

Abstract
The incidence of hepatocellular carcinoma (HCC) is on the rise worldwide. Although the incidence of HCC in males is considerably higher than in females, the projected rates of HCC incidence are increasing for both sexes. A recently appreciated risk factor for HCC is the growing problem of non-alcoholic fatty liver disease (NAFLD), which is usually associated with obesity and the metabolic syndrome. In this study, we showed that under conditions of fatty liver, female mice were more likely to develop HCC than expected from previous models. Using an inducible knockout model of the tumor-suppressive isoform of hepatocyte nuclear factor 4 alpha (“P1-HNF4α”) in the liver in combination with prolonged high fat (HF) diet, we found that HCC developed equally in male and female mice as early as 38 weeks of age. Similar sex-independent HCC occurred in the “STAM” model of mice, in which severe hyperglycemia and HF feeding results in rapid hepatic lipid deposition, fibrosis, and ultimately HCC. In both sexes, reduced P1-HNF4α activity, which also occurs under chronic high-fat diet feeding, increased hepatic lipid deposition and produced a greatly augmented circadian rhythm in interleukin 6 (Il6), a factor previously linked with higher HCC incidence in males. Loss of HNF4α combined with HF feeding induced epithelial-mesenchymal transition in an IL-6-dependent manner. Collectively, these data provide a mechanism-based working hypothesis that could explain the rising incidence of aggressive HCC.
Keywords: HNF4α, hepatocellular carcinoma, HCC, NAFLD, IL-6, circadian
Introduction
Hepatocellular carcinoma (HCC) is one of the deadliest forms of cancer worldwide and the most common primary liver cancer to present in the clinic (1). Recently, a significant rise in the incidence of HCC has been observed, which has been attributed to the increasing prevalence of non-alcoholic fatty liver disease (NAFLD), known to be associated with obesity and the metabolic syndrome (2,3). Progression of NAFLD to HCC usually occurs in stages, starting with NAFLD, followed by nonalcoholic steatohepatitis (NASH), increasing fibrosis ending in cirrhosis, and, finally, HCC (4). However, NAFLD can lead to HCC independent of fibrosis in up to 20% of patients (5). Primary liver cancer risk is correlated with increasing body mass index (BMI), with a pronounced association for individuals with a BMI above 32 kg/m2 (6) (7–9). Unfortunately, HCC is often discovered at a late stage when surgical resection is not an option. Understanding the mechanisms by which NAFLD leads to HCC will be essential to combat the growing incidence of this deadly cancer.
HCC incidence has been reported to be three-to-five times more frequent in males than in females (10). A similar gender disparity has been reported under conditions of chemical mutagen-induced HCC in mice (11). In contrast, National Cancer Institute Surveillance, Epidemiology and End Results Program (SEER) data reveal increasing incidence of HCC in both men and women, with the percent change in burden being greater in women of some races compared to their male counterparts (12). Several factors contribute to the sex specificity of HCC, though sex hormones are a primary factor. Inhibition of interleukin 6 (IL-6) production by estrogen protects against HCC in female mice, and estrogen receptor agonists can prevent IL-6 production in male mice treated with diethylnitrosamine (DEN) (13). This is consistent with human data showing an increased risk of HCC in females following menopause (reviewed in (14)). Increasing rates of HCC in both sexes suggest that additional mechanisms are at work. One such mechanism is NAFLD, which we propose partially bypasses the traditional mechanisms that provide protection from HCC in females. Additional studies of HCC in female rodent models are needed to examine the pathophysiology underlying the rising HCC incidence in females; however, established mouse models of HCC wherein females manifest HCC with similar temporal progression as their male counterparts are lacking.
Hepatocyte nuclear factor 4 alpha (HNF4α) is a liver-enriched transcription factor also found in the kidney, pancreas, stomach and intestine (15,16). Several mutations in the Hnf4a gene induce an early-onset form of non-insulin-dependent diabetes mellitus (maturity-onset diabetes of the young, MODY1), involving progressive loss of insulin secretion and ultimately, moderate to severe hyperglycemia (17). A major role of HNF4α is regulating liver-specific gene expression, which drives hepatocyte cell fate (18–21); though it also plays prominent roles in hepatic gluconeogenesis and lipid metabolism. The HNF4α gene encodes different isoforms, which result from alternate promoter usage of the “P1” and “P2” promoters, as well as differential splicing (16). Isoform expression differs across development, differentiation, and tissue. P1-HNF4α is highly expressed in the adult liver and has potent tumor suppressor activity, in part via its repression at genes that promote proliferation (22–25). Global knockout of HNF4α in mice results in embryonic lethality, while liver-specific loss results in death within six weeks of age (20). Loss of hepatic P1-HNF4α expression in the adult rodent results in a fatty liver, possibly due to reduced apolipoprotein gene expression (23). Rodent models of insulin resistance show reduced hepatic nuclear P1-HNF4α (25,26), suggesting that it may play a central role in connecting NAFLD to HCC. P2-HNF4α is not observed in healthy adult liver; instead its expression is observed in approximately 50% of hepatocellular carcinomas (25,27), where it represses the circadian gene Aryl Hydrocarbon Receptor Nuclear Translocator Like, Arntl1 (Bmal1) gene, and promotes cytoplasmic export of P1-HNF4α (25). Aberrant expression of the P2-driven isoform also occurs in colon cancer (28,29). In DEN-treated mice, ablation of P1-HNF4α promotes tumor growth (30), while its loss in immortalized human hepatocytes causes transformation (31). These data are consistent with the proposed role of the P1 isoform as a tumor suppressor, and suggest that suppression of P1-HNF4α expression or activity over time, may cause malignant transformation.
While an inverse correlation between Hnf4a and Il6 has been reported (23,31), this study reveals a sex-independent and robust circadian induction of IL-6 in the absence of P1-HNF4α, which under prolonged HF diet challenge results in early-onset HCC to an equal extent in male and female mice. Furthermore, loss of HNF4α combined with HF diet induces epithelial-mesenchymal transition (EMT) in an IL-6-dependent manner. These findings implicate liver lipid metabolism and circadian rhythm dysregulation as mechanisms through which HCC risk and aggressiveness may increase irrespective of gender.
Materials and Methods
Animals.
Animal use was approved by the UT Health Institutional Animal Care and Use Committee. Mice were group housed in pathogen-free conditions and fed ad libitum with chow (PicoLab Rodent Diet 5053). Animals were entrained in 12-h light/12-h dark cycles. Hnf4aF/Fand SA+/Cre-ERT2 (Albtm1(cre/ERT2)Mtz) mice were provided by Dr. Frank Gonzalez (23). To generate conditional Hnf4F/F;AlbERT2cre mice, Hnf4aF/F mice were crossed with the tamoxifen-inducible hepatocyte-specific Cre recombinase expressing mouse SA+/Cre-ERT. Cre+ mice with inducible loss of Hnf4a are referred to as “H4LivKO”. For long-term experiments, Cre− and Cre+ littermate mice were administered tamoxifen at six weeks of age and maintained on chow or fed high fat diet (HF, Research Diets D12492).
Tamoxifen injections.
Hnf4aF/F; AlbERT2cre and wildtype (WT) littermate mice were injected intraperitoneally with tamoxifen (10 mg ml−1) in corn oil for 5 consecutive days (days 1–5).
STAM Mouse Model.
The NASH-HCC model (“STAM” mouse) was generated by a single subcutaneous injection of 200 μg streptozotocin (STZ) (Sigma, MO, USA) two days after birth. At four weeks, mice were placed on HF diet (D12492).
Metabolic phenotyping.
Mice were placed in metabolic cages (Comprehensive Lab Animal Monitoring System-CLAMS, Columbus Instruments) at 15 weeks of age and provided ground diet ad libitum. Data was collected and averaged over four days, and analyazed using Oxymax V 4.87. Energy expenditure data is normalized to body weight.
MRI.
Body composition was measured by EchoMRI-100 T (Echo Medical Systems) at 20 weeks of age. Results shown were averaged from three independent measurements.
Basal glucose measurement.
Mice were fasted for five hours and tail blood was measured using a glucose meter (ACCU-CHEK Nano).
Hepatic triglyceride measurements.
Liver triglycerides (TG) were assayed using a kit from Cayman (cat. #10010303) according to the manufacturer’s protocol. Briefly, 400 mg of liver tissue was homogenized in NP40 substrate assay reagent containing protease inhibitors. Ten μl of liver samples or standards were added to reactions.
RNA extraction, and quantitative RT- PCR.
Total RNA was isolated using TRIzol reagent according to the manufacturer’s protocol (Thermo Fisher Scientific). One μg of total RNA was used for cDNA synthesis using an iScript cDNA synthesis kit (Bio-Rad). For primers and PCR details, see Supplementary Materials and Table S1.
miRNA measurements.
Total RNA was purified using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol with the exception of an overnight RNA precipitation at −80°. cDNA synthesis was performed using 5 ng RNA and a miRCURY RT kit (QIAGEN cat. #339320) according to manufacturer’s protocol. miRCURY LNA miRNA PCR SYBER Green (QIAGEN cat. #339320) was used for qPCR amplification using Mm-miR-124-3P (YP00206026) and mm-miR-24-3P (YP00204260) primer mixes. miR-103-3p served as control.
Expression profiling (RNA-seq) and analysis.
Livers were rinsed in PBS and immediately frozen in liquid nitrogen. RNA was extracted using a miRNeasy Mini Kit (Qiagen) and submitted to Novogene for library preparation and sequencing. Data was deposited into NCBI GEO, GSE137051.
Cell culture.
AML12 cells were purchased from the American Tissue Culture Collection (ATCC) and used at low passage (<10). (See Supplementary Materials.) Due to recent purchase, no mycoplasma testing was performed.
Transient transfection and siRNA.
Transient transfection of plasmids was performed with Lipofectamine 2000 (Thermo Fisher Scientific). Briefly, 1 × 106 cells were transfected with 1 μg of plasmid using 5 μl of Lipofectamine 2000. HNF4α knockdown was performed using siRNA (Sense: CGCUUGAGGAAGACCUACUdTdT, Antisense: UCAUCCAGAAGGAGUUCGCdTdT). Approximately 1 × 106 cells were transfected with 25 pmol of siRNA and 5 μl of Lipofectamine RNAiMAX Transfection reagent. Scrambled siRNA with similar GC content served as control. High titer pilenti-siRNAIL6 lentiviral vector stock (Applied Biological Materials) was produced in HEK 293T cells and titrated as previously described (25). AML12 cells were transduced with lentiviral stock at ~50 multiplicity of infection in the presence of 8 mg ml−1 polybrene for 8 h at 37 °C. Two μg ml−1 puromycin was added to eliminate uninfected cells. pLenti scrambled oligonucleotides served as control.
MTT assay.
Cell viability was assessed by MTT proliferation assay using a CellTiter 96® Non-Radioactive Cell Proliferation Assay (MTT) kit (Promega) according to manufacturer’s instructions.
ELISA.
Serum IL-6 was measured using a mouse IL-6 uncoated ELISA kit (Invitrogen) per the manufacturer’s instructions. Optical density (OD) was measured at 450 nm and concentrations determined based on reference standards.
Transwell migration assays.
Cells (50,000/well) were seeded into the upper chamber of a Transwell insert in serum-free medium. Medium containing 20% FBS was placed in the lower chamber, and cells were incubated for 24-h in a CO2 incubator. Migrated cells were fixed with methanol for 10-min and stained with 0.1% crystal violet. Cells were imaged using a Cytation5 and four randomly chosen fields were used for quantification.
Histology.
Liver sections (5 μM) were fixed in formalin and embedded in paraffin. Sections were washed twice with xylene (3-min) and rehydrated in ethanol gradients (100%, 2 × 5 min; 90%, 1 × 5 min; 80%, 1 × 5 min; and 70%, 1 × 5 min). Sections were stained with hematoxylin and eosin (H&E) and imaged using a Cytation5.
Immunohistochemistry.
Sections were subjected to heat-induced antigen retrieval in high pH Target Retrieval Solution (Dako/Agilent), and incubated at room temperature for 15 min with 1:3000 dilution of anti-glutamine synthetase antibody or for 30 min with 1:4000 dilution of anti-alpha-fetoprotein antibody (For antibodies, Table S2). Bound GS antibody was detected using Agilent’s Animal Research Kit- Avidin Biotin HRP (ARK catalog #K3954), and AFP antibody was detected using Biocare’s Mach 4 detection kit (Biocare Medical). Sections were counterstained in hematoxylin, dehydrated, mounted and cover-slipped. (For scoring, see Table S3 and S4.)
Quantification and statistical analysis.
Results are expressed as the mean ± SEM. Experiments involving two variables were analyzed by two-way ANOVA using Sidak’s multiple comparisons test (Prism 8.0). Significance was defined as a P < 0.05. A sample size calculator (https://clincalc.com/stats/samplesize.aspx) assisted with number approximations. JTK_Cycle was applied for rhythmicity, using a window of 20–28 h to capture circadian oscillations (Table S5).
For additional methods, see Supplementary Material.
Results
P1-HNF4α ablation in the liver results in sex-independent changes in liver metabolism
To assess whether the loss of P1-HNF4α tumor suppressor activity affects hepatic metabolism similarly in male and female livers, AlbCreERT2 mice were crossed with floxed Hnf4a mice to generate an inducible model of HNF4α knockout in the liver (Hnf4aF/F;AlbERT2cre) as previously described (23). Cre Recombinase-positive and -negative mice (Cre+ and Cre−, H4LivKO and WT, respectively) were treated with tamoxifen for five days, to delete HNF4α in Cre+ hepatocytes (Fig. 1A). Male and female H4LivKO livers became pale in appearance several days after the first tamoxifen injection (Fig. 1B), and hematoxylin and eosin (H&E) staining revealed excess lipid accumulation in hepatocytes in both sexes (Fig. 1C). H4LivKO mice developed fasting/resting phase (zeitgeber time 4, ZT4) hypoglycemia following a 4-h fast, though feeding/active phase (ZT16) blood glucose was normal in both genders and under ad libitum feeding conditions (Fig. S1A). MRI data taken from animals six weeks after the onset of HF diet feeding revealed a trend towards higher fat and lower lean mass in H4LivKO mice, though it was not significant (Fig. S1B). We previously demonstrated that in male mice, P1-HNF4α provides circadian restraint at a number of genes involved in proliferation and epithelial-to-mesenchymal transition (25). To determine whether HNF4α target genes varied similarly during the circadian cycle in male and female mice after HNF4α loss, livers were collected from tamoxifen-treated WT and littermate H4livKO mice and analyzed for Hnf4a and HNF4α target gene expression. While the circadian clock gene Arntl1 (Bmal1) oscillated similarly in both sexes and genotypes, cyclin D1 RNA (Ccnd1) was induced and robustly oscillatory in both male and female H4LivKO mice throughout the 24-h cycle (Fig. 1D). Apolipoprotein C2, ApoC2, which HNF4α is known to activate (32), was similarly affected by the absence of HNF4α in both sexes as was the steroidogenic cytochrome P450 gene (Cyp17a1) (Fig. 1D, S1C). CCND1 was robustly expressed following the loss of HNF4α in both sexes, consistent with qPCR results (Fig. S1D).
Figure 1. Loss of hepatic Hnf4a with HF feeding results in hepatocellular carcinoma (HCC) in male and female mice.
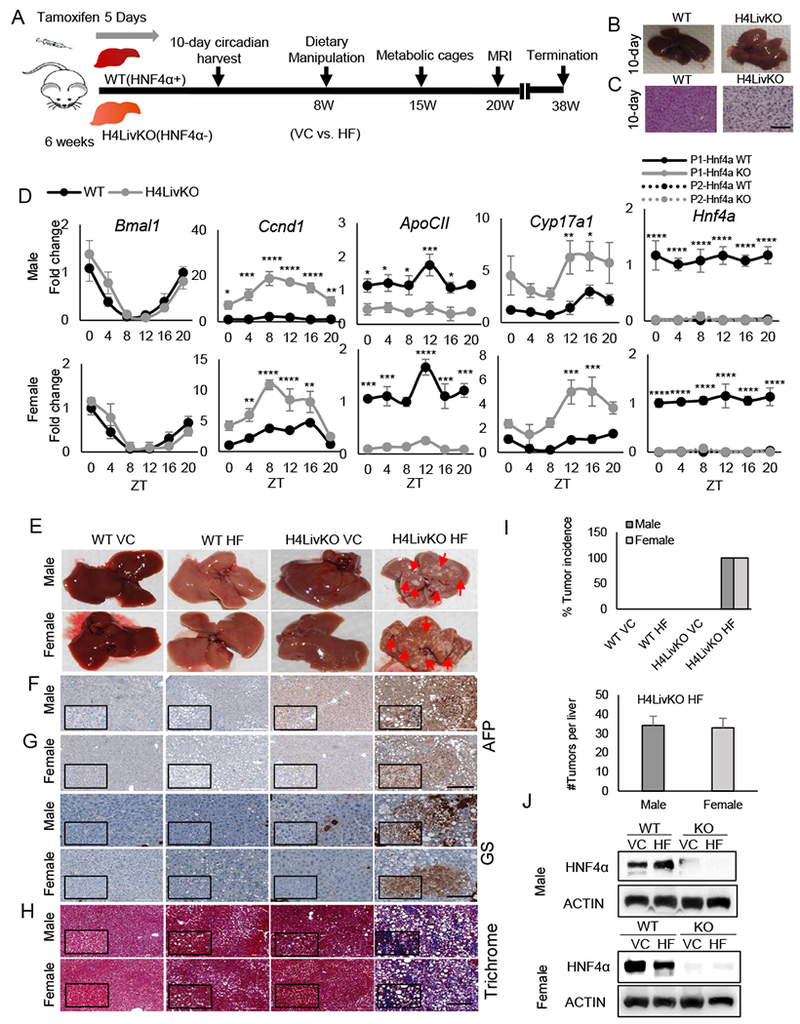
A. Experimental timeline. B. Livers of WT (Cre−) and H4LivKO (Cre+) mice, 10 days after initial tamoxifen injection. C. Hematoxylin and eosin (H&E) staining of WT and Hnf4a knockout (H4LivKO) liver (Scale bar=100 μm.) D. Hepatic gene expression in WT and H4LivKO mice 10 days after initial tamoxifen injection as measured by RT-PCR. E. Livers taken both sexes of WT and H4LivKO mice fed vivarium chow (VC) or high fat (HF) diet for 30 weeks. F-H. Staining of livers using antibodies to Alpha Fetoprotein (AFP), or glutamine synthetase (GS) (G) (Scale bar=200 μm.), and trichrome stain (H) (Scale bar=100 μm.) I. Percent tumor incidence and number of tumors per liver. (N = 8-16) J. Western blot of HNF4α in male and female H4LivKO livers under conditions of VC or HF feeding. Two-way ANOVA, Sidak’s multiple comparisons test, relative to WT, *P < 0.03, **P < 0.005, ***P < 0.0005, ****P < 0.0001. (N = 4-5) Error bars = SEM. (Table S5 for JTK_Cycle Rhythmicity Statistics.)
Liver-specific deletion of HNF4α promotes weight gain after prolonged high fat feeding
Prolonged HF feeding can induce spontaneous HCC in male mice (33). To determine whether HF diet and loss of hepatic P1-HNF4α could provide a model of accelerated liver disease and HCC, tamoxifen-treated male and female WT (Cre−) and H4LivKO (Cre+) mice were injected with tamoxifen at six weeks and either maintained on vivarium chow (VC) or placed on a HF diet under ad libitum feeding conditions two weeks following injections (Fig. 1A). Mice were weighed weekly throughout, starting with tamoxifen injections (Fig. S1E). While H4LivKO males and females on HF gained weight relative to controls, male H4LivKO on HF gained more weight compared to their female counterparts (Fig. S1E, right panel). Consistent with previous reports, WT females were protected from HF diet-induced obesity (34,35). Metabolic phenotyping seven weeks following the switch to HF diet revealed that total food intake was similar between genotypes during the light/rest and dark/active phases (Fig. S2A). As expected, low and non-oscillating respiratory exchange ratio (RER) was observed for all HF diet-fed animals (36); however, no significant difference was observed in H4LivKO mice compared to WT littermates (Fig. S2B). Energy expenditure rhythms were also comparable across genotypes with reduced amplitude in HF diet-fed mice (Fig. S2C).
Loss of HNF4α combined with HF feeding results in early onset and sex-independent HCC
Based on the similar hepatic response to loss of P1-HNF4α in male and female livers, we wondered whether, in combination with diet-induced obesity, the H4LivKO liver would provide a permissive environment for spontaneous HCC in both sexes. Tamoxifen-treated, Cre− (WT) and Cre+ (H4LivKO) mice on VC or HF feeding were euthanized at 38 weeks of age (Fig. 1A). In contrast to DEN exposure in mice, where males develop HCC much more frequently than females (13), we observed that male and female H4LivKO mice subjected to HF diet developed tumors with a 100% incidence (Fig. 1E). In contrast, no HCC was detected in WT mice of either sex on HF diet, though variable steatosis, fibrosis, lobular inflammation, hepatocyte ballooning, and macrophage infiltration was noted in some WT livers (Table S3). Livers of H4LivKO mice exposed to vivarium chow diet (VC) also showed increased lipid accumulation and were paler in appearance compared to controls (Fig. 1E ), though there was no evidence of HCC. Some H4LivKO mice on VC also showed variable lobular inflammation (Table S3). Though female WT and H4LivKO mice on HF feeding were more resistant to weight gain than males, H&E staining revealed lipid deposition in both sexes on HF diet (Fig. S2D), and hepatic triglyceride (TG) levels were also increased, which was further exacerbated in H4LivKO mice on HF diet (Fig. S2E). While some steatohepatitis and hepatic fibrosis were observed in WT mice subjected to HF diet, alpha fetoprotein (AFP) and glutamine synthetase (GS), both markers of HCC, were not detected (Fig. 1F–G, Table S3 and Table S4). Further analysis revealed robust GS and AFP expression only in HF-fed H4LivKO mice regardless of sex, concomitant with the presence of severe fibrosis as measured by Trichrome staining (Fig. 1F–H). Tumor nodules appeared as aggregates of hepatocytes with clear cytoplasm that were distinct clones from the surrounding liver parenchyma (Tables S3 and S4). Tumors were similar in size between males and females (Fig. 1I). P1-HNF4α expression remained low at the 38-week time point (Fig. 1J), though some was detectable (potentially due to proliferation of hepatocytes that escaped Cre-driven excision, or the differentiation of other cells into hepatocytes over 38 weeks). These data reveal that loss of P1-HNF4α combined with HF diet provides an equally permissive environment for HCC in both male and female mice.
HNF4α deletion predisposes to HCC-signature gene expression in male and female mice
To investigate the sex-independent and early onset HCC in H4LivKO mice on HF diet, we analyzed the tumor environment compared to controls. Consistent with other studies assessing proliferative capacity at single time points in male H4LivKO mice (23), expression of Ki67, a marker of proliferation, was elevated in the absence of HNF4α (Fig. 2A). Elevated expression of Interleukin 17 (IL17) and its receptor (IL17Ra) correlates with poor prognosis of HCC (37) and interleukin 23 (IL23) signaling promotes HCC progression, its levels correlating with elevated IL-17A and MMP9 (38,39). Analysis of hepatic Il17Ra and Il23 expression in H4LivKO mice revealed a significant induction in the absence of HNF4α, which was further increased in the context of HF feeding in both sexes (Fig. 2A). Transforming growth factor-alpha (TGFα), a strong mitogen in the context of HCC (40) (41) was also increased in the liver of H4LivKO mice exposed to HF diet regardless of sex (Fig. 2A). Importantly, HCC-associated gene expression was generally augmented by HF diet regardless of genotype.
Figure 2. Prolonged loss of hepatic Hnf4a with high fat feeding results in the induction of proliferation and inflammation genes in male and female mice.
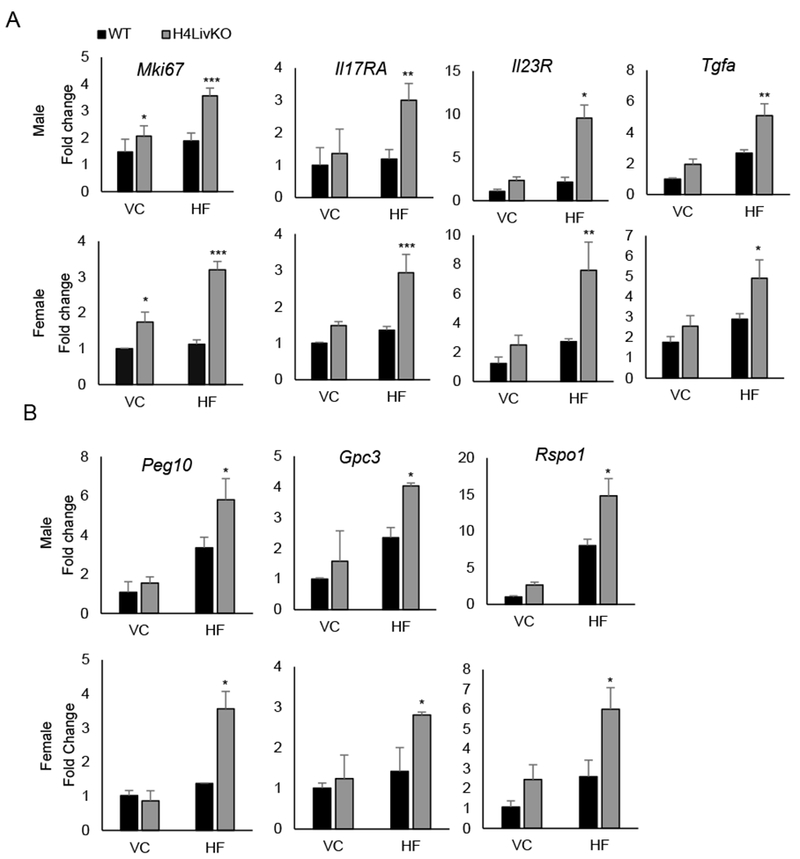
A. Quantitative PCR reveals the hepatic expression of proliferation marker Mki67 and HCC-associated inflammatory markers in WT and H4LivKO mice of both sexes after prolonged feeding on vivarium chow (VC) or high fat (HF) diet. B. Expression of HCC-specific genes in corresponding livers as measured by quantitative RT-PCR. Two-way ANOVA, Sidak’s multiple comparisons test, compared to VC control, *P < 0.03, **P < 0.005, ***P < 0.0005 (N = 4-6) Error bars = SEM.
HCC and hepatoblastoma (HPBL) have distinct patterns of gene expression, with insulin-like growth factor 2 (Igf2), fibronectin (Fn1), delta like non-canonical notch ligand 1 (Dlk1), transforming growth factor beta 1 (Tgfb1), and ERBB receptor feedback inhibitor 1 (Errfl1 or Mig6) being over-expressed in HPBL, while Glypican 3 (Gpc3), spondin-2 (Spon2), and paternally expressed 10 (Peg10) are overexpressed in HCC (42). To validate whether tumors in H4LivKO livers on HF diet are HCC, we examined the expression of HCC and HPBL markers. HCC-associated genes were elevated in H4LivKO livers on HF diet, while the HPBL-associated markers showed no change in control vs. H4LivKO livers (Fig. 2B and S3).
Loss of HNF4α with HF feeding alters the tumor environment similarly in male and female mice
To determine how gene expression changes in surrounding tumor-bearing liver tissue of H4LivKO mice fed a HF diet (H4LivKOHF), we performed RNA-seq on non-tumourous regions of WT and H4LivKO liver on both diets. RNA-seq revealed vastly different gene expression profiles in tumor-bearing livers (“H4LivKOHF”) compared to the other groups as revealed by heat maps of pooled data from each condition (Fig. 3A). Comparison of H4LivKOHF to the H4LivKO on vivarium chow (“H4LivKOVC”) revealed the largest changes, with almost equal distribution of the 1353 gene expression changes being up- vs. down-regulated (Fig. 3B and S4A). Annotation analysis revealed that genes involved in beta-catenin independent WNT signaling, degradation of AXIN, and auto degradation of E-cadherin were upregulated in the H4LivKOHF livers (Fig. 3C). In contrast, the tumor suppressor PTEN and several genes involved in TNF signaling and regulation of tumor suppressor p53 (Tp53) were repressed in H4LivKO liver in both sexes (Fig. 3C–D). Since WT mice on HF diet did not develop tumors, we also analyzed genes altered in the H4LivKOHF vs. WTHF males. Interestingly, genes involved in the Unfolded Protein Response (UPR), degradation of the extracellular matrix, and activation of matrix metalloproteinases were upregulated in H4LivKOHF livers compared to the WTHF (Fig. 3C), while a significant down regulation of AKT signaling was observed in H4LivKOHF livers compared to WTHF. To determine whether RNA-seq results were consistent in female livers not subjected to sequencing, we performed qPCR on select targets (Fig. 3D–E). Down-regulation of PTEN plays a significant role in tumorigenesis and progression of HCC (43–45). Consistent with the RNA-seq data, we observed loss of Pten expression in H4LivKOHF livers in both sexes (Fig. 3D). Deregulation of the Wnt signaling pathway is a primary event in hepatocarcinogenesis and linked to the epithelial to mesenchymal transition (EMT) (46–48). Axin2 and Apc, known inhibitors of WNT signaling were downregulated in H4LivKOHF livers of both sexes (Fig. 3D), not attributable solely to the loss of HNF4α, however, as livers harvested from young mice 10 days following HNF4α knockout revealed no change in the expression of Apc, Pten, or Axin2 (Fig. S4B).
Figure 3. Loss of HNF4α followed by high fat feeding results in a permissive hepatic environment for HCC in male and female mice.
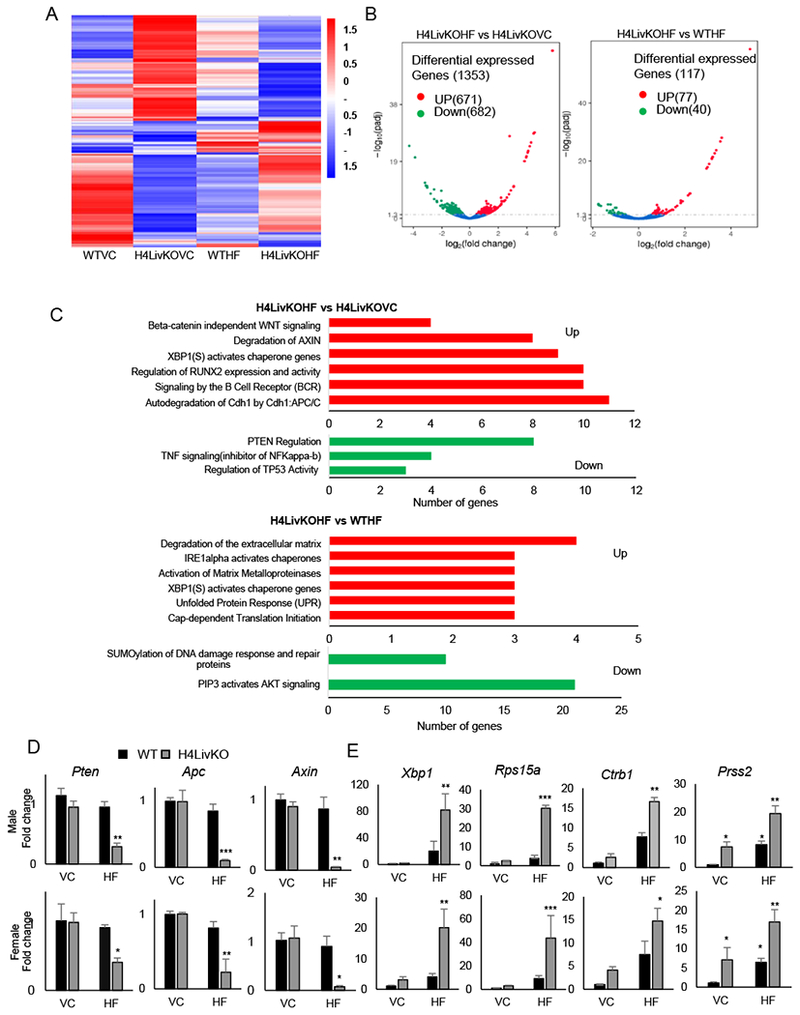
A. Heat maps generated from RNA-seq data reveal changes in gene expression between male WT vs. H4LivKO mice fed prolonged vivarium chow (VC) or high fat (HF) diet. B. Volcano plots reveal differentially expressed genes in H4LivKO fed HF diet (H4LivKOHF) or vivarium chow (H4LivKOVC), (left); or KOHF and WT mice fed HF diet for prolonged (30 weeks) (WTHF) (right) (N=3). C. Gene annotation reveals pathways altered in H4LivKOHF vs. H4LivKOVC (top) and H4LivKOHF vs. WTHF (bottom). D-E. RT-PCR reveals altered expression of genes involved in tumor suppression or inhibition of WNT/β catenin signaling (D) and genes involved in translation and protein folding (E) in WT and H4LivKO livers from animals fed VC or HF. Two-way ANOVA, Sidak’s multiple comparisons test (N=4-6) *P < 0.03, **P < 0.005, ***P < 0.0005, ****P < 0.0001.
Based on the expression changes in genes regulating cap-dependent translation, we analyzed expression of Ribosomal protein s15a (Rps15a), a gene which plays a role in cap dependent translation initiation and is induced in HCC (49,50). Rsp15a expression was robustly increased in both sexes in the H4LivKOHF livers (Fig. 3E), though no significant changes in expression occurred 10 days following HNF4α loss in young animals (Fig. S4C).
Catenin Beta 1 (CTNNB1) (51), a transcription factor downstream of WNT, is over-activated in up to 50% of HCC (52). While targeted sequencing of exon 3, codon 41 of the murine Ctnnb1 gene (a Glycogen Synthase Kinase-3 beta [GSK3β] target site commonly mutated in HCC) (53) revealed no obvious changes in tumor vs. control tissue, several CTNNB1 target genes, including Ccnd1, Myc, Matrix Metallopeptidase 14 (Mmp14), Lymphoid Enhancer Binding Factor 1 (Lef-1), Matrix Metallopeptidase 7 (Mmp7), and Cyclooxygenase 2b (Cox2) were increased in the context of H4LivKO under HF feeding (Fig. S5), suggesting elevated CTNNB1 activity. Thus, the loss of HNF4α with prolonged HF feeding was necessary for the HCC-permissive environment at 38 weeks of age.
Matrix metalloproteinases (MMPs), regulators of the tumor microenvironment (54), showed altered expression by RNA-seq. Thus we tested the expression of chymotrypsinogen B1 (Ctrb1) and serine protease 2 (Prss2), which play roles in the activation of MMPs and ECM degradation. While both genes were elevated in the H4LivKOHF tumor-bearing livers, Prss2 but not Ctrb1 appears to be controlled in a circadian manner by HNF4α, as its loss produced a pronounced de novo oscillation in the liver 10 days after hepatic HNF4α loss (Fig. S4C). X-Box Binding Protein 1 (XBP1) functions as an essential transcription factor regulating the UPR, which is often induced in cancer. Our data revealed significant induction of Xbp1 by RNA-seq and qPCR only in the H4LIVKOHF livers, though transient ablation of HNF4α in young animals did not affect its circadian expression in either sex (Fig. 3E and S4C).
IL-6 induction in HNF4α-deficient liver is not sex-dependent
IRE1α-XBP1 signaling induces HCC progression via IL-6 induction or activation of ATF6 (55,56). Elevated IL-6 is involved in the pathogenesis of HCC and ablation of IL-6 can prevent hepatocarcinogenesis (13). Because IL-6 has been implicated in the sex specificity of HCC (13), we sought to determine whether, in our model, IL-6 was affected independent of sex, similar to proliferation genes. When livers of young WT and H4LivKO mice were analyzed for markers of proliferation and inflammation, we noted that Ki67 expression oscillated over the 24-h cycle in the absence of HNF4α as early as 10 days post tamoxifen injection in male mice, whereas H4LivKO female mice maintained elevated levels consistently over 24-h (Fig. S6A). However, HNF4α loss produced a significant circadian induction of Il6 in both male and female livers (Fig. 4A). Following prolonged feeding on VC or HF diet, IL6 remained greatly induced in H4LivKO liver, particularly under HF feeding conditions (Fig. 4B). Though P1-HNF4α expression remained low in aged animals (Fig. 1J and S6B), comparison of P1-HNF4α and Il6 expression in healthy livers of both sexes, revealed that levels of P1-HNF4α were elevated in females compared to males, which held true regardless of zeitgeber time or diet (Fig. S6C–D). In young adult mice, hepatic P1-Hnf4a expression was inverse to the levels of Il6, and reduced and non-oscillatory Il6 mRNA levels were observed in female livers (Fig. S6C), which may partially explain the delay in liver disease and HCC in DEN-treated females compared to males usually reported. To determine whether tumor-bearing H4LivKO mice showed higher levels of circulating IL-6, we performed ELISAs to measure circulating IL-6 in young and old WT and H4LivKO serum. While young H4LivKO mice of both sexes showed increases in circulating IL-6 (particularly at certain ZTs) (Fig. S6E), 38-week H4LivKO mice showed a pronounced induction of circulating IL-6, which was exacerbated by nutrient insult in the form of HF diet (Fig. 4C). Finally, to assess whether Il6 and Hnf4a expression are correlated in human HCC, we accessed R2 genomics data on human HCC https://hgserver1.amc.nl/cgi-bin/r2/main.cgi. These data revealed that in the context of human HCC, Il6 and Hnf4a are negatively correlated (r=−.231, P=6.7e-06) (Fig. 4D), even though Il6 levels were not correlated with sex in the context of HCC.
Figure 4. Inducible loss of hepatic HNF4α results in a sex-independent circadian induction of Il6.
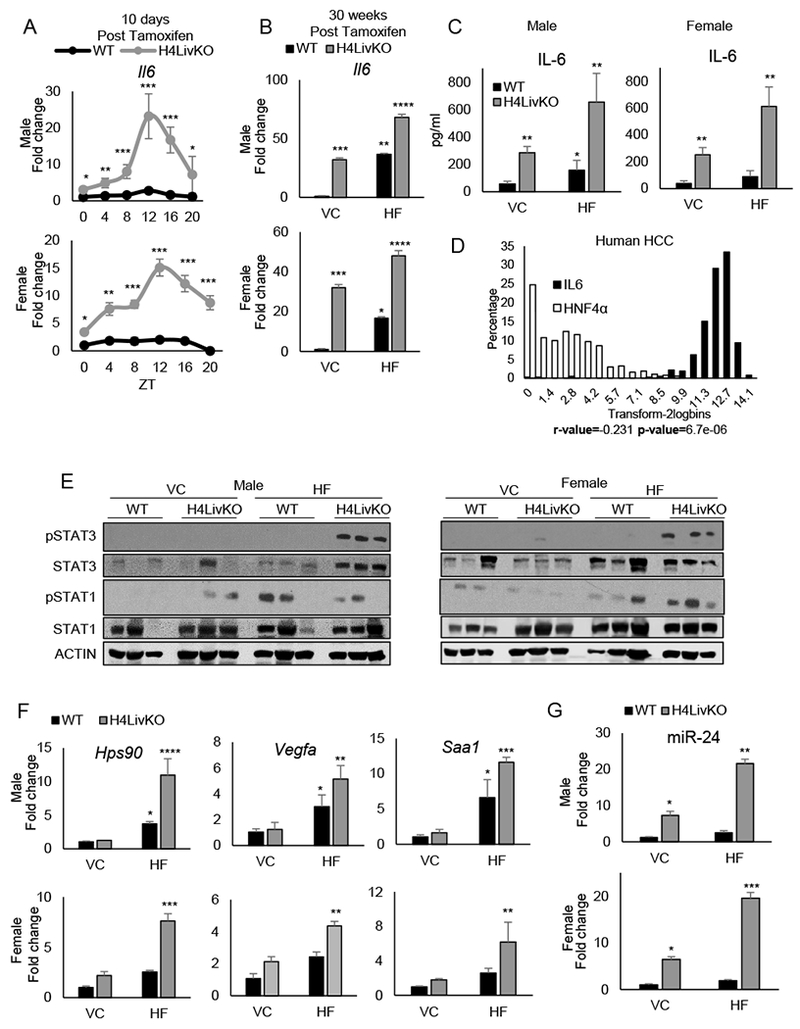
A. RT-PCR showing 24-h hepatic Il6 expression in young WT and H4LivKO mice following tamoxifen treatment. B-C. RT-PCR of Il6 in livers of WT or H4LivKO mice fed vivarium chow (VC) or high fat diet (HF) for 30 weeks (B) and corresponding serum IL-6 (C). D. Human TCGA data showing inverse correlation between IL6 and HNF4a in HCC (P=6.7e-06; r=−0.231). E. Western blot of liver lysates using antibodies to phosphorylated STAT1 and STAT3, total STAT proteins, and ACTIN. F. RT-PCR shows expression of STAT3 target genes. G. Fold change in miR-24 expression in male and female WT and H4LivKO livers from animals fed VC or HF diet. Two-way ANOVA, Sidak’s multiple comparisons test, *P < 0.03, **P < 0.005, ***P < 0.0005, ****P < 0.0001 (N=4-6) (Table S5 for JTK_Cycle Rhythmicity Statistics.)
Recent studies performed in mice have revealed that the transcription factors STAT1 and STAT3 have distinct function in the context of NAFLD vs. HCC (57). Specifically, obesity promotes HCC in a STAT3-dependent manner, while STAT1 is necessary for NASH and fibrosis. Because Il6 signaling induces STAT3 phosphorylation, we assessed phosphorylated STAT1 and STAT3 protein in both sexes and genotypes on both diets. Consistent with STAT3 phosphorylation being responsible for HCC, a pronounced induction of STAT3 phosphorylation was observed only in HCC-containing livers of H4LivKO mice fed HF diet, regardless of sex (Fig. 4E). However, STAT1 phosphorylation was considerably more variable, with a general increase in most animals under HF feeding regardless of genotype (Fig. 4E). To verify that increases in STAT3 phosphorylation resulted in increased STAT3 target gene expression, we analyzed select targets of STAT3 that were upregulated in our RNA-seq data set, including the heat shock protein 90 (Hsp90), vascular endothelial growth factor A (Vegfa), and serum amyloid A1 (Saa1). Hsp90, Vegfa, and Saa1 were all elevated in tumor bearing livers, regardless of sex (Fig. 4F). Because STAT3 has been shown to regulate miR-24, a microRNA previously implicated in transformation and in promoting an inflammatory feedback circuit with miR-124 (which has an opposite role to miR-24 in its ability to directly target IL-6) in HCC (31), we wondered whether a similar mechanism was present in male and female H4LivKO in which tumors were present. Analysis of hepatic miRNA-24 revealed an induction in H4LivKO mice on both VC and HF diet, though levels were the highest in H4LivKO mice on HF diet, consistent with their tumor-bearing state (Fig. 4G). Alternatively, the tumor suppressive miR-124 was significantly lower in H4LivKO mice on HF compared to all other groups, consistent with the high levels of hepatic and circulating IL-6 found in this group (Fig. S6F).
Induction of EMT in HNF4α-deficient liver is IL-6 dependent
Our previous studies revealed a circadian repression of genes involved in EMT by P1-HNF4α (25). Thus, we examined the expression of Ctnnb1, E-Cadherin (Cdh1) and Snail Family Transcriptional Repressor 1 (Snai1) in H4LivKO mice. Similar to the effects of acute loss of P1-HNF4α on EMT genes, we observed a significant increase in the expression of Ctnnb1 and Snai1 (both of which increase the EMT phenotype), while Cdh1 was reduced in H4LivKO mice fed HF (Fig. 5A). Since IL-6 has previously been associated with EMT (58), we tested whether P1-HNF4α-induced changes in Il6 expression were responsible for changes in EMT in hepatocytes. To this end, we transiently transfected non-transformed hepatocyte AML12 cells with empty vector (EV) or expression vector for Il6 (IL6) with siRNA to HNF4α (HNF4αsiRNA) or scrambled oligonucleotides (SC) (Fig. 5B, left). In addition, we knocked down IL-6 using siRNA with or without siRNA to Hnf4a (Fig. 5B, right). A gain in Il6 expression increased cell proliferation, as measured by MTT assay at 24- and 48-h following plating, with knockdown of Hnf4a and overexpression of Il6 together having the most pronounced effect (Fig. 5C, left). In contrast, while knockdown of Hnf4a alone increased cell proliferation at 24- and 48-hr, the concomitant knockdown of Il6 resulted in a blunting of proliferation at both time points tested (Fig. 5C, right). Similarly, when testing the invasive potential of cells overexpressing Il6, increased invasion was observed after Il6 overexpression, and augmented by the absence of Hnf4a (Fig. 5D, left panels). Conversely, inhibition of both Il6 and Hnf4a partially rescued the invasiveness of cells compared to the knockdown of Hnf4a alone (Fig. 5D, right panels).
Figure 5. Loss of HNF4α combined with high fat diet induces epithelial-mesenchymal transition in an IL-6-dependent manner.
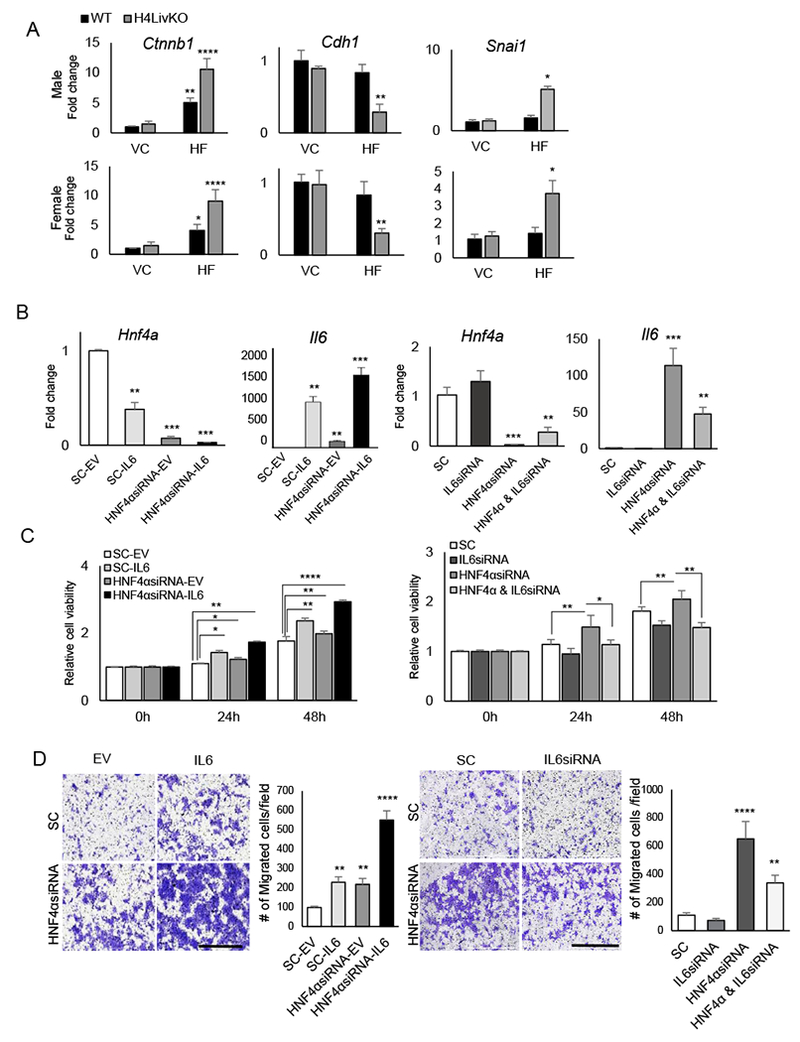
A. RT-PCR reveals mRNA abundance of genes affecting EMT in livers of 38 week-old WT or H4LivKO mice fed vivarium chow (VC) or high fat diet (HF) (N=4-6). B. RT-PCR reveals levels of Hnf4a and Il6 after transfection of AML12 cells with empty vector (SC-EV), scrambled oligonucleotides (SC-IL6), siRNA to Hnf4a (HNF4αsiRNA-EV) or siRNA to Hnf4a with overexpression of Il6 (HNF4αsiRNA-IL6). C. MTT assay reveals proliferating AML12 cells after transfection with empty vector (EV) or Il6 in combination with scrambled or siRNA for Hnf4a (left panel) or with scrambled or siRNA for Hnf4a in combination with scrambled or siRNA for Il6 (right panel) at 24- and 48- h following transfection. D. Migration assay reveals migration of AML12 cells expressing scrambled or siRNA for Hnf4a in combination with empty vector or Il6 transfection (left panel) or with scrambled or siRNA for Hnf4a in combination with scrambled or siRNA for Il6 (right panel), 24-h after plating (N=6). Quantification (right). . Two-way ANOVA, Sidak’s multiple comparisons test, *P < 0.03, **P < 0.005, ***P < 0.0005, ****P < 0.0001.
Previous studies reveal that HF diet reduces hepatic nuclear P1-HNF4α in diet-induced obesity models as well as in a genetic model of obesity (db/db mice) (25,26). Thus, we wanted to test whether another model of HCC utilizes P1-HNF4α loss to induce HCC irrespective of sex. We used the STAM model (59), which follows the general progression of human disease, from NASH to HCC in a minimal amount of time (Fig. 6A). Mice treated with streptozotocin (STZ) or vehicle at 2 days of age were placed on HF diet four weeks later, resulting in severe hyperglycemia (Fig. S7). By 17 weeks of age, STZ-treated mice showed greatly deteriorated liver health, with severe fibrosis. Indiscriminately, both male and female mice showed HCC, ranging from small lesions to large tumor nodules (Fig. 6B). Based on the sex-independent manifestation of HCC, we analyzed expression of the P1 isoform of HNF4α. In both male and female mice treated with STZ, P1-HNF4α was severely depressed, while the P2 isoform showed an opposite pattern of expression (Fig. 6C). Consistent with the de-repression of Il6 in the context of HNF4α loss, Il6 levels were greatly induced in both male and female mice treated with STZ (Fig. 6C), while Bmal1 levels decreased, in congruence with our previous report of transcriptional repression at the Bmal1 locus by the P2 isoform of HNF4α in the context of HCC (25). While the EMT-promoting genes Ctnnb1 and Snai1 were elevated in STZ-treated mice, the epithelial gene Cdh1 was depressed in livers of tumor-bearing mice of both sexes (Fig. 6D). To determine whether elevated Il6 translated to increases in STAT3 phosphorylation similarly in male and female mouse livers of STZ-treated mice, we performed western blot analysis of liver homogenate. While total STAT1 and STAT3 levels varied minimally in STZ vs. vehicle-treated livers, STAT3 phosphorylation increased in both male and female STZ-treated livers, as did the expression of STAT3 target genes, consistent with their tumor bearing state (Fig. 6E–F).
Figure 6. The accelerated model of fatty liver-induced HCC (STAM model) results in HCC in male and female mice.
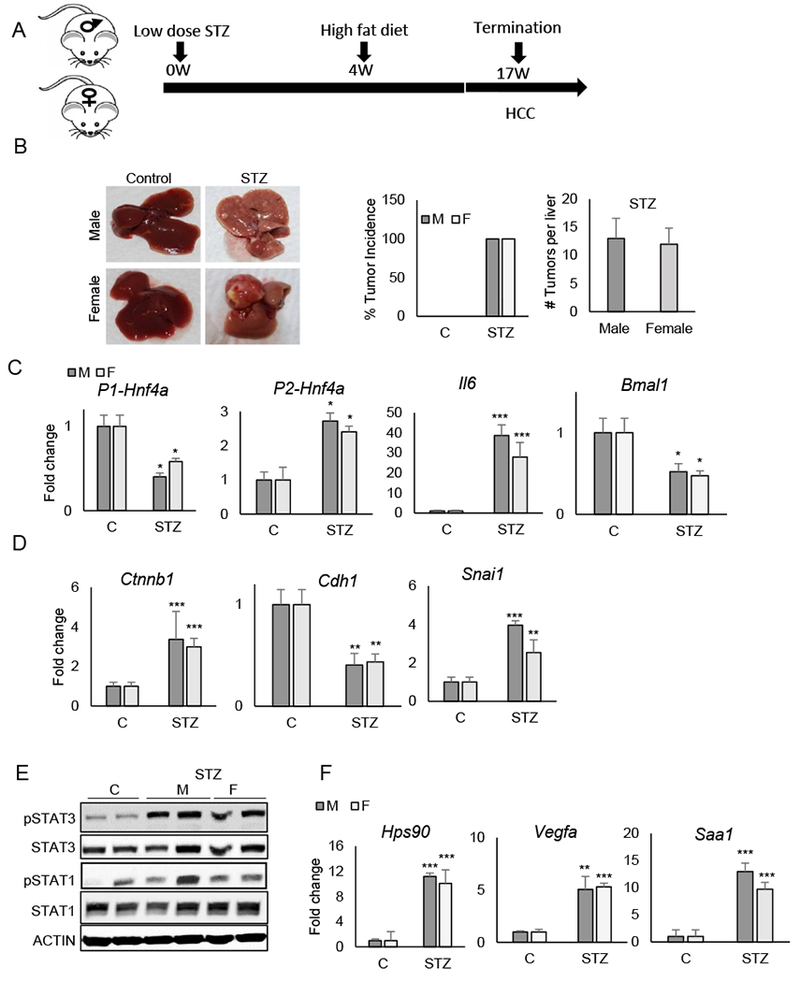
A. Timeline of experimentation. B. Livers of control- and STZ-treated STAM mice at 17 weeks of age (left). Percent tumor incidence and number of tumors per liver in control and STAM mice (right). C-D. RT-PCR reveals mRNA abundance of P1-Hnf4a, P2-Hnf4a, Il6 and Bmal1 (C) and genes affecting epithelial-mesenchymal transition in control vs. STAM mice (D). E. Western blot of liver lysates from mice treated with STZ or Vehicle (C) using antibodies to phosphorylated STAT1 and STAT3, total STAT proteins, and ACTIN. F. RT-PCR reveals mRNA levels of STAT3 target genes (N=4-5). Two-way ANOVA, Sidak’s multiple comparisons test, *P < 0.03, **P < 0.005, ***P < 0.0005, ****P < 0.0001.
Discussion
The prevalence of HCC is increasing worldwide, despite new anti-viral hepatitis treatments. This increase has been attributed to increasing NAFLD caused by obesity and type 2 diabetes (60). According to the U.S. Center for Disease Control and Prevention, liver and intrahepatic bile duct cancers are among the top five cancers in terms of death rates in males; there is also an increase in liver cancer deaths in females. Chemical carcinogens commonly used to mimic HCC in rodent models induce HCC either selectively or much more aggressively in males. As a result, studies of HCC in females have been fewer, although progress is being made to model the disease in both sexes (61). Our data provide mechanistic insight into the association of lipid metabolism dysregulation and HCC risk for both sexes, an association that will likely be important in the decades to come, considering the prevalence of NAFLD and its association with HCC development.
Our study demonstrates that loss of the tumor suppressive isoform of HNF4α in combination with HF diet provides an equally permissive environment for HCC in male and female mice, inducing 100% penetrance at only 38 weeks of age for all mice. Furthermore, in male and female mice made diabetic through STZ followed by HF feeding, a similar sex-independent HCC is observed, coincident with a reduction in P1-Hnf4a expression and a sex-independent induction of Il6 (Fig. 7). These data highlight the increased risk of HCC associated with diabetes, in particular, which is true for both sexes (62). We have previously published that there is a partial shift of P1-HNF4α from the nucleus to the cytoplasm in hepatocytes following prolonged HF feeding and under conditions of whole body insulin resistance (25). The loss of nuclear P1-HNF4α is not due merely to HF feeding, however, as a genetic model of obesity and diabetes (the db/db mouse) also shows a loss of P1-HNF4α in hepatocytes (25,26). Furthermore, the STAM model is severely hyperglycemic due to the destruction of pancreatic beta cells soon after birth by STZ, a compound routinely used to mimic human diabetes mellitus in rodents. We observe a similar loss of P1-Hnf4a expression in the liver of male and female STAM mice, which results in a similar sex-independent induction of Il6 and phosphorylation of STAT3.
Figure 7.
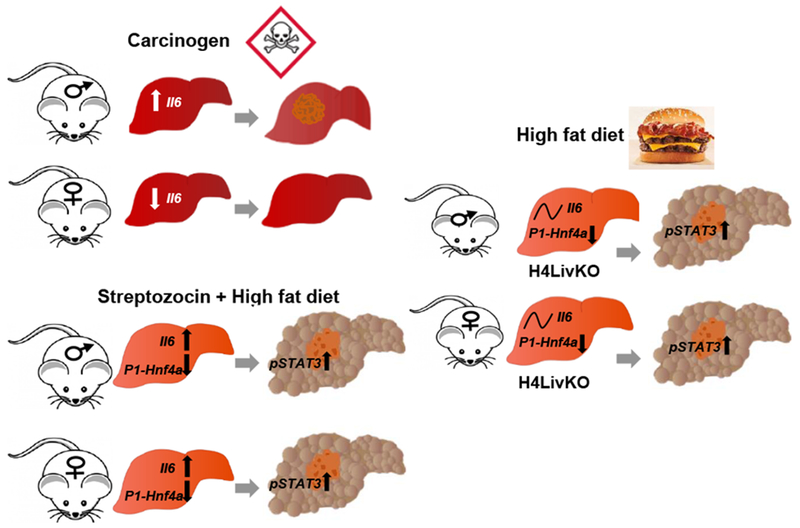
Model of sex-independent HCC under conditions of P1-Hnf4a loss and high fat feeding. Males are more susceptible to spontaneous and chemical carcinogen-induced HCC in part due to sex-specific induction of Il6 in existing models of the disease. Under conditions of P1-HNF4α loss, which occurs in many spontaneous liver cancers, a sex-independent circadian induction of Il6 occurs. The STAM and liver-inducible Hnf4a knockout models both provide evidence that loss of nuclear P1-Hnf4a induces proliferation and inflammation leading to similar HCC incidence in both sexes. Since reduced P1-HNF4α activity also occurs in diet-induced obesity and models of insulin resistance, P1-Hnf4a may be a central player linking NAFLD and HCC risk.
Interestingly, the first mutation in the Hnf4a gene identified as causing MODY1, was identified as having altered subcellular localization in the liver, losing its ability to extract with the soluble nuclear portion of liver cells (63). Thus, while sex-specific factors are known to provide protection for female livers from HCC (26), reduced nuclear P1-HNF4α may bypass the normal protective mechanisms in females, inducing IL-6 and STAT3 phosphorylation and activity similarly in both sexes. Considering the growing incidence of NAFLD, this mechanism may prove to be a pivotal junction for prevention and treatment. Understanding the mechanisms behind NAFLD-induced HCC will be essential to reducing HCC-associated mortality in both males and females, whether by finding better ways for early detection, better risk stratification of patients with NAFLD, improving prevention strategies, or finding new treatment options.
Supplementary Material
Statement of Significance:
This study provides a mechanism for the growing incidence of hepatocellular carcinoma in both men and women that is linked to non-alcoholic fatty liver disease.
Acknowledgments
We thank Dr. Chris Janssen and Cneshia Traylor for assistance with mouse experiments, Zhengmei Mao for help with tissue processing, and Dr. Frank Gonzalez, Dr. Pierre Chambon, and Dr. Daniel Metzger for providing Hnf4aF/F and Albtm1(cre/ERT2)Mtz mice.
This study was supported by the following grants: National Institutes of Health (NIH) DK114037 (K.E.M.), American Cancer Society (ACS) grant RSG-17-215-01-C (K.E.M.), and USDA National Institute of Food and Agriculture CA-R-NEU-5680 (F.M.S.). Additional funds provided by the UT Health Science Center at Houston.
Footnotes
The authors declare no conflict of interest.
References
- 1.Massarweh NN, El-Serag HB. Epidemiology of Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Cancer Control 2017;24:1073274817729245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seyda Seydel G, Kucukoglu O, Altinbasv A, Demir OO, Yilmaz S, Akkiz H, et al. Economic growth leads to increase of obesity and associated hepatocellular carcinoma in developing countries. Annals of hepatology 2016;15:662–72 [DOI] [PubMed] [Google Scholar]
- 3.Wong RJ, Cheung R, Ahmed A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology (Baltimore, Md) 2014;59:2188–95 [DOI] [PubMed] [Google Scholar]
- 4.Cholankeril G, Patel R, Khurana S, Satapathy SK. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Current knowledge and implications for management. World journal of hepatology 2017;9:533–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kanwal F, Kramer JR, Mapakshi S, Natarajan Y, Chayanupatkul M, Richardson PA, et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018;155:1828–37 e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Wang B, Shen F, Fan J, Cao H. Body mass index and risk of primary liver cancer: a meta-analysis of prospective studies. Oncologist 2012;17:1461–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steele CB, Thomas CC, Henley SJ, Massetti GM, Galuska DA, Agurs-Collins T, et al. Vital Signs: Trends in Incidence of Cancers Associated with Overweight and Obesity - United States, 2005-2014. MMWR Morbidity and mortality weekly report 2017;66:1052–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El-Serag HB, Mason AC. Rising incidence of hepatocellular carcinoma in the United States. The New England journal of medicine 1999;340:745–50 [DOI] [PubMed] [Google Scholar]
- 9.Singh S, Singh PP, Roberts LR, Sanchez W. Chemopreventive strategies in hepatocellular carcinoma. Nature reviews Gastroenterology & hepatology 2014;11:45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology 2004;127:S5–S16 [DOI] [PubMed] [Google Scholar]
- 11.Prieto J, Inflammation HCC and sex: IL-6 in the centre of the triangle. Journal of hepatology 2008;48:380–1 [DOI] [PubMed] [Google Scholar]
- 12.Petrick JL, Kelly SP, Altekruse SF, McGlynn KA, Rosenberg PS. Future of Hepatocellular Carcinoma Incidence in the United States Forecast Through 2030. J Clin Oncol 2016;34:1787–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naugler WE, Sakurai T, Kim S, Maeda S, Kim K, Elsharkawy AM, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 2007;317:121–4 [DOI] [PubMed] [Google Scholar]
- 14.Brady CW. Liver disease in menopause. World J Gastroenterol 2015;21:7613–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sladek FM, Zhong WM, Lai E, Darnell JE, Jr. Liver-enriched transcription factor HNF-4 is a novel member of the steroid hormone receptor superfamily. Genes Dev 1990;4:2353–65 [DOI] [PubMed] [Google Scholar]
- 16.Yusuf D, Butland SL, Swanson MI, Bolotin E, Ticoll A, Cheung WA, et al. The transcription factor encyclopedia. Genome Biol 2012;13:R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonald TJ, Ellard S. Maturity onset diabetes of the young: identification and diagnosis. Ann Clin Biochem 2013;50:403–15 [DOI] [PubMed] [Google Scholar]
- 18.Kaestner KH. Making the liver what it is: the many targets of the transcriptional regulator HNF4alpha. Hepatology (Baltimore, Md) 2010;51:376–7 [DOI] [PubMed] [Google Scholar]
- 19.Bolotin E, Liao H, Ta TC, Yang C, Hwang-Verslues W, Evans JR, et al. Integrated approach for the identification of human hepatocyte nuclear factor 4alpha target genes using protein binding microarrays. Hepatology (Baltimore, Md) 2010;51:642–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Hepatocyte nuclear factor 4alpha (nuclear receptor 2A1) is essential for maintenance of hepatic gene expression and lipid homeostasis. Mol Cell Biol 2001;21:1393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeLaForest A, Nagaoka M, Si-Tayeb K, Noto FK, Konopka G, Battle MA, et al. HNF4A is essential for specification of hepatic progenitors from human pluripotent stem cells. Development 2011;138:4143–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walesky C, Gunewardena S, Terwilliger EF, Edwards G, Borude P, Apte U. Hepatocyte-specific deletion of hepatocyte nuclear factor-4alpha in adult mice results in increased hepatocyte proliferation. Am J Physiol Gastrointest Liver Physiol 2013;304:G26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonzo JA, Ferry CH, Matsubara T, Kim JH, Gonzalez FJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. The Journal of biological chemistry 2012;287:7345–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saandi T, Baraille F, Derbal-Wolfrom L, Cattin AL, Benahmed F, Martin E, et al. Regulation of the tumor suppressor homeogene Cdx2 by HNF4alpha in intestinal cancer. Oncogene 2013;32:3782–8 [DOI] [PubMed] [Google Scholar]
- 25.Fekry B, Ribas-Latre A, Baumgartner C, Deans JR, Kwok C, Patel P, et al. Incompatibility of the circadian protein BMAL1 and HNF4alpha in hepatocellular carcinoma. Nat Commun 2018;9:4349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie X, Liao H, Dang H, Pang W, Guan Y, Wang X, et al. Down-regulation of hepatic HNF4alpha gene expression during hyperinsulinemia via SREBPs. Mol Endocrinol 2009;23:434–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cai SH, Lu SX, Liu LL, Zhang CZ, Yun JP. Increased expression of hepatocyte nuclear factor 4 alpha transcribed by promoter 2 indicates a poor prognosis in hepatocellular carcinoma. Therap Adv Gastroenterol 2017;10:761–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chellappa K, Jankova L, Schnabl JM, Pan S, Brelivet Y, Fung CL, et al. Src tyrosine kinase phosphorylation of nuclear receptor HNF4alpha correlates with isoform-specific loss of HNF4alpha in human colon cancer. Proc Natl Acad Sci U S A 2012;109:2302–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chellappa K, Deol P, Evans JR, Vuong LM, Chen G, Briancon N, et al. Opposing roles of nuclear receptor HNF4alpha isoforms in colitis and colitis-associated colon cancer. Elife 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walesky C, Edwards G, Borude P, Gunewardena S, O’Neil M, Yoo B, et al. Hepatocyte nuclear factor 4 alpha deletion promotes diethylnitrosamine-induced hepatocellular carcinoma in rodents. Hepatology 2013;57:2480–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, et al. An HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011;147:1233–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garrison WD, Battle MA, Yang C, Kaestner KH, Sladek FM, Duncan SA. Hepatocyte nuclear factor 4alpha is essential for embryonic development of the mouse colon. Gastroenterology 2006;130:1207–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J Hepatol 2016;65:579–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C, He Y, Xu P, Yang Y, Saito K, Xia Y, et al. TAp63 contributes to sexual dimorphism in POMC neuron functions and energy homeostasis. Nat Commun 2018;9:1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi H, Seeley RJ, Clegg DJ. Sexual differences in the control of energy homeostasis. Front Neuroendocrinol 2009;30:396–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell metabolism 2012;15:848–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liao R, Sun J, Wu H, Yi Y, Wang J-X, He H-W, et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. Journal of experimental & clinical cancer research : CR 2013;32:3- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J, Lau G, Chen L, Yuan Y-F, Huang J, Luk JM, et al. Interleukin 23 promotes hepatocellular carcinoma metastasis via NF-kappa B induced matrix metalloproteinase 9 expression. PloS one 2012;7:e46264–e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ngiow SF, Teng MW, Smyth MJ. A balance of interleukin-12 and −23 in cancer. Trends in immunology 2013;34:548–55 [DOI] [PubMed] [Google Scholar]
- 40.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene 2006;25:3787–800 [DOI] [PubMed] [Google Scholar]
- 41.Harada K, Shiota G, Kawasaki H. Transforming growth factor-alpha and epidermal growth factor receptor in chronic liver disease and hepatocellular carcinoma. Liver 1999;19:318–25 [DOI] [PubMed] [Google Scholar]
- 42.Luo J-H, Ren B, Keryanov S, Tseng GC, Rao UNM, Monga SP, et al. Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas. Hepatology (Baltimore, Md) 2006;44:1012–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. The Journal of clinical investigation 2004;113:1774–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He C, Dong X, Zhai B, Jiang X, Dong D, Li B, et al. MiR-21 mediates sorafenib resistance of hepatocellular carcinoma cells by inhibiting autophagy via the PTEN/Akt pathway. Oncotarget 2015;6:28867–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Z, Yi J, Li X, Long W. Correlation between loss of PTEN expression and PKB/AKT phosphorylation in hepatocellular carcinoma. Journal of Huazhong University of Science and Technology Medical sciences = Hua zhong ke ji da xue xue bao Yi xue Ying De wen ban = Huazhong keji daxue xuebao Yixue Yingdewen ban 2005;25:45–7 [DOI] [PubMed] [Google Scholar]
- 46.Vilchez V, Turcios L, Marti F, Gedaly R. Targeting Wnt/β-catenin pathway in hepatocellular carcinoma treatment. World journal of gastroenterology 2016;22:823–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cavard C, Colnot S, Audard V, Benhamouche S, Finzi L, Torre C, et al. Wnt/beta-catenin pathway in hepatocellular carcinoma pathogenesis and liver physiology. Future oncology (London, England) 2008;4:647–60 [DOI] [PubMed] [Google Scholar]
- 48.Takigawa Y, Brown AMC. Wnt signaling in liver cancer. Current drug targets 2008;9:1013–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guo P, Wang Y, Dai C, Tao C, Wu F, Xie X, et al. Ribosomal protein S15a promotes tumor angiogenesis via enhancing Wnt/beta-catenin-induced FGF18 expression in hepatocellular carcinoma. Oncogene 2018;37:1220–36 [DOI] [PubMed] [Google Scholar]
- 50.Xu M, Wang Y, Chen L, Pan B, Chen F, Fang Y, et al. Down-regulation of ribosomal protein S15A mRNA with a short hairpin RNA inhibits human hepatic cancer cell growth in vitro. Gene 2014;536:84–9 [DOI] [PubMed] [Google Scholar]
- 51.Herbst A, Jurinovic V, Krebs S, Thieme SE, Blum H, Goke B, et al. Comprehensive analysis of beta-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/beta-catenin signaling. BMC Genomics 2014;15:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khalaf AM, Fuentes D, Morshid AI, Burke MR, Kaseb AO, Hassan M, et al. Role of Wnt/beta-catenin signaling in hepatocellular carcinoma, pathogenesis, and clinical significance. J Hepatocell Carcinoma 2018;5:61–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ogawa K, Yamada Y, Kishibe K, Ishizaki K, Tokusashi Y. Beta-catenin mutations are frequent in hepatocellular carcinomas but absent in adenomas induced by diethylnitrosamine in B6C3F1 mice. Cancer Res 1999;59:1830–3 [PubMed] [Google Scholar]
- 54.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010;141:52–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, Mori K, et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. Journal of hepatology 2003;38:605–14 [DOI] [PubMed] [Google Scholar]
- 56.Fang P, Xiang L, Huang S, Jin L, Zhou G, Zhuge L, et al. IRE1alpha-XBP1 signaling pathway regulates IL-6 expression and promotes progression of hepatocellular carcinoma. Oncology letters 2018;16:4729–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018;175:1289–306 e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bharti R, Dey G, Mandal M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: A snapshot of IL-6 mediated involvement. Cancer Lett 2016;375:51–61 [DOI] [PubMed] [Google Scholar]
- 59.Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol 2013;46:141–52 [DOI] [PubMed] [Google Scholar]
- 60.Kulik L, El-Serag HB. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019;156:477–91 e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Romualdo GR, Prata GB, da Silva TC, Fernandes AAH, Moreno FS, Cogliati B, et al. Fibrosis-associated hepatocarcinogenesis revisited: Establishing standard medium-term chemically-induced male and female models. PLoS One 2018;13:e0203879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mantovani A, Targher G. Type 2 diabetes mellitus and risk of hepatocellular carcinoma: spotlight on nonalcoholic fatty liver disease. Ann Transl Med 2017;5:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sladek FM, Dallas-Yang Q, Nepomuceno L. MODY1 mutation Q268X in hepatocyte nuclear factor 4alpha allows for dimerization in solution but causes abnormal subcellular localization. Diabetes 1998;47:985–90 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.