

Abstract
5-methylcytosine (mC) is an epigenetic mark that is written by methyltransferases, erased through passive and active mechanisms, and impacts transcription, development, diseases including cancer, and aging. Active DNA demethylation involves TET-mediated stepwise oxidation of mC to 5-hydroxymethylcytosine, 5-formylcytosine (fC), or 5-carboxylcytosine (caC), excision of fC or caC by thymine DNA glycosylase (TDG), and subsequent base excision repair. Many elements of this essential process are poorly defined, including TDG excision of caC. To address this problem, we solved high-resolution structures of human TDG bound to DNA with cadC (5-carboxyl-2′-deoxycytidine) flipped into its active site. The structures unveil detailed enzyme-substrate interactions that mediate recognition and removal of caC, many involving water molecules. Importantly, two water molecules contact a carboxylate oxygen of caC and are poised to facilitate acid-catalyzed caC excision. Moreover, a substrate-dependent conformational change in TDG modulates the hydrogen bond interactions for one of these waters, enabling productive interaction with caC. An Asn residue (N191) that is critical for caC excision is found to contact N3 and N4 of caC, suggesting a mechanism for acid-catalyzed base excision that features an N3-protonated form of caC but would be ineffective for C, mC, or hmC. We also investigated another Asn residue (N140) that is catalytically essential and strictly conserved in the TDG-MUG enzyme family. A structure of N140A-TDG bound to cadC DNA provides the first high-resolution insight into how enzyme-substrate interactions, including water molecules, are impacted by depleting the conserved Asn, informing its role in binding and addition of the nucleophilic water molecule.
Graphical Abstract
INTRODUCTION
Methylation of cytosine by DNA methyltransferases (DNMT) produces 5-methylcytosine (mC), a prevalent mark for epigenetic regulation that is erased through passive and active mechanisms.1 An essential pathway for active DNA demethylation in vertebrates includes mC oxidation by a TET (ten-eleven translocation) enzyme, excision of oxidized mC by thymine DNA glycosylase (TDG) and base excision repair (BER) (Figure 1).2 TET enzymes oxidize mC to give 5-hydroxymethyl-C (hmC) and catalyze additional oxidation reactions to yield 5-formyl-C (fC) or 5-carboxyl-C (caC).3-6 While mC and hmC are not excised by a DNA glycosylase, TDG removes fC and caC and subsequent BER steps yield unmodified cytosine.7-10 DNA demethylation is required for regulating cell differentiation during vertebrate development, for transcriptional regulation in post-mitotic cells including neurons, and for suppressing tumor development,11 but many aspects of this key epigenetic process are poorly defined.
Figure 1.
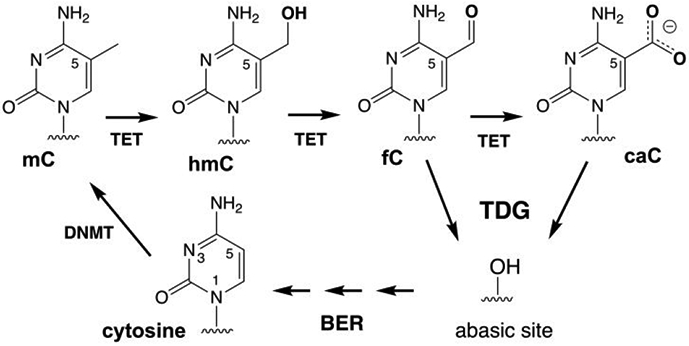
Methylation of cytosine and active demethylation by the TET-TDG-BER pathway in vertebrates.
In particular, our understanding of how TDG recognizes and excises fC and caC, but not their precursors (C, mC, hmC), remains poor. TDG was discovered as an enzyme that excises thymine from G·T mispairs, which can arise via mC deamination and cause C→T mutations.12-13 Depletion of TDG in mice causes embryonic lethality,14-15 a phenotype that likely reflects the inability of other mammalian glycosylases to excise fC or caC, an essential step of active DNA demethylation. Chromatin structure appears to have a role in regulating TDG activity, at least for G·T and G·fC pairs.16-17 Like other monofunctional glycosylases, TDG excises nucleobases from DNA by hydrolyzing the N-glycosyl bond of a deoxynucleotide flipped into its active site.18-19 TDG employs acid catalysis to activate the nucleobase leaving group (LG) for excision of caC but not for fC or other target bases (U, T).20-21 Acid catalysis is effective for caC because it exists as a monoanion at physiological pH,22 and N-glycosyl bond cleavage is hindered by the poor LG quality of a departing caC dianion (Figure 2).21 Protonation yields a neutral form of caC (amino, zwitterion, or imino) and provides catalysis because a neutral base will depart as a monoanion.21 However, the mechanism of acid-catalyzed caC excision remains obscure, due in part to insufficient structural information.
Figure 2.
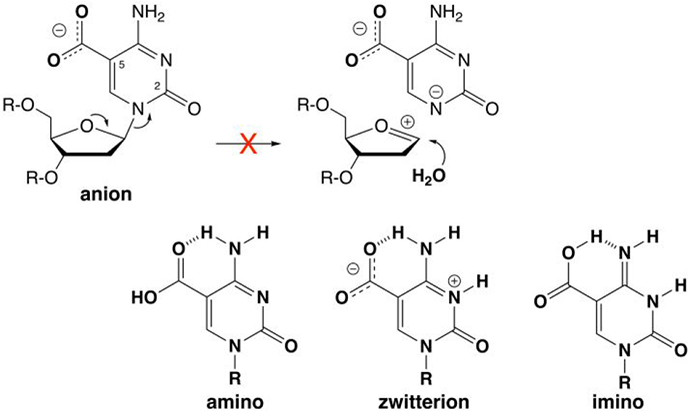
The caC base is a monoanion at physiological pH and N-glycosyl bond hydrolysis is likely precluded by the poor leaving-group (LG) quality of a departing caC dianion. For clarity, the focus is on LG departure in a stepwise mechanism (oxacarbenium ion intermediate), without showing details for nucleophile addition (see text). Leaving group quality is improved by protonation of the caC anion to give a neutral species (amine, zwitterion, or imino), as shown by previously calculated N1 acidities, where the free energy of deprotonation (kcal mol−1) in water is 27.7 for the caC anion and 20.5, 13.3, and 16.0 for the amino, zwitterion, and imino forms of neutral caC, respectively.21
The mechanism for addition of the nucleophilic water molecule in the hydrolytic reaction also remains unclear, for TDG and the related bacterial MUG (mismatch uracil glycosylase) enzymes. Previous studies indicate that nucleophile addition requires an Asn residue that is strictly conserved in the large TDG-MUG enzyme family (Asn140 in human TDG).23-26 Stepping back, we note that the overall reaction for these enzymes could involve a stepwise mechanism, where LG departure gives a discrete (short-lived) oxacarbenium ion intermediate followed by nucleophile addition.18-19, 27 A stepwise mechanism, with rate-limiting LG departure, is indicated for UNG (uracil DNA glycosylase),28-29 which is of the same superfamily as TDG-MUG enzymes. Alternatively, the TDG-MUG reaction could follow a concerted mechanism with a single oxacarbenium ion-like transition state (TS) in which LG departure is greatly advanced over nucleophile addition (a “loose” or “highly dissociative” TS). Such a mechanism was indicated by a QM/MM study of the TDG reaction (for fC excision).30 Previous structures of TDG reveal that the side chain (carboxamide) oxygen of the conserved Asn contacts the nucleophile,31-32 suggesting a key role in positioning the nucleophile. Our understanding of how the conserved Asn promotes nucleophile addition for TDG-MUG enzymes could be advanced by structures for wild-type enzyme and a variant that lacks the conserved Asn, bound to a common substrate and solved at sufficient resolution to observe water molecules in the active site, but such structures have not been reported to date.
To address these problems, we solved high resolution structures of TDG and N140A-TDG bound to DNA with cadC (5-carboxyl-2′-deoxycytidine) flipped into the active site. The structures redefine the molecular basis for recognition and hydrolytic excision of caC by TDG, including key roles for water molecules that were not observed in previous structures.33 Our new structures also reveal a substrate-dependent conformational switch that alters the hydrogen bonding properties for one of these water molecules, enabling it to interact productively with the carboxylate group of caC. The results suggest a new mechanism for acid-catalyzed base excision that is consistent with previous experimental findings and would be specific for caC and ineffective for C, mC, or hmC. Our results also reveal, for the first time, the structural effects of depleting the carboxamide group of the conserved Asn residue for TDG-MUG enzymes, including the effect on active-site water molecules, informing the role of the Asn in mediating nucleophile addition during N-glycosyl bond hydrolysis.
RESULTS
High-resolution structures of TDG bound to cadC DNA.
Determining the structure of an enzyme-substrate complex requires an approach to halt catalytic activity while minimally perturbing substrate interactions, which typically involves a nonreactive substrate analogue or a catalytically inactive enzyme variant. We used both approaches to solve high-resolution structures of TDG bound to DNA with cadC flipped into its active site (Table S1). One structure, solved at 1.55 Å resolution, includes wild-type TDG82-308 and DNA containing 2′-fluoroarabino-cadC (cadCF) (Figure 3a; PDB ID: 6U17). The subtle 2′-F substitution fully precludes TDG cleavage of the N-glycosyl bond for cadC and other substrates (dT, dU, fdC), likely by destabilizing the putative oxacarbenium ion-like transition state in the reaction for N-glycosyl bond cleavage.18, 24, 30, 32-35 We also determined a high-quality structure of N140A-TDG82-308 bound to DNA with cadC in its active site (1.60 Å resolution; PDB ID: 6U16). The N140A substitution halts catalytic activity of TDG while not diminishing its binding affinity for many substrates.24-25, 36 The mutation prevents cadC cleavage in our crystallization conditions, consistent with single turnover kinetics experiments showing that N140A-TDG82-308 lacks significant caC excision activity in solution (reaction half-life 70 days; Figure S1A).36
Figure 3.
High-resolution structure of TDG bound to G·caC DNA. (a) Crystal structure of TDG82-308 bound to DNA with cadCF in its active site (1.55 Å; PDB ID 6U17). TDG82-308 is cyan, water molecules are red spheres, the target DNA strand (with cadCF) is yellow and the complementary strand green (with N, blue; O, red; P, orange). The 2Fo–Fc electron density map, contoured at 1.0σ, is shown for DNA. (b) Alignment of our new structure and a previous structure of TDG111-308 bound to cadCF DNA (3.01 Å; PDB ID 3UOB). Coloring for the new structure is the same except that residues 107-122 are dark blue. The previous structure exhibited 2:1 binding, one TDG subunit (pink) bound at a G·caC site and the adjacent subunit (dirty violet) at a nonspecific site.
Our results provide a new structural paradigm for how TDG recognizes and excises caC from DNA. Previous structures of TDG bound to cadCF DNA, and of N140A-TDG bound to cadC DNA, were solved at moderate resolution (3.0 Å), lack water molecules, and were obtained from crystals produced with DNA that yields 2:1 binding (TDG:DNA), with one TDG subunit bound at a specific site (G·caC) and the other at a nonspecific site (Figure 3b).33, 35, 37 The 2:1 complex has an interface between TDG subunits that disrupts or alters their DNA interactions when compared to structures of TDG-DNA complexes that feature 1:1 binding,31-32, 38 the stoichiometry that is likely to be physiologically relevant.37, 39-40 Our new structures were generated using DNA that gives 1:1 binding and under conditions that yield high resolution and feature hundreds of ordered water molecules, including some that mediate key enzyme-substrate interactions, as detailed below. In addition, the structures were produced using TDG82-308, a construct that fully recapitulates the substrate binding affinity and glycosylase activity of full length TDG (410 residues),31-32 unlike the smaller construct (TDG111-308) used for the previous structures.
Owing to the use of improved constructs for both TDG and the DNA, our new structures feature 16 N-terminal residues (107-122) that were absent in the original structures, due likely to disorder associated with 2:1 binding (Figure 3b). These N-terminal residues, which include an alpha helix (α0), interact with DNA and with other regions of TDG, including a loop (β2-α4) that contains an important catalytic residue (Thr197) and a helix (α1) that contains residues which contact the flipped base. A comparison of protein backbone conformation in the new and the previous structure of TDG bound to cadCF DNA reveals substantial variation, as indicated by a percentile-based spread (pbs) of 0.51 Å.41 The variance is most pronounced in two helices (α3, α6), one strand (β4), and two loops (α1-α2, β2-α4) and is likely attributable to differences in structural resolution as wells as binding stoichiometry.31 Indeed, smaller variances are observed when comparing our new structure to structures of TDG82-308 bound to other substrates or to product DNA, including G·U (pbs 0.14 Å; PDB ID 5HF7), G·fC (pbs 0.23 Å; PDB ID 5T2W), or G·AP (pbs 0.14 Å; PDB ID 5FF8), which all feature 1:1 binding and resolution of 1.54 Å to 2.2 Å.31-32
TDG interactions with the caC base.
Our structures reveal detailed interactions with the caC base, including contacts involving water molecules that have not been previously observed (Figure 4). As such, our results provide a robust structural accounting for findings that TDG binds tightly to G·caC DNA, with a dissociation constant (Kd = 2.3 nM) that is about 100-fold greater than that for nonspecific DNA.39 It is important to note that while the discussion below focuses largely on wild-type TDG82-308 bound to cadCF DNA, the same suite of interactions with the caC base, including water-mediated contacts, are observed for N140A-TDG82-308 bound to cadC DNA (Figure S2a). Notably, these structural observations account for findings that the N140A mutation has little effect on the binding affinity of TDG for G·caC and other substrates.24, 36
Figure 4.
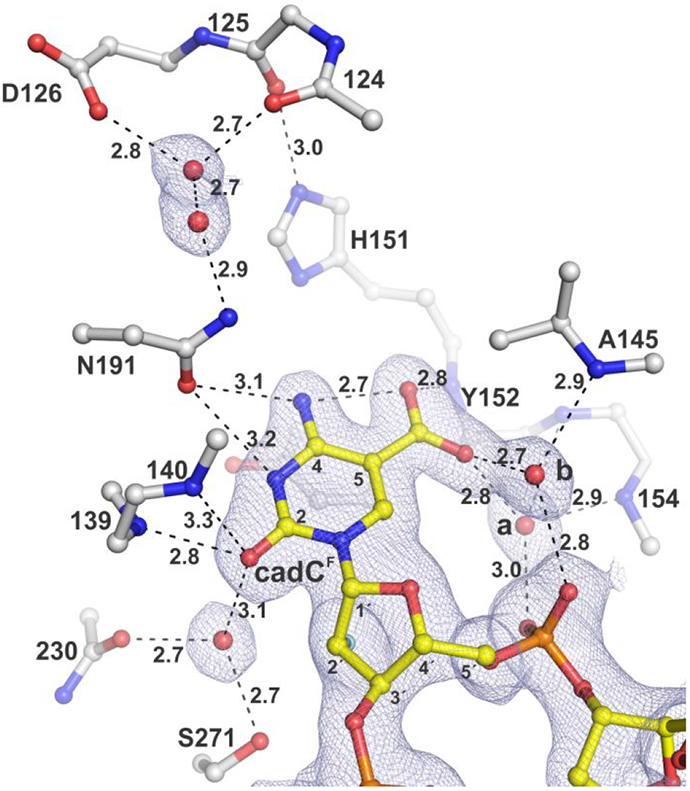
Structure of TDG82-308 bound to DNA with cadCF flipped into the active site. The 2Fo–Fc electron density map, contoured at 1.0σ, is shown for cadCF DNA and water molecules. Dashed lines represent hydrogen bonds, with interatomic distances (Å) shown. Water molecules contacting the carboxylate of cadC are labeled (a, b). The 2′-F substituent, colored cyan, is partially obscured by C2′, though its electron density is visible.
Our structures reveal that the caC O2 oxygen is contacted by two backbone amides (139, 140) and a water molecule that is tightly bound by the hydroxyl of S271 and a backbone oxygen. Together, these contacts could promote catalysis by stabilizing negative charge developing on the O2 carbonyl oxygen, helping to activate the substrate in the ground state or stabilize the departing caC base in the transition-state of the reaction.
The structures also show that the N191 carboxamide oxygen is within hydrogen-bonding distance to the N3 and exocyclic N4 of caC. Notably, the N191 carboxamide nitrogen interacts with the carboxylate of D126 through two ordered water molecules, a hydrogen bond network that could contribute to activating the caC leaving group, as discussed below. Both of these residues are conserved in TDG from vertebrates. Previous studies indicate that N191 could be essential for caC excision, possibly by facilitating the formation of a neutral form of the caC base that is protonated at N3, such as the zwitterion or the imino species (Figure 2).21 Thus, N191A-TDG was shown to lack substantial caC excision activity (in a 3 h reaction) while its binding affinity for G·caC substrate and its fC excision activity were equivalent to that of wild-type TDG.21 To advance these findings we quantified the caC excision activity of N191A-TDG82-308 using single-turnover kinetics, finding a rate constant of kmax = (3.7 ± 0.3) × 10−5 min−1 (Figure S1b). This large 3750-fold reduction in activity, relative to wild-type TDG, indicates a critical catalytic role for the N191 carboxamide, one that is unique to caC excision.
A striking observation in the new structures is that two water molecules contact a carboxylate oxygen of caC (Figures 4, 5, S2). The waters are each coordinated by contacts with the 5′ phosphate of the flipped cadC nucleotide and a backbone amide nitrogen of TDG, such that each water can donate a hydrogen bond to the carboxylate of caC. As such, these waters likely contribute to substrate binding by stabilizing the flipped conformation of cadC. Moreover, they are poised to facilitate acid-catalysis by helping to transfer a proton to the carboxylate of the anionic caC base. Importantly, the structures also reveal a channel from the TDG surface to its active site that contains many ordered water molecules and a hydrogen bonding network that could potentially shuttle a proton from solvent to the carboxylate of caC. Notably, the water molecules revealed here to interact with the caC carboxylate have been observed in previous structures of TDG82-308 bound to other DNA substrates (dU, fdC),31-32, 38 but they do not contact U or fC and are thus unlikely to mediate excision of these bases, which are removed without acid catalysis.
Figure 5.
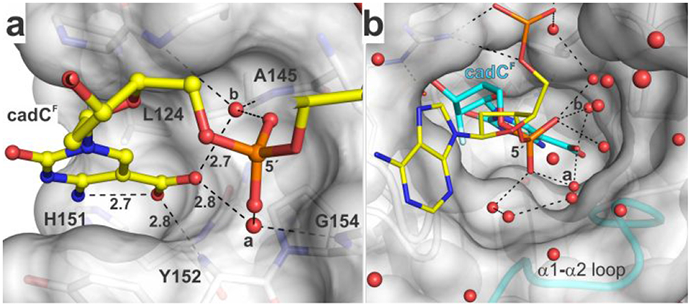
Binding pocket for the caC carboxylate and water molecules that contact this group and populate a channel to the surface. (a) Binding pocket for carboxyl of caC as seen in the structure of TDG82-308 bound to cadCF DNA. Select residues of TDG are shown in surface and stick format, with formatting otherwise similar to that in Figure 4. The 2′-F substituent is not shown (for clarity). (b) Water-filled channel from the TDG active site to its surface. The hydrogen bonding network (dashed lines) could facilitate transfer of a solvent proton to caC. Water molecules that contact the carboxylate of cadC are labeled (a, b).
Our structures also provide new definition regarding the role of other TDG residues that interact with the caC base. We confirm the previous finding that a backbone nitrogen (Y152) forms a relatively short contact with a caC carboxylate oxygen,33 which could contribute substantially to substrate binding and caC excision. By contrast, our structures do not support previous proposals that the caC carboxylate is contacted by the side chain of either N157 or N230.33, 42 Notably, the pocket surrounding the caC carboxylate includes hydrophobic moieties of several residues (L124, A145, H151, Y152, P153) (Figure 5a). Such an environment could favor a neutral relative to an ionized group and could thereby elevate the pKa of the caC carboxylate, which could facilitate acid-catalyzed caC excision.
A substrate-dependent conformational switch.
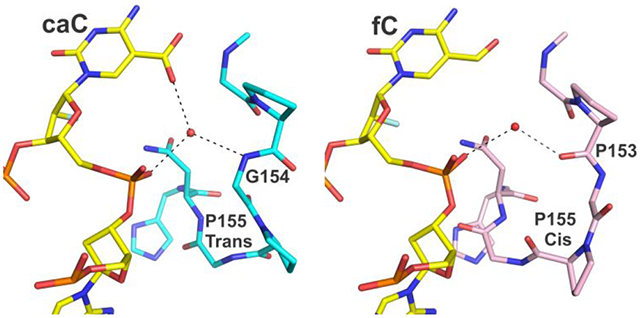
The new structures also reveal a remarkable substrate-dependent conformational switch involving a TDG loop (α1-α2), which modulates the hydrogen bond interactions of a water molecule and thus its capacity to promote caC binding and excision (Figure 6a, water “a”; shown also in Figures 4, 5). The conformational change involves residues 153-156, as revealed by comparing structures of TDG82-308 bound to DNA containing cadCF or fdCF, and its magnitude is remarkably large given that cadC and fdC differ by a single atom. For cadC DNA, the water molecule donates a hydrogen bond to the 5′ phosphate and receives a hydrogen bond from a backbone nitrogen (G154), such that it can donate a hydrogen bond to the caC carboxylate (Figure 6). By contrast, for fdC DNA, the water molecule donates a hydrogen bond to the 5′ phosphate and to a backbone oxygen. If the loop conformation observed for fdC DNA were adopted for cadC DNA, the water molecule would likely present a lone pair to the carboxylate of caC, which would be unfavorable for binding or excision of caC. Importantly, the conformation of this loop is identical in the high-resolution structures reported here for wild-type- and N140A-TDG82-308 bound to cadC DNA (not shown).
Figure 6.
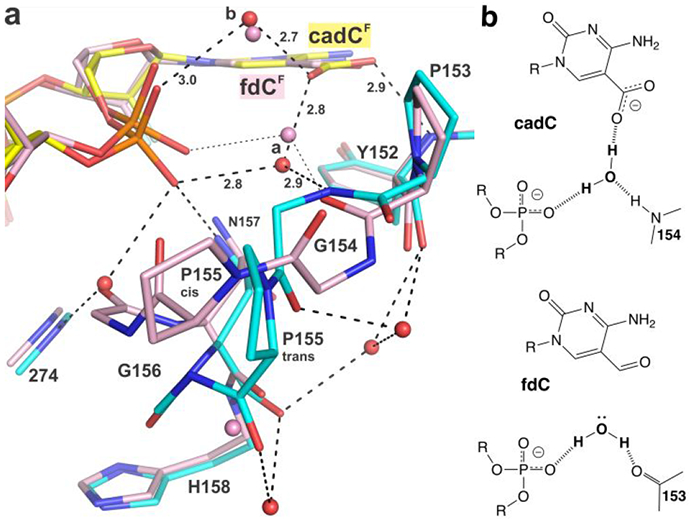
Substrate dependent conformational switch. (a) The new structure of TDG82-308 (cyan) bound to cadCF DNA (yellow) superimposed with the previous structure of TDG82-308 bound to fdCF DNA (pink) (PDB ID 5T2W). Red (or pink) spheres represent water molecules. For cadC DNA, the water molecule of interest receives a hydrogen bond from a backbone nitrogen (G154), while for fdC DNA, the corresponding water (pink) donates a hydrogen bond to a backbone oxygen (P153). Cis-trans isomerization is shown for P155 (b) Effect of the conformational switch on hydrogen bond interactions of the ordered water molecule and its capacity to provide a hydrogen bond to the carboxylate of caC.
The switch involves cis-trans isomerization for P155 and large conformational changes for the flanking Gly residues (G154, G156). The trans isomer of P155 is seen for TDG bound to cadC DNA, and the cis isomer is adopted for fdC DNA. Notably, both P155 isomers are populated in a 1.54 Å structure of TDG82-308 bound to DNA with dU in its active site, but the α1-α2 loop conformation is otherwise quite similar to fdC DNA (Figure S3). As such, the hydrogen bond interactions involving the water molecule (“a”) are identical for dU and fdC DNA (Figures 6, S3). It is also notable that the conformational switch enlarges the solvent-filled channel from the TDG surface to its active site, for cadC DNA (Figure 5b) relative to fdC DNA (not shown). Several high-resolution structures TDG bound to abasic DNA product reveal an α1-α2 loop conformation identical to that for G·caC DNA (not shown), which could potentially facilitate departure of the excised base, a possibility that warrants additional studies.
Nucleophile binding and addition.
A critical element of N-glycosyl bond hydrolysis is coordination and addition of the nucleophilic water molecule. Previous studies indicate that this step requires an Asn that is strictly conserved in TDG-MUG enzymes (Asn140 in human TDG),24-26 but its role in TDG hydrolysis of cadC remains unclear, due in part to insufficient structural information. We addressed this problem using structural and biochemical approaches. Single turnover kinetics experiments show that N140A-TDG82-308 has exceedingly low caC excision activity; kmax = (6.8 ± 0.7) × 10−6 min−1 (Figure S1A).36 Thus, depleting the carboxamide of N140 causes a 20500-fold loss in caC activity, similar to the massive effects observed for other substrates (G·fC, G·T, G·U).24, 32 The structure of TDG82-308 bound to caCF DNA reveals that the putative nucleophilic water molecule is coordinated by the carboxamide oxygen of N140 and the backbone oxygen of T197 (Figure 7). Assignment of this water as the nucleophile is supported by the effect of removing the N140 carboxamide on caC activity and findings of an identical nucleophile binding mechanism in structures of TDG82-308 bound to DNA with dUF or fdCF DNA.31-32 In addition, the position of the nucleophile and its distance from the electrophile (C1′) are similar in all three structures (with caCF, fdCF, dUF). Notably, the N140 carboxamide group is positioned by close contacts from its nitrogen to the hydroxyl of T197 and a backbone oxygen (195). The catalytic importance of T197 is indicated by previous findings that the T197A mutation causes a 32-fold loss in glycosylase activity for a G·T substrate, and its strict conservation in TDG-MUG enzymes.35 Importantly, the new structure reveals a second water molecule that contacts the N140 carboxamide oxygen, in addition to contacts with the nucleophile and the 3′ oxygen of cadC (“W2”, Figure 7). This second N140-interacting water molecule, not seen in previous structures, could potentially contribute to nucleophile addition, as discussed below.
Figure 7.
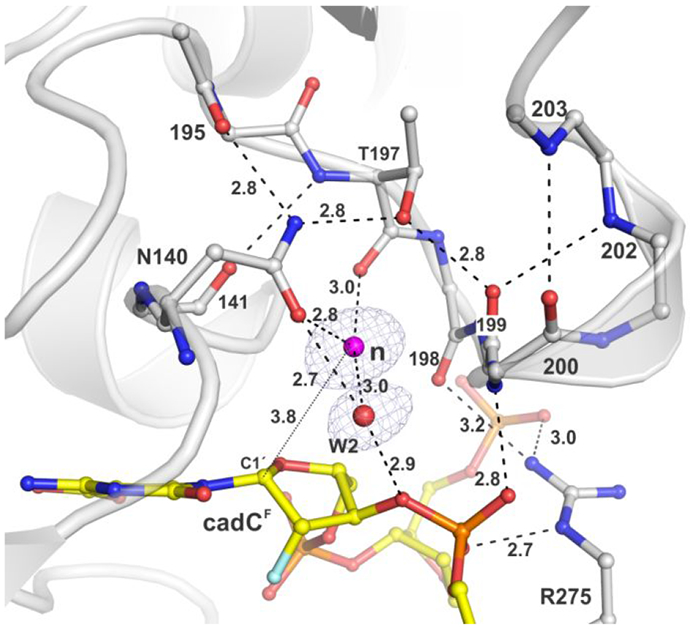
Coordination of the nucleophilic water molecule for TDG82-308 bound to cadCF DNA (PDB ID: 6U17). The 2Fo–Fc electron density map, contoured at 1.0σ, is shown for water molecules only (some omitted for clarity). Dashed lines represent hydrogen bonds with interatomic distances (in Å) shown (otherwise ≤3.3 Å). The nucleophile (magenta sphere) and electrophile (C1′) are separated by 3.8 Å (thin dotted line).
Effect of removing the carboxamide of N140.
The mechanism of nucleophile binding and addition for TDG-MUG enzymes is also informed by our structure of N140A-TDG82-308 bound to cadC DNA (Figure 8). Importantly, this is the first high-resolution structure of any TDG-MUG enzyme that reveals the effect of depleting the carboxamide group of the essential Asn, including effects on water molecules. Remarkably, N140A-TDG binds a water molecule (“*”, Figure 8) using two of the three contacts observed for the nucleophilic water of wild-type TDG (197 backbone oxygen, water W2). The third contact, to the N410 carboxamide oxygen for wild-type TDG, is replaced by a contact to the A140 backbone oxygen for N140A-TDG. While the water molecule for N140A-TDG exhibits a similar displacement (3.6 Å) from the electrophile (C1′ of cadC), its relative position in the active site is altered by about 1 Å (for aligned structures). The absence of significant caC excision activity for N140A-TDG indicates that this water molecule (*) cannot serve as the nucleophile, due likely to improper positioning and possibly additional factors, as discussed below. Our structure also reveals that the N140A substitution does not alter the binding of the second water molecule (W2) that contacts the N140 carboxamide oxygen in wild-type TDG; its position and two of its interactions are retained for N140A-TDG (Figure 8). This observation supports the conclusion that, although this water molecule contacts the N140 carboxamide, it is not the nucleophile.
Figure 8.
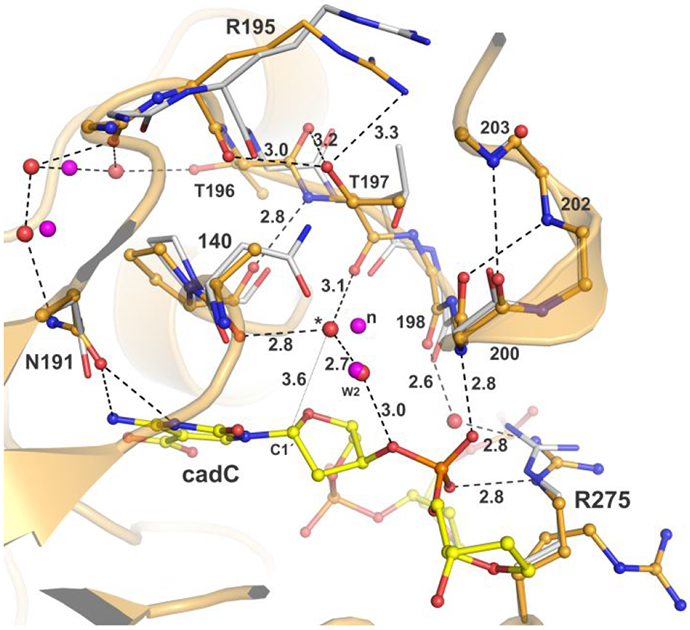
Effects of removing the N140 carboxamide. Structure of N140A-TDG82-308 (wheat) bound to DNA (light green) with cadC flipped into its active site (PDB ID: 6U16). Aligned to this is the structure of TDG82-308 (white) bound to cadCF DNA (PDB ID: 6U17). Water molecules are red spheres for N140A-TDG82-308 and magenta for TDG82-308. The putative nucleophile for TDG82-308 is labeled “n”; a corresponding water displaced by 1 Å for N140A-TDG82-308 is labeled “*” and its distance from the electrophile (C1′) is 3.6 Å (thin dotted line). The second water that contacts N140 of TDG82-308 is labeled “W2”.
Removing the N140 carboxamide also exerts effects beyond coordination of the nucleophile. The effects on TDG backbone conformation are relatively small at the substitution site but more pronounced for a loop (β2-α4; 192-204) that includes T197, due to loss of two contacts between the loop and the N140 carboxamide nitrogen. In turn, this alters a contact involving a backbone oxygen (198) and R275, the “plug” residue that fills the DNA void generated by nucleotide flipping (Figure 8).35, 37 R275 contacts the phosphate of nucleotides adjacent to the flipped nucleotide (cadC) and likely stabilizes the flipped conformation. N140A-TDG exhibits a second conformation for R275, which does not contact the DNA backbone and would likely not hinder retrograde flipping of cadC, raising the possibility that the N140A mutation could impact nucleotide flipping. However, findings that the N140A mutation has little effect on substrate binding affinity suggest a relatively small effect on flipping.24, 36
Minimal effects of 2′-F on enzyme-substrate interactions.
2′-F-substituted deoxynucleotides have long been used to generate a stable enzyme-substrate (E-S) complex for structural, biophysical, and biochemical studies of many DNA glycosylases.24, 26, 32, 34, 40, 43-45 The 2′-F substitution is subtle and is thought to not dramatically alter the conformation of a nucleotide flipped into a glycosylase active site or perturb enzyme interactions. However, this has not been directly investigated by structural methods (to our knowledge), due perhaps to experimental impediments. Such studies require an enzyme mutation that halts base excision activity but does not perturb interactions with the flipped nucleotide, and structures of that variant enzyme bound to two DNA duplexes, with and without a 2′-F in the flipped nucleotide. Our structures show that the N140A mutation satisfies the first requirement, as it does not substantially perturb interactions with the flipped nucleotide (Figures 4, S2). To address the second requirement, we solved a structure of N140A-TDG82-308 bound to cadCF DNA at 2.40 Å resolution (PDB ID: 6U15). Comparing our structures of N140A-TDG82-308 bound to DNA with either cadC or cadCF indicates that TDG interactions with the caC base, including water-mediated contacts to O2 and the carboxyl group, are not substantially altered by the 2′-F substitution, aside from minor differences that could be attributed to the large difference in structural resolution (1.60 Å, 2.40 Å). In addition, the conformation of the flipped nucleotides in the active site are remarkably similar (Figure 9). One minor difference is that a water molecule seen in the structure of N140A-TDG82-308 bound to cadC DNA is missing in the corresponding structure with cadCF, due likely to steric clash with the 2′-F substituent, but the water seems unlikely to contribute substantially to substrate binding or catalysis. These structural observations are consistent with findings that the affinity of N140A-TDG for binding G·U or G·T DNA is not substantially altered by a 2′-F substitution in the target nucleotide (dU or dT).24 More broadly, our results provide the first direct structural evidence that a 2′-F-arabino substitution does not substantially alter the conformation of a nucleotide flipped into a DNA glycosylase active site or perturb enzyme-substrate interactions.
Figure 9.
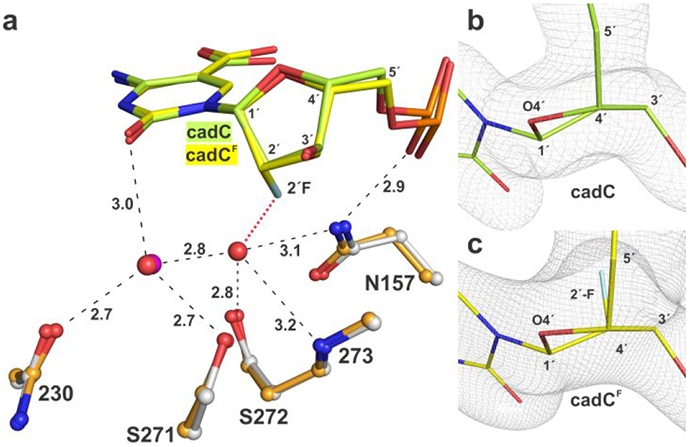
Effect of 2′-F substituent on the conformation of cadC flipped into the active site of N140A-TDG82-308. (a) Alignment of structures of N140A-TDG82-308 bound to DNA containing cadC (lime) or cadCF (yellow) reveals little difference in cadC conformation. Water molecules are red spheres for cadC and magenta for cadCF; one water is seen for cadC but not cadCF, due likely to steric hindrance with 2′-F (red dotted line). Dashed lines represent hydrogen bonds with interatomic distances (Å). (b, c) Close-up view of cadC and cadCF nucleotides, flipped into the N140A-TDG82-308 active site, indicate that 2′-F has a minor effect on sugar pucker (C1′-exo, slight O4′-exo). View is along a C4′-C2′ axis with C4′ in foreground. 2Fo–FC electron density maps are contoured at 1.0σ.
DISCUSSION
Optimal approach for studying an E-S complex.
Our results provide new information that could help guide decisions regarding the optimal approach for studying the enzyme-substrate complex of a DNA glycosylase, where the goal is to halt catalysis in a manner that minimally perturbs substrate interactions. We took two common approaches, using wild-type enzyme with a nonreactive substrate analogue (2′-F-cadC) and mutant enzyme with natural substrate (cadC). Regarding the second approach, our results provide the first robust structural evidence that removing the carboxamide of the catalytic Asn in TDG-MUG enzymes hinders productive binding of the essential nucleophilic water molecule while preserving interactions with the flipped nucleotide. Indeed, we show that this approach can be highly informative for studying the E-S complex for TDG and we suspect it could be similarly productive for bacterial MUG enzymes. However, the approach does have some drawbacks. Removing the N140 carboxamide causes structural changes in TDG that are unrelated to nucleophile binding, as detailed above. Also, N140A-TDG exhibits some residual activity, depending on the substrate (e.g., G·U DNA) such that base excision might occur during crystal growth.24 As noted above, this is not a problem for G·caC DNA under our crystallization conditions. By contrast, the other approach for studying an E-S complex, using wild-type enzyme and DNA containing a 2′-F-substituted substrate analogue, has no major shortcomings. Indeed, our structures indicate that the 2′-F substitution has little effect on the structure of the flipped nucleotide or its enzyme interactions. Thus, our results show that for TDG, and perhaps most other DNA glycosylases, using wild-type enzyme and DNA with a 2′-F-substituted nucleotide is the most robust approach for studying the E-S complex. Previous structural studies of MutY led to a similar conclusion.46 Notably, the 2′-F substitution fully precludes base excision activity for most DNA glycosylases, which is important for crystallography given the incubation time needed for crystal growth. Availability of the phosphoramidite form of the 2′-F deoxynucleotide can be an issue, as they can be challenging to produce, though some are commercially available.
Mechanism of acid-catalyzed caC excision.
Previous studies show that TDG employs acid catalysis to activate the nucleobase leaving group (LG) for excision of caC but not for excision of fC or other substrate bases (U, T).21 The unique need of caC for LG activation is explained by its anionic ionization state at physiological pH, which likely hinders N-glycosyl bond cleavage due to the poor LG quality of a departing caC dianion (Figure 2). Protonation of the caC anion yields a neutral form of caC, including the amino, zwitterion, or imino species, which are all predicted to have dramatically enhanced LG quality when compared to the caC anion, as judged by N1 acidity (Figure 2).21 Previous findings that candidate general acid residues (His151, Tyr152) do not contribute to caC excision indicate that acid-catalyzed excision does not involve direct protonation of caC by a general acid.21 Thus, the mechanism of acid-catalyzed caC excision by TDG has remained unknown.
Our discovery here that two ordered water molecules contact a caC carboxylate oxygen suggests a mechanism for transfer of a proton to the carboxylate group of the caC anion to yield the neutral amino form of caC, a much better leaving group relative to the caC anion (Figure 10a). Notably, our structures also reveal a channel from the protein surface to the active site which contains ordered water molecules and a network of hydrogen bonds that could potentially shuttle a proton from solvent to one of the two waters that contacts the caC carboxylate (Figure 5b). Together, these findings suggest one potential mechanism for acid-catalyzed activation of the caC anion (Figure 10a).
Figure 10.
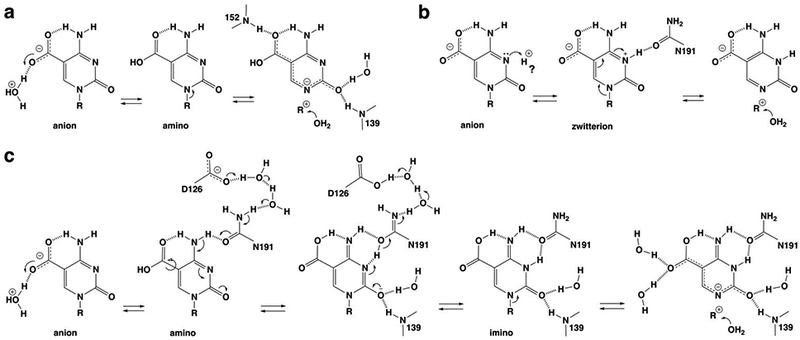
Potential mechanisms for acid-catalyzed excision of caC by TDG, with a focus on steps leading to N-glycosyl bond cleavage. We note that subsequent steps, including potential mechanisms for nucleophile addition and product release, are not shown. (a) Activation of the caC anion through protonation at its carboxylate to give the neutral caC amino; the mechanism involves proton shuttling through one of the two water molecules shown here to contact the caC carboxylate. (b) Activation of the caC anion through protonation at N3 to give the neutral zwitterion; the “?” indicates that the source of the requisite proton is unclear. (c) A new mechanism for activation of the caC anion involves water-mediated protonation of the carboxylate to give the neutral caC amino and its conversion to the caC imino, mediated by N191 and its interaction (through water molecules) with a general base, D126. For all three mechanisms, some interactions with water molecules or TDG groups are shown only in the most relevant steps or not at all (for clarity).
However, such a mechanism does not account for biochemical and structural findings that point to an essential role for N191 in acid catalysis of caC excision. Depleting the carboxamide of N191 (via N191A mutation) reduces the caC excision activity of TDG by 3750-fold (Figure S1b) but does not alter its binding affinity for a G·caC substrate.21 The N191A mutation does not alter TDG activity for G·fC substrates and only modestly reduces activity for G·U and G·T substrates (by 5- and 13-fold), and these three substrates are processed without acid-catalysis.21, 24, 35 These observations, together with structural findings that the N191 carboxamide oxygen contacts N3 and N4 of caC, suggest that N191 could mediate the formation of an N3-protonated form of caC in the active site, thereby activating the caC anion for excision.
Notably, direct protonation of the caC anion at N3 yields a zwitterion, which is predicted to be a good leaving group and could be another potential mechanism for acid-catalyzed activation of caC excision (Figure 10b).21 However, our high-resolution structures, which include hundreds of ordered water molecules, do not reveal a water molecule that contacts N3 or N4 of caC or the carboxamide oxygen of N191. As such, the source of the proton that would be needed for direct protonation at N3 is unclear. Given the low pKa of 4.2 for converting the anionic caC base to a neutral species,21, 47 it seems unlikely that the N3-protonated form of caC is substantially populated in the TDG-DNA complex prior to nucleotide flipping, but this possibility cannot be excluded. Another concern regarding a mechanism involving activation of the caC anion by direct protonation at N3 has to do with specificity. TDG does not excise C, mC, or hmC from DNA, yet the N3 acidity for these bases is similar to that for the caC anion (assuming a mechanism that protonates exclusively at N3 to give a zwitterion).21 Thus, if TDG were to catalyze caC excision through direct protonation at N3, how would it avoid acid-catalyzed excision of the other three bases? One potential answer is that the corresponding nucleotides (dC, dmC, dhmC) do not flip productively into the active site, which is consistent with findings that TDG binds with reduced affinity to DNA containing these nucleotides relative to DNA containing fdC and cadC.33, 36, 39 Still, this seems unlikely to fully account for the absence of TDG activity for these nucleotides.
We propose a new mechanism for acid-catalyzed caC excision that would be specific for caC and is consistent with previous findings and the results reported here (Figure 10c). It begins with protonation of the caC anion at a carboxylate oxygen to give the neutral amino form of caC, through a proton transfer mechanism as described above. Accounting for its essential role, the next step involves N191-mediated conversion of the caC amino to its imino form, facilitated by ordered water molecules and the carboxylate of D126, a residue that is conserved in vertebrate TDG. As noted above, leaving group quality is expected to be substantially improved for the imino relative to the amino of caC (Figure 2).21 The amino to imino conversion would begin with abstraction of a proton from N4 of caC by the N191 carboxamide, facilitated by proton shuttling to a general base, D126, through hydrogen bonds involving two ordered water molecules (Figure 4). The resulting negative charge on the caC base could be accommodated through delocalization to the O2 oxygen, stabilized by hydrogen bonds from two backbone amides and an ordered water. Subsequent transfer of a proton back through the D126-water-N191 network to N3 of caC would yield the imino species. Notably, abstraction of the caC N4 proton would likely be enabled by the initial protonation step, which converts the carboxylate, an electron-donating group, to carboxylic acid, a strongly electron-withdrawing substituent (σm is −0.10 and 0.37 for -COO− and -COOH, respectively).48 Because this mechanism begins with protonation at the carboxylate group to give an electron-withdrawing substituent, it would not likely be effective for acid-catalyzed excision of C, mC, or hmC. Additional studies will be needed to test this and other potential mechanisms for acid-catalyzed caC excision by TDG.
Nucleophile binding and addition.
Together, our biochemical and structural results inform the mechanism of nucleophile binding and addition in the hydrolytic reaction catalyzed by TDG enzymes, and by extension, the related bacterial MUG enzymes. This central catalytic function requires an Asn that is strictly conserved in the TDG-MUG family (N140 for human TDG).24-26 Our structure of TDG82-308 bound to cadCF DNA reveals an ordered water molecule that contacts the N140 carboxamide oxygen and a backbone oxygen (197) and likely represents the nucleophile for cadC hydrolysis. Importantly, the same interactions and relative position are observed for the putative nucleophile in high-resolution structures of TDG82-308 bound to DNA with fdCF or dUF flipped into the active site.31-32 Our structures also provide the first robust structural information regarding the effects of depleting the carboxamide of the conserved Asn for any enzyme in the TDG-MUG family. As noted above, the structures reveal that the nucleophile for wild-type TDG, and a corresponding water molecule for N140A-TDG, are each coordinated by three hydrogen bonds, two of which are conserved. However, the water molecules occupy different relative positions in the active site and are separated by 1 Å (in aligned structures). These observations provide a structural context for findings that the N140A mutation reduces caC activity by 20500-fold (Figure S1a) and causes similar activity reductions for other substrates.24, 32
The massive effect of the N140A mutation could reflect a critical role for the conserved Asn in nucleophile positioning, as suggested by the perturbed nucleophile location revealed here for N140A-TDG. It seems unlikely that the activity loss caused by the N140A mutation reflects a major role for the conserved Asn in nucleophile activation, given that the N140D mutation, which introduces a potential general base, also reduces TDG activity dramatically, albeit to a far smaller extent than the N140A mutation.25, 49 While N140D-TDG could potentially bind the nucleophile using a carboxylate oxygen in a manner analogous to that for the carboxamide oxygen of TDG, the carboxylate would likely disrupt contacts with T197 and the 195 backbone oxygen that are formed by the carboxamide nitrogen of wild-type TDG, which could perturb nucleophile positioning. Moreover, it seems unlikely that strong nucleophile activation would be needed for a reaction that is likely to be either stepwise, with a highly electrophilic oxacarbenium ion intermediate, or concerted with a loose transition state in which LG departure is greatly advanced over nucleophile approach.18-19, 27, 30
Nevertheless, along with a key role in nucleophile positioning, the conserved Asn could potentially stabilize positive charge associated with nucleophile addition and facilitate transfer of the released proton to a water molecule (Figure 11). As shown by our structures, the other active-site water (W2) contacts the N140 carboxamide, the nucleophilic water, and the 3′-oxygen of the flipped cadC nucleotide (Figure 7). Stabilization of positive charge by N140 could involve a hydrogen bond that is strengthened by delocalization of negative charge to its carboxamide oxygen, stabilized by hydrogen bonds from its nitrogen to T197 and a backbone oxygen (105). A recent QM/MM study of the TDG reaction suggests the positive charge could be delocalized to the side chains of N140 and T197 through relatively strong hydrogen bonds.30 A key catalytic role for T197 is indicated by findings that the T197A mutation causes a 32-fold loss in G·T activity,35 though this could also reflect impaired nucleophile positioning. Our structures suggest the positive charge arising upon nucleophile addition could also be stabilized by the second water molecule. Moreover, this water is poised to be the initial recipient of the proton that is released by the nucleophile. It is assumed the proton would then be transferred to solvent or possibly to the excised anionic base, depending on conformational changes and interactions that arise in the product complex. Additional studies will be needed to test this and other possible mechanisms for nucleophile addition during N-glycosyl bond hydrolysis by enzymes in the TDG-MUG family.
Figure 11.
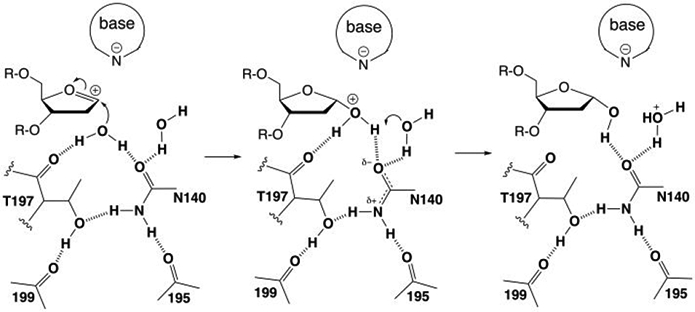
Potential role for the conserved Asn and Thr residues of TDG-MUG enzymes in positioning the nucleophilic water molecule, stabilizing positive charge upon nucleophile addition, and facilitating proton transfer to another water molecule. The structural interactions are shown in Figure 7. Labels denote residues in human TDG.
MATERIALS AND METHODS
Materials.
We expressed and purified TDG82-308, comprising residues 82-308 of human TDG (410 total residues), as described.31 Expression vectors for N140A- and N191A variants were generated by site-directed mutagenesis24 and the vector for the C276A variant was obtained from DNA 2.0 (Newark, CA). The variants were purified using the same methods as for wild-type TDG82-308. Enzyme preparations were >99% pure, as judged by SDS-PAGE (Coomassie-stained gel) and concentration was determined by absorbance at 280 nm,39, 50 using ε280 = 17.4 mM−1cm−1 for TDG82-308 (and the variants). Standard oligodeoxynucleotides (ODNs) were obtained from IDT and those containing 5-carboxyl-dC (cadC) were synthesized by the Keck Foundation Biotechnology Research Laboratory at Yale University. Also produced at Yale were ODNs containing 2′-fluoroarabino-substituted cadC (cadCF), using a phosphoramidite that was synthesized as described.33, 51 ODNs were purified by reverse phase HPLC,38, 45 exchanged into 0.02 M Tris-HCl pH 7.5, 0.04 M NaCl, and quantified by absorbance.52 The DNA included a 28mer target strand, 5′-AGC TGT CCA TCG CTC AxG TAC AGA GCT G, where x is cadC or cadCF, and the complementary strand, 5′-CAG CTC TGT ACG TGA GCG ATG GAC AGC T, such that cadCF is paired with dG and located in a CpG dinucleotide context.52-53
X-ray Crystallography.
Samples used for crystallization contained 0.35 mM TDG82-308 (or N140A-TDG82-308) and 0.42 mM DNA in a buffer of 5 mM Tris-HCl pH 7.5, 0.13 M NaCl, 0.2 mM DTT, 0.2 mM EDTA. Crystals were grown at room temperature (~22 °C) by sitting drop vapor diffusion, using 1 μl of the TDG-DNA sample and 1 or 2 ul of mother liquor, which was 30% (w/v) PEG 4000, 0.2 M ammonium acetate, 0.1 M sodium acetate, pH 6.0. Crystals were cryoprotected using mother liquor supplemented with 18% ethylene glycol and flash cooled in liquid nitrogen. X-ray diffraction data were collected at the Stanford Synchrotron Radiation Lightsource (SSRL beamlines 12-2). Images were processed using XDS and MOSFLM and scaled with Aimless from the CCP4 program suite,54-57 assisted by autoxds (http://smb.slac.stanford.edu/facilities/software/xds). Resolution cutoff was determined based on CC1/2 values.58 Structures were solved by molecular replacement using Phaser,59 and a prior structure of DNA-bound TDG82-308 as the search model (PDB ID: 5T2W). Refinement was performed using BUSTER-TNT,60 and model building was performed using Coot.61 TLS refinement utilized the TLSMD server,62-63 as described.38 The data collection and refinement statistics are provided in Supplementary Table S1. Structural figures were made with PyMOL (http://www.pymol.org).
Enzyme activity assays.
Glycosylase activity was determined for TDG82-308 variants acting on DNA containing a G·caC pair using single turnover kinetics experiments performed under saturating enzyme conditions.31, 64 Detailed methods are given in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENT
The studies were supported by a grant from the National Institutes of Health, National Institute of General Medical Sciences (R01-GM072711, to ACD) and the National Human Genome Research Institute (K01-HG006699, to QD). X-ray diffraction data were collected at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and the National Institutes of Health, National Institute of General Medical Sciences (P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
Supporting Information
Accession Codes
Coordinates and structure factor amplitudes have been deposited with the Protein Data Bank (accession codes 6U15, 6U16, 6U17).
The authors declare no competing financial interest.
REFERENCES
- 1.Kohli RM; Zhang Y, TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502 (7472), 472–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drohat AC; Coey CT, Role of Base Excision "Repair" Enzymes in Erasing Epigenetic Marks from DNA. Chem Rev 2016, 116 (20), 12711–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tahiliani M; Koh KP; Shen Y; Pastor WA; Bandukwala H; Brudno Y; Agarwal S; Iyer LM; Liu DR; Aravind L; Rao A, Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324 (5929), 930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.He YF; Li BZ; Li Z; Liu P; Wang Y; Tang Q; Ding J; Jia Y; Chen Z; Li L; Sun Y; Li X; Dai Q; Song CX; Zhang K; He C; Xu GL, Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science 2011, 333 (6047), 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ito S; Shen L; Dai Q; Wu SC; Collins LB; Swenberg JA; He C; Zhang Y, Tet Proteins Can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333 (6047), 1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfaffeneder T; Hackner B; Truss M; Munzel M; Muller M; Deiml CA; Hagemeier C; Carell T, The Discovery of 5-Formylcytosine in Embryonic Stem Cell DNA. Angew Chem Int Ed Engl 2011, 50 (31), 7008–7012. [DOI] [PubMed] [Google Scholar]
- 7.Maiti A; Drohat AC, Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J Biol Chem 2011, 286 (41), 35334–35338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song CX; Szulwach KE; Dai Q; Fu Y; Mao SQ; Lin L; Street C; Li Y; Poidevin M; Wu H; Gao J; Liu P; Li L; Xu GL; Jin P; He C, Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 2013, 153 (3), 678–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen L; Wu H; Diep D; Yamaguchi S; D'Alessio AC; Fung HL; Zhang K; Zhang Y, Genome-wide analysis reveals TET- and TDG-dependent 5-methylcytosine oxidation dynamics. Cell 2013, 153 (3), 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber AR; Krawczyk C; Robertson AB; Kusnierczyk A; Vagbo CB; Schuermann D; Klungland A; Schar P, Biochemical reconstitution of TET1-TDG-BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat Commun 2016, 7, 10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schomacher L; Niehrs C, DNA repair and erasure of 5-methylcytosine in vertebrates. BioEssays 2017, 39 (3). [DOI] [PubMed] [Google Scholar]
- 12.Neddermann P; Jiricny J, The purification of a mismatch-specific thymine-DNA glycosylase from HeLa cells. J Biol Chem 1993, 268 (28), 21218–24. [PubMed] [Google Scholar]
- 13.Neddermann P; Gallinari P; Lettieri T; Schmid D; Truong O; Hsuan JJ; Wiebauer K; Jiricny J, Cloning and expression of human G/T mismatch-specific thymine-DNA glycosylase. J Biol Chem 1996, 271 (22), 12767–12774. [DOI] [PubMed] [Google Scholar]
- 14.Cortazar D; Kunz C; Selfridge J; Lettieri T; Saito Y; Macdougall E; Wirz A; Schuermann D; Jacobs AL; Siegrist F; Steinacher R; Jiricny J; Bird A; Schar P, Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 2011, 470 (7334), 419–423. [DOI] [PubMed] [Google Scholar]
- 15.Cortellino S; Xu J; Sannai M; Moore R; Caretti E; Cigliano A; Le Coz M; Devarajan K; Wessels A; Soprano D; Abramowitz LK; Bartolomei MS; Rambow F; Bassi MR; Bruno T; Fanciulli M; Renner C; Klein-Szanto AJ; Matsumoto Y; Kobi D; Davidson I; Alberti C; Larue L; Bellacosa A, Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 2011, 146 (1), 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tarantino ME; Dow BJ; Drohat AC; Delaney S, Nucleosomes and the three glycosylases: High, medium, and low levels of excision by the uracil DNA glycosylase superfamily. DNA Repair (Amst) 2018, 72, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deckard CE 3rd; Banerjee DR; Sczepanski JT, Chromatin Structure and the Pioneering Transcription Factor FOXA1 Regulate TDG-Mediated Removal of 5-Formylcytosine from DNA. J. Am. Chem. Soc 2019, 141 (36), 14110–14114. [DOI] [PubMed] [Google Scholar]
- 18.Drohat AC; Maiti A, Mechanisms for enzymatic cleavage of the N-glycosidic bond in DNA. Org. Biomol. Chem 2014, 12 (42), 8367–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stivers JT; Jiang YL, A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev 2003, 103 (7), 2729–59. [DOI] [PubMed] [Google Scholar]
- 20.Maiti A; Drohat AC, Dependence of substrate binding and catalysis on pH, ionic strength, and temperature for thymine DNA glycosylase: Insights into recognition and processing of G.T mispairs. DNA Repair 2011, 10 (5), 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maiti A; Michelson AZ; Armwood CJ; Lee JK; Drohat AC, Divergent mechanisms for enzymatic excision of 5-formylcytosine and 5-carboxylcytosine from DNA. J. Am. Chem. Soc 2013, 135 (42), 15813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sumino M; Ohkubo A; Taguchi H; Seio K; Sekine M, Synthesis and properties of oligodeoxynucleotides containing 5-carboxy-2'-deoxycytidines. Bioorg Med Chem Lett 2008, 18 (1), 274–7. [DOI] [PubMed] [Google Scholar]
- 23.Gallinari P; Jiricny J, A new class of uracil-DNA glycosylases related to human thymine-DNA glycosylase. Nature 1996, 383 (6602), 735–8. [DOI] [PubMed] [Google Scholar]
- 24.Maiti A; Morgan MT; Drohat AC, Role of two strictly conserved residues in nucleotide flipping and N-glycosylic bond cleavage by human thymine DNA glycosylase. J Biol Chem 2009, 284 (52), 36680–36688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardeland U; Bentele M; Jiricny J; Schar P, Separating substrate recognition from base hydrolysis in human thymine DNA glycosylase by mutational analysis. J Biol Chem 2000, 275 (43), 33449–33456. [DOI] [PubMed] [Google Scholar]
- 26.Barrett TE; Scharer OD; Savva R; Brown T; Jiricny J; Verdine GL; Pearl LH, Crystal structure of a thwarted mismatch glycosylase DNA repair complex. EMBO J 1999, 18 (23), 6599–6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berti PJ; McCann JA, Toward a detailed understanding of base excision repair enzymes: transition state and mechanistic analyses of N-glycoside hydrolysis and N-glycoside transfer. Chem. Rev 2006, 106 (2), 506–55. [DOI] [PubMed] [Google Scholar]
- 28.Werner RM; Stivers JT, Kinetic isotope effect studies of the reaction catalyzed by uracil DNA glycosylase: evidence for an oxocarbenium ion-uracil anion intermediate. Biochemistry 2000, 39 (46), 14054–14064. [DOI] [PubMed] [Google Scholar]
- 29.Dinner AR; Blackburn GM; Karplus M, Uracil-DNA glycosylase acts by substrate autocatalysis. Nature 2001, 413 (6857), 752–755. [DOI] [PubMed] [Google Scholar]
- 30.Naydenova E; Dietschreit JCB; Ochsenfeld C, Reaction Mechanism for the N-Glycosidic Bond Cleavage of 5-Formylcytosine by Thymine DNA Glycosylase. J Phys Chem B 2019, 123 (19), 4173–4179. [DOI] [PubMed] [Google Scholar]
- 31.Coey CT; Malik SS; Pidugu LS; Varney KM; Pozharski E; Drohat AC, Structural basis of damage recognition by thymine DNA glycosylase: Key roles for N-terminal residues. Nucleic Acids Res 2016, 44 (21), 10248–10258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pidugu LS; Flowers JW; Coey CT; Pozharski E; Greenberg MM; Drohat AC, Structural Basis for Excision of 5-Formylcytosine by Thymine DNA Glycosylase. Biochemistry 2016, 55 (45), 6205–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L; Lu X; Lu J; Liang H; Dai Q; Xu GL; Luo C; Jiang H; He C, Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol 2012, 8 (4), 328–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scharer OD; Kawate T; Gallinari P; Jiricny J; Verdine GL, Investigation of the mechanisms of DNA binding of the human G/T glycosylase using designed inhibitors. Proc Natl Acad Sci U S A 1997, 94 (10), 4878–4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maiti A; Noon MS; Mackerell AD Jr.; Pozharski E; Drohat AC, Lesion processing by a repair enzyme is severely curtailed by residues needed to prevent aberrant activity on undamaged DNA. Proc Natl Acad Sci U S A 2012, 109 (21), 8091–8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coey CT; Drohat AC, Defining the impact of sumoylation on substrate binding and catalysis by thymine DNA glycosylase. Nucleic Acids Res 2018, 46 (10), 5159–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maiti A; Morgan MT; Pozharski E; Drohat AC, Crystal Structure of Human Thymine DNA Glycosylase Bound to DNA Elucidates Sequence-Specific Mismatch Recognition. Proc Natl Acad Sci USA 2008, 105 (26), 8890–8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malik SS; Coey CT; Varney KM; Pozharski E; Drohat AC, Thymine DNA glycosylase exhibits negligible affinity for nucleobases that it removes from DNA. Nucleic Acids Res 2015, 43 (19), 9541–9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morgan MT; Maiti A; Fitzgerald ME; Drohat AC, Stoichiometry and affinity for thymine DNA glycosylase binding to specific and nonspecific DNA. Nucleic Acids Res 2011, 39 (6), 2319–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buechner CN; Maiti A; Drohat AC; Tessmer I, Lesion search and recognition by thymine DNA glycosylase revealed by single molecule imaging. Nucleic Acids Res 2015, 43 (5), 2716–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pozharski E, Percentile-based spread: a more accurate way to compare crystallographic models. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 9), 970–978. [DOI] [PubMed] [Google Scholar]
- 42.Hashimoto H; Zhang X; Cheng X, Activity and crystal structure of human thymine DNA glycosylase mutant N140A with 5-carboxylcytosine DNA at low pH. DNA Repair (Amst) 2013, 12 (7), 535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stivers JT; Pankiewicz KW; Watanabe KA, Kinetic mechanism of damage site recognition and uracil flipping by Escherichia coli uracil DNA glycosylase. Biochemistry 1999, 38 (3), 952–63. [DOI] [PubMed] [Google Scholar]
- 44.Chepanoske CL; Porello SL; Fujiwara T; Sugiyama H; David SS, Substrate recognition by Escherichia coli MutY using substrate analogs. Nucleic Acids Res 1999, 27 (15), 3197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dow BJ; Malik SS; Drohat AC, Defining the Role of Nucleotide Flipping in Enzyme Specificity Using (19)F NMR. J. Am. Chem. Soc 2019, 141 (12), 4952–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee S; Verdine GL, Atomic substitution reveals the structural basis for substrate adenine recognition and removal by adenine DNA glycosylase. Proc Natl Acad Sci U S A 2009, 106 (44), 18497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai Q; Sanstead PJ; Peng CS; Han D; He C; Tokmakoff A, Weakened N3 Hydrogen Bonding by 5-Formylcytosine and 5-Carboxylcytosine Reduces Their Base-Pairing Stability. ACS Chem Biol 2016, 11 (2), 470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansch C; Leo A; Taft RW, A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem. Rev 1991, 91 (2), 165–195. [Google Scholar]
- 49.Hashimoto H; Hong S; Bhagwat AS; Zhang X; Cheng X, Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation. Nucleic Acids Res 2012, 40 (20), 10203–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gill SC; von Hippel PH, Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem 1989, 182 (2), 319–26. [DOI] [PubMed] [Google Scholar]
- 51.Dai Q; Lu X; Zhang L; He C, Synthesis of DNA oligos containing 2'-deoxy-2'-fluoro-D-arabinofuranosyl-5-carboxylcytosine as hTDG inhibitor. Tetrahedron 2012, 68 (26), 5145–5151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgan MT; Bennett MT; Drohat AC, Excision of 5-halogenated uracils by human thymine DNA glycosylase: Robust activity for DNA contexts other than CpG. J Biol Chem 2007, 282 (38), 27578–27586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waters TR; Swann PF, Kinetics of the action of thymine DNA glycosylase. J Biol Chem 1998, 273 (32), 20007–20014. [DOI] [PubMed] [Google Scholar]
- 54.Kabsch W, Xds. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Evans PR, An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr 2011, 67 (Pt 4), 282–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AG; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS, Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011, 67 (Pt 4), 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Battye TG; Kontogiannis L; Johnson O; Powell HR; Leslie AG, iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 2011, 67 (Pt 4), 271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karplus PA; Diederichs K, Linking crystallographic model and data quality. Science 2012, 336 (6084), 1030–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCoy AJ; Grosse-Kunstleve RW; Storoni LC; Read RJ, Likelihood-enhanced fast translation functions. Acta Crystallogr D 2005, 61, 458–464. [DOI] [PubMed] [Google Scholar]
- 60.Bricogne G; Blanc E; Brandl M; Flensburg C; Keller P; Paciorek W; Rovers i. P.; Sharff A; Smart OS; Vonrhein C; Womack TO BUSTER version 2.10.2, Global Phasing Ltd.: Cambridge, United Kingdom, 2011. [Google Scholar]
- 61.Emsley P; Cowtan K, Coot: model-building tools for molecular graphics. Acta Crystallogr D 2004, 60 (Pt 12 Pt 1), 2126–2132. [DOI] [PubMed] [Google Scholar]
- 62.Painter J; Merritt EA, Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr 2006, 62 (Pt 4), 439–50. [DOI] [PubMed] [Google Scholar]
- 63.Painter J; Merritt EA, TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr 2006, 39, 109–111. [Google Scholar]
- 64.Coey CT; Drohat AC, Kinetic Methods for Studying DNA Glycosylases Functioning in Base Excision Repair. Methods in enzymology 2017, 592, 357–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.