

Abstract
Sterol 14α-demethylases (CYP51) are the cytochrome P450 enzymes required for biosynthesis of sterols in eukaryotes, the major targets for antifungal agents and prospective targets for treatment of protozoan infections. Human CYP51 could be and, for a while, was considered as a potential target for cholesterol-lowering drugs (the role that is now played by statins, which are also in clinical trials for cancer) but revealed high intrinsic resistance to inhibition. While microbial CYP51 enzymes are often inhibited stoichiometrically and functionally irreversibly, no strong inhibitors have been identified for human CYP51. In this study, we used comparative structure/functional analysis of CYP51 orthologs from different biological kingdoms and employed site-directed mutagenesis to elucidate the molecular basis for the resistance of the human enzyme to inhibition and also designed, synthesized, and characterized new compounds. Two of them inhibit human CYP51 functionally irreversibly with their potency approaching the potencies of azole drugs currently used to inhibit microbial CYP51.
Graphical Abstract
INTRODUCTION
Despite chemotherapeutic advances and declines in certain death rates, cancer remains a major public health problem worldwide.1 Different types of cancer cells have different mechanisms reprogramming their metabolic and signaling pathways,2 and they often require different highly selective therapeutic approaches for effective targeted treatment.2 The accumulation of cholesterol appears to be a common phenomenon in cancer cells.3–6 The majority of the cholesterol in the human body is synthesized in the liver7 and then distributed into the bloodstream, reaching other tissues in the form of lipoproteins.8 Cancer cells must synthesize cholesterol endogenously (for rapid proliferation and for formation of lipid rafts) for signal transduction facilitating metastasis, increasing cell migration and invasion, and reducing apoptosis.3,5,9,10 Growing experimental evidence has accumulated that cholesterol may play critical roles in the progression of cancers, making the cholesterol biosynthetic pathway an attractive potential drug target because it might be general for many types of cancer.
Cholesterol-lowering statin drugs, currently in clinical trials for cancer,11 support this overall notion. Statins inhibit the enzyme 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase (EC 1.1.1.88), which acts upstream of the pathway converting HMG-CoA to mevalonate. Multiple reports indicate that statins can display strong antiproliferative effects in various cancer cell lines, exhibit robust selectivity for tumor cells over normal cells, slow down the growth of several tumor types, and reduce risk of cancer recurrence.5,6,12–14 The in vivo results have been highly variable, however, ranging from statistically significant positive effects to none at all.4,13–16 Recently it has been suggested that clinical trials should be reorganized because the doses of statins required to kill cancer cells are much higher (at least 10-fold) than those used to control hypercholesterolemia.16 At the standard doses, most statins target the liver17 with their maximal concentrations in plasma being approximately 10–100 nM,18 much lower than the EC50 values in cancer cells (0.5–30 μM).19–23 Increasing the dosage increases the risk of adverse side effects, which, at standard regimens, are rather rare (mostly musculoskeletal20), though their incidence rises with the time of therapy and interactions with some other drugs.24 The full spectrum of possibilities for anticancer activity of statins is still unclear because, in addition to cholesterol-lowering effects, they can affect other branches of the mevalonate pathway (e.g., the production of isoprenoids/protein prenylation) and thus influence other regulatory processes involved in cancer.3,10,13,14,25
Alternative targets in the cholesterol pathway can be considered, and cytochrome P450 51 (CYP51, also known as CYP51A1) lanosterol 14α-demethylase (EC 1.14.13.70) is an attractive candidate for several reasons. (i) This enzyme initiates the branch of the pathway that is committed solely to cholesterol production. (ii) Its orthologs from pathogenic fungi and protozoa are known to be perfectly druggable. (iii) Their inhibitors (both systemic clinical antifungal drugs26 and experimental antiprotozoal drug candidates, such as VNI, VFV,27 and their analogs28) have broad tissue distribution, including the ability to penetrate the brain with plasma concentrations reaching 25–50 μM.28
Human CYP51, however, is highly resistant to inhibition. While protozoan and fungal sterol 14α-demethylases can be inhibited functionally irreversibly under the assay conditions29 at an equimolar ratio of the inhibitor to the enzyme,30 human CYP51 converts all lanosterol into the 14α-demethylated product even at a 50-fold molar excess of an inhibitor.31 Our molecule VFV was initially the first compound that was able to inhibit lanosterol 14α-demethylation by 50%. Although it is not a very potent human CYP51 inhibitor, we found that VFV can slow proliferation of various cancer cells, including acute myeloid leukemia and different types of solid cancers (lung, skin, and hormone-responsive and nonresponsive breast cancer) with EC50 values of 10–30 μM31 within the range of those of statins.
High-resolution X-ray structures of VFV-bound human CYP51 [4UHI, 4UHL]31 revealed the topology of the enzyme–inhibitor complex and outlined possible options for its further stabilization. The purpose of the present work was to apply the structural information for the directed design of human CYP51 inhibitors that would approach potencies of the drugs that target the microbial sterol 14α-demethylases.
RESULTS AND DISCUSSION
Synthesis.
The synthesis of the first generation of VFV analogs (Figure 1) was performed using the previously described procedure28 except that an additional step, a palladium-catalyzed Stille cross-coupling reaction,32 was carried out to replace the ortho-chlorine atom (in the biphenyl ring) with the desired substituents (compounds 1, 3, and 4) (red box, Scheme 1). 1-(3-Chloro-4′-fluoro-[1,1′-biphenyl]-4-yl)ethan-1-one (intermediate 1) and the selected organotin reagent were added to a mixture of Pd/P(t-Bu)3 and CsF in dioxane and stirred at 100 °C under anhydrous conditions. The reaction was monitored by LC–MS, and after 2–4 h, the desired product (intermediate 2) was formed in good yields (60–98%). Following the bromination of the acetyl group and subsequent condensation with imidazole, the asymmetric hydrogenation of 1-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethan-1-one (intermediate 3) into (S)-1-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethan-1-ol (intermediate 4) was not successful using previously described conditions (blue box, Scheme 1).33 Presumably, the steric effects of hindered aromatic/heteroaromatic substituents influenced the enantioselective reduction of the ketone by RuCl(p-cymene)[(R,R)-Ts-DPEN]. Using 1-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethan-1-one/TEA/HCOOH (1:5:5 v/v/v) (in dichloromethane under argon for 24 h at 40 °C), the alcohol formation was only 10%. Therefore, the hydrogenation of the ketone could be completed only by increasing the concentrations of the catalyst, triethylamine, and formic acid.
Figure 1.

First generation of VFV derivatives. Although log P > 5 and mol wt > 500 are “violations of Lipinski’s rule of five”, there are numerous examples of successful drugs with larger values, especially those aimed at intracellular membrane-bound targets (e.g., cinacalcet/Sensipar at a log P of 6.5 and systemic clinical drug inhibitors of microbial CYP51 such as itraconazole (log P = 6.8, mol wt = 704) and posaconazole (log P = 5.5, mol wt = 701)).
Scheme 1. General Synthetic Method To Prepare the ortho-Cl-Substituted VFV Analogsa,b.

a(R)-N-(1-(4-(4-fluorophenyl)naphthalen-1-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide (compound 2) was prepared using 1-(4-(4-fluorophenyl)naphthalen-1-yl)ethan-1-one as a starting material, and the synthesis was performed as previously reported.28
bRed box: Stille cross-coupling reaction. Blue box: enantioselective reduction of ketone to an (S)-alcohol.
The VNI analogs used to test the inhibition of the T318I mutant of human CYP51 were from our in-house collection of fungal CYP51 inhibitors.34 The subsequent subset of VFV-Cl analogs (Figure 2) was synthesized by coupling of intermediate 5 (Scheme 1) and variously substituted side chains of the corresponding VNI derivatives.28
Figure 2.

Second generation of VFV derivatives.
Structural Background for Human CYP51 Resistance to Inhibition.
With <10% of residues identical across the biological kingdoms, the protozoan, fungal, and human CYP51 enzymes catalyze the same three-step stereospecific reaction of oxidative removal of the 14α-methyl group from the cyclized sterol precursors.35 On average, the evolutionarily younger enzymes are catalytically faster, and their susceptibility to inhibition is lower. Comparative crystallographic analysis indicates that, while preserving strict catalytic conservation via high similarity at the secondary and tertiary structural levels, they differ in some structural segments that might imply biologically relevant increased molecular flexibility. Thus, protozoan CYP51 enzymes (kcat = 2–7 min−1) have the longest FG loop, which, in addition, carries two short helical regions (F″ and G′) (Figure 3A). The loop covers the entry into the substrate access channel and closes upon substrate binding,36 thus serving as a “trapdoor”. In the structures of fungal CYP51 enzymes (kcat = 12–25 min−1), this loop is shorter and has only one helical region (F″) (Figure 3B). We observed that the potencies of many fungal inhibitors correlate with the number of interactions they form around the entrance, presumably restricting the loop movements.34 The FG loop in human CYP51 (kcat = 45–64 min−1) is fungal-like. In addition, its I-helix (the core helix in the cytochrome P450 fold) contains a loop-like area in the middle, directly in front of the heme plane (Figure 3C). This low-energy segment in the catalytically relevant portion of the active site makes the human enzyme more flexible, presumably unable to hold inhibitors tightly enough to prevent their replacement with the substrate.
Figure 3.

Phylum-specific structural segments that might imply an evolutionary increase in the CYP51 flexibility. (A) Protozoan (3G1Q), (B) fungal (5TZ1), and (C) human (3I4K) CYP51. Distal P450 view.
First-Generation VFV Analogs.
Binding of VFV somewhat “repairs” the human CYP51 I-helix, partially restoring the main-chain helical H-bonding and thus restricting flexibility of this area.31 Although the overall picture is complicated by the fact that we found not one but two VFV molecules bound, butterfly-like, in the human CYP51 active site (Figure 4A), we first decided to attempt structure-directed VFV modification based on the heme-coordinated molecule. In the structure, the ortho-fluorine atom at the biphenyl arm of the heme-bound VFV is directed toward the I-helix groove (Figure 4B), forming van der Waals contacts with L310 and A311 and thus pushing the backbone 0.6 Å closer to T315 so that the main-chain carbonyl oxygen of A311 is now H-bonded with the side-chain hydroxyl oxygen of T315. We assumed that substituents larger than fluorine here might shift the upstream backbone even further: the main-chain H-bonds will be restored, flexibility will be decreased, and the inhibitory effect will be stronger.
Figure 4.

VFV in the human CYP51 structure (4UHI). (A) “Butterfly-like” binding mode of the inhibitor: the imidazole ring of one VFV molecule is coordinated to the catalytic heme iron, and the biphenyl arm of the other VFV molecule is protruding from the substrate entry channel (A′, F″, and the β4 hairpin are marked). (B) Enlarged view. The biphenyl ortho-F atom of the heme-coordinated VFV molecule is located 3 Å from the I-helical backbone, pushing it downstream and thus shortening the loop-like region.
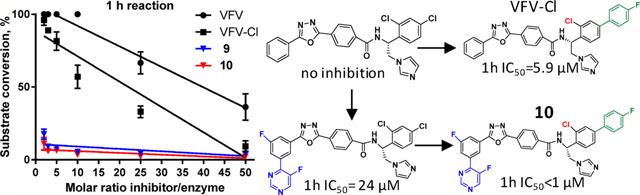
This approach has been successful with VFV-Cl (a 4-fold decrease in the concentration required to prevent conversion of 50% of the substrate in a 1 h reaction (1 h IC50, Table 1 and Figure S1)), but we were unable to achieve further improvement. Efforts to replace the fluorine with bromine or with a cyclopropyl ring were unsuccessful, and all bulkier substituents in this position were too large to serve the purpose. With the increase in the size of the substituent, both the inhibitory effects and spectral binding affinities decreased. The smaller red shift in the Soret band maximum indicated weaker Fe–N coordination, suggesting steric hindrance in the heme proximity (Table 1). On the other hand, the compound devoid of any substituents in the ortho-position of the proximal ring of the biphenyl arm (H instead of F) did not inhibit human CYP51 activity (results not presented).
Table 1.
First-Generation VFV Derivatives as Inhibitors of Human CYP51a
| Inhibitor | ||||||
|---|---|---|---|---|---|---|
 |
 |
 |
 |
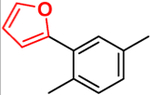 |
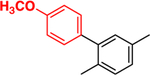 |
|
|
Kd, μM (fold change) |
0.09 | 0.06 (↓1.5) |
1.65 (↑18) |
1.15 [↑13] |
3.90 [↑43] |
2.32 [↑26] |
| Soret band maximum, nm | 425 | 425 | 424 | 423 | 423 | 421 |
| 1 min reaction: IC50, μM | <0.5* | <0.5 | 1.5 | 3.0 | 5.2 | 12 |
| 1h IC50, μM | 24.4 | 5.9 | >25 | >25 | >25 | >25 |
The P450 concentration in the reaction mixture was 0.5 μM; the concentration range of the inhibitors was 1–25 μM.
T318I Mutant: Dynamics-Affecting Mutation Increases Susceptibility of Human CYP51 to Inhibition.
Multiple sequence alignment suggested that the loop-like region in the human CYP51 I-helix might be formed because of the sequence of six polar (alcohol) residues (threonines and serines) that follows the CYP51 signature histidine (proton relay residue, H314 in human CYP51,36 and begins with the “conserved P450 threonine” (T315)). This -TSSTTS- sequence is unique for CYP51 enzymes from animals and invariant across the vertebrate classes (Figure 5A). In the orthologs from other biological kingdoms, it is interrupted with at least one nonpolar residue. The side-chain oxygens of these Thr/Ser residues serve as H-bond acceptors, disrupting the backbone H-bonding upstream of the I-helix (Figure 5B). To probe if interruption of this polar sequence alters CYP51 properties, Thr318 in human CYP51 was changed to isoleucine because protozoan and plant CYP51’s always contain the latter residue in this position.
Figure 5.

Middle portion of the CYP51 I-helix. (A) Fragment of sequence alignment; the six polar residues in human CYP51 are underlined in blue, and T318 and the corresponding protozoan/plant Ile are marked in red. (B) Backbone H-bonding in human vs Trypanosoma cruzi (T. cruzi) CYP51’s.
The mutant protein was successfully expressed in Escherichia coli (E. coli) and purified from the bacterial membranes with the oxidized and reduced CO-complexed absorbance spectra displaying the characteristic Soret band maxima at 417 and 448 nm, respectively. The purified protein also produced typical type 1 difference spectra upon the addition of lanosterol (Figure 6A). Both the Kd and ΔAmax values were ~2-fold higher, indicating that the mutation did not change the substrate binding efficiency [ΔAmax/Kd], though the maximal iron spin-state low-to-high transition in the mutant (45% vs 27% for the WT) occurred at a 2-fold higher lanosterol concentration, ~2 μM versus 1 μM. The mutant was catalytically active, yet approximately one-half compared to the WT (25 vs 45 min−1 (Figure 6B)). The decrease in the rates of 14α-demethylation of obtusifoliol and eburicol was very similar to that observed for lanosterol (data not shown). There was no effect on the mutant stability (Figure 6C).
Figure 6.

Functional characterization of the T318I mutant. (A) Spectral response to the substrate binding. Left: difference absorbance spectra upon T318I titration with lanosterol; right: comparative titration curves for the mutant and the WT (fit to one site–total, binding–saturation), ~2 μM P450. (B) Michaelis–Menten plots, 0.25 μM P450, 1 min reaction. (C) Stability at 42 °C monitored as the decrease in the P450 concentration; the initial P450 concentration was 2 μM.
As anticipated, the T318I mutation increased the susceptibility of the enzyme to inhibition both by the antifungal drugs and by our experimental inhibitors, VFV-Cl and the antiprotozoal drug candidate VNI, which does not inhibit the WT human CYP51 at all (Figure S2). The concentration of VFV-Cl required to prevent conversion of 50% of the substrate in a 1 h reaction (1 h IC50) was approximately 3-fold lower(2.1 vs 5.9 μM for the WT enzyme (Figure S3)). The 1 h IC50 value for VNI was still >25 μM (I/E = 50/1), but the binding affinity for the mutant increased by approximately 12-fold (Kd = 0.4 vs 4.7 μM for the WT enzyme (Figure S4)). We screened the mutant enzyme against our in-house collection of VNI derivatives, designed to inhibit fungal sterol 14α-demethylases. Similar to what was earlier observed for Candida albicans CYP51 and Aspergillus fumigatus CYP51B, the “long-tail” analogs (Figure 7A), the compounds that form a larger number of interactions with the enzyme substrate entrance residues, including the FG loop,34 were much more potent than VNI (Figure 7B). Thus, the T318I mutation indeed made human CYP51 more “fungal-like”.
Figure 7.

Inhibition of human CYP51 with the VNI derivatives that are more potent than VNI as inhibitors of fungal CYP51. (A) Structural formulas. (B) The T318I mutation makes human CYP51 “more fungal-like”. (C) Three strongest T318I inhibitors display a clear inhibitory effect on the WT enzyme (1 h reaction, 0.5 μM P450, 50 μM lanosterol, and inhibitor/enzyme molar ratio = 50/1). Graphs represent mean ± SD of two independent experiments in duplicate.
Second-Generation VFV Analogs and the Issue of the Inhibitor/Enzyme Stoichiometry.
A direct correlation was observed between the inhibition of the T318I mutant and the WT human CYP51 enzyme, in particular with compounds d, e, and f (Figure 7C). Therefore, the same modifications (Figure 2) were introduced into the tail of the VFV-Cl molecule, which was the strongest reversible human CYP51 inhibitor. Four of the six compounds synthesized (7, 8, 9, and 10) were more potent than VFV-Cl (Table 2, 1 h IC50 values of <5.9 μM). Two of these, 9 and 10 (with the “tails” of the two most potent VNI derivatives, e and f in Figure 7A), were functionally irreversible. The substrate was unable to replace them in the enzyme active center during the time of the assay, and there was no inhibitory concentration-dependent increase in the substrate conversion over the range of concentrations tested (1–25 μM, corresponding to a 2–50-fold molar excess of the inhibitor over the enzyme).
Table 2.
Second-Generation VFV Derivatives as Inhibitors of Human CYP51a
| Inhibitor | ||||||
|---|---|---|---|---|---|---|
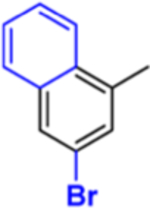 |
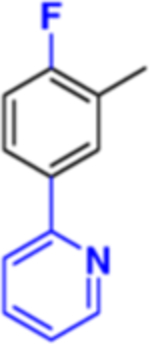 |
 |
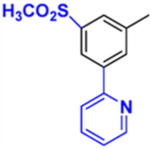 |
 |
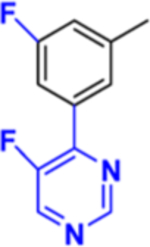 |
|
| Kd, μM | 0.47 | 2.50 | 0.13 | 0.02 | 0.15 | 0.09 |
| Soret band maximum, nm | 425 | 425 | 425 | 425 | 425 | 425 |
| 1 min reaction: IC50, μM | <0.5* | <0.5 | <0.5 | <0.5 | <0.5 | <0.5 |
| 1h IC50, μM** | 15 | 19 | 3.6 | 4.4 | <1 | <1 |
The P450 concentration in the reaction mixture was 0.5 μM; the concentration range of the inhibitors was 1–25 μM.
This inhibitory concentration range was used in all our experiments because we expected that (as it was observed in the case of VFV31) the human CYP51 active site must accommodate not one but two analogous molecules. Repetition of the experiments at an equimolar enzyme/inhibitor ratio resulted in a 100% substrate conversion (Figure 8), indicating that the probability of two bound inhibitor molecules is very high.
Figure 8.

Inhibition of human CYP51 at I/E molar ratios 2.5/1 vs 1/1 implied binding of two inhibitor molecules. 1 h reaction.
Molecular modeling (assuming that the inhibitors adopt the same orientation as that of the parent compound VFV) suggested that the strong potency and functionally irreversible effects of compounds 9 and 10 might be connected with the intermolecular H-bond formation, both between the two inhibitor molecules and the protein residue Y107 (Figure 9).
Figure 9.

4UHI1 structures-based molecular model of compound 10 bound in the human CYP51 active site. (A) Position of the inhibitor molecules relative to the I-helix; the backbone helical H-bonds are red. The H-bonds predicted to have been formed by the inhibitor are presented as violet dashed lines. The participating atoms of the inhibitor molecules are shown in the corresponding colors in their structural formulas. (B) Inhibitor contacting residues. The complete list that relates the residues to the corresponding secondary structural elements (marked in Figure 10B) in the model and in the X-ray structures of human CYP51 is presented in Table S1.
Crystallographic Analysis of Human CYP51 in a Complex with Compound 10.
The crystals formed in the tetragonal space group I4 and diffracted to 2.80 Å, the asymmetric unit containing two protein molecules (Table S2). Only one molecule of the inhibitor remained bound inside the CYP51 active site (Figure S5). However, the density for the second molecule was clearly seen near the substrate entrance, and the entrance itself was occupied by one of the eight molecules of the detergent (deoxycholic acid). The inhibitor and the other seven deoxycholate molecules form a channel, which goes through the whole unit cell (Figure S5A), suggesting that the detergent, perhaps due to its structural similarity to the CYP51 substrates (Figure S5C), may have displaced the second inhibitor molecule upon crystal packing. This “run-away” inhibitor plus the surrounding detergent molecules may have served as crystallization centers because the crystals grew very rapidly, crystallization required a very small amount of the precipitant (see Experimental Section), and the use of other detergents did not produce diffracting crystals.
Comparison of this structure with the structure of VFV-bound human CYP5131 (Figure 10A) supports the possibility that the human CYP51 active site initially contained two molecules of compound 10. First, although crystallized in different space groups (tetragonal I4 vs monoclinic C121 (4UHI)), both complexes are very similar, the RMSD for all Cα atoms being only 0.45 Å. For comparison, the RMSD for all Cα atoms between the two 6Q2T molecules in the asymmetric unit (molecular breathing) is 0.28 Å, which is only1.6-fold smaller. The only substantial rearrangement occurs in the F″ helix, which “pushes down” the tail of the inhibitor (Figure 10B). Second, the active-site volume remains roughly the same, decreasing only from 2200 to 2120 Å3. For comparison, the active-site volume of ketoconazole-bound human CYP51 (one ketoconazole molecule) is 1760 Å3,31 and the active-site volume of VFV-bound Trypanosoma brucei CYP51 (one VFV molecule) is 1840 Å.27 The inhibitor-contacting residues are also mostly the same (Table S1 and Figure S6). Third, in comparing the model with the structure, there are only changes in the positions of the side chains of three residues: H236, W239, and M487, which again might have happened because of crystal packing (Figure 11A). Finally, the observation that binding of compound 10 “repairs” the main-chain H-bonding in the I-helix (Figure 11B) also advocates two molecules of the inhibitor bound within the human CYP51 active site before crystallization.
Figure 10.

Superimposed structures of VFV-bound (4UHI) and compound 10-bound (6Q2T) human CYP51, tan and blue ribbons, respectively. The carbon atoms of VFV, compound 10, and the detergent are orange, magenta, and yellow, respectively. (A) Overall (distal P450) view. The active site area is within the black square. A portion of the second molecule of compound 10 is seen at the top. (B) Enlarged view of the active site. The secondary structural elements that provide ligand-contacting residues (listed in Table S1) are marked.
Figure 11.

Compound 10 in the human CYP51 active site. (A) Rearrangements of H236, W239, and M487 push the tail of the inhibitor (magenta) down relative to its position predicted by the VFV-structure-based model (gray). The arrows show the directions of the rearrangements. (B) Overall (distal P450) view of the compound 10-bound human CYP51 structure showing that the I-helix is straightened.
CONCLUSIONS
In summary, the results support the proposal that human CYP51 resistance to inhibition is due at least in part to the lack of the main-chain H-bonds in the central portion of the I-helix (the loop-like region directly in front of the catalytic center) and the shorter fungal-like FG loop (a “trapdoor”, the gating area controlling the entrance into the substrate access channel). The peculiarities of these two essential structural segments make the human CYP51 molecule more dynamic, resulting in higher catalytic rates and a more facile replacement of the inhibitory molecules in the enzyme active site with a substrate. Consideration of both these structural features afforded the design of stoichiometric, functionally irreversible inhibitors. To conclude, although larger molecular chemotypes may be required to inhibit human CYP51, the enzyme is druggable and can serve as a target for cholesterol-related diseases, particularly as drugs used as adjuvants in postcancer chemotherapy to prevent/alleviate metastatic growth. VFV-Cl and compounds 9 and 10 can be further tested in different types of cancer cells. Minor modifications can be introduced to adjust the lipophilicity if this is an issue.28 Inhibitors of human CYP51 that penetrate the blood–brain barrier might also be helpful for treatment of various neurodegenerative processes, including Alzheimer’s disease, by lowering cholesterol in the brain where it is only of an endogenous origin. Moreover, as we have found recently using VFV,37 inhibitors of human CYP51 can be used to treat human infections with herpes viruses where the production of host cholesterol is required for replication and infectivity.
EXPERIMENTAL SECTION
Chemical Synthesis.
All reagents and solvents were of commercial grade and were purchased from Fisher Scientific, Sigma-Aldrich, Santa Cruz, or Alpha Aesar. 1H NMR spectra were acquired on a Bruker AVANCE-400 MHz spectrometer in CDCl3 and DMSO-d6 at 25 °C or using a 14.0 T Bruker magnet equipped with a Bruker AV-III console operating at 600.13 MHz. Spectra were acquired in 3 mm NMR tubes using a Bruker 5 mm TCI cryogenically cooled NMR probe. Chemical shift values are given in δ (ppm) relative to TMS as an internal standard. Coupling constants are given in Hertz. The following abbreviations are used to designate multiplicities: s = singlet, d = doublet, dd = double of doublets, and m = multiplet. Compounds were purified by flash chromatography using silica gel (high purity grade, pore size of 60 Å, 230–400 mesh particle size, 40–63 μm) (Sigma-Aldrich). The final compounds were filtered through HF Bond Elut-SCX cartridges (500 mg, 6 mL, Agilent Technologies) using a 2.0 M solution of ammonia in methanol (Sigma-Aldrich). LC–MS (UV and ESI-MS) and reversed-phase HPLC were used for analyzing the purity of the compounds (>95%). Analytical LC–MS was performed on an Agilent 1200 series with UV detection at 214 and 254 nm along with ELSD detection. General LC–MS parameters were as follows: Phenomenex-C18 Kinetex column, 50 mm × 2.1 mm, 2 min gradient, 5% (0.1% TFA/CH3CN)/95% (0.1% TFA/H2O) to 100% (0.1% TFA/CH3CN) (all v/v). High-resolution mass spectrometry was performed by direct liquid infusion using an Orbitrap mass spectrometer (Thermo-Finnigan, San Jose, CA) equipped with an Ion-Max source housing and a standard electrospray ionization (ESI) probe operating in the positive ionization mode at a resolving power of 30,000 (at m/z = 400). ESI source parameters were as follows: spray voltage, 5 kV; N2 sheath gas, 8; auxiliary gas, 0; capillary voltage, 35 V; tube lens voltage, 108 V at 675 m/z. The reversed-phase HPLC system (Waters) was equipped with a dual-wavelength UV 2489 detector set at 250 and 291 nm (VNI, ε291 = 36 mM−1 cm−1) and a Symmetry C18 (3.5 μm)4.6 mm × 75 mm column. The mobile phase was 55% 15 mM ammonium acetate solution at pH 7.2 and 45% CH3CN (v/v) with an isocratic flow rate of 1.0 mL/min. The enantiomeric excess (e.e.) of the final compounds was evaluated by analytical chiral chromatography on a Chiralpak IB-3 column, 250 mm × 4.6 mm i.d. (Daicel Corporation, Japan) using a Waters HPLC system with the detector set at 250 and 291 nm. As eluent, a solution of hexane, ethanol, diethylamine, and ethylenediamine, (ratio of 70:30:0.1:0.1, v/v/v/v) was used, and the flow rate was 0.7 mL/min.
General Procedure for Stille Cross-Coupling Reaction.
Pd/P(t-Bu)3 (0.3 equiv) and CsF (2.2 equiv) were mixed together in an oven-dried Schlenk tube under argon. Then, 1-(3-chloro-4′-fluoro-[1,1′-biphenyl]-4-yl)ethan-1-one (1 equiv), the corresponding organotin reagent (1.06 equiv), and dioxane (1 mM) were added. The reaction mixture was stirred at 100 °C and the formation of the desired product was monitored by LCMS. The final product was detected after 2 or 4 h, showing a shorter reaction time in comparison with the examples reported in the literature.32 The crude residue was filtered on Celite, the solvent evaporated under reduced pressure, and the cross-coupled product was purified by silica gel flash chromatography using hexane/EtOAc (9:1 v/v) as the mobile phase. A white solid was obtained, and the yield was from 60 to 98%.
Compound 1: (R)-N-(1-(4-Fluoro-[1,1′:3′,1″-terphenyl]-4′-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide.
Yield: 50%. 1H NMR (400 MHz, CDCl3): δ ppm 9.70 (s, 1H), 9.50 (d, J = 8.6 Hz, 1H), 8.14 (m, 4H), 7.94 (m, 3H),7.57 (m, 12H), 7.12 (m, 3H), 6.22 (s, 1H), 5.90 (m, 1H), 5.20 (m, 1H), 4.00 (dd, J = 14.0, 2.6 Hz, 1H). LCMS, found [M + H]+ m/z 606.0, Rt = 1.611 min. Mass calcd for C38H28FN5O2 [M + H]+, 606.2300; found, Mobs = 606.2296, Δppm = 0.65. HPLC, tR = 47.4 min, purity of 97%. Enantiomeric excess: >99%.
Compound 2: (R)-N-(1-(4-(4-Fluorophenyl)naphthalen-1-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide.
Yield: 30%. 1H NMR (400 MHz, CDCl3): δ ppm 9.90 (s, 1H), 9.47 (d, J = 9 Hz, 1H), 8.37 (d, J = 8.6 Hz, 1H), 8.11–8.03 (m, 5H), 7.89–7.85 (m, 3H), 7.64–7.60 (m, 1H), 7.56–7.44 (m, 6H), 7.41–7.35 (m, 3H), 7.19–7.15 (m, 2H), 6.59–6.54 (m, 1H), 5.54 (t, J = 25, 12.6 Hz, 1H), 4.58 (dd, J = 14.0, 2.8 Hz, 1H). LCMS, found [M + H]+ m/z 579.90, Rt = 1.605 min. Mass calcd for C36H26FN5O2 [M + H]+, 580.2143; found, Mobs = 580.2141, Δppm = 0.34. HPLC, tR = 23.6 min, purity of >99%. Enantiomeric excess: 99%.
Compound 3: (R)-N-(1-(4′-Fluoro-3-(furan-2-yl)-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide.
Yield: 20%. 1H NMR (400 MHz, CDCl3): δ ppm 9.90 (s, 1H), 8.12–8.10 (m, 5H), 7.97–7.93 (m, 1H), 7.68 (dd, J = 8.6, 2.0 Hz, 1H), 7.65 (dd, J = 8.6, 2.0 Hz, 1H),7.55–7.54 (m, 8H), 7.19–7.12 (m, 3H), 6.21–6.19 (m, 1H), 5.35–5.30 (m, 1H), 4.54–4.50 (m, 1H). LCMS, found [M + H]+ m/z 595.9 tR = 1.63 min. Mass calcd for C36H26FN5O3 [M + H]+, 596.2092; found, Mobs = 596.2094, Δppm = −0.34, HPLC, tR = 21.7 min, purity of 99%. Enantiomeric excess: 98.7%.
Compound 4: (R)-N-(1-(4-Fluoro-4″-methoxy-[1,1′:3′,1″-terphenyl]-4′-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide.
Yield: 45%. 1H NMR (400 MHz, CDCl3): δ ppm 9.69 (s, 1H), 9.49 (d, J = 8.5 Hz, 1H), 8.12 (m, 5H), 7.97 (d, J = 8.04 Hz, 1H), 7.90 (d, J = 8.4 Hz, 1H), 7.63 (dd, J =8.12, 1.8 Hz, 1H), 7.58–7.52 (m, 8H), 7.46 (d, J = 1.9 Hz, 1H),7.15–7.10 (m, 5H), 6.37 (s, 1H), 5.96–5.92 (m, 1H), 5.22–5.15 (m, 1H), 4.04 (dd, J = 13.7, 2.7 Hz, 1H), 3.92 (s, 3H). LCMS, found [M + H]+ m/z 637.8, tR = 1.83 min. Mass calcd for C39H30FN5O3 [M + H]+, 636.2405; found, Mobs = 636.2402, Δppm = 0.47, HPLC, tR =37.8 min, purity of >99%. Enantiomeric excess: 99.8%.
Compound 5: (R)-4-(5-(3-Bromonaphthalen-1-yl)-1,3,4-oxadiazol-2-yl)-N-(1-(3-chloro-4′-fluoro-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethyl)benzamide.
Yield: 20%. 1H NMR (400 MHz, CDCl3): δ ppm 10.07 (s, 1H), 9.54 (d, J = 8.7 Hz, 1H), 9.24 (d, J = 8.7 Hz, 1H), 8.27 (s, 1H), 8.15 (d, J = 8.5 Hz, 3H), 7.90 (d, J =8.0 Hz, 3H), 7.83 (d, J = 8.06 Hz, 1H), 7.72–7.69 (m, 1H), 7.63 (d, J = 7.46 Hz, 1H), 7.58–7.57 (m, 2H), 7.51–7.47 (m, 3H), 7.12 (t, J =8.62 Hz, 2H), 6.19–6.15 (m, 1H), 5.39–5.32 (m, 1H), 4.52 (d, J =11.5 Hz, 1H). LCMS, found [M + H]+ m/z 693.7, tR = 1.90 min. Mass calcd for C36H24BrClFN5O2 [M + H]+, 692.0859; found, Mobs = 692.0848, Δppm = 1.58, HPLC, tR = 12.7 min, purity of 97%. Enantiomeric excess: 99%.
Compound 6: (R)-4-(5-(2-Fluoro-5-(pyridin-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)-N-(1-(3-chloro-4′-fluoro-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethyl)benzamide.
Yield: 10%. LCMS, found [M + H]+ m/z 659.8, Rt = 1.738 min. Mass calcd for C37H25ClF2N6O2 [M + H]+, 659.1768; found, Mobs = 659.1784, Δppm = −2.4, HPLC, tR = 23.2 min, purity of 98%. Enantiomeric excess: 97.7%.
Compound 7: (R)-4-(5-(3-Bromo-5-(methylsulfonyl)phenyl)-1,3,4-oxadiazol-2-yl)-N-(1-(3-chloro-4′-fluoro-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethyl)benzamide.
Yield: 40%. 1H NMR (600 MHz, CDCl3): δ ppm 10.2 (s, 1H), 9.57 (br, 1H), 8.68 (s, 1H), 8.55 (s, 1H), 8.30 (s, 1H), 8.02–7.98 (m, 1H), 7.88 (d, J =7.6 Hz, 1H), 7.68–7.76 (m,1H), 7.60 (s, 1H), 7.52–7.44 (m, 3H),7.14–7.11 (m, 2H), 6.32 (m, 1H), 5.39–5.35 (m, 1H), 4.66 (d, J =7.6 Hz, 1H), 3.34 (s, 3H). LCMS, found [M + H]+ m/z 720.7, Rt =1.703 min. Mass calcd for C37H27ClFN7O4S [M + H]+, 720.0478; found, Mobs = 720.0483, Δppm = −0.69, HPLC, tR = 21.9 min, purity of 96%. Enantiomeric excess: 98%.
Compound 8: (R)-N-(1-(3-Chloro-4′-fluoro-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(3-(methylsulfonyl)-5-(pyridin-2-yl)phenyl)-1,3,4-oxadiazol-2-yl)benzamide.
Yield: 20%. 1H NMR (600 MHz, CDCl3): δ ppm 10.3 (d, J = 8.4 Hz, 1H), 9.68 (s, 1H), 9.03 (d, J = 1.5 Hz, 1H), 8.83 (s, 1H), 8.80 (d, J =5.0 Hz, 1H), 8.72–8.71 (m, 1H), 8.36 (d, J = 8.4 Hz, 1H), 8.20 (d, J = 7.0 Hz, 1H), 8.17–8.14 (m, 2H), 7.97 (d, J = 8.2 Hz, 1H), 7.70 (s, 1H), 7.64–7.57 (m, 2H), 7.53–7.52 (m, 1H), 7.50 (s, 1H), 7.46–7.41 (m, 3H), 7.30–7.28 (m, 1H), 7.07–7.05 (m, 2H), 6.03–5.99 (m, 1H), 5.44–5.39 (m, 1H), 4.43 (d, J = 14 Hz, 1H), 3.54 (s, 3H). LCMS, found [M + H]+ m/z 719.8, Rt = 1.626 min. Mass calcd for C38H28ClFN6O4S [M + H]+, 719.1638; found, Mobs = 719.1647, Δppm = −1.25, HPLC, tR = 16.3 min, purity of 96%. Enantiomeric excess: >98%.
Compound 9: (R)-N-(1-(3-Chloro-4′-fluoro-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(3-fluoro-5-(pyridin-2-yl)-phenyl)-1,3,4-oxadiazol-2-yl)benzamide.
Yield: 10%. 1H NMR (600 MHz, CDCl3): δ ppm 9.95 (s, 1H), 9.86 (br, 1H), 8.96 (s, 1H),8.70 (s, 1H), 8.34 (br, 1H), 8.23 (br, 1H), 8.14 (d, J = 7.3 Hz, 2H),8.04 (d, J = 7.6 Hz, 1H), 7.95–7.90 (m, 4H), 7.84 (br, 1H), 7.56 (d, J = 16 Hz, 2H), 7.49–7.46 (m, 4H), 7.11 (t, J = 8.4 Hz, 2H), 6.14–6.11 (m, 1H), 5.44–5.40 (m, 1H), 4.50 (d, 10.4 Hz, 1H). LCMS, found [M + H]+ m/z 659.8, Rt = 1.738 min. Mass calcd for C37H25ClF2N6O2 [M + H]+, 659.1768; found, Mobs = 659.1799, Δppm = −4.70, HPLC, tR = 44.1 min, purity of 97%. Enantiomeric excess: 99%.
Compound 10: (R)-N-(1-(3-Chloro-4′-fluoro-[1,1′-biphenyl]-4-yl)-2-(1H-imidazol-1-yl)ethyl)-4-(5-(3-fluoro-5-(5-fluoropyrimidin-4-yl)phenyl)-1,3,4-oxadiazol-2-yl)benzamide.
Yield: 20%. 1H NMR (600 MHz, CDCl3): δ ppm 10.0 (s, 1H), 9.55 (d, J = 8.6 Hz 1H), 9.15 (d, J = 2.8 Hz), 8.75 (d, J = 2.8 Hz, 1H), 8.72 (s, 1H), 8.12–8.09 (m, 3H), 8.00 (d, J = 2.8 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 1.7 Hz, 1H), 7.56 (s, 1H), 7.51–7.47 (m, 3H), 7.44 (s, 1H), 7.12 (t, J = 8.6 Hz, 2H),6.15–6.11 (m,1H), 5.36–5.31 (m, 1H), 4.48 (dd, J = 14.3, 3.0 Hz, 1H). LCMS, found [M + H]+ m/z 678.9, Rt = 1.792 min. Mass calcd for C36H23ClF3N7O2 [M + H]+, 678.1627; found, Mobs = 678.1633, Δppm = −0.88, HPLC, tR = 27.4 min, purity of 99%. Enantiomeric excess: 98%.
Site-Directed Mutagenesis.
Site-directed mutagenesis of human CYP51 Thr 318 (ACT) to Ile (ATT) was performed in the pET17b plasmid using PfuTurbo DNA polymerase (Agilent Technologies) and confirmed by DNA sequencing. The sequences of the forward and reverse primers were 5′-GCATACATCCTCAATTACTAGTGCTTGG-3′ and 5′-CCAAGCACTAGTAATTGAGGATGTATGC-3′, respectively. The mutated Cyp51 cDNA was excised by digestion with NdeI (5′–) and HindIII (3′–) (New England Biolabs), subcloned into the pCW expression vector at the same restriction sites using T4 DNA ligase (New England Biolabs) and sequenced again to ensure the proper ligation. For protein expression, the plasmid was transformed into E. coli HMS174 (DE3) (Novagen) competent cells.
CYP51 Activity Assay.
Full-length human CYP51, the wild type (WT) and the T318I mutant, and rat NADPH-cytochrome P450 reductase (CPR) were expressed and purified as described previously.31 The standard reaction mixture contained 0.5 μM P450 and 1.0 μM CPR, 100 μM L-α−1,2-dilauroyl-sn-glycerophosphocholine, 0.4 mg/mL isocitrate dehydrogenase, and 25 mM sodium isocitrate in 50 mM potassium phosphate buffer (pH 7.2) containing 10% glycerol (v/v). After addition of the radiolabeled ([3-3H]) lanosterol (~4000 dpm/nmol; dissolved in 45% HPCD, w/v, final concentration of 50 μM), the mixture was preincubated for 30 s at 37 °C in a shaken water bath, and the reaction was initiated by the addition of 100 μM NADPH and stopped by extraction of the sterols with EtOAc. The extracted sterols were analyzed by a reversed-phase HPLC system (Waters) equipped with a β-RAM detector (INUS Systems) using a NovaPak octadecylsilane (C18) column (particle size of 4 μm, 3.9 mm × 150 mm) and a linear gradient of H2O/CH3CN:CH3OH (1.0:4.5:4.5, v/v/v) (solvent A) to CH3OH (solvent B), increasing from 0 to 100% B for 30 min at a flow rate of 1.0 mL/min. The activity was monitored as the percentage of the product formation (4,4-dimethylcholesta-8,14,24-trien-3β-ol). For steady-state kinetic analysis, the reactions were run for 60 s at 37 °C, and the P450 concentration was 0.25 μM. Michaelis–Menten parameters were calculated using GraphPad Prism 6 (GraphPad, La Jolla, CA) with the reaction rates (nmol product formed/nmol P450/min) being plotted versus the total substrate concentration and using nonlinear regression (hyperbolic fits).
Evaluation of Inhibitory Potency.
The inhibitory effects of compounds on human CYP51 activity were initially compared on the basis of decreases in substrate conversion in 60 min reactions at a 50-fold molar excess of an inhibitor over the enzyme (final concentrations of 0.5 μM P450 and 25 μM inhibitor).30 The effects of VFV analogs were further evaluated in more detail by determining their IC50 (concentration required to decrease the initial rate of the reaction by 50%) and “1 h IC50” (concentration required to prevent conversion of 50% of the substrate in a 1 h reaction) values. The values were calculated using GraphPad Prism 6 with the percentage of lanosterol converted being plotted against the inhibitor concentration and the curves fitted with nonlinear regression (log(inhibitor) vs the normalized response).
CYP51 Stability Assay.
Stabilities of human CYP51, WT and the T314I mutant, were monitored as the decrease in the P450 content upon incubation of the 2 μM protein samples in 200 mM potassium phosphate buffer, pH 7.2, 10% glycerol (v/v) and 0.1% Triton X-100 (v/v) in a Thermal Cycler 480 (Perkin Elmer) at 42 °C.36 The aliquots were taken after 2, 5, 10, and 20 min. The spectra were recorded on a dual-beam Shimadzu UV-240IPC spectrophotometer. The experiments were performed in triplicate.
CYP51 Substrate Binding Assay.
Spectral titration of 2 μM human CYP51, WT and the T314I mutant, with lanosterol was carried out in 50 mM phosphate buffer, pH 7.4, containing 100 mM NaCl (1 cm of optical path length). The high-spin form content was estimated from the absolute spectra as the ratio (ΔA393–470/ΔA417–470) and from the difference spectra with the response of 0.11 absorbance units/nmol of P450 corresponding to a 100% low-to-high spin transition.36 The Kd values of the enzyme–substrate complex were calculated as blue shifts in the P450 Soret band maximum (from 417 to 393 nm) in GraphPad Prism 6 by fitting the data for the substrate-induced absorbance changes in the difference spectra Δ(Amax − Amin) versus the substrate concentration to a one site-total binding equation (binding–saturation).
CYP51 Inhibitor Binding Assay.
Spectral titrations with VFV derivatives and selected VNI derivatives were carried out in 5 cm optical path length cuvettes at 0.5 μM P450 concentration.28 The ligands were added from 0.2 mM stock solutions in DMSO, and the titration range was varied depending on the ligand affinities. The Kd values of the enzyme–inhibitor complex were calculated as red shifts in the P450 Soret band maximum (from 417 to 421–425 nm) in GraphPad Prism 6 by fitting the hyperbolic data for the ligand-induced absorbance changes in the difference spectra Δ(Amax − Amin) versus the ligand concentration to the Morrison (quadratic) equation.
Crystallization, Data Collection, Structure Refinement, and Analysis.
For crystallographic experiments, the previously described construct of truncated human CYP51 (with the N-terminal membrane anchor sequence replaced for the MAKKTSSKGKL fragment) was expressed in E. coli and purified in two steps including affinity chromatography on Ni2+-NTA agarose and cation exchange chromatography on CM Sepharose.31 A 10 μM solution of compound 10 was added to all the buffers at the CM Sepharose stage of the purification procedure. The absorbance spectra of the collected protein–inhibitor complexes were taken to make sure that the Soret maximum was at 425 nm. The initial screening of crystallization conditions was performed using Hampton Research crystallization kits. The diffracting crystals were obtained by the hanging drop vapor diffusion technique and were grown at 16 °C. In final experiments, the 300 μM solution of the human CYP51–compound 10 complex was preincubated for 2 h at 4 °C with 15 mM detergent (deoxycholic acid, sodium salt, Hampton Research) and 5.5 mM tris(carboxyethyl)-phosphine (TCEP) and centrifuged at 16,000g for 10 min. The supernatant was mixed with various volumes of the mother liquor (0.2 M potassium formate (pH 7.4) with 8% PEG 3350 (w/v)) and equilibrated against the reservoir solution. Crystals appeared within a few hours and were cryoprotected in 30% glycerol (v/v) and flash-cooled in liquid nitrogen. The best diffraction was observed in the drop that contained 3 μL of the human CYP51–compound 10- detergent mixture and 0.3 μL of the mother liquor.
The data were collected at 100 K using synchrotron radiation on the insertion device of the Life Sciences Collaborative Access Team at the Advanced Photon Source, Argonne National Laboratory (Argonne, IL), beamline 21-ID-G at a wavelength of 0.9787 Å and using a MAR 300 CCD detector. The diffraction images were processed with HKL-2000 to maximum resolution of 2.80 Å, and the structure was solved in Phaser MR (CCP4 Program Suite,38 version7.0.050) using atomic coordinates of the VFV-bound human CYP51 (PDB ID 4UHI). Iterative models of the protein–inhibitor complexes were built with Coot 0.8.9.239 and refined with Refmac5 in the CCP4 Suite. Structure superimposition and RMSD calculation was performed in lsqcab (CCP4 Suite). Active-site volumes were calculated in BioVia Discovery Studio Visualizer 2019, which was also used to prepare Figures 3, 4, and 11B. The other structural figures were prepared in Chimera 1.13.1. The electron density maps are from Coot (Figure S9).
Supplementary Material
ACKNOWLEDGMENTS
We thank Dr. John D. York (Vanderbilt University) for the helpful discussion. The research was supported by funding from the National Institutes of Health (no. GM067871, G.I.L.). Vanderbilt University is a member institution of the Life Sciences Collaborative Access Team at Sector 21 of the Advanced Photon Source (Argonne, IL). Use of the Advanced Photon Source at the Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02-06CH11357.
ABBREVIATIONS
- CYP
cytochrome P450
- CYP51
sterol 14α-demethylase
- IC50
inhibitor concentration that decreases the enzyme turnover rate by 50%
- 1 h IC50
inhibitor concentration that prevents conversion of 50% of the substrate in a 1 h reaction
- DMF
N,N-dimethylformamide
- DPPA
diphenyl phosphoryl azide
- DBU
1,5-diazabiciclo[5.4.0]undec-7-ene
- THF
tetrahydrofuran
- HPLC
high-performance liquid chromatography
- Pd-(dppf)Cl2
[1,1′-bis(diphenylphosphino)ferrocene]-dichloropalladium(II)
- LC–MS
liquid chromatography-mass spectrometry
- ESI
electrospray ionization
- DMSO
dimethyl sulfoxide
- TFA
trifluoroacetic acid
- ELSD
evaporative light scattering detector
- rt.
room temperature
- equiv.
equivalent
- Hz
Hertz
- J
coupling constant (NMR)
- tR
retention time (chromatography)
- TMS
tetramethylsilane
- MHz
megahertz
- DMAP
4-(N,N-dimethylamino)pyridine
- MW
microwave reactor
- EDC/HCl
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b01485.
Table S1: ligand-contacting residues (≤4.5 Å) in human CYP51 complexes, Table S2: data collection and refinement statistics, Figure S1: inhibitory effects of VFV and VFV-Cl on human CYP51 activity, Figure S2: T318I mutation increases susceptibility of human CYP51 to inhibition, Figure S3: inhibitory effects of VFV-Cl on the activity of the T318I and WT human CYP51, Figure S4: T318A mutation increases the binding affinity of human CYP51 to VNI, Figure S5: X-ray structure of human CYP51 cocrystallized with compound 10, Figure S6: active site forming secondary structural residues and ligand contacting residues in the superimposed compound 10-bound and VFV-bound human CYP51 structures, Figure S7: HRMS spectra, Figure S8: 1H NMR spectra, and Figure S9: electron density maps for the heme-coordinated inhibitor, the active site entering deoxycholate, and the run-away inhibitor (PDF)
Molecular formula strings (CSV)
Accession Codes
The coordinates and structure factors of human CYP51 in a complex with compound 10 have been deposited to PDB under accession code 6Q2T. The atomic coordinates and experimental data will be released upon acceptance of the article for publication.
The authors declare no competing financial interest.
REFERENCES
- (1).Siegel RL; Miller KD; Jemal A Cancer statistics, 2018. CA: Cancer J. Clin 2018, 68, 7–30. [DOI] [PubMed] [Google Scholar]
- (2).Hanahan D; Weinberg RA Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- (3).Chimento A; Casaburi I; Avena P; Trotta F; De Luca A; Rago V; Pezzi V; Sirianni R Cholesterol and its metabolites in tumor growth: therapeutic potential of statins in cancer treatment. Front. Endocrinol 2018, 9, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kuzu OF; Noory MA; Robertson GP The role of cholesterol in cancer. Cancer Res. 2016, 76, 2063–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Murai T Cholesterol lowering: role in cancer prevention and treatment. Biol. Chem 2015, 396, 1–11. [DOI] [PubMed] [Google Scholar]
- (6).Dutta A; Sharma-Walia N Curbing lipids: impacts on cancer and viral infection. Int. J. Mol. Sci 2019, 20, 644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Brown MS; Goldstein JL Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J. Lipid Res 1980, 21, 505–517. [PubMed] [Google Scholar]
- (8).Simons K; Ikonen E How cells handle cholesterol. Science 2000, 290, 1721–1726. [DOI] [PubMed] [Google Scholar]
- (9).Sezgin E; Levental I; Mayor S; Eggeling C The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat. Rev. Mol. Cell Biol 2017, 18, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ding X; Zhang W; Li S; Yang H The role of cholesterol metabolism in cancer. Am. J. Cancer Res 2019, 9, 219–227. [PMC free article] [PubMed] [Google Scholar]
- (11).Gu L; Saha ST; Thomas J; Kaur M Targeting cellular cholesterol for anticancer therapy. FEBS J. 2019, DOI: 10.1111/febs.15018. [DOI] [PubMed] [Google Scholar]
- (12).Freeman MR; Solomon KR Cholesterol and prostate cancer. J. Cell. Biochem 2004, 91, 54–69. [DOI] [PubMed] [Google Scholar]
- (13).Altwairgi AK Statins are potential anticancerous agents. Oncol. Rep 2015, 33, 1019–1039. [DOI] [PubMed] [Google Scholar]
- (14).Mei Z; Liang M; Li L; Zhang Y; Wang Q; Yang W Effects of statins on cancer mortality and progression: a systematic review and meta-analysis of 95 cohorts including 1,111,407 individuals. Int. J. Cancer 2017, 140, 1068–1081. [DOI] [PubMed] [Google Scholar]
- (15).Li Y; He X; Ding YE; Chen H; Sun L Statin uses and mortality in colorectal cancer patients: an updated systematic review and meta-analysis. Cancer Med. 2019, 6, 3305–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Abdullah MI; de Wolf E; Jawad MJ; Richardson A The poor design of clinical trials of statins in oncology nay explain their failure – lessons for drug repurposing. Cancer Treat. Rev 2018, 69, 84–89. [DOI] [PubMed] [Google Scholar]
- (17).Hamelin BA; Turgeon J Hydrophilicity/lipophilicity: relevance for the pharmacology and clinical effects of HMG-CoA reductase inhibitors. Trends Pharmacol. Sci 1998, 19, 26–37. [DOI] [PubMed] [Google Scholar]
- (18).Luo Z; Zhang Y; Gu J; Feng P; Wang Y Pharmacokinetic properties of single- and multiple-dose pitavastatin calcium tablets in healthy chinese volunteers. Cur. Ther. Res 2015, 77, 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lynch C; Zhao J; Huang R; Kanaya N; Bernal L; Hsieh JH; Auerbach SS; Witt KL; Merrick BA; Chen S; Teng CT; Xia M Identification of estrogen-related receptor alpha agonists in the Tox21 compound library. Endocrinology 2018, 159, 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Simic I; Reiner Z Adverse Effects of statins - myths and reality. Cur. Pharm. Des 2015, 21, 1220–1226. [DOI] [PubMed] [Google Scholar]
- (21).Abdullah MI; Abed MN; Richardson A Inhibition of the mevalonate pathway augments the activity of pitavastatin against ovarian cancer cells. Sci. Rep 2017, 7, 8090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).You HY; Zhang WJ; Xie XM; Zheng ZH; Zhu HL; Jiang FZ Pitavastatin suppressed liver cancer cells in vitro and in vivo. OncoTargets Ther. 2016, Volume 9, 5383–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Robinson E; Nandi M; Wilkinson LL; Arrowsmith DM; Curtis ADM; Richardson A Preclinical evaluation of statins as a treatment for ovarian cancer. Gynecol. Oncol 2013, 129, 417–424. [DOI] [PubMed] [Google Scholar]
- (24).Akimoto H; Negishi A; Oshima S; Okita M; Numajiri S; Inoue N; Ohshima S; Kobayashi D Onset timing of statin-induced musculoskeletal adverse events and concomitant drug-associated shift in onset timing of MAEs. Pharmacol. Res. Perspect 2018, 6, No. e00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Murai T The role of lipid rafts in cancer cell adhesion and migration. Int. J. Cell Biol 2012, 2012, 763283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lepesheva GI; Friggeri L; Waterman MR CYP51 as drug targets for fungi and protozoan parasites: past, present and future. Parasitology 2018, 145, 1820–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lepesheva GI; Hargrove TY; Rachakonda G; Wawrzak Z; Pomel S; Cojean S; Nde PN; Nes WD; Locuson CW; Calcutt MW; Waterman MR; Daniels JS; Loiseau PM; Villalta F VFV as a new effective CYP51 structure-derived drug candidate for Chagas disease and visceral leishmaniasis. J. Infect. Dis 2015, 212, 1439–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Friggeri L; Hargrove TY; Rachakonda G; Blobaum AL; Fisher P; de Oliveira GM; da Silva CF; Soeiro MNC; Nes WD; Lindsley CW; Villalta F; Guengerich FP; Lepesheva GI Sterol 14α-demethylase structure-based optimization of drug candidates for human infections with the protozoan Trypanosomatidae. J. Med. Chem 2018, 61, 10910–10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Holdgate GA; Meek TD; Grimley RL Mechanistic enzymology in drug discovery: a fresh perspective. Nat. Rev. Drug Discovery 2018, 17, 115–132. [DOI] [PubMed] [Google Scholar]
- (30).Lepesheva GI; Ott RD; Hargrove TY; Kleshchenko YY; Schuster I; Nes WD; Hill GC; Villalta F; Waterman MR Sterol 14α-demethylase as a potential target for antitrypanosomal therapy: enzyme inhibition and parasite cell growth. Chem. Biol 2007, 14, 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hargrove TY; Friggeri L; Wawrzak Z; Sivakumaran S; Yazlovitskaya EM; Hiebert SW; Guengerich FP; Waterman MR; Lepesheva GI Human sterol 14α-demethylase as a target for anticancer chemotherapy: towards structure-aided drug design. J. Lipid Res 2016, 57, 1552–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Littke AF; Schwarz L; Fu GC Pd/P(t-Bu)3: A mild and general catalyst for stille reactions of aryl chlorides and aryl bromides.J. Am. Chem. Soc 2002, 124, 6343–6348. [DOI] [PubMed] [Google Scholar]
- (33).Friggeri L; Hargrove TY; Rachakonda G; Williams AD; Wawrzak Z; Di Santo R; De Vita D; Waterman MR; Tortorella S; Villalta F; Lepesheva GI Structural basis for rational design of inhibitors targeting Trypanosoma cruzi sterol 14α-demethylase: two regions of the enzyme molecule potentiate its inhibition. J. Med. Chem 2014, 57, 6704–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Friggeri L; Hargrove TY; Wawrzak Z; Blobaum AL; Rachakonda G; Lindsley CW; Villalta F; Nes WD; Botta M; Guengerich FP; Lepesheva GI Sterol 14α-demethylase structure-based design of VNI ((R)- N-(1-(2,4-dichlorophenyl)-2-(1 H-imidazol-1-yl)ethyl)-4-(5-phenyl-1,3,4-oxadiazol-2-yl)benzamide) derivatives to target fungal infections: synthesis, biological evaluation, and crystallographic analysis. J. Med. Chem 2018, 61, 5679–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lepesheva GI; Waterman MR Sterol 14α-demethylase cytochrome P450 (CYP51), a P450 in all biological kingdoms. Biochim. Biophys. Acta 2007, 1770, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hargrove TY; Wawrzak Z; Fisher PM; Child SA; Nes WD; Guengerich FP; Waterman MR; Lepesheva GI Binding of a physiological substrate causes large-scale conformational reorganization in cytochrome P450 51. J. Biol. Chem 2018, 293, 19344–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mercorelli B; Luganini A; Celegato M; Palú G; Gribaudo G; Lepesheva GI; Loregian A Trafficking and de novo synthesis of cholesterol are required for human cytomegalovirus replication and infectivity and are new antiviral targets. PLoS Pathog., submitted for publication, 2019. [Google Scholar]
- (38).Potterton E; Briggs P; Turkenburg M; Dodson E A Graphical user interface to the CCP4 program suite. Acta Crystallogr., Sect. D: Biol. Crystallogr 2003, 59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- (39).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.