chromodomain-helicase-DNA-binding protein 3 (CHD3) and CHD4 catalyze ATP-dependent chromatin remodeling in association with activity of histone deacetylase 1 (HDAC1) and HDAC2 subunits. Non-enzymatic NuRD components include, among others, the (i) methyl-CpG-binding domain 2 (MBD2) and MBD3, (ii) metastasis-associated gene 1 (MTA1), MTA2 and MTA3, and (iii) retinoblastoma-binding protein 4 (RBBP4) and RBBP7 proteins. MBD3 links the histone deacetylase subcomplex containing MTA1, HDAC and RBBP proteins with the chromatin-remodeling subcomplex containing CHD4 and GATAD2A/B (11). Of these NuRD components, MTA1 is widely overexpressed in human cancers by mechanisms associated, at least in part, with activation of the TGFβ1 and WNT/β-catenin pathways (13,14). MTA1 acts as a corepressor by interacting with ERα and repressing ERα-activated gene transcription (15,16). MTA1 associates with MBD3 and recruits HDAC1 to deacetylate TFs, such as HIF1α, and regulates their transactivation functions (17). In addition, MTA1 interacts with the CHD subunits in integrating their chromatin remodeling functions (18). The pleotropic roles of MTA1 in regulating gene transcription extend to induction of EMT in cancer cells (13,14) and promotion of invasion and metastases (19). MTA1 has also been linked to chromatin remodeling during DNA repair by recruitment to sites of DNA lesions and interaction with poly (ADP ribose) polymerase 1 (PARP1) (20).
MUC1-C activates EZH2 and BMI1 in the epigenetic reprogramming of TNBC cells; however, there is no known link between MUC1-C and chromatin remodeling complexes, such as NuRD. The present studies demonstrate that MUC1-C is necessary for induction of the MTA1, MBD3 and CHD4 subunits of the NuRD complex in TNBC cells. The results show that MUC1-C interacts directly with MYC and thereby (i) activates the MTA1 and MBD3 genes, and (ii) induces post-transcriptional expression of the CHD4 protein. Of functional significance, we demonstrate that the MUC1-C→MYC→NuRD pathway drives luminal→basal dedifferentiation of TNBC cells.
Materials and Methods
Cell culture.
Human BT-549 TNBC (ATCC) and T47D luminal BC (ATCC) cells were cultured in RPMI1640 medium (Corning Life Sciences, Corning, NY, USA) containing 10% heat-inactivated fetal bovine serum (FBS; GEMINI Bio-Products, West Sacramento, CA, USA), 100 μg/ml streptomycin, 100 U/ml penicillin and 10 μg/ml insulin. SUM149 TNBC cells (21) were grown in Ham’s F-12 medium (Corning) supplemented with 10 mM HEPES, 5% FBS, 100 μg/ml streptomycin, 100 U/ml penicillin, 5 μg/ml insulin and 1 μg/ml hydrocortisone. MCF-7 luminal BC cells (ATCC) were cultured in Eagle’s MEM medium (Corning) containing 10% FBS, 100 μg/ml streptomycin, 100 U/ml penicillin and 10 μg/ml insulin. ZR-75-1 luminal BC cells (ATCC) were grown in RPMI1640 medium containing 10% FBS, 100 μg/ml streptomycin and 100 U/ml penicillin. MCF-10A cells (ATCC) were cultured in Mammary Epithelial Growth Medium (Lonza, Walkersville, MD, USA). Cells were treated with 5 μM GO-203 (22,23) and 5 μM JQ1 (24). Cell growth and viability were assessed by trypan blue exclusion. Cells were used in the described experiments for ~6 months. Authentication of cells was performed every 4-6 months by short tandem repeat (STR) analysis (Genetic Analysis Facility, Hospital for Sick Children, Toronto, Canada) of genomic DNA purified with the GenElute Mammalian Genomic DNA Miniprep Kit (Millipore Sigma, Burlington, MA, USA). Cells were monitored for mycoplasma contamination using the MycoAlert Mycoplasma Detection Kit (Lonza, Rockland, ME, USA).
Tetracycline-inducible and stable gene silencing and expression.
MUC1shRNA (MISSION shRNA TRCN0000122938 and shRNA#2 TRCN0000430218), MYCshRNA (MISSION shRNA TRCN0000039642 and shRNA#2 TRCN0000039640), MTA1shRNA (MISSION shRNA TRCN0000230496), MBD3shRNA (MISSION shRNA TRCN0000274441), CHD4shRNA (MISSION shRNA TRCN0000021361) or a control scrambled shRNA (CshRNA, Millipore Sigma) was used in pLKO-puro vector or inserted into the pLKO-tet-puro vector (Plasmid #21915; Addgene, Cambridge, MA, USA). The MUC1-C cDNA was inserted into the pinducer20 vector (Plasmid #44012; Addgene). The viral vectors were produced in 293T cells as described (9). Cells transduced with the vectors were selected for growth in 1-3 μg/ml puromycin or 100 μg/ml geneticin. Cells were treated with 500 ng/ml doxycycline (DOX; Millipore Sigma).
Quantitative reverse-transcription PCR (qRT-PCR).
Total cellular RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). cDNAs were synthesized using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Grand Island, NY, USA). The cDNA samples were amplified using the Power SYBR Green PCR Master Mix (Applied Biosystems) and the CFX96 Real-Time PCR System (BIO-RAD, Hercules, CA, USA) as described (9). Primers used for qRT-PCR are listed in Supplemental Table S1.
Immunoblotting and immunoprecipitation.
Total and nuclear lysates prepared from subconfluent cells were subjected to immunoblot analysis using anti-MUC1-C (#HM-1630-P1ABX; Thermo Fisher Scientific, Waltham, MA, USA), anti-MYC (#ab32072; Abcam), anti-ERα (#ab108398; Abcam) anti-β-actin (#A5441; Sigma), anti-MTA1 (#5647), anti-MBD3 (#14540), anti-CHD4 (#11912), anti-HDAC1 (#5356), anti-SOX2 (#D6D9), anti-KLF4 (#D1F2), anti-BMI1 (#D20B7), anti-CD44 (#156-3C11), anti-OCT4 (#2750S; Cell Signaling Technology, Danvers, MA, USA). Nuclear proteins were immunoprecipitated in the absence and presence of 50 μg/ml ethidium bromide (EtBr; #15585-011, Thermo Fisher Scientific) as described (25).
Direct protein binding studies.
GST, GST-MYC (full length; 1-439), GST-MUC1-CD (full length; 1-72), GST-MUC1-CD(1-45), GST-MUC1-CD(46-72) and GST-MUC1-CD(AQA) were prepared as described (9). Purified GST-MUC1-CD was cleaved with thrombin to remove GST. Binding assays with GST fusion proteins and MUC1-CD or His-MYC were performed for 2 h at room temperature. Adsorbates to glutathione-conjugated beads were detected by immunoblotting.
Chromatin immunoprecipitation (ChIP) assays.
Soluble chromatin was precipitated with anti-MUC1-C (#HM-1630-P1ABX; Thermo Fisher Scientific), anti-MTA1 (#5647), anti-MBD3 (#14540), anti-CHD4 (#11912), anti-HDAC1 (#5356; Cell Signaling Technology), anti-MYC (#ab56), anti-H3K27ac (#ab4729; Abcam) or a control non-immune IgG (Santa Cruz Biotechnology). For re-ChIP analysis, anti-MUC1-C complexes from the primary ChIP were eluted and reprecipitated with anti-MYC. The precipitates were analyzed by qPCR using the Power SYBR Green PCR Master Mix and the ABI Prism 7300 sequence detector (Applied Biosystems). Data are reported as relative-fold enrichment compared to IgG (9). Primers used for ChIP qPCR are listed in Supplemental Table S2.
Promoter luciferase reporter assays.
The MTA1(−777 to +249) and MBD3(−977 to +187) promoter regions contained in the LightSwitch Promoter Reporter vectors (Active Motif, Carlsbad, CA, USA) were cloned into pGL3-basic to generate pMTA1-Luc and pMBD3-Luc. Cells were transfected with (i) an empty pGL3-basic vector, (ii) pMTA1-Luc, (iii) pMBD3-Luc, and (iv) SV-40-Renilla-Luc in the presence of Lipofectamine 3000 Reagent (Invitrogen). At 48 h after transfection, cells were lysed using passive lysis buffer (Promega, Madison, WI, USA). Luminescence was detected with the Dual-Luciferase Reporter Assay System (Promega).
RNA-seq analysis.
Total RNA from cells cultured in triplicates was isolated using Trizol reagent (Invitrogen). TruSeq Stranded mRNA (Illumina, San Diego, CA, USA) was used for library preparation. Raw sequencing reads were aligned to the human genome (GRCh38.74) using STAR. Raw feature counts were normalized and differential expression analysis using DESeq2. Differential expression rank order was utilized for subsequent Gene Set Enrichment Analysis (GSEA), performed using the fgsea (vl.8.0) package in R. Gene sets queried included those from the Hallmark Gene Sets available through the Molecular Signatures Database (MSigDB). The accession number for the RNA-seq data is GEO Submission GSE134147.
Analysis of TCGA-BRCA cohort data.
Normalized gene expression counts and clinical annotations for the TCGA-BRCA cohort were obtained from FireBrowse. To assess associations in gene expression, Pearson correlation was calculated on normalized, log2 transformed expression counts.
TNBC xenograft tumor model studies.
Six-week old female nude mice (Taconic Farms, Hudson, NY, USA) were injected subcutaneously in the flank with 3 x 106 SUM149/tet-MUC1shRNA cells suspended in 50% Matrigel as described (21). When tumor size reached ~100 mm3, the mice were pair-matched into groups of 6 mice each and fed here without or with 625 ppm DOX daily for 56 days. Tumor measurements were calculated by the formula: (width)2 x length/2. These experiments were conducted in accordance with and approved by the Dana-Farber Cancer Institute Animal Care and Use Committee (IACUC) under protocol 03-029.
Statistical analysis.
Each experiment was performed at least three times. Data are expressed as the mean±SD. The unpaired Student’s t-test was used to determine differences between means of groups. A p-value of <0.05 was considered statistically significant.
Results
MUC1-C induces expression of MTA1, MBD3 and CHD4.
MUC1-C activates PRC1 and PRC2; however, there is no known link between MUC1-C and the NuRD complex. To determine whether MUC1-C regulates expression of NuRD components, we analyzed BT-549 TNBC cells expressing a tet-inducible control shRNA (tet-CshRNA) or a tet-MUC1shRNA. We found that DOX-induced MUC1-C silencing (Fig. 1A) is associated with decreases in MTA1 and MBD3, but not CHD4 or HDAC1, mRNA levels (Fig. 1B; Supplemental Fig. S1A). Targeting MUC1-C in SUM149 TNBC cells (Fig. 1C) was similarly associated with downregulation MTA1 and MBD3 mRNA (Fig. 1D; Supplemental Fig. S1B). In concert with these results, we found that DOX-inducible (Figs. 1E and 1F) and stable (Supplemental Figs. S2A and S2B) silencing of MUC1-C suppresses expression of MTA1 and MBD3 protein in BT-549 and SUM149 cells. Targeting MUC1-C also decreased CHD4 protein (Figs. 1E and 1F; Supplemental Figs. S2A and S2B), suggesting that, in contrast to MTA1 and MBD3, MUC1-C induces CHD4 expression at a post-transcriptional level. For confirmation, silencing MUC1-C with a different MUC1shRNA#2 also resulted in downregulation of MTA1, MBD3 and CHD4 expression (Supplemental Fig. S2C). Notably, targeting MUC1-C had no effect on expression of these NuRD components in luminal type MCF-7, T47D and ZR-75-1 cells (Supplemental Figs. S3A–S3D), indicating that MUC1-C drives MTA1, MBD3 and CHD4 in TNBC cells.
Figure 1. MUC1-C drives MTA1, MBD3 and CHD4, and not HDAC1, expression.
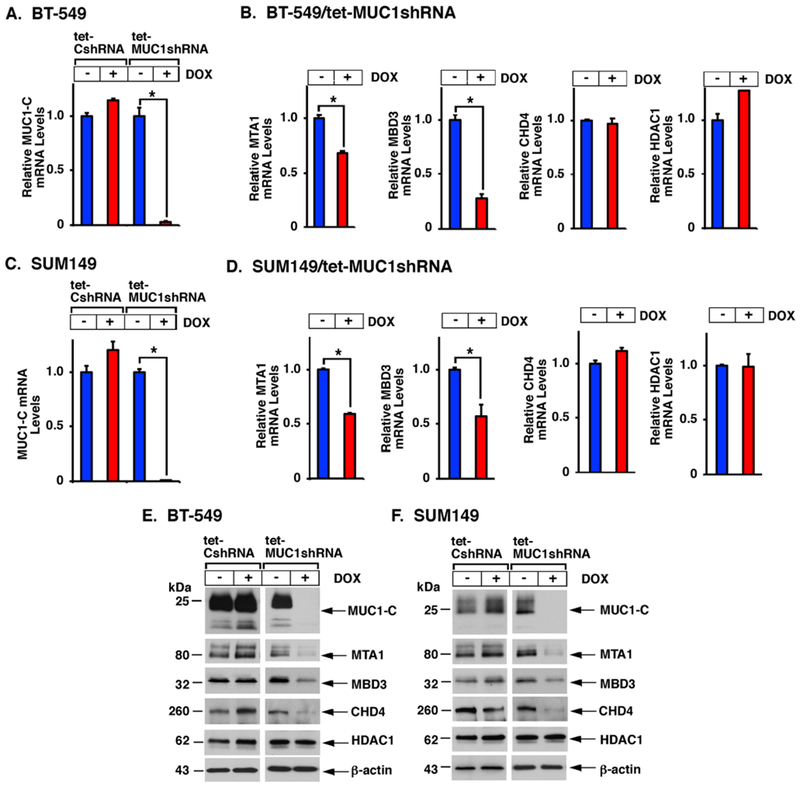
A-D. BT-549 (A,B) and SUM149 (C,D) cells stably expressing a tet-CshRNA or tet-MUC1shRNA were left untreated or treated with 500 ng/ml DOX for 7 days. Cells were analyzed for MUC1-C, MTA1, MBD3, CHD4 and HDAC1 mRNA levels using primers listed in Supplemental Table S1. The results (mean±SD) are expressed as relative mRNA levels compared to that obtained for untreated cells (assigned a value of 1). The asterisk (*) denotes a p-value of <0.05. E-F. Lysates were immunoblotted with antibodies against the indicated proteins.
MUC1-C→MYC signaling induces MTA1, MBD3 and CHD4 expression.
MUC1-C regulates expression of PRC1 and PRC2 proteins by both NF-κB p65- and MYC-dependent mechanisms (4). Here, targeting NF-κB p65 genetically or pharmacologically had no effect on MTA1 or MBD3 expression. However, silencing MUC1-C was associated with decreases in MYC expression (Fig. 2A). Moreover, GSEA analysis of RNA-seq data demonstrated that silencing MUC1-C correlates significantly with downregulation of MYC target genes (Fig. 2B). In extending these results, we found that silencing MYC in DOX-treated BT-549/tet-MYCshRNA and SUM149/tet-MYCshRNA cells is associated with decreases in (i) MTA1 and MBD3, but not CHD4, mRNA levels (Fig. 2C and 2D), and (ii) MTA1, MBD3 and CHD4 protein (Figs. 2E and 2F). As confirmation of MYC involvement, we used a different MYCshRNA#2 and treated cells with the BRD4 inhibitor JQ1, which downregulates MYC (24), and found suppression of MTA1, MBD3 and CHD4 expression (Supplemental Figs. S4A–S4C). These findings supported involvement of a MUC1-C→MYC pathway in inducing MTA1, MBD3 and CHD4 expression.
Figure 2. MUC1-C induces MTA1, MBD3 and CHD4 by a MYC-mediated pathway.
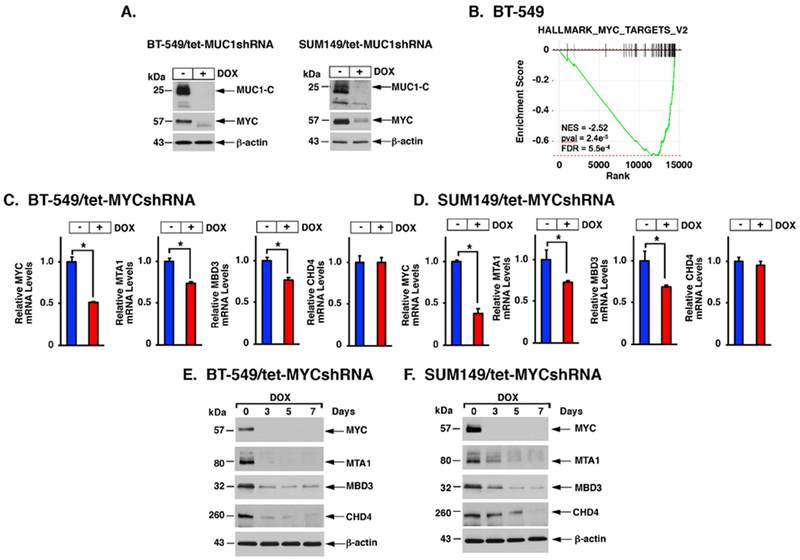
A. BT-549/tet-MUC1shRNA (left) and SUM149/tet-MUC1shRNA (right) cells were left untreated or treated with 500 ng/ml DOX for 7 days. Lysates were immunoblotted with antibodies against the indicated proteins. B. RNA-seq was performed in triplicate on BT-549/CshRNA and BT-549/MUC1shRNA cells. The datasets were analyzed with GSEA, using the Hallmark gene signature collection. Silencing MUC1 was significantly associated with suppression of MYC target gene expression. C-D. BT-549 (C) and SUM149 (D) cells expressing a tet-MYCshRNA were left untreated or treated with 500 ng/ml DOX for 7 days. Cells were analyzed for MYC, MTA1, MBD3, CHD4 and HDAC1 mRNA levels. The results (mean±SD) are expressed as relative mRNA levels compared to that obtained for untreated cells (assigned a value of 1). E-F. BT-549/tet-MYCshRNA (E) and SUM149/tet-MYCshRNA (F) cells were treated with 500 ng/ml DOX for the indicated days. Lysates were immunoblotted with antibodies against the indicated proteins.
MUC1-C associates with MYC in driving MTA1, MBD3 and CHD4 expression.
MUC1-C and MYC are aberrantly expressed in diverse cancers; however, there is no available evidence that these oncoproteins interact with each other. Coimmunoprecipitation studies performed using nuclear lysates demonstrated that MUC1-C forms a complex with MYC (Figs. 3A and 3B). These results were confirmed by including ethidium bromide as a control to inhibit non-specific association of proteins and nucleic acids (25) (Supplemental Figs. S5A and S5B), invoking a potential direct interaction. The MUC1-C cytoplasmic domain is a 72 aa intrinsically disordered protein with the capacity to interact with diverse effectors (Fig. 3C, upper panel) (23). MYC is a 439-aa protein that includes a C-terminal helix-loop-helix leucine zipper (HLH-LZ) domain (Fig. 3C, lower panel). To determine if MUC1-C binds directly to MYC, we incubated GST or GST-MYC(FL; full length) with purified MUC1-CD. Analysis of the adsorbates showed that GST-MYC, and not GST, binds to MUC1-CD (Fig. 3D). Incubation of MUC1-CD deletion mutants further demonstrated that this interaction is mediated by MUC1-CD(1-45) and not MUC1-CD(46-72) (Supplemental Fig. S6A). In addition, mutation of the MUC1-CD CQC motif to AQA blocked binding to MYC (Supplemental Fig. S6B), indicating that the CQC motif confers the interaction. In extending this analysis, incubation of MUC1-CD with GST-MYC deletion mutants demonstrated binding to (i) MYC(294-439), and not MYC(1-156) or MYC(157-293) (Fig. 3E), and (ii) MYC(355-439) which contains the HLH-LZ domain (Fig. 3F). MUC1-CD(1-45) and not MUC1-CD(46-72) conferred binding to MYC(355-439) (Supplemental Fig. S6C). Moreover, mutation of the MUC1-CD CQC motif to AQA abrogated the interaction with MYC(355-439) (Supplemental Fig. S6D), demonstrating that MUC1-C CQC interacts directly with the MYC HLH-LZ domain. Based on these findings, we treated cells with the GO-203 inhibitor, which targets the CQC motif (22,23) (Fig. 3C), and found decreases in MUC1-C, MYC, MTA1, MBD3 and CHD4 proteins (Figs. 3G and 3H), confirming similar effects when targeting MUC1-C genetically and pharmacologically on the regulation of these NuRD components.
Figure 3. MUC1-C forms a direct complex with MYC to activate MTA1, MBD3 and CHD4 expression.
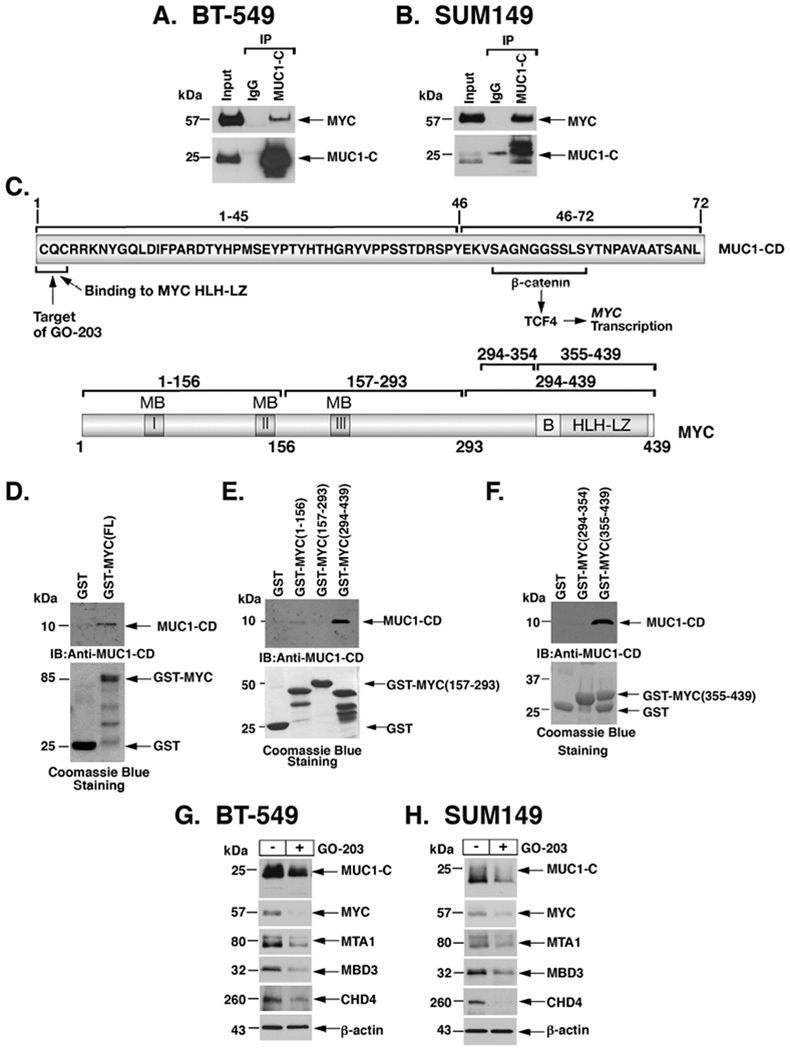
A-B. Nuclear lysates from BT-549 (A) and SUM149 (B) cells were incubated with anti-MUC1-C or a control IgG. The input and precipitates were analyzed by immunoblotting with anti-MYC and anti-MUC1-C. C. Upper panel: Schema of MUC1-C (ED, extracellular domain; TM, transmembrane domain) with the sequence of the 72-aa cytoplasmic domain (CD). Highlighted is the MUC1-C SAGNGGSSLS motif that binds directly to β-catenin and in a complex with TCF4 activates the MYC gene. Also highlighted is the MUC1-C CQC motif that is necessary for MUC1-C homodimerization and confers direct binding to the MYC helix-loop-helix-leucine zipper (HLH-LZ) domain. The MUC1-C CQC motif is the target of the GO-203 inhibitor, which blocks that site and thereby interactions with binding partners. Lower panel: Schema of the MYC protein. The MYC transactivation function is regulated by an unstructured N-terminal domain that contains MYC boxes (MBs) and a nuclear localization signal. The MYC C-terminal region includes a basic (B) region and a helix-loop-helix-leucine zipper (HLH-LZ) domain, which confers binding to HLH-containing partner proteins. D. GST and GST-MYC (full length; 1-439) were incubated with purified MUC1-CD. The adsorbates were immunoblotted with anti-MUC1-CD. Input of the GST proteins was assessed by Coomassie Blue staining. E and F. GST, GST-MYC (1-156), GST-MYC(157-293) and GST-MYC(294-439) were incubated with purified MUC1-CD (E). GST, GST-MYC(294-354) and GST-MYC(355-439) were incubated with purified MUC1-CD (F). The adsorbates were immunoblotted with anti-MUC1-CD. Input of the GST proteins was assessed by Coomassie Blue staining. G-H. BT-549 (G) and SUM149 (H) cells were left untreated or treated with 5 μM GO-203 for 48 h. Lysates were immunoblotted with antibodies against the indicated proteins.
MUC1-C/MYC complexes occupy and activate the MTA1 and MBD3 promoters.
Our results show that, in contrast to CHD4, MUC1-C drives increases in MTA1 and MBD3 mRNA levels, supporting a potential transcriptional mechanism. Along these lines, the MTA1 and MBD3 promoters include putative MYC binding motifs (Fig. 4A). ChIP-PCR analysis of the MTA1 promoter demonstrated occupancy by MUC1-C and MYC (Fig. 4B, left). As a control, there was no detectable MUC1-C or MYC occupancy on a region ~5-kb upstream to the MTA1 promoter (Supplemental Fig. S7A). Re-ChIP studies further showed that MUC1-C associates with MYC on the MTA1 promoter (Fig. 4B, right). Similar results were obtained with ChIP and re-ChIP analyses of the MBD3 promoter (Figs. 4C, left and right; Supplemental Fig. S7B). Silencing MUC1-C was also associated with decreases in H3K27 acetylation on the MTA1 (Fig. 4D) and the MBD3 (Fig. 4E) promoters. In addition, we found that silencing MUC1-C and MYC is associated with activation of pMTA1-Luc (Fig. 4F) and pMBD3-Luc (Fig. 4G) promoter-reporters, supporting a role for MUC1-C→MYC signaling in driving transcription of the MTA1 and MBD3 genes.
Figure 4. MUC1-C activates the MTA1 and MBD3 promoters.
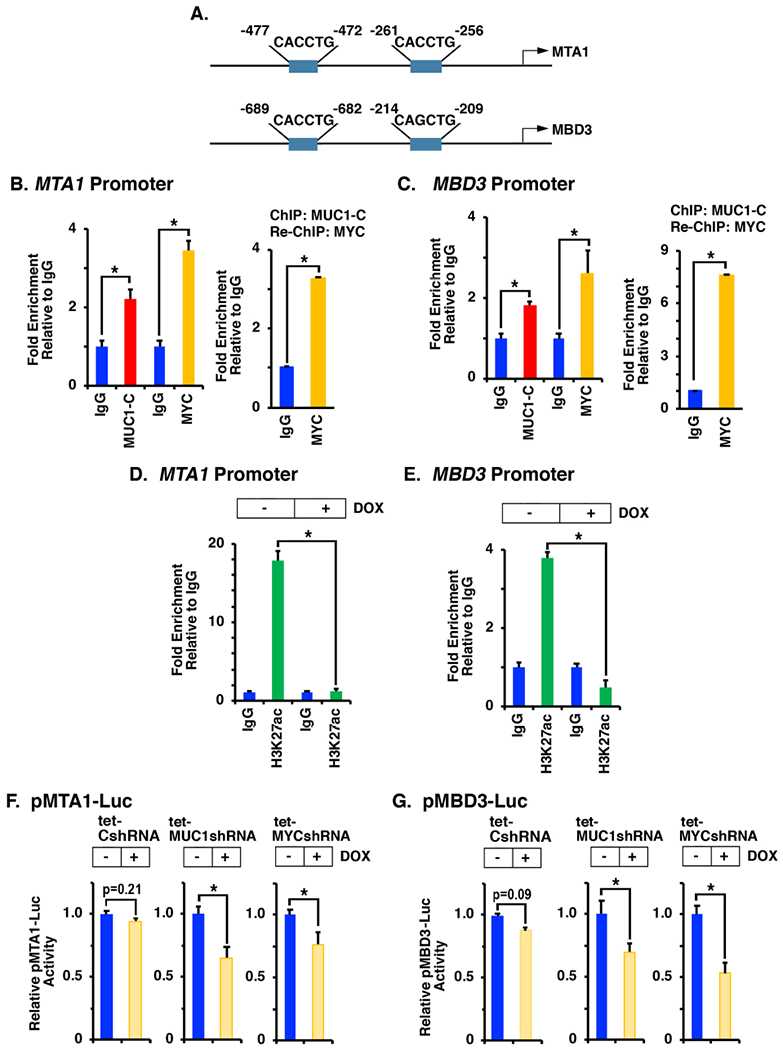
A. Schemas of the MTA1 and MBD3 promoters highlighting the putative MYC binding sites. B and C. Chromatin from BT-549 (B) and SUM149 (C) cells was precipitated with (i) anti-MUC1-C, anti-MYC or a control IgG (left) or (ii) anti-MUC1-C (ChIP) and then reprecipitated with anti-MYC or a control IgG (re-ChIP) (right). D and E. BT-549/tet-MUC1shRNA (D) and SUM149/tet-MUC1shRNA (E) cells were left untreated or treated with 500 ng/ml DOX for 7 days. Chromatin was precipitated with anti-H3K27ac or a control IgG. The DNA samples were amplified by qPCR with primers for the MTA1 and MBD3 promoters (Supplemental Table S2). The results (mean±SD of three determinations) are expressed as the relative fold enrichment compared to that obtained with the IgG control (assigned a value of 1). F and G. BT-549/tet-CshRNA (left panels), BT-549/tet-MUC1shRNA (middle panels) and BT-549/tet-MYCshRNA (right panels) cells left untreated or treated with 500 ng/ml DOX for 5 days were transfected with pGL3-Basic Luc, pMTA1-Luc (F) or pMBD3-Luc (G) vectors for 48 h and then analyzed for luciferase activity. The results (mean±SD of three determinations) are expressed as relative luciferase activity as compared to that obtained for control untreated cells (assigned a value of 1).
MUC1-C drives NuRD occupancy on the ESR1 promoter and represses ESR1 expression.
MUC1-C plays a role in the regulation of the PRC1 and PRC2 subunits by interacting directly with BMI1 and EZH2 (4). In the present work, nuclear coimmunoprecipitation studies demonstrated that MUC1-C forms complexes with MTA1, MBD3, CHD4 and HDAC1 (Figs. 5A and 5B; Supplemental Figs. S5A and S5B). MTA1 has been linked to the differential regulation of the ESR1 promoter in a cell context-dependent manner (26). In concert with the findings that MUC1-C drives MTA1/MBD3/CHD4, we found that MUC1-C occupies the ESR1 promoter with MTA1 (Fig. 5C, left) and silencing MUC1-C decreases MTA1 occupancy (Figs. 5C, right). By extension, we found that silencing MUC1-C similarly decreases MBD3 (Fig. 5D, left) and CHD4 (Fig. 5D, right) occupancy on the ESR1 promoter. MBD3 links the histone deacetylase subcomplex containing MTA1 and HDAC1 with the chromatin-remodeling subcomplex containing CHD4 (11). Consistent with MUC1-C-induced recruitment of MBD3, we found that silencing MUC1-C decreases HDAC1 occupancy on the ESR1 promoter (Fig. 5E, left) and is associated with increases in H3K27 acetylation (Fig. 5E, right). These results were extended by demonstrating that targeting MUC1-C with MUC1shRNA#2 (Supplemental Figs. S8A–S8C) or the GO-203 inhibitor decreases MUC1-C, MTA1, MBD3 and CHD4 occupancy on the ESR1 promoter (Supplemental Figs. S8D–S8F). The significance of these findings was supported by the demonstration that silencing MUC1-C induces ESR1 mRNA and ERα protein levels (Figs. 5F, left and right, and 5G, left and right).
Figure 5. MUC1-C confers NuRD component occupancy on the ESR1 promoter.
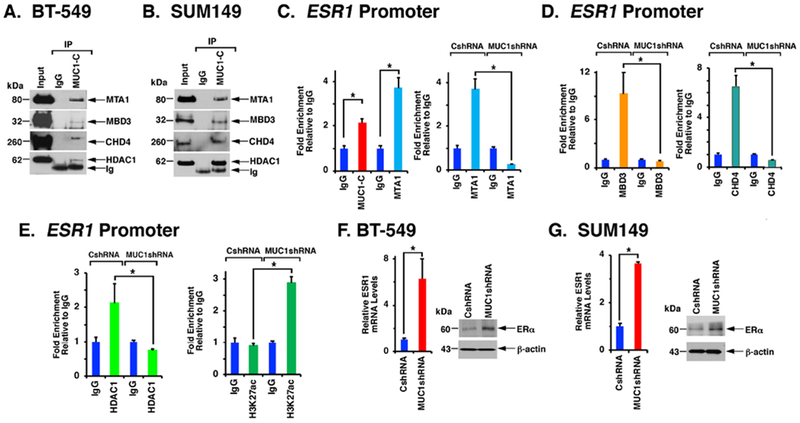
A and B. Nuclear lysates from BT-549 (A) and SUM149 (B) cells were incubated with anti-MUC1-C or a control IgG. The precipitates and input were analyzed by immunoblotting with antibodies against the indicated proteins. C. Chromatin from BT-549 cells was precipitated with anti-MUC1-C, anti-MTA1 or a control IgG (left). Chromatin from BT-549/CshRNA and BT-549/MUC1shRNA cells was precipitated with anti-MTA1 or a control IgG (right). D. Chromatin from BT-549/CshRNA and BT-549/MUC1shRNA cells was precipitated with anti-MBD3 (left), anti-CHD4 (right) or a control IgG. E. Chromatin from BT-549/CshRNA and BT-549/MUC1shRNA cells was precipitated with anti-HDAC1 (left), anti-H3K27ac (right) or a control IgG. The DNA samples were amplified by qPCR with primers for the ESR1 promoter (Supplemental Table S2). The results (mean±SD of three determinations) are expressed as the relative fold enrichment compared to that obtained with the IgG control (assigned a value of 1). F and G. BT-549 (F) and SUM149 (G) cells expressing a CshRNA or MUC1shRNA were analyzed for ESR1 mRNA levels (left). The results (mean±SD) are expressed as relative mRNA levels compared to that obtained for CshRNA expressing cells (assigned a value of 1). Lysates were immunoblotted with antibodies against the indicated proteins (right).
Effects of silencing MUC1-C on ESR1 induction are recapitulated by targeting MTA1, MBD3 and CHD4.
To extend the findings that MUC1-C suppresses a luminal phenotype, RNA-seq was performed on cells with and without MUC1-C silencing. Analysis of the data using the Hallmark Molecular Signature Database (MSigDB) showed that MUC1-C plays a significant role in regulation of the Estrogen Response Early and Estrogen Response Late gene sets (Fig. 6A; Supplemental Figs. S9 and S10. We also found that silencing MUC1-C is associated with induction of (i) the luminal-specific cytokeratins CK8/18 (27), and (ii) NRIP1, SPEDF, ERG3, and TOB1, which have been linked with the luminal phenotype (28–30) (Supplemental Fig. S11). These responses were associated with downregulation of (i) the basal-phenotype associated AXL, TIMP1, FBN1, DSC3 and CK16/17 markers (29,31) (Supplemental Fig. S11). We therefore investigated the effects of targeting MUC1-C on the expression of other genes, such as FOXA1 and GATA3, associated with the luminal phenotype. FOXA1 is a forkhead family member that activates lineage-specific transcription and contributes to the luminal phenotype by facilitating estrogen responsiveness (32,33). GATA3 is a zinc finger DNA binding protein that promotes luminal differentiation of breast epithelial cells (34,35). As found for the ESR1 gene, silencing MUC1-C was associated with induction of FOXA1 and GATA3 expression (Fig. 6B). To determine if these effects of MUC1-C suppression are recapitulated by targeting NuRD components, we analyzed cells with MTA1 silencing and found induction of ESR1, FOXA1 and GATA3 expression (Fig. 6C). In addition, silencing MBD3 (Fig. 6D) and CHD4 (Fig. 6E) was associated with induction of ESR1, FOXA1 and GATA3, confirming that the effects of targeting MUC1-C are recapitulated by suppression of NuRD function. In concert with these findings, analysis of the breast cancer cohort from TCGA demonstrated that MTA1 and MBD3 are negatively associated with ESR1 (Fig. 6F), FOXA1 (Supplemental Fig. S12A) and GATA3 (Supplemental Fig. S12B) expression.
Figure 6. Effects of silencing MUC1-C on ESR1 activation are recapitulated by targeting MTA1, MBD3 and CHD4.
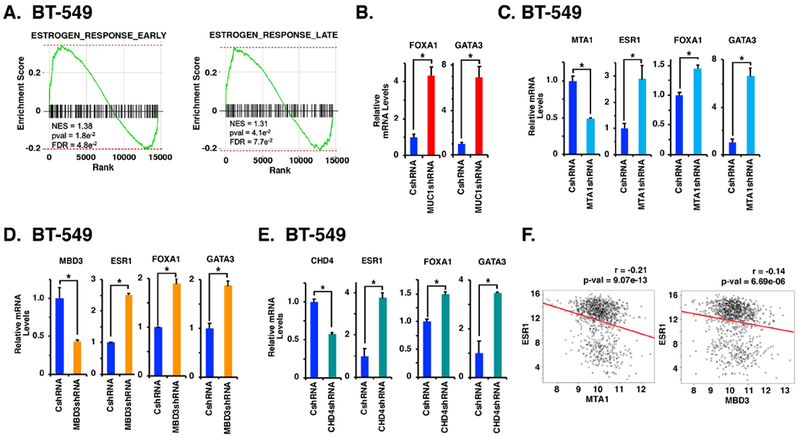
A. RNA-seq was performed in triplicate on BT-549/CshRNA and BT-549/MUC1shRNA cells. The BT-549 datasets were analyzed by GSEA, using the Hallmark gene signature collection. Silencing MUC1-C expression was significantly associated with induction of the Estrogen Response Early and Estrogen Response Late gene sets. B-E. BT-549 cells expressing a CshRNA, MUC1shRNA (B), MTA1shRNA (C), MBD3shRNA (D) or CHD4shRNA (E) were analyzed for the indicated mRNA levels. The results (mean±SD) are expressed as relative mRNA levels compared to that obtained for CshRNA expressing cells (assigned a value of 1). F. Normalized, log2 transformed expression data were obtained for the TCGA-BRCA cohort and correlations were determined. Pearson correlation coefficient and significance are shown.
MUC1-C drives TNBC dedifferentiation.
In gain-of-function studies, MCF-10A breast epithelial cells transduced with a tet-inducible MUC1-C vector (MCF-10A/tet-MUC1-C) responded to DOX treatment with (i) induction of MTA1, MBD3 and CHD4 (Fig. 7A), and (ii) no apparent effect on the already constitutively low to undetectable levels of ESR1, FOXA1 and GATA3. The NuRD complex has been linked to driving reprogramming and pluripotency of stem cells (36,37). In this line of thinking, we found that MUC1-C also induces the stemness-associated MYC, KLF4, OCT4, BMI1, ALDH1 and CD44 proteins (Fig. 7B), of which MYC, KLF4 and OCT4 are among the reprogramming factors that induce pluripotency and dedifferentiation of somatic cells (38). In accord with the results in MCF-10A cells, silencing MUC1-C in BT-549 cells was associated with suppression of MYC, SOX2, KLF4 and OCT4 (Fig. 7C). These findings were extended in an in vivo SUM149/tet-MUC1shRNA model in which targeting MUC1-C with DOX treatment over 3 weeks was associated with suppression of tumor growth (21). Here, DOX-induced MUC1-C silencing for 3 weeks resulted in (i) suppression of MTA1, MBD3 and CHD4 (Figs. 7D and 7E). Moreover, prolonged DOX treatment over 8 weeks was associated with (i) recurrence of growth, (ii) increases in MUC1-C, consistent with overriding of the tet-MUC1shRNA, and (iii) induction of MTA1, MBD3 and CHD4 expression (Figs. 7D and 7E). We also found that MUC1-C expression is associated with upregulation of the MYC, SOX2, KLF4 and OCT4 reprogramming factors (Fig. 7F). These findings collectively supported a program in which MUC1-C drives TNBC dedifferentiation and plasticity in vitro and in vivo.
Figure 7. MUC1-C drives dedifferentiation of TNBC cells.
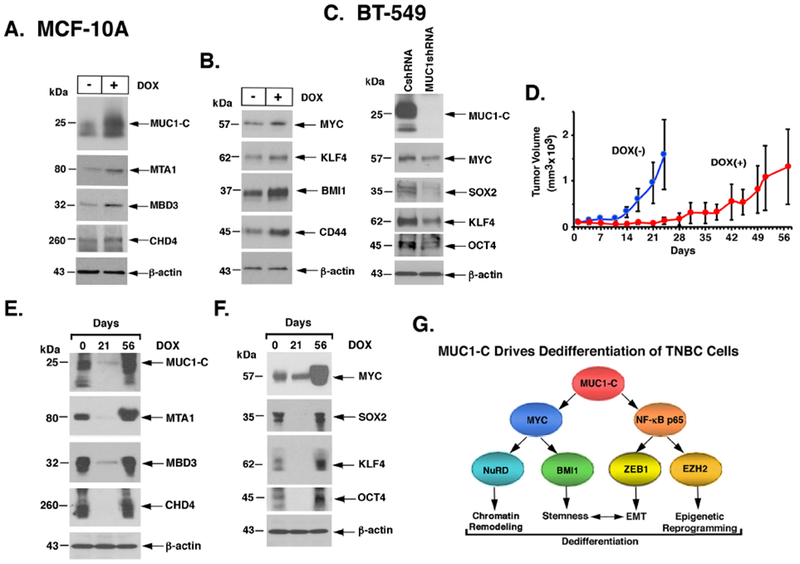
A and B. MCF-10A cells expressing tet-MUC1-C (MCF-lOA/tet-MUC1-C) were left untreated or treated with 500 ng/ml DOX for 7 days. Lysates were immunoblotted with antibodies against the indicated proteins. C. Lysates from BT-549 cells expressing a CshRNA or MUC1shRNA#2 were immunoblotted with antibodies against the indicated proteins. D-F. Female nude mice were injected subcutaneously with SUM149/tet-MUC1shRNA cells. Mice pair-matched into two groups when tumor volumes reached 100 mm3 were fed without (blue) and with DOX (red) for 56 days when the study was terminated. Tumor volumes are expressed as the mean±SD for 6 mice (D). Lysates from tumors harvested on days 0, 21 and 56 were immunoblotted with antibodies against the indicated proteins (E and F). G. Proposed model depicting the role of MUC1-C in integrating NuRD activation with dedifferentiation and plasticity of TNBC cells. MUC1-C has been linked to activation of the IKK→NF-κB p65 and MYC pathways. The disordered MUC1-C cytoplasmic domain binds directly to NF-κB p65 and thereby induces expression of (i) ZEB1 with the induction of EMT and (ii) EZH2 with induction of H3K27 methylation. The present studies further demonstrate that the MUC1-C cytoplasmic domain binds to the MYC HLH-LZ domain, which contributes to regulation of the MYC transactivation function. MUC1-C→MYC signaling induces expression of the BMI1 stem cell factor and, as shown in the present work, the NuRD components MTA1, MBD3 and CHD4. In turn, MUC1-C associates with the NuRD complex and integrates EMT, stemness and epigenetic reprogramming with chromatin remodeling in the dedifferentiation of TNBC cells.
Discussion
The oncogenic MUC1-C protein is aberrantly expressed in TNBC cells and drives epigenetic reprogramming in association with induction of the EMT program and the CSC state (3,4). In this way, MUC1-C induces BMI1 (Fig. 7G) and activation of the PRC1 complex with downregulation of TSG expression (4). MUC1-C also activates EZH2 (Fig. 7G) and other components of the PRC2 complex, including SUZ12 and EED, with repression of TSGs, such as CDH1, CDKN2A, PTEN, BRCA1 and miR-200c (4,39). The present studies extend the involvement of MUC1-C in reprogramming of the cancer cell epigenome by demonstrating that MUC1-C is necessary for inducing expression of the MTA1, MBD3 and CHD4 components of the NuRD ATP-dependent chromatin-remodeling complex (Fig. 7G). MTA1 is overexpressed in breast and other types of cancers, and has been linked to EMT, aggressiveness, metastatic disease and a poor prognosis (19,40). MTA1 functions in part by recruiting HDAC1 with deacetylation of TFs and histones, and the downregulation of certain TSGs, such as BRCA1 and Gai2 (15–17,41,42). The NuRD histone deacetylase subcomplex containing MTA1, HDAC1 and RBBP proteins associates with MBD3, which plays a role in the activation of pluripotency-associated genes and the response to differentiation signals (43). In this context, MBD3 links deacetylation of H3K27ac with recruitment of PRC2 and, in turn, H3K27 trimethylation and gene repression (43). The present findings that MUC1-C is necessary for inducing expression of NuRD components uncovers new insights into a role for MUC1-C in integrating EMT, the CSC state and epigenetic reprogramming (4,39) with chromatin remodeling of TNBC cells (Fig. 7G). In extending this work, additional studies will be needed to determine whether MUC1-C plays a role in the regulation of other ATP-dependent chromatin-remodeling complexes, such as SWI/SNF, which functions in the control of chromatin access (10).
MUC1-C has been linked to induction of the WNT/β-catenin/TCF4 pathway and thereby activation of MYC in cancer cells (6). In the present work, we found that MUC1-C also binds directly to MYC and that MUC1-C/MYC complexes drive transcription of the MTA1 and MBD3 genes. The intrinsically disordered MUC1-C cytoplasmic domain has the capacity to interact with multiple TFs, including NF-κB p65, STAT1/3 and ZEB1, as well as the BMI1 and EZH2 effectors of epigenomic reprogramming (4,39). The present findings extend this paradigm by showing that MUC1-C directly interacts with the MYC HLH-LZ domain and that silencing MUC1-C is associated with the suppression of multiple MYC target genes (Fig. 7G). MYC had been associated with induction of MTA1 (44), but not to our knowledge with the MBD3 gene. We also found that MUC1-C upregulates expression the ATP-dependent CHD4 helicase by a MYC-dependent mechanism. CHD4 functions in nucleosome remodeling and in the repression of TSGs by recruitment of epigenetic effectors, such as DNMTs (11,45). As found for targeting MUC1-C (4), silencing CHD4 reactivates hypermethylated TSGs (45), supporting the potential importance of MUC1-C in integrating induction of DNMT3b and the CHD4 protein in the hypermethylation of TSGs. In contrast to activation of the MTA1 and MBD3 genes, our results indicate that the MUC1-C→MYC pathway increases CHD4 expression by a post-transcriptional mechanism. In this regard, MUC1-C and MYC have been separately linked to induction of LIN28B and the downregulation of let-7 (46,47), which may potentially target the CHD4 3’UTR. Subsequent studies will thus be needed to determine whether let-7 is indeed responsible for MUC1-C→MYC-mediated CHD4 regulation. Additional experimentation will also be needed to determine whether the interactions of MUC1-C with MYC and other Yamanaka reprogramming TFs, such as SOX2, KLF4 and OCT4, as shown here in the TNBC model, are of importance for plasticity and dedifferentiation of stem cells.
NuRD is a multifaceted complex that is also involved in chromatin remodeling necessary for the differentiation of mouse embryonic stem cells to specific lineages (11). In cancer cells and like MUC1-C (4), NuRD is associated with the induction of EMT (11); however, less is known about the role of this complex in TNBC dedifferentiation and progression. Of potential interest in this regard, MTA1 activates the ESR1 promoter in ERα-positive BC cells and represses it in TNBC cells (26). The present studies further demonstrate that, in addition to inducing their expression, MUC1-C associates with MTA1/MBD3/CHD4 and that these nuclear complexes are detectable on the ESR1 promoter. Targeting MUC1-C and thereby downregulation of MTA1/MBD3/CHD4 in TNBC cells resulted in ESR1 induction and, as determined by RNA-seq analysis, activation of the estrogen response early and late pathways. MUC1-C had been linked to the regulation of ERα function (48); but not to activation of the ESR1 gene. The previously unrecognized findings that silencing MUC1-C in TNBC cells induces ESR1, FOXA1 and GATA3, which are functionally associated with the luminal phenotype of breast epithelial cells (32–35), thus invoked the possibility that MUC1-C→MYC signaling contributes to a program of dedifferentiation by activating the NuRD complex. Indeed, we found that silencing MTA1, MBD3 or CHD4 similarly induces ESR1, FOXA1 and GATA3 expression, confirming that suppression of the NuRD complex recapitulates the effects of targeting MUC1-C and MYC. In further support for involvement of MUC1-C in activating NuRD, gain-of-function studies in MCF-10A breast epithelial cells demonstrated that MUC1-C induces MTA1, MBD3 and CHD4. In addition, we found that MUC1-C is associated with expression of MYC, KLF4, SOX2 and OCT4 in this model, linking MUC1-C-induced activation of the NuRD complex with reprogramming and providing further support for involvement of MUC1-C in driving a program of dedifferentiation (Fig. 7G).
In summary, the present work identifies a previously unrecognized pathway in which MUC1-C and MYC drive expression of the MTA1/MBD3/CHD4 components of the NuRD complex in association with dedifferentiation of TNBC cells. These findings and the demonstration that overexpression of MYC in mammary epithelial cells represses lineage-specific TFs, such as ERα and GATA3, and induces a stem cell-like state (49) will be of importance for determining whether MUC1-C→MYC signaling contributes to enhancer reprogramming and thereby cancer cell plasticity (50). MUC1-C has been linked to the EMT program by inducing EMT-TFs, such as TWIST1, ZEB1 and SNAIL (21). Other studies have demonstrated that as part of the NuRD complex, MTA1 regulates EMT and metastasis of cancer cells (13,14,19,40). Based on the present work, the effects of targeting MUC1-C with inhibitors, such as GO-203, and thereby downregulating MTA1, would suppress the EMT program and, in turn, the CSC state, drug resistance and plasticity. The interconnectivity of MUC1-C with the NuRD complex thus extends the role for MUC1-C in integrating EMT, stemness and epigenetic reprogramming (4) with chromatin remodeling in TNBC progression (Fig. 7G). Furthermore, in regard to the significant unmet clinical need for identifying specific molecular targets in TNBC, our results provide support for development of the available MUC1-C-targeted agents in therapeutic strategies for TNBC patients.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under grant numbers CA97098, CA166480, CA229716 and CA233084 awarded to D. Kufe and CA232979 awarded to S. Liu.
Abbreviations:
- MUC1
mucin 1
- MUC1-C
MUC1 C-terminal transmembrane subunit
- TF
transcription factor
- TSG
tumor suppressor gene
- DNMT
DNA methyltransferase
- PRC
polycomb repressive complex
- EMT
epithelial-mesenchymal transition
- CSC
cancer stem cell
- TNBC
triple negative breast cancer
- NuRD
nucleosome remodeling and histone deacetylase
- CHD
chromodomain-helicase-DNA-binding protein
- HDAC1
histone deacetylase 1
- MBD
methyl-CpG-binding domain
- MTA1
metastasis-associated gene 1
- RBBP
retinoblastoma-binding protein
- ERα
estrogen receptor α
- PARP1
poly(ADP ribose) polymerase 1
- MSigDB
Molecular Signatures Database
- ESR1
estrogen receptor 1
Footnotes
Conflict of Interest Statement
DK has equity interests in Genus Oncology, Reata Pharmaceuticals, Hillstream BioPharma, Nanogen Therapeutics and Victa BioTherapeutics, serves as a member of the board of directors of Nanogen and Victa, and is a paid consultant to Reata, CanBas and Victa. The other authors disclosed no potential conflicts of interest.
References
- 1.Kufe D. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer 2009;9:874–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kufe D. MUC1-C oncoprotein as a target in breast cancer: activation of signaling pathways and therapeutic approaches. Oncogene 2013;32:1073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajabi H, Kufe D. MUC1-C oncoprotein integrates a program of EMT, epigenetic reprogramming and immune evasion in human carcinomas. BBA Reviews on Cancer 2017;1868:117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajabi H, Hiraki M, Kufe D. MUC1-C activates polycomb repressive complexes and downregulates tumor suppressor genes in human cancer cells. Oncogene 2018;37:2079–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajabi H, Ahmad R, Jin C, Kosugi M, Alam M, Joshi M, et al. MUC1-C oncoprotein induces TCF7L2 transcription factor activation and promotes cyclin D1 expression in human breast cancer cells. J Biol Chem 2012;287:10703–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bouillez A, Rajabi H, Pitroda S, Jin C, Alam M, Kharbanda A, et al. Inhibition of MUC1-C suppresses MYC expression and attenuates malignant growth in KRAS mutant lung adenocarcinomas. Cancer Res 2016;76:1538–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alam M, Bouillez A, Tagde A, Ahmad R, Rajabi H, Maeda Ύ , et al. MUC1-C represses the Crumbs complex polarity factor CRB3 and downregulates the Hippo pathway. Mol Cancer Res 2016;14:1266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad R, Raina D, Joshi MD, Kawano T, Kharbanda S, Kufe D. MUC1-C oncoprotein functions as a direct activator of the NF-κB p65 transcription factor. Cancer Res 2009;69:7013–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajabi H, Alam M, Takahashi H, Kharbanda A, Guha M, Ahmad R, et al. MUC1-C oncoprotein activates the ZEB1/miR-200c regulatory loop and epithelial-mesenchymal transition. Oncogene 2014;33:1680–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clapier CR, Iwasa J, Cairns BR, Peterson CL. Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat Rev Mol Cell Biol 2017;18:407–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lai AY, Wade PA. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat Rev Cancer 2011;11:588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bornelov S, Reynolds N, Xenophontos M, Gharbi S, Johnstone E, Floyd R, et al. The nucleosome remodeling and deacetylation complex modulates chromatin structure at sites of active transcription to fine-tune gene expression. Mol Cell 2018;71:56–72 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pakala SB, Singh K, Reddy SD, Ohshiro K, Li DQ, Mishra L, et al. TGF-beta1 signaling targets metastasis-associated protein 1, a new effector in epithelial cells. Oncogene 2011;30:2230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan D, Avtanski D, Saxena NK, Sharma D. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires beta-catenin activation via Akt/GSK3- and MTA1/Wnt1 protein-dependent pathways. J Biol Chem 2012;287:8598–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazumdar A, Wang RA, Mishra SK, Adam L, Bagheri-Yarmand R, Mandal M, et al. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nat Cell Biol 2001;3:30–7. [DOI] [PubMed] [Google Scholar]
- 16.Molli PR, Singh RR, Lee SW, Kumar R. MTA1-mediated transcriptional repression of BRCA1 tumor suppressor gene. Oncogene 2008;27:1971–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoo YG, Kong G, Lee MO. Metastasis-associated protein 1 enhances stability of hypoxia-inducible factor-1alpha protein by recruiting histone deacetylase 1. EMBO J 2006;25:1231–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torrado M, Low JKK, Silva APG, Schmidberger JW, Sana M, Sharifi Tabar M, et al. Refinement of the subunit interaction network within the nucleosome remodelling and deacetylase (NuRD) complex. FEBS J 2017;284:4216–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malisetty VL, Penugurti V, Panta P, Chitta SK, Manavathi B. MTA1 expression in human cancers - Clinical and pharmacological significance. Biomed Pharmacother 2017;95:956–64. [DOI] [PubMed] [Google Scholar]
- 20.Li DQ, Yang Y, Kumar R. MTA family of proteins in DNA damage response: mechanistic insights and potential applications. Cancer Metastasis Rev 2014;33:993–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hata T, Rajabi H, Yamamoto M, Jin C, Ahmad R, Zhang Y, et al. Targeting MUC1-C inhibits TWIST1 signaling in triple-negative breast cancer. Mol Cancer Ther 2019. July 15; [Epub ahead of print]:pii: molcanther.0156.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raina D, Kosugi M, Ahmad R, Panchamoorthy G, Rajabi H, Alam M, et al. Dependence on the MUC1-C oncoprotein in non-small cell lung cancer cells. Mol Cancer Ther 2011;10:806–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raina D, Agarwal P, Lee J, Bharti A, McKnight C, Sharma P, et al. Characterization of the MUC1-C cytoplasmic domain as a cancer target. PLoS One 2015;10:e0135156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011;146:904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandoval GJ, Pulice JL, Pakula H, Schenone M, Takeda DY, Pop M, et al. Binding of TMPRSS2-ERG to BAF chromatin remodeling complexes mediates prostate oncogenesis. Mol Cell 2018;71:554–66 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang HJ, Lee MH, Kang HL, Kim SH, Ahn JR, Na H, et al. Differential regulation of estrogen receptor alpha expression in breast cancer cells by metastasis-associated protein 1. Cancer Res 2014;74:1484–94. [DOI] [PubMed] [Google Scholar]
- 27.Walker LC, Harris GC, Holloway AJ, McKenzie GW, Wells JE, Robinson BA, et al. Cytokeratin KRT8/18 expression differentiates distinct subtypes of grade 3 invasive ductal carcinoma of the breast. Cancer Genet Cytogenet 2007;178:94–103. [DOI] [PubMed] [Google Scholar]
- 28.Yu-Rice Y, Jin Y, Han B, Qu Y, Johnson J, Watanabe T, et al. FOXC1 is involved in ERalpha silencing by counteracting GATA3 binding and is implicated in endocrine resistance. Oncogene 2016;35:5400–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dai X, Cheng H, Bai Z, Li J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J Cancer 2017;8:3131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaudhary S, Krishna BM, Mishra SK. A novel FOXA1/ESR1 interacting pathway: A study of Oncomine breast cancer microarrays. Oncol Lett 2017;14:1247–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Badola S, Parker A, Campbel M, Boedigheimer M, Grubinska B, Turci S, et al. Epigenetic signature of human breast cancer subtypes. Cancer Res 2009;69:23 Suppl:Abstract B45. [Google Scholar]
- 32.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, et al. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 2008;132:958–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laganiere J, Deblois G, Lefebvre C, Bataille AR, Robert F, Giguere V. From the Cover: Location analysis of estrogen receptor alpha target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc Natl Acad Sci U S A 2005;102:11651–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell 2006;127:1041–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asselin-Labat ML, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC, et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat Cell Biol 2007;9:201–9. [DOI] [PubMed] [Google Scholar]
- 36.Kaji K, Caballero IM, MacLeod R, Nichols J, Wilson VA, Hendrich B. The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat Cell Biol 2006;8:285–92. [DOI] [PubMed] [Google Scholar]
- 37.dos Santos RL, Tosti L, Radzisheuskaya A, Caballero IM, Kaji K, Hendrich B, et al. MBD3/NuRD facilitates induction of pluripotency in a context-dependent manner. Cell Stem Cell 2014;15:102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takahashi K, Yamanaka S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol 2016;17:183–93. [DOI] [PubMed] [Google Scholar]
- 39.Rajabi H, Hiraki M, Tagde A, Alam M, Bouillez A, Christensen CL, et al. MUC1-C activates EZH2 expression and function in human cancer cells. Sci Rep 2017;7:7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sen N, Gui B, Kumar R. Role of MTA1 in cancer progression and metastasis. Cancer Metastasis Rev 2014;33:879–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo J, Su F, Chen D, Shiloh A, Gu W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000;408:377–81. [DOI] [PubMed] [Google Scholar]
- 42.Ohshiro K, Rayala SK, Wigerup C, Pakala SB, Natha RS, Gururaj AE, et al. Acetylation-dependent oncogenic activity of metastasis-associated protein 1 co-regulator. EMBO Rep 2010;11:691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reynolds N, Salmon-Divon M, Dvinge H, Hynes-Allen A, Balasooriya G, Leaford D, et al. NuRD-mediated deacetylation of H3K27 facilitates recruitment of Polycomb Repressive Complex 2 to direct gene repression. EMBO J 2012;31:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang XY, DeSalle LM, Patel JH, Capobianco AJ, Yu D, Thomas-Tikhonenko A, et al. Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc Natl Acad Sci U S A 2005;102:13968–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia L, Huang W, Bellani M, Seidman MM, Wu K, Fan D, et al. CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell 2017;31:653–68 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci USA 2009;106:3384–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alam M, Ahmad R, Rajabi H, Kufe D. MUC1-C induces the LIN28B→LET-7→HMGA2 axis and self-renewal in NSCLC cells. Mol Cancer Res 2015;13:449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei X, Xu H, Kufe D. MUC1 oncoprotein stabilizes and activates estrogen receptor α. Mol Cell 2006;21:295–305. [DOI] [PubMed] [Google Scholar]
- 49.Poli V, Fagnocchi L, Fasciani A, Cherubini A, Mazzoleni S, Ferrillo S, et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nature communications 2018;9:1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fagnocchi L, Poli V, Zippo A. Enhancer reprogramming in tumor progression: a new route towards cancer cell plasticity. Cell Mol Life Sci 2018;75:2537–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.