

Abstract
目的
探讨Necroptosis与凋亡在激素诱导成骨细胞死亡中的相互作用。
方法
向MC3T3-E1细胞内添加浓度为10-6 mol/L的地塞米松诱导细胞死亡,随后在本组细胞中分别添加凋亡抑制剂z-VAD-fmk(40 μmol/L)和Necroptosis抑制剂Necrostatin-1(40 μmol/L),作用2 h后,AV/PI双染观察细胞死亡的变化情况,并对细胞进行Hoechst染色计算凋亡率,用透射电镜观察细胞超微结构的改变,测定细胞线粒体膜电位和ATP水平,对Necroptosis和凋亡通路的相关蛋白进行Western blot检测。
结果
10-6 mol/L地塞米松可以诱导MC3T3-E1细胞同时发生凋亡和Necroptosis。AV/PI双染的结果发现,当凋亡受到抑制后,更多的细胞发生坏死(P < 0.01),而透射电镜结果也证实细胞发生了坏死样改变,Hoechst染色结果发现,凋亡细胞数明显减少(P < 0.01);当使用Necroptosis特异性抑制剂Necrostatin-1将这种作用阻断后,包括Hoechst染色和AV/PI双染的结果都证实凋亡的细胞数明显增多(P < 0.01),同时,在这一作用过程中伴随着MMP及ATP的明显变化(P < 0.01)。
结论
在激素诱导成骨细胞死亡过程中,Necroptosis和凋亡在一定条件下可以相互转化,这一转化过程中伴随着线粒体功能的明显改变。
Keywords: 糖皮质激素, MC3T3-E1细胞, 细胞凋亡, 程序性坏死, Necrostatin-1, caspase
Abstract
Objective
To investigate the relationship between necroptosis and apoptosis in MCET3-E1 cell death induced by glucocorticoids.
Methods
MC3T3-E1 cells were incubated with 10-6 mol/L dexamethasone followed by treatment with the apoptosis inhibitor z-VAD-fmk (40 μmol/L) or the necroptosis inhibitor necrostatin-1 (40 μmol/L) for 2 h. At 72 h after incubation with dexamethasone, the cells were harvested to determine the cell viability using WST-1 assay and the rate of necrotic cells using annexin V/PI double staining; the percentage of apoptotic cells was determined using Hoechst staining. The mitochondrial membrane potential and the level of ATP in the cells were also evaluated. Transmission electron microscopy was used to observe the microstructural changes of the cells. The expressions of RIP-1 and RIP-3 in the cells were detected by Western blotting.
Results
At a concentration of 10-6 mol/L, dexamethasone induced both apoptosis and necroptosis in MC3T3- E1 cells. Annexin V/PI double staining showed that inhibition of cell apoptosis caused an increase in cell necrosis manifested by such changes as mitochondrial swelling and plasma membrane disruption, as shown by electron microscopy; Hoechst staining showed that the percentage of apoptotic cells was significantly reduced. When necroptosis was inhibited by necrostatin-1, MC3T3-E1 cells showed significantly increased apoptosis as shown by both AV/PI and Hoechst staining, and such changes were accompanied by changes in mitochondrial membrane potential and ATP level in the cells.
Conclusion
In the process of dexamethasone-induced cell death, necroptosis and apoptosis can transform reciprocally accompanied by functional changes of the mitochondria.
Keywords: glucocorticoids, MC3T3-E1 cells, apoptosis, necroptosis, necrostatin-1, caspase
随着糖皮质激素(GCs)在多种临床疾病中的广泛应用,因长期服用激素引起的骨骼系统并发症,如股骨头缺血性坏死(SONFH)和骨质疏松(GIOP)的发病率逐年升高[1]。大量体外实验证实,GCs对骨细胞和成骨细胞的毒性作用是导致GIOP和SONFN后期股骨头塌陷的一个重要原因[2]。以往认为GCs对成骨细胞的毒性作用以诱导细胞凋亡为主[3],但之后的研究发现,单纯细胞凋亡无法解释GIOP伴随的骨量丢失[4]以及SONFH体内出现的股骨头塌陷等问题[5]。这些研究提示我们,在激素对骨细胞和成骨细胞的作用过程中,很可能存在其他未知的死亡方式。
Necroptosis是近年来发现的细胞生命周期中除坏死和凋亡之外的第三种关键性的死亡形式。它具备有类似凋亡的上下游完整的分子信号调控机制,但是最终发生Necroptosis的细胞却表现出细胞肿胀、细胞器溶解等坏死样的改变[6]。已有大量研究证实,Necroptosis作为一种可控的细胞死亡方式,参与了多种疾病的病理进程[7],而且,在某些细胞内,外界刺激诱导的Necroptosis与细胞凋亡在一定条件下可以相互转换,这也是引起细胞功能改变及相关疾病发生的重要机制[8]。我们之前通过动物实验已经证实,Necroptosis很可能参与了GIOP的病理进程[9]。同时,我们通过体外实验也发现,在GCs诱导成骨细胞死亡的过程中,除了凋亡,也有Necroptosis的参与[10]。那么,我们是否可以通过抑制Necroptosis从而彻底阻断GCs对成骨细胞的毒性作用呢?Necroptosis与凋亡在GCs对成骨细胞的致死机制中有着怎样的相互作用目前未见报道。本研究通过体外实验,对这一问题进行了进一步的研究。
1. 方法
1.1. 细胞和试剂来源
成骨细胞株MC3T3-E1(ATCC),细胞血清及培养基(Gibco),z-VAD-fmk和Necrostatin-1(Santa cruz),地塞米松和DMSO(Sigma),RIP-1、RIP-3、caspase-3及caspase-8抗体(Abcam),其余试剂盒(碧云天生物技术)。
1.2. 实验分组
将MC3T3-E1细胞(ATCC:CRL-1427)按照5×105/mL的密度接种至培养瓶内24 h后,换用含0.2% FBS的α-DMEM继续培养24 h,使细胞同步化于G0期,经胰酶消化后,按照每孔200 µL密度接种于96孔板上,随机分为4组:C组:空白对照组;D组:细胞内加入10-6 mol/L地塞米松;Z+D组:细胞内加入z-VAD-fmk 40 µmol/L,作用2 h后加入10-6 mol/L地塞米松;N+D组:细胞内加入Necrostatin-140 µmol/L,作用2 h后加入10-6 mol/L地塞米松;至72 h后,胰酶消化收集细胞悬液,进行以下相关结果的检测。
1.3. AnnexinV/PI双染
分别用胰酶消化各实验组细胞,制备成单细胞悬液,800 r/min离心5 min后弃去上清液,用PBS缓冲液冲洗2次,1000 r/min下再次离心5 min,之后按照AnnexinV-FITC/PI试剂盒说明书加入200 µL结合缓冲液轻轻重悬细胞,加入10 µL PI溶液和5 µL Annexin Ⅴ,混匀后冰浴避光下室温染色30 min,每管内补足结合缓冲液至500 µL后上流式细胞仪检测。
1.4. Hoechst染色
选取各组细胞置于6孔板内,吸尽培养液,加入0.5 mL固定液,固定10 min,用PBS或0.9% NaCl洗2遍,加入0.5 mL Hoechst染色液,染色5 min,之后去染色液,用PBS或0.9% NaCl洗2遍,最后滴一滴抗荧光淬灭封片液于载玻片上,盖上贴有细胞的盖玻片,于荧光显微镜下检测,激发波长350 nm左右,发射波长460 nm左右。
结果判定:正常细胞呈均匀蓝染圆形颗粒,凋亡细胞可见致密白色细胞碎片。在盖玻片的四个象限,随机拍摄三个视野(200倍下),总共有12个视野。统计12个视野的细胞总数和凋亡细胞数,得出凋亡比率;凋亡比率=(细胞总数-白色细胞数)/细胞总数×100%。
1.5. 透射电镜
0.25%胰酶消化收集各实验组细胞,PBS液冲洗2次,之后向细胞内加入3%戊二醛,固定1 h,PBS液冲洗2次,再向细胞内添加1%锇酸固定1 h,丙酮酸梯度脱水;包埋,超薄切片,铀铅双染色;于透射电镜80 kV下观察细胞形态,并选择典型切片图像拍照。
1.6. 线粒体膜电位水平检测
将各组细胞用胰酶消化,PBS重悬,2000 r/min下离心5 min,培养液中加入血清和酚红;按照线粒体膜电位(MMP)水平检测试剂盒说明书,制备反应缓冲液备用,吸取500 µL的JC-1工作液,将细胞均匀悬浮,置于培养箱内20 min,取出细胞,2000 r/min离心5 min,加入稀释好的反应缓冲液洗涤2次,再用500 µL JC-1染色缓冲液(1×)重悬细胞,用流式细胞仪分析结果。
1.7. 三磷酸腺甙水平检测
0.25%胰酶消化收集各实验组细胞,用PBS液冲洗2次;吸取约10 µL细胞悬液放入96孔白板内,按照三磷酸腺甙(ATP)水平检测试剂盒说明书首先添加100 µL的裂解液进行充分裂解,之后4 ℃下12 000 r/min离心5~10 min,取上清液内添加ATP检测工作液10 µL,移液器混匀后即上机检测,用酶标仪的Luminometer模式进行测定。
1.8. Western blot
因为激素作用后有部分细胞会脱落,为确保实验的准确性,我们分两部分提取总蛋白进行测定。首先将培养液倒入15 mL离心管内,2500 r/min离心5 min,弃上清并加入4 mL PBS洗涤后继续2500 r/min离心5 min,然后加入裂解液100 µL在冰上裂解30 min,最后4 ℃下12 000 r/min离心5 min,提取上清液至0.5 mL离心管内-20 ℃下保存。随后于瓶内加入3 mL在4 ℃下预冷的PBS液进行冲洗,然后向瓶内加入300 µL含有PMSF的裂解液,置于冰上30 min进行细胞裂解,然后用刮棒将细胞碎片和裂解液移入1.5 mL离心管内,4 ℃下12 000 r/min离心5 min,提取上清液至0.5 mL离心管内-20 ℃下保存。
取出所需测定的样品,沸水中煮5 min使蛋白变性,然后置入0.5 mL的离心管内,加入5×SDS的缓冲液,电泳槽内加入足够电泳液,40 V条件下进行电泳4~5 h,参考试剂说明书将样品经转膜后行免疫反应,最后用DAB显色法显影,将胶片拍照,用Image Pro Plus 6.0对结果进行统计学分析。
1.9. 统计学方法
为确保所有实验的可重复性,我们对上述各实验均分别独立进行3次。实验所得的计量数据均采用均数±标准差来表示,实验所得的所有数据全部录入统计学软件SPSS 20.0,然后再进行进一步的处理和分析,采用统计分析中的单因素方差分析one-way ANOVA进行多组之间的均数比较,组间差异应用SNK-q法进行比较。以P < 0.05认为差异有统计学意义。
2. 结果
2.1. AV-PI双染结果
我们首先分别选取48和72 h两个时间点对各组细胞进行AV/PI双染,观察MC3T3-E1细胞在不同时间点的变化情况,结果如下。
C组细胞在48~72 h的过程中变化不大,有少量AV+和PI+的细胞增多,提示我们随着培养时间的延长,有少部分细胞发生了死亡;D组细胞在48 h时,PI+细胞较对照组开始增多,而到72 h时,我们发现有很大部分细胞表现为AV-PI+(P < 0.01,图 1),提示我们随着时间进展,凋亡细胞由早期向晚期进展,到72 h时有部分细胞的细胞膜破损,发生了坏死。Z+D组到72 h时PI+细胞较对照组明显增多(P < 0.01),而且与D组相比也有显著增多(P < 0.05),N+D组72 h时,可见PI+细胞与Z+D组相比明显减少(P < 0.01),而且少于D组(P < 0.01),AV+细胞增多。
1.
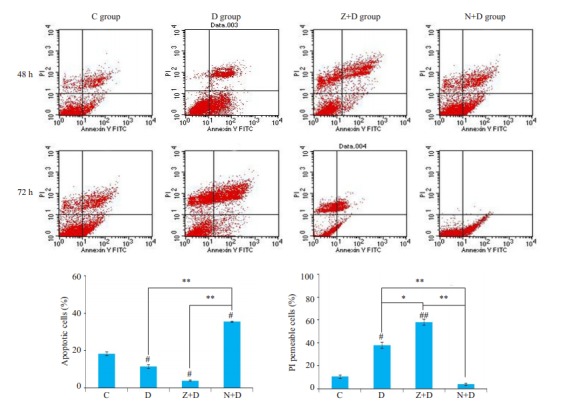
各实验组AV/PI双染结果图
Results of AV/PI double staining in each group. C: Control group; D: Dexamethasone group; Z+D: z-VAD-fmk+ dexamethasone group; N+D: Necrostatin-1+dexamethasone group (compared with controls #P < 0.05; ##P < 0.01; compared with others *P < 0.05; **P < 0.01).
2.2. Hoechst凋亡染色
由图 2中可以看出,C组细胞染色均匀,胞核呈现蓝色,凋亡细胞少见;D组细胞数量明显减少,可见明显固缩为白色的细胞核及大量显示白色的细胞碎片,提示有大量的凋亡细胞和坏死细胞;Z+D组细胞数量明显减少,凋亡细胞较D组少见(P < 0.05)而细胞碎片明显增多;N+D组可见较多细胞恢复为正常的蓝染,同时伴随部分固缩为白色的细胞核,提示Nec-1的作用下,坏死细胞数明显减少,同时,凋亡的细胞数相比D组有所增多,统计学分析显示,这部分增多并无统计学意义(P>0.05)。
2.
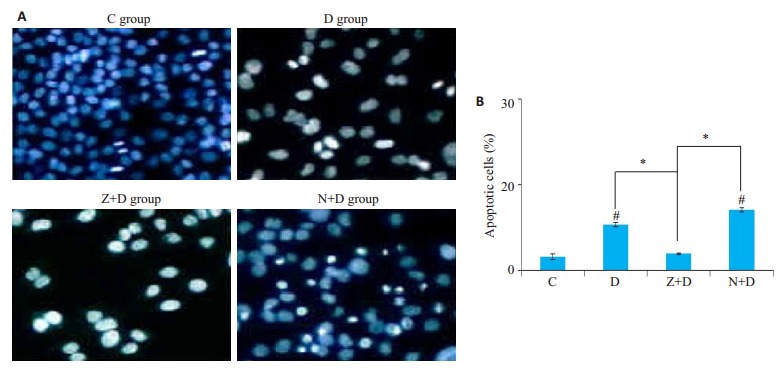
各实验组Hoechst染色结果及凋亡细胞数对比
Hoechst staning (Original magnification: × 200) (A) and the percentage of apoptotic cells (B) at 72 h in each group (compared with controls #P < 0.05; compared with others *P < 0.05).
2.3. 透射电镜检测
对照组正常MC3T3-E1细胞胞膜完整,各细胞器清晰可见,细胞核结构完整,核内染色质无异常;而D组细胞胞膜不连续,细胞内可见大量肿胀的线粒体,部分线粒体内部电子密度降低,嵴消失,呈空泡状,同时细胞核极度固缩,内可见染色质聚集成团;Z+D组细胞可见胞膜膜破裂,胞质内大量溶解的细胞器等坏死样改变,细胞核尚正常;N+D组细胞胞膜完整,胞内仅有少量轻度肿胀的线粒体,但细胞核的固缩与D组相比无明显改善(图 3)。
3.
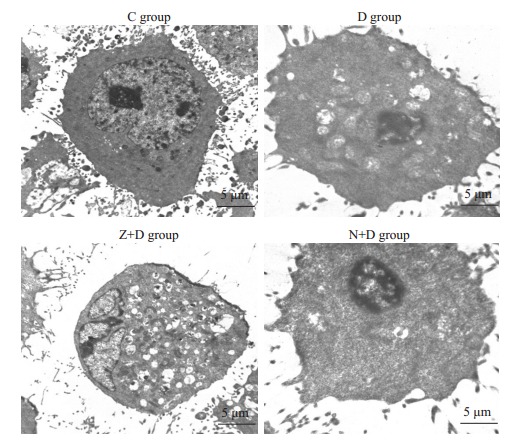
应用透射电镜观察各实验组细胞微结构的改变
Changes in organelle structures of the cells observed under trasnmission electron microscopy (scale bar=5 μm).
2.4. ATP测定
图 4中,在D组中,当我们用Dex诱导细胞后,ATP水平较对照组明显下降(P < 0.05),而当我们用z-VADfmk阻断凋亡后,ATP水平下降更加明显(P < 0.01),反之,当我们用Nec-1阻断这种坏死后,可见ATP水平较C组明显增高(P < 0.01),与D组相比也有明显改善(P < 0.05)。
4.

72 h细胞内ATP水平变化
Changes of ATP level at 72 h in each group (compared with controls #P < 0.05; ##P < 0.01; *P < 0.05; **P < 0.01).
2.5. MMP水平检测
为了解线粒体在此过程中所起的作用,我们对细胞的MMP水平进行了测定(图 5),D组细胞的MMP水平较正常组出现明显下降(P < 0.05),当我们用zVAD-fmk干预后,MMP水平较D组下降更明显(P < 0.05),而使用Nec-1干预后,MMP水平较Z+D组有明显提高(P < 0.05)。
5.
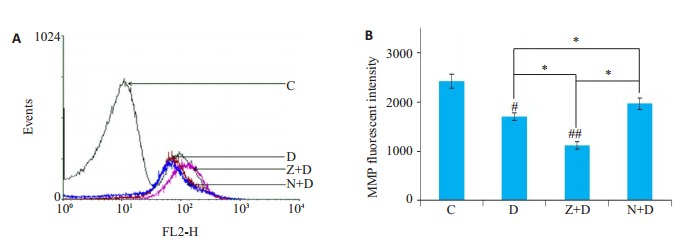
各实验组细胞MMP水平变化
Change of mitochondrial membrane potential (MMP) in each group. A: The waveforms of each group by flow cytometry; B: The MMP fluorescent intensity of each group (compared with controls #P < 0.05; ##P < 0.01; *P < 0.05).
2.6. Western blot检测
为进一步验证Necroptosis与凋亡之间的关系,我们分别对Necroptosis通路的关键蛋白RIP-1、RIP-3还有凋亡通路的蛋白进行了Western blot检测,结果显示(图 6),与对照组相比,Dex可显著增强RIP-1和RIP-3的表达活性(P < 0.05),同时增强caspase-3和caspase-8的活性(P < 0.05);添加z-VAD-fmk进行干预后,可见caspase-3和caspase-8的活性明显下降(P < 0.05),同时,RIP-3的表达都明显增强(P < 0.01),RIP-1的表达也有增高,但并无统计学意义(P>0.05);而用Necrostatin-1干预后,可见RIP-1活性下降明显(P < 0.05),caspase-3活性明显增强(P < 0.05),RIP-3和caspase-8变化不大(P>0.05)。
6.
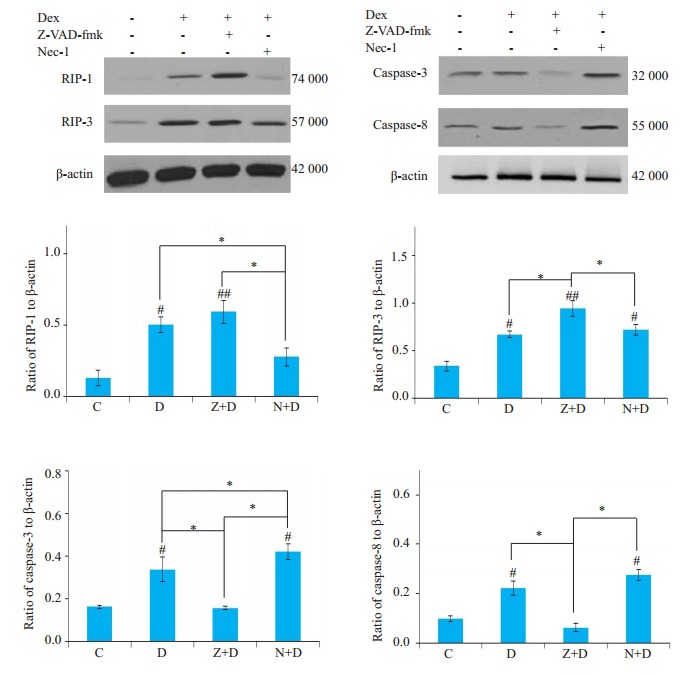
各实验组细胞RIP-1、RIP-3、caspase-3、caspase-8的表达活性
Expression of RIP-1, RIP-3, caspase-3 and caspase-8 in each group (compared with controls #P < 0.05; ##P < 0.01; *P < 0.05).
3. 讨论
我们前面的研究已经证实,不同浓度梯度的地塞米松可以诱导MC3T3-E1细胞出现不同的表现,MC3T3-E1细胞在高浓度地塞米松诱导条件下会发生Necroptosis[10],那么,在较低浓度下的地塞米松对MC3T3-E1细胞的作用过程中是否也有Necroptosis的参与,凋亡和Necroptosis这两种死亡方式各自充当了怎样的角色,他们二者之间有无相互作用呢?为此,我们在之前细胞实验研究的基础上,试图探讨凋亡与Necroptosis在激素诱导成骨细胞死亡中的相互作用。
正常细胞膜外表面存在大量中性的磷脂,而内表面存在大量含负电荷的磷脂,主要是磷脂酰丝酸(PS)。而Annexin V作为Ca2+离子依赖性的磷脂结合蛋白,可以与PS特异性相结合。因为在细胞凋亡的早期,细胞膜发生一系列改变,使得PS从膜内翻转到膜外,所以早期凋亡的细胞可被Annexin V染色。碘化丙啶(PI)是一种核酸染料,可以与DNA结合染色,它不能透过完整的细胞膜,所以只有当细胞膜受到损伤的时候,PI才能进入细胞内进行着色[12]。因此,AV/PI双染可以作为鉴别凋亡和Necroptosis的有效手段[13]。本实验结果发现,MC3T3-E1细胞在受到10-6 mol/L地塞米松作用后,开始表现出的是凋亡,但是,随着地塞米松作用时间的延长,我们通过透射电镜发现,细胞的线粒体开始表现为空泡化,说明线粒体受到了损伤,紧接着,细胞膜开始受到破坏,这样使得PI开始进入细胞内并着色,同时,我们还发现,细胞核此时表现出的是类似凋亡的固缩和染色质的集聚。而Western blot的结果显示,此时RIP-1和RIP-3的表达开始增强,同时caspase-3和caspase-8的表达较正常组也有增强。这些结果告诉我们,很可能此时的细胞内存在不止一种死亡形式。
之前就有研究发现,机械应力诱导的软骨细胞死亡中除了细胞凋亡,也有Necroptosis的参与[14]。而通过同时抑制Necroptosis与细胞凋亡还可以对心肌细胞产生显著保护作用[15]。这些研究证实了Necroptosis与凋亡在细胞死亡中的共存性[16]。但是,二者之间是否存在相互抑制或相互转化的关系目前还存在较大的争议,在某些病理过程中,Necroptosis与细胞凋亡是相辅相成的[17],但在某些病理状态下,又存在相互抑制的作用[18]。那么,在激素诱导成骨细胞死亡的过程中,二者的相互作用是怎样的呢?在肿瘤细胞中,用z-VAD-fmk抑制人外周血白血病T细胞(Jurkat)凋亡后并未使得细胞复活,而是走向了坏死[19]。而且,后续研究发现,这种坏死样改变可以被Necrostatin-1抑制[20]。我们通过研究证实,在激素诱导MC3T3-E1细胞凋亡的过程中,抑制凋亡后细胞也将向坏死样改变进展。AV/PI结果发现,用z-VAD-fmk抑制凋亡后,PI+细胞开始增多。Hoechst染色证实,z-VAD-fmk作用后凋亡细胞明显减少,可见这部分细胞正是因为发生了坏死而导致PI+的。透射电镜也发现,细胞出现明显的胞膜破裂,同时线粒体受损严重,几乎全部呈现空泡化。Western blot的结果显示,这种改变伴随着caspase-3和caspase-8活性的下降,RIP-1和RIP-3的表达明显增强,所以这种坏死样改变可能正是Necroptosis [21]。
之前有人发现Necrostatin-1可以将由紫草素诱导的Necroptosis转变为凋亡[22]。我们之前的研究也证实,Necrostatin-1可以阻断地塞米松诱导MC3T3-E1细胞发生的Necroptosis。通过本部分研究,我们发现在使用Necrostatin-1干预细胞后,AV+/PI-细胞明显增多。这部分细胞一直被认为是早期的凋亡细胞,但是,最新的研究发现,AV+PI-的细胞并非都代表凋亡,而是包含早期的凋亡细胞和非caspase依赖的坏死细胞在内[23]。所以并不能说明细胞在Necrostatain-1的作用下转向了凋亡。为了进一步探讨细胞的走向,我们首先通过Hoechst染色对凋亡细胞进行计数,证实Necrostatin-1的干预确实使凋亡细胞数目增多,同时,电镜观察也发现有部分细胞确实出现了包括核固缩,染色质集聚在内的一些专属于凋亡的微观表现[24]。Western blot的结果提示在RIP-1活性受到抑制的同时,caspase-3和caspase-8的表达活性明显增强。以上结果证实,当细胞受到Necrostatin-1作用后,起码有一部分转向了凋亡。
线粒体在细胞死亡的过程中起着很关键的作用[25]。线粒体膜通透性的改变会引起线粒体膜电位的降低,使得线粒体内呼吸链受阻,ATP合成减少,最终出现坏死样改变[26]。本实验中发现,10-6 mol/L地塞米松诱导MC3T3-E1细胞出现死亡后伴随着MMP的下降和ATP水平的下降;而z-VAD-fmk的干预使得细胞转向Necroptosis,这时伴随出现的线粒体改变时MMP水平的明显下降和ATP的严重合成障碍;而当我们用Necrostatin-1阻断Necroptosis后,细胞的MMP水平开始回升,同时ATP合成和明显增多,很可能正是为细胞下一步走向凋亡储存能量[27]。
总之,10-6 mol/L地塞米松可以诱导MC3T3-E1细胞发生凋亡,也可诱导Necroptosis的发生,而且二者之间在一定条件下可以相互转换,这与其他的细胞系中的发现相一致[28]。而正是它们之间的这种相互转换关系,使得在某种特定情形下可以通过诱导细胞发生Necroptosis从而产生保护作用[29]。同时,在凋亡与Necroptosis的相互转换的过程中,线粒体很可能起着关键的作用[30]。但是,这种相互作用中到底是哪种因子起了关键性作用,最终是通过哪种信号的转导实现的还需要我们更进一步的研究证实。
Biography
冯敏,博士,主治医师,E-mail:syo0817@163.com
Funding Statement
国家自然科学基金(81672173);陕西省人民医院孵化基金(2018YXQ-05)
Supported by National Natural Science Foundation of China (81672173)
Contributor Information
冯 敏 (Min FENG), Email: syo0817@163.com.
强 辉 (Hui QIANG), Email: qianghui359@163.com.
References
- 1.Weinstein RS. Clinical practice. Glucocorticoid-induced bone disease. N Engl J Med. 2011;365(1):62–70. doi: 10.1056/NEJMcp1012926. [DOI] [PubMed] [Google Scholar]
- 2.Weinstein RS. Glucocorticoid-induced osteoporosis and osteonecrosis. Endocrinol Metab Clin NorthAm. 2012;41(3):595. doi: 10.1016/j.ecl.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frenkel B, White W, Tuckermann J. Glucocorticoid-induced osteoporosis. http://d.old.wanfangdata.com.cn/Periodical/zgsyfjxzz201116039. Adv Exp Med Biol. 2015;872:179–215. doi: 10.1007/978-1-4939-2895-8_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lane NE. Glucocorticoid-induced osteoporosis: new insights into the pathophysiology and treatments. https://link.springer.com/article/10.1007%2Fs11914-019-00498-x. Curr Osteoporos Rep. 2019;17(1):1–7. doi: 10.1007/s11914-019-00498-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinstein RS, Hogan EA, Borrelli MJ, et al. The pathophysiological sequence of Glucocorticoid-induced osteonecrosis of the femoral head in male mice. Endocrinology. 2017;158(11):3817–31. doi: 10.1210/en.2017-00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–21. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galluzzi L, Kepp O, Chan FK, et al. Necroptosis: mechanisms and relevance to disease. http://d.old.wanfangdata.com.cn/Periodical/zgblslzz201012041. Annu Rev Pathol. 2017;24(12):103–30. doi: 10.1146/annurev-pathol-052016-100247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikseresht S, Khodagholi F, Dargahi L, et al. Necroptosis resumes apoptosis in hippocampus but not in frontal cortex. J Cell Biochem. 2017;118(12):4628–38. doi: 10.1002/jcb.26127. doi: 10.1002/jcb.26127. [DOI] [PubMed] [Google Scholar]
- 9.Feng M, Zhang R, Gong F, et al. Protective effects of necrostatin-1 on glucocorticoid-induced osteoporosis in rats. https://www.ncbi.nlm.nih.gov/pubmed/25220755. J Steroid Biochem Mol Biol. 2014;144(Pt B):455–62. doi: 10.1016/j.jsbmb.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Feng M, Qiang H, Zhang RR, et al. (2018) Necrostatin-1 inhibits the cell death of osteoblasts induced by glucocorticoid. Int J Clin Exp Pathol. 2018;11:675–84. [PMC free article] [PubMed] [Google Scholar]
- 11.Sheridan C, Martin SJ. Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion. 2010;10(6):640–8. doi: 10.1016/j.mito.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Crowley LC, Marfell BJ, Scott AP, et al. Quantitation of apoptosis and necrosis by Annexin V binding, propidium iodide Uptake, and flow cytometry. Cold Spring HarbProtoc. 2016;(11) doi: 10.1101/pdb.prot087288. [DOI] [PubMed] [Google Scholar]
- 13.Sawai H, Domae N. Discrimination between primary necrosis and apoptosis by necrostatin-1 in Annexin V-positive/propidium iodidenegative cells. Biochem Biophys Res Commun. 2011;411(3):569–73. doi: 10.1016/j.bbrc.2011.06.186. [DOI] [PubMed] [Google Scholar]
- 14.Zhang C, Lin S, Li T, et al. Mechanical force-mediated pathological cartilage thinning is regulated by necroptosis and apoptosis. Osteoarthritis Cartilage. 2017;25(8):1324–34. doi: 10.1016/j.joca.2017.03.018. [DOI] [PubMed] [Google Scholar]
- 15.Guo XY, Yin HF, Li L, et al. Cardioprotective role of tumor necrosis factor receptor-associated factor 2 by suppressing apoptosis and necroptosis. Circulation. 2017;136(8):729. doi: 10.1161/CIRCULATIONAHA.116.026240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol. 2018;15(12):738–52. doi: 10.1038/s41575-018-0065-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Orzalli MH, Kagan JC. Apoptosis and necroptosis as host defense strategies to prevent viral infection. Trends Cell Biol. 2017;27(11):800–9. doi: 10.1016/j.tcb.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang CX, Sun P, Deng MH, et al. Gasdermin D protects against noninfectious liver injury by regulating apoptosis and necroptosis. Cell Death Dis. 2019;10(7):481. doi: 10.1038/s41419-019-1719-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsumura H, Shimizu Y, Ohsawa Y, et al. Necrotic death pathway in Fas receptor signaling. J Cell Biol. 2000;151(6):1247–55. doi: 10.1083/jcb.151.6.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Degterev A, Huang ZH, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–9. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 21.Shi G, Jia P, Chen H, et al. Necroptosis occurs in osteoblasts during tumor necrosis factor-alpha stimulation and caspase-8 inhibition. Brazilian J Med Biol Res. 2019;52(1):e7844. doi: 10.1590/1414-431X20187844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han WD, Xie JS, Li L, et al. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis. 2009;14(5):674–86. doi: 10.1007/s10495-009-0334-x. [DOI] [PubMed] [Google Scholar]
- 23.Pietkiewicz S, Schmidt JH, Lavrik IN. Quantification of apoptosis and necroptosis at the single cell level by a combination of imaging flow cytometry with classical Annexin V/propidium iodide staining. http://www.ncbi.nlm.nih.gov/pubmed/25975759. J Immunol Methods. 2015;423(SI):99–103. doi: 10.1016/j.jim.2015.04.025. [DOI] [PubMed] [Google Scholar]
- 24.Liu L, Fan JY, Ai GH, et al. Berberine in combination with cisplatin induces necroptosis and apoptosis in ovarian cancer cells. Biol Res. 2019;52(1):37. doi: 10.1186/s40659-019-0243-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchi S, Patergnani S, Missiroli SA, et al. Mitochondrial and endoplasmic reticulum Calcium homeostasis and cell death. http://d.old.wanfangdata.com.cn/NSTLQK/NSTL_QKJJ0213127869/ Cell Calcium. 2018;69(SI):62–72. doi: 10.1016/j.ceca.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Carraro M, Bernardi P. Calcium and reactive Oxygen species in regulation of the mitochondrial permeability transition and of programmed cell death in yeast. Cell Calcium. 2016;60(2, SI):102–7. doi: 10.1016/j.ceca.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai PP, Mao YX, Sun XY, et al. Attenuation of oxidative stressinduced osteoblast apoptosis by curcumin is associated with preservation of mitochondrial functions and increased Akt-GSK3 beta signaling. Cell Physiol Biochem. 2017;41(2):661–77. doi: 10.1159/000457945. [DOI] [PubMed] [Google Scholar]
- 28.Meng HY, Liu Z, Li XY, et al. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc Natl Acad Sci USA. 2018;115(9):E2001–9. doi: 10.1073/pnas.1722013115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safferthal C, Rohde K, Fulda S. Therapeutic targeting of necroptosis by Smac mimetic bypasses apoptosis resistance in acute myeloid leukemia cells. Oncogene. 2017;36(11):1487–502. doi: 10.1038/onc.2016.310. [DOI] [PubMed] [Google Scholar]
- 30.Zhao WW, Feng HT, Sun W, et al. Tert-butyl hydroperoxide (t-BHP) induced apoptosis and necroptosis in endothelial cells: Roles of NOX4 and mitochondrion. https://www.ncbi.nlm.nih.gov/pubmed/28088644. Redox Biol. 2017;11(11):524–34. doi: 10.1016/j.redox.2016.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]