

Abstract
5-Fluoro-2′-deoxycytidine was synthesized by treating 5-fluoro-2′-deoxyuridine with 2,4,6-trimethylphenol in the presence of 1-methylpyrrolidine and trifluoroacetic anhydride, followed by aminolysis. Among N-acetyl, pivaloyl, and benzoyl, N-acetyl was found to be suitable for the protection of the exocyclic amine of 5-fluoro-2′-deoxycytidine because of the stability of the N4-protected nucleoside under acidic conditions and its ease of removal after solid-phase synthesis. This modified nucleoside was incorporated into d(CG)6 sequences through the phosphoramidite chemistry-based solid-phase synthesis. Circular dichroism experiments suggest that replacement of 2′-deoxycytidine with 5-fluoro-2′-deoxycytidine does not lead to detectable conformational changes, either in the B- or Z-form. 19F NMR spectroscopy of d(CG)6 containing 5-fluoro-2′-deoxycytidine revealed that B/Z-DNA transition induced by sodium chloride is likely initiated at terminal ends, leading to unwinding at the middle of duplexes, and eventual switch of handedness when sodium chloride concentration reaches a threshold value.
Introduction
Among the chemical modifications of nucleic acids, fluorination has attracted immense interest because of the small size and electronegativity of the fluorine atom and frequently favored pharmacological properties.1−4 While most efforts have focused on nucleosides with fluorine-containing sugars, modification of purine and pyrimidine with fluorine has also yielded useful properties.5 Of particular note, introduction of fluorine into nucleic acids has enabled structural studies via 19F NMR spectroscopy. On the one hand, fluorine is small in size, and it is possible to predict conformational differences with these modified nucleic acids; on the other hand, 19F is the only stable fluorine isotope with a large gyromagnetic ratio, and fluorine chemical shifts are quite sensitive to conformational changes. These features allow for the monitoring of conformational changes in nucleic acids using 19F NMR spectroscopy.6 Of particular interest, Fujimoto and co-workers demonstrated that incorporation of 5-trifluoromethyl-2′-deoxycytidine into oligonucleotides allowed for the tracking of B-/Z-DNA transition by 19F NMR spectroscopy.7 In the present work, we wish to report the synthesis of 5-fluoro-2′-deoxycytidine (FdC 5) and its incorporation into oligonucleotides through the phosphoramidite chemistry-based solid-phase synthesis, and subsequent study of DNA handedness transition, that is, B- and Z-DNA, using 19F NMR spectroscopy.
Results and Discussion
Synthesis of 5-Fluoro-2′-deoxycytidine 5
While FdC was previously synthesized from 5-fluoro-2′-deoxyuridine (FdU 1) in 56% yield via a tetrazolide intermediate,8 the chemistry requires the use of 1H-tetrazole, which can be limited in practical synthesis because of the thermal sensitivity of 1H-tetrazole and consequent shipping restrictions.
Conversion of uracil nucleosides to the corresponding cytosine nucleosides is well documented in the literature. Two common approaches involve the formation of uracil 4-arylether (such as 4-(p-nitrophenyl)ether 2) or 4-triazolide 3.9−11 Attempted syntheses of FdC 5 from FdU 1 through intermediates 2 or 3, however, were unsuccessful, leading only to the recovery of starting material 1 (Scheme 1).
Scheme 1.

Reagents and conditions: (i). TMSCl, 1-methylpyrrolidine, p-nitrophenol, (CF3CO)2O, CH3CN, 0 °C, 4 h; (ii). Ac2O, C5H5N, r.t., 12 h; (iii). POCl3, 1,2,3-triazole (for 3a, 2 h) or 1,2,4-triazole (for 3b, 5 h); (iv). TMSCl, 1-methylpyrrolidine, 2,4,6-trimethylphenol, (CF3CO)2O, CH3CN, 0 °C, 4 h; v). aq. NH4OH, 65 °C, 24 h.
Conversion of FdU 1 into the transiently protected 2,4,6-trimethylphenyl ether 4 followed by aminolysis, on the other hand, led to the isolation of the desired FdC 5 in 50% yield, with recovery of the starting material FdU 1 in 36% yield.
Choice of the Protecting Group for the Exocyclic Amine of FdC
While Fritz and co-workers8 incorporated FdC into oligonucleotides using N-benzoyl-protected FdC (N-Bz-FdC 6b), it was found by us that N-Bz-FdC is unstable under the acidic conditions required for the detritylation reaction in solid-phase synthesis. Thus, N-acetyl-6a, N-benzoyl-6b, and N-pivaloyl-FdC 6c were prepared (Scheme 2) and evaluated for their suitability for solid-phase synthesis.
Scheme 2.

Reagents and conditions: (i). a. TMSCl, C5H5N; b. acyl chloride; c. aq. NEt3.
In order to incorporate FdC into oligonucleotides, a suitable protecting group for exocyclic amine is required. Thus, the stability of N-acyl FdC 6 under acidic and basic conditions was evaluated. As detritylation reactions in solid-phase synthesis are typically carried out by the treatment with di- or trichloroacetic acid (DCA and TCA, respectively), the stability of N-4-protected FdC under these conditions is crucial. Such an N-4-acyl protecting group should also ensure the stability of the oligonucleotide during the cleavage and deprotection stages of solid-phase synthesis, typically under alkaline conditions. In addition, the deprotection conditions should not lead to structural modifications of oligonucleotides.
As solutions of DCA and TCA are the most commonly used in the detritylation step of solid-phase synthesis of oligonucleotides, the stability of N-acyl-FdC 6 was determined in 3% solutions of each acid in methanol. Using an aqueous solution of KF sealed in a capillary insert as an NMR internal standard, the progress of decomposition, if any, was measured by the ratio of integrals of the two 19F signals in 19F NMR at appropriate intervals.
It was found that N-Bz-FdC 6b and N-Piv-FdC 6c decompose instantaneously in DCA and TCA (Table 1), making N-benzoyl and pivaloyl groups unsuitable for solid-phase synthesis. N-Ac-FdC 6a, on the other hand, was found to be more stable, with a half-life of 4.13 h in 3% TCA, but stable in DCA after 24 h. As such, N-Ac-FdC 6a is compatible with the detritylation conditions used in solid-phase synthesis, where the exposure time to TCA is as little as 20 s per cycle on a 1 μmol scale.12
Table 1. Stability of N-Acyl-FdC under Acidic Conditions.
| substance | acid (3% in MeOH) | stability |
|---|---|---|
| N-Bz-FdC 6b | TCA | instant decomposition |
| N-Bz-FdC 6b | DCA | instant decomposition |
| N-Piv-FdC 6c | TCA | instant decomposition |
| N-Piv-FdC 6c | DCA | instant decomposition |
| N-Ac-FdC 6a | TCA | 4.13 h (half-life) |
| N-Ac-FdC 6a | DCA | stable after 24 h |
Next, the lability of the N-acyl-protecting groups for FdC under basic conditions was determined by HPLC experiments. Thus, N-acyl-FdC 6 was incubated in appropriate alkaline solutions containing a suitable internal standard. Aliquots were withdrawn at appropriate intervals for HPLC analysis. The ratios of peak areas of the internal standard and N-acyl-FdC were used to determine the half-life of each N-acyl protecting group under respective alkaline conditions.
As can be seen in Table 2, N-Bz-FdC 6b undergoes deglycosylation at 55 °C at pH 11. N-Piv-FdC 6c, however, is quite stable at pH 9 and 11 but almost completely deprotected in concentrated aqueous ammonium hydroxide at room temperature in 5 min. N-Ac-FdC 6a is more labile than N-Piv-FdC 6c, with a half-life of 51.3 and 21.8 h at pH 9 and 11, respectively, and virtually completely deprotected in concentrated aqueous ammonium hydroxide at room temperature in 5 min. To our surprise, N-Ac-FdC 6a was deprotected by methanol quite effectively, with a half-life of 26.7 h at room temperature. Exposure to ethanol, propanol, and butanol, however, did not lead to deacetylation of N-Ac-FdC. Because of the instability of N-Ac-FdC in methanol, ethanol was used, wherever necessary, in column chromatography for the purification of N-Ac-FdC analogues.
Table 2. Lability of N-Acetyl-, Benzoyl-, and Pivaloyl-FdC under Various Alkaline Conditions.
| substance | pH | temp. (°C) | half-life |
|---|---|---|---|
| N-Bz-FdC 6b | 11 | 55 | deglycosylation |
| N-Piv-FdC 6c | 9 | 55 | >60 h |
| N-Piv-FdC 6c | 11 | 55 | 39.2 h |
| N-Piv-FdC 6c | conc. aq. NH4OH | 22 | <5 min (complete deprotection) |
| N-Ac-FdC 6a | 9 | 55 | 51.3 h |
| N-Ac-FdC 6a | 11 | 55 | 21.8 h |
| N-Ac-FdC 6a | conc. aq. NH4OH | 22 | <5 min (complete deprotection) |
| N-Ac-FdC 6a | MeOH | 22 | 26.7 h |
Considering the stability of N-acetyl-FdC 6a in acidic conditions and its ease of deprotection under alkaline conditions, N-Ac-FdC 6a was used in subsequent transformations.
Solid-Phase Synthesis of d(CG)6
To demonstrate the utility of FdC in the structural elucidation of oligonucleotides, N-acetyl-FdC 6a was incorporated into oligonucleotides by the phosphoramidite chemistry-based solid-phase synthesis. First, 5′-OH of N-Ac-FdC 6a was protected with the dimethoxytrityl (DMTr) group, followed by the conversion of 7 to the corresponding phosphoramidite 8 as a mixture of two diastereomers (Scheme 3), as indicated by 31P NMR spectroscopy. It was found during the purification of 5′-O-DMTr-N-4-acetyl-FdC 7 by column chromatography that the N-acetyl group in 6a and 7 is labile in the presence of triethylamine; thus, diisopropylethylamine (Hünig’s base), instead of triethylamine, was used to ensure the stability of 7 and 8 during column chromatography.
Scheme 3.

Reagents and conditions: (i). DMTr-Cl, C5H5N; (ii). (CNCH2CH2O)P(Cl)N(i-Pr)2, (i-Pr)2NEt.
Six d(CG)6 sequences containing single FdC modification at different positions were subsequently prepared by solid-phase synthesis on an ABI DNA synthesizer on 1 μmol scales. After cleavages from solid supports and deprotection, the oligonucleotides were characterized by anion exchange HPLC and electrospray mass spectroscopy (Table 3).
Table 3. Characterization of Modified d(CG)6 DNA Sequences by Mass Spectroscopy (ESI, Detected for Anions), Midpoint Concentrations of NaCl for the B/Z-DNA Transition, Melting Temperatures (Tm), and HPLC.
| mass |
|||||
|---|---|---|---|---|---|
| name/sequence (5′ → 3′) | expecteda | foundb | midpoint NaCl (M) | Tm (°C, 20 mM NaCl) | purity (%, denaturing anion exchange HPLC) |
| d(CG)6/d(CGCGCGCGCGCG) | c | 2.6 | 82 | c | |
| d(CG)6-1F/d(FCGCGCGCGCGCG) | 3665.37 | 3665.5 | 2.5 | 84 | 95.3 |
| d(CG)6-3F/d(CGFCGCGCGCGCG) | 3665.37 | 3665.6 | 2.6 | 86 | 96.3 |
| d(CG)6-5F/d(CGCGFCGCGCGCG) | 3665.37 | 3665.6 | 2.6 | 85 | 93.2 |
| d(CG)6-7F/d(CGCGCGFCGCGCG) | 3665.37 | 3665.5 | 2.4 | 84 | 94.4 |
| d(CG)6-9F/d(CGCGCGCGFCGCG) | 3665.37 | 3665.5 | 2.6 | 84 | 96.4 |
| d(CG)6-11F/d(CGCGCGCGCGFCG) | 3665.37 | 3665.0 | 2.4 | 83 | 92.1 |
Calculated for C114H143FN48O70P11–.
Found for mono-anion based on multiply-charged anionic species.
Purchased from IDT. FC: FdC.
Characterization of the B/Z-DNA Transition in FdC-modified d(CG)6 by Circular Dichroism
As one of the assumed advantages of substitution at C-5 of deoxycytidine with fluorine is its small size, minimal steric alteration is anticipated. As such, the FdC-modified d(CG)6 sequences were annealed in low to high concentrations of sodium chloride (0.1–4 M) and evaluated for their ability to adopt B- and Z-duplexes and transition from B- to Z-form by circular dichroism (CD), in comparison with the unmodified d(CG)6. As shown in Figure 1, the FdC-modified d(CG)6 sequences show the typical CD profile for B-DNA in low concentration of sodium chloride but undergo transition gradually to the Z-form with the increase of sodium chloride concentration. In samples containing 4 M sodium chloride, all the sequences show characteristic CD profiles for Z-DNA.13,14
Figure 1.

CD profiles of (a) unmodified d(CG)6; (b) d(CG)6-11F; and (c) d(CG)6-7F in varying concentrations of NaCl.
Furthermore, the absorbances of these duplexes at 260 and 290 nm were recorded, and the A260/A290 ratios were plotted to determine the midpoint concentration of sodium chloride required for the B → Z DNA transition. As shown in Table 3, d(CG)6 sequences containing a single FdC modification have midpoint concentrations of NaCl required for B → Z-DNA transition very similar to that of the unmodified d(CG)6 (2.6 M), which is close to the literature value (2.5 M).15 These results are in agreement with the above CD observation, confirming that FdC introduces minimal distortion to the overall DNA structures. Incorporation of FdC into d(CG)6 appears to slightly increase the duplex stability in the B-form, as indicated by the slightly higher melting temperature (Tm in 20 mM NaCl, Table 3).
Use of FdC as a Probe to Study B → Z-DNA Transition
While B-type duplex is the dominant form of DNA in biological systems, the left-handed Z-DNA has been found to also exist under certain conditions, such as elevated salt concentrations and torsional stress.14−16 Despite over 40 years of investigation, numerous questions about Z-DNA remain. Of these, concrete evidence is still lacking with respect to the detailed structural changes in B → Z-transition, despite various hypotheses put forward primarily through theoretical articulation.17 As 19F NMR shifts are quite sensitive to structural changes, this form of spectroscopy has been found to be useful in the investigation of protein and nucleic acid structures and interactions.6
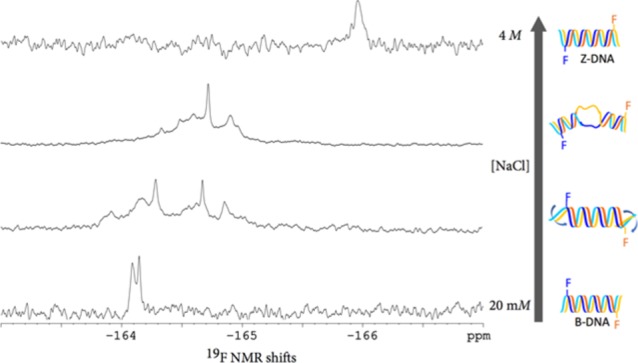
Examination of the 19F NMR spectra (Figure 2a–f) of d(CG)6-11F reveals a clear trend of chemical shifts. In 20 mM sodium chloride, where the DNA duplex is presumed to adopt B-form DNA as indicated by the CD spectrum, two close 19F signals at −164.1 ppm are observed. As the concentration of sodium chloride reaches 4 M, a condition under which this duplex was established by CD spectroscopy to exist in the Z-DNA form, the 19F NMR signals shifted toward up-field. A gradual shift of the 19F resonance from −164 to −166 pm corroborates the B- to Z-transition with the increase of sodium chloride concentration from 20 mM to 4 M.
Figure 2.

19F NMR spectra of d(CG)6 modified with FdC [(a–f): d(CG)6-11F; (a′–f′): d(CG)6-7F] in the presence of varying concentrations of NaCl. (a). 4 M; (a′). 4.5 M; (b and b′). 3.0 M; (c and c′). 2.5 M; (d and d′). 2.0 M; (e and e′). 1 M; (f and f′). 20 mM. The color-coded models correspond to DNA duplexes. The bold blue arrows in the cartoons indicate unwinding of duplexes and the “bubbles” correspond to unpaired regions. Concentrations of the modified d(CG)6 were between 0.23 and 0.58 mM.
The changes in 19F NMR chemical shifts depicted in Figure 2a–f support a model for the B → Z-DNA transition, where B-DNA duplex unwinds from termini as the salt concentration increases, leading to eventual switching of handedness when the salt concentration reaches a threshold value. This model is in agreement with the 19F NMR spectra of the d(CG)6-7F: d(CG)6 duplex (Figure 2a′–f′), which was annealed by mixing d(CG)6-7F and d(CG)6 in a 1:5 ratio. In this case, changes in 19F shifts only became significant when sodium chloride concentration reaches 3 M (Figure 2c′), further supporting the scenario where unwinding of interior occurs after the duplex unwinds at the termini.
Further investigation of the FdC-modified d(CG)6 by 1H NMR revealed that incorporation of FdC appears to affect hydrogen bonds. As shown in the 1H NMR of unmodified d(CG)6 and those modified with FdC at 7 and 11 positions (Figure 3, samples were prepared in H2O–D2O, 9:1 v/v, at 0.4 mM), the NH protons in the most downfield region that are typically found in hydrogen bonding display different chemical shifts, suggesting disruptions to hydrogen bonding by FdC.
Figure 3.

1H NMR of (a). d(CG)6-11F, (b). (d). d(CG)6-7F, and (c). d(CG)6 in H2O/D2O (9:1, v/v). Oligonucleotides were annealed in Tris-HCl (10 mM, pH 7.5 containing 20 mM NaCl) at a 0.4 mM (ssDNA) concentration.
Conclusions
FdC 5 was successfully synthesized from FdU 1. N-Acetyl was found to be a suitable protecting group for FdC for the incorporation of this modified nucleoside into oligonucleotides through the phosphoramidite chemistry-based solid-phase synthesis. Compared with unmodified d(CG)6, oligonucleotides containing a single FdC modification showed very little difference in CD profiles both in the B- and Z-forms, indicating minimal distortions of DNA duplexes by this fluorinated nucleoside. 19F NMR spectroscopy of d(CG)6 duplexes containing FdC at varying sodium chloride concentrations suggested that B → Z-DNA transition is likely initiated by the unwinding of duplex at termini, leading to switches of handedness with the increase of sodium chloride concentration.
Overall, FdC has the potential to be used as a versatile probe for DNA structures and interactions with other molecules. The possibility of disruptions to hydrogen bonding by FdC should, however, be considered.
Methods
Instrumentation
1H NMR spectra were recorded at 400 or 600 MHz with Bruker AVANCE 400 and 600 NMR spectrometers with 14.1 and 7.05 T Ultrashield magnets, respectively. 13C NMR spectra were measured at 100.6 MHz, 31P NMR at 162.0 MHz, and 19F at 376.6 or 564.7 MHz with the same spectrometers. Coupling constants (J) and chemical shifts are given in ppm and hertz (Hz), respectively. Deuterated solvents were purchased from C/D/N Inc. EI (electron impact) and FAB (fast atom bombardment) mass spectra were obtained with a Thermo Scientific DFS mass spectrometer; ESI (electrospray) spectra were measured with a Bruker HCT Plus ion-trap mass spectrometer. Circular Dichroism spectra were recorded using a Jasco J-600 Circular Dichroism Spectrometer.
Chromatography
Desican silica gel (230–400 mesh) was used for flash chromatography. Thin layer chromatography (TLC) was performed on Silicycle Silica plate F-254 TLC plates using the following solvent mixtures:
Solvent A: methanol/dichloromethane (5:95, v/v).
Solvent B: methanol/dichloromethane (10:90, v/v).
Solvent C: methanol/dichloromethane (20:80, v/v).
HPLC was conducted with a Dionex 3000 Ionic Chromatography system. Reverse-phase Acclaim PA columns (4.6 × 150 mm, C18, 3 μm) were eluted with water/acetonitrile mixtures at a flow rate of 0.7 mL/min. Anion-exchange high-performance liquid chromatography was carried out on a DNAPac 200 column (4 × 250 mm) using a denaturing elution condition (containing 4 M urea at pH 12.4 and linear gradient elution with NaCl).18
Solvents and Chemicals
Toluene was dried by heating under reflux over sodium metal in the presence of benzophenone for 4 h and then distilled. N,N-Diisopropylethylamine, N,N-dimethylformamide, N-methylpyrrolidine, pyridine, and acetonitrile were dried by heating over calcium hydride and then distilled. Dichloromethane was dried by heating under reflux over phosphorus pentoxide and then distilled. All other reagents were purchased from Sigma-Aldrich or TCI without further purification prior to use unless stated otherwise.
Oligonucleotide Synthesis
Oligonucleotides were synthesized using an ABI 3400 DNA synthesizer on 1 μmol scales using the standard 1 μmol cycle conditions. Phosphoramidites were prepared as 100 mM solutions in dry acetonitrile. 5-(Ethylthio)-1H-tetrazole (250 mM in dry acetonitrile) was used as the activator and coupling reactions were allowed for 60 s. Detritylation was carried out by delivering trichloroacetic acid (3% in dichloromethane) for 110 s. After the solid-phase synthesis was complete, the products were incubated with concentrated aqueous ammonium hydroxide (28%) at 55 °C for 14 h. After lyophilization, the products were passed through Amberlite IR120 resin in the sodium form. The products were found by anion exchange HPLC to contain 92–96% full length sequences.
Synthesis of 5-Fluoro-2′-deoxycytidine 5
5-Fluoro-2′-deoxyuridine 1 (5.00 g, 20.3 mmol) was dried under vacuum at 70 °C for 3 h and then suspended in dry acetonitrile (150 mL). Chlorotrimethylsilane (7.76 mL, 60.9 mmol), followed by N-methylpyrrolidine (16.86 mL, 0.163 mol) were added, and the reaction mixture was stirred for 1 h at room temperature; upon cooling (ice-water bath), trifluoroacetic anhydride (8.47 mL, 60.9 mmol) was added dropwise over 5 min. After 30 min, 2,4,6-trimethylphenol (8.30 g, 60.9 mmol) was added and the reaction mixture was stirred at 0 °C for 3 h. Aqueous ammonium hydroxide (25 mL, 28%) was added and the flask was sealed and heated at 65 °C for 24 h. Upon cooling, the products were evaporated to dryness under reduced pressure, and the residue was purified by column chromatography on silica gel. The appropriate fractions, which were eluted with dichloromethane/methanol (93:7 v/v), were combined and concentrated under reduced pressure to give the title compound as a pale orange solid (2.98 g, 60%). The starting material 1 (1.80 g, 36%) was also recovered. Rf (System C): 0.28. HR-MS (EI) found 245.0806, C9H12FN3O4 required 245.0812. δH (DMSO-d6): 1.98 (1H, H-2′, ddd, J = 6.0, 7.0 and 13.3), 2.12 (1H, H-2″, ddd, J = 3.5, 6.0 and 13.3), 3.58 (2H, H-5′ and H-5″, m), 3.77 (1H, H-4′, ddd, J = 3.4, 3.4 and 3.4), 4.21 (1H, H-3′, m), 5.09 (1H, 5′-OH, dd, J = 4.9 and 4.9, ex), 5.20 (1H, 3′-OH, d, J = 4.2, ex), 6.11 (1H, H-1′, ddd, J = 2.0 (5JF,H), 6.0 and 7.4), 7.49 (1H, NH2, s, ex), 7.73 (1H, NH2, s, ex), 8.08 (1H, H-6, d, 3JF,H = 7.3). δC (DMSO-d6): 40.9, 61.5, 70.6, 85.6, 87.7, 125.6, 126.0, 135.3, 135.7, 153.8, 157.7, 157.9. δF (DMSO-d6): −167.3.
Synthesis of N-Acetyl-5-fluoro-2′-deoxycytidine 6a
To a solution of 5-fluoro-2′-deoxycytidine 5 (1.00 g, 4.08 mmol) in dry pyridine (20 mL), chlorotrimethylsilane (2.0 mL, 15.8 mmol) was added. After 30 min, acetyl chloride (0.44 mL, 6.2 mmol) was added and the reaction mixture was stirred at room temperature for 4 h. The products were chilled (ice-water bath). Water was added until all salts dissolved, followed by addition of triethylamine (2.85 mL, 20.4 mmol). The products were stirred at room temperature for 30 min and then evaporated to dryness under reduced pressure. The residue was purified by column chromatography on silica gel. The appropriate fractions, which were eluted with dichloromethane/ethanol (97:3 v/v), were combined and concentrated under reduced pressure to give the title compound as a yellow solid (0.85 g, 73%). Rf (System B): 0.12. HR-MS (EI) found 287.0909, C11H14FN3O5 required 287.0918. δH (DMSO-d6): 2.08 (1H, H-2′, ddd, J = 6.2, 6.2 and 13.3), 2.22 (3H, N–Ac, s) 2.28 (1H, H-2″, ddd, J = 4.4, 6.3 and 13.3), 3.59 (1H, H-5′, ddd, J = 3.4, 3.8 and 12.0), 3.67 (1H, H-5″, ddd, J = 3.4, 3.8 and 12.0), 3.85 (1H, H-4′, ddd, J = 3.4, 3.4 and 3.4), 4.24 (1H, H-3′, m), 5.20 (1H, 5′-OH, t, J = 4.4, ex), 5.27 (1H, 3′-OH, d, J = 3.8, ex), 6.05 (1H, H-1′, ddd, J = 1.2, 6.2 and 6.2), 8.51 (1H, H-6, d, J = 6.6), 10.59 (1H, NH, s, br, ex). δC (DMSO-d6): 25.1, 41.2, 61.0, 69.9, 86.7, 88.3, 130.2 (d, C-6, 2JF,C = 34.5), 137.5 (d, C-5, 1JF,C = 240.7), 152.7, 153.7, 170.3. δF (DMSO-d6): −158.3.
Synthesis of N-Benzoyl-5-fluoro-2′-deoxycytidine 6b
To a solution of 5-fluoro-2′-deoxycytidine 5 (0.200 g, 0.82 mmol) in dry pyridine (4 mL), chlorotrimethylsilane (0.42 mL, 3.31 mmol) was added. After 30 min, benzoyl chloride (0.17 mL, 1.46 mmol) was added and the reaction mixture was stirred at room temperature for 4 h. The products were chilled (ice-water bath). Water was added until all salts dissolved, followed by addition of triethylamine (0.57 mL, 4.1 mmol). The mixture was stirred at room temperature for 30 min and then evaporated under reduced pressure. The residue was purified by column chromatography on silica gel. The appropriate fractions, which were eluted with dichloromethane/ethanol (97:3 v/v), were combined and concentrated under reduced pressure to give the title compound as a white solid (194 mg, 68%). Rf (System B): 0.24. HR-MS (EI) found 349.1066, C16H16FN3O5 required 349.1074. δH (D2O): 2.28 (1H, H-2′, ddd, J = 6.1, 6.2 and 14.1), 2.46 (1H, H-2″, ddd, J = 4.7, 6.2 and 14.1), 3.71 (1H, H-5′, dd, J = 4.8 and 12.5), 3.80 (1H, H-5″, dd, J = 3.4 and 12.5), 4.03 (1H, H-4′, ddd, J = 4.3, 4.4 and 4.7), 4.37 (1H, H-3′, ddd, J = 4.4, 4.4 and 6.2), 6.17 (1H, H-1′, dd, J = 6.2 and 6.2), 7.46 (2H, t, J = 7.3), 7.56 (1H, t, J = 7.3), 7.85 (2H, d, J = 7.3), 8.14 (1H, H-6, br). δF (D2O): −156.7 (br).
Synthesis of N-Pivaloyl-5-fluoro-2′-deoxycytidine 6c
To a solution of 5-fluoro-2′-deoxycytidine 5 (0.200 g, 0.82 mmol) in dry pyridine (4 mL), chlorotrimethylsilane (0.42 mL, 3.31 mmol) was added. After 30 min, pivaloyl chloride (0.15 mL, 1.22 mmol) was added and the reaction mixture was stirred at room temperature for 4 h. The products were cooled (ice-water bath). Water was added until all salts dissolved, followed by addition of triethylamine (0.57 mL, 4.1 mmol). After stirring at room temperature for 30 min, the products were evaporated to dryness under reduced pressure. The residue was purified by column chromatography on silica gel. The appropriate fractions, which were eluted with dichloromethane/ethanol (98:2 v/v), were combined and concentrated under reduced pressure to give the title compound as a pale green solid (190 mg, 70%). Rf (system B): 0.30. HR-MS (EI) found 329.1381, C14H20FN3O5 required 329.1387. δH (DMSO-d6): 1.19 (9H, −(CH3)3, s), 2.11 (1H, H-2′, ddd, J = 6.2, 6.2 and 12.2), 2.27 (1H, H-2″, m), 3.59 (1H, H-5′, ddd, J = 3.7, 4.7 and 12.0), 3.67 (1H, H-5″, ddd, J = 3.6, 5.1 and 12.0), 3.85 (1H, H-4′, ddd, J = 3.5, 3.5 and 3.5), 4.25 (1H, H-3′, m), 5.21 (1H, 5′-OH, t, J = 4.9, ex), 5.29 (1H, 3′-OH, d, J = 4.4, ex), 6.05 (1H, H-1′, ddd, J = 1.3, 6.2 and 6.2), 8.48 (1H, H-6, s, br), 10.5 (1H, −NH, s, br, ex). δC (DMSO-d6) include the following signals: 27.2, 41.1, 61.1, 70.0, 86.6, 88.3, 138.0, 140.4. δF (DMSO-d6): −153.3.
Synthesis of N-Acetyl-5-fluoro-5′-(4,4′-dimethoxytrityl)-2′-deoxycytidine 7
N-Acetyl-5-fluoro-2′-deoxycutidine 6a (1.70 g, 5.92 mmol) was co-evaporated with dry pyridine (3 × 10 mL) and then taken up in dry pyridine (15 mL), followed by addition of 4,4′-dimethoxytrityl chloride (3.00 g, 8.85 mmol). After the reaction mixture was stirred at room temperature for 45 min, a triethylamine/ethanol (1:1 v/v) mixture (3.5 mL) was added. The products were diluted with dichloromethane (100 mL) and extracted with saturated aqueous sodium bicarbonate (35 mL). The layers were separated and the aqueous layer was back-extracted with dichloromethane (30 mL). The combined organic layers were dried (MgSO4) and evaporated to dryness. The oily residue was purified by column chromatography on silica gel. The appropriate fractions, which were eluted with dichloromethane/ethanol/triethylamine (98.2:1.5:0.3 v/v/v), were combined and concentrated under reduced pressure to give the title compound as a light pink solid (2.96 g, 85%). Rf (system A): 0.28. HR-MS (EI) found 589.2217, C32H32FN3O7 required 589.2224. δH (DMSO-d6): 2.16–2.23 (4H, H-2′ and −CH3 (acetyl), m), 2.34 (1H, H-2″, ddd, J = 4.8, 6.5 and 11.5), 3.20 (1H, H-5′, dd, J = 2.7 and 10.2), 3.27 (1H, H-5″, dd, J = 5.4 and 10.6), 3.74 (6H, −(OCH3)2, s), 3.98 (1H, H-4′q, J = 4.5), 4.27 (1H, H-3′, m), 5.37 (1 H, 3′-OH, d, J = 4.6, ex), 6.06 (1H, H-1′, t, J = 5.8), 6.90 (4H, d, J = 8.8), 7.23–7.40 (9H, m), 8.15 (1H, H-6, d, J = 6.3), 10.5 (1H, −NH, s, br, ex). δF (DMSO-d6): −158.6.
Synthesis of N-Acetyl-5-fluoro-5′-(4,4′-dimethoxytrityl)-2′-deoxycytidine-3′-(2-cyanoethyl)-N,N-diisopropylphosphoramidite 8
N-Acetyl-5-fluoro-5′-(4,4′-dimethoxytrityl)-2′-deoxycitidine 7 (1.00 g, 1.70 mmol) was co-evaporated with dry toluene (3 × 10 mL) and then dissolved in dry dichloromethane (20 mL). Dry N,N-diisopropylethylamine (1.5 mL, 8.6 mmol), followed by (2-cyanoethyl)-N,N-diisopropyl phosphochloridite (0.66 g, 2.8 mmol) were added, and the reaction mixture was stirred at room temperature for 30 min. N,N-Diisopropylethylamine (1.0 mL) was added and the products were evaporated under reduced pressure. The residue was purified by column chromatography on silica gel. The appropriate fractions, which were eluted with hexane/acetone/N,N-diisopropylethylamine (70:28:2 v/v), were combined and concentrated under reduced pressure. The residue was dissolved in dichloromethane (2 mL) and added dropwise to pentane (20 mL) at −40 °C under stirring. Cloudy suspension was centrifuged for 20 min and the supernatant was decanted. The white solid was collected and dried to give the title compound as a colorless solid (0.86 g, 64%). Rf (System A): 0.41. HR-MS (ESI) found 790.3381 (M – H+), C41H49FN5O8P required 789.3303. δH (CDCl3): 1.12 (3H, CH3, (i-Pr), d, J = 6.8), 1.19–1.21 (9H, −CH3 (i-Pr), m), 2.38 (1H, H-2′, ddd, J = 6.8, 6.8 and 13.8), 2.50 (1H, CH2, t, J = 6.3), 2.65 (1H, CH2, J = 8.3), 2.71 (3H, CH3 (acetyl) s), 2.66 (4H, P(CH2)2CN, m), 2.81 (1H, H-2″, m), 3.42–3.47 (2H, H-5′, H-5″, m), 3.57–3.65 (2H, 2xCH (i-Pr), m), 3.67–3.78 (1H, CH2, m), 3.81 (3H, −OMe, s), 3.82 (3H, −OMe, m), 3.84–3.92 (1H, CH2, m), 4.21–4.27 (1H, H-4′, m), 4.66–4.74 (1H, H-3′, m), 6.17–6.22 (1H, H-1′, m), 6.83–6.87 (4H, m), 7.23–7.33 (Ar, m, overlap with CHCl3 residue signals), 7.40–7.42 (2H, m), 7.62 (1H, NH, s, br), 8.19 and 8.24 (integrate 1H, H-6, each as d, 3JF,H = 5.5). δF (CDCl3): −166.4. δP (CDCl3): 148.8 and 149.2.
Circular Dichroism Spectroscopy
CD samples were prepared by dissolving oligonucleotides (10 μM) in Tris-HCl buffer (10 mM, pH 7.5) and NaCl (0.02–4 M) in autoclaved miliQ water. Oligonucleotides were annealed on a Mastercycler Nexus gradient thermal cycler by heating to 95 °C over 10 min followed by slow cooling to 25 °C over a period of 50 min.
UV/Vis Spectroscopy
The absorbance of annealed oligonucleotides was measured at 260 and 290 nm on a Genesys 10S UV–Vis Spectrophotometer. Samples were prepared in the same manner as those used for CD measurement.
Tm Measurement
Oligonucleotides were prepared at 5 μM (ssDNA) in Tris-HCl buffer (10 mM, pH 7.5, containing 20 mM NaCl) annealed in a Mastercycler Nexus gradient thermal cycler by first heating to 95 °C, followed by slow cooling to 25 °C over a period of 35 min. Melting temperatures were measured using a Cary 3000 UV/vis spectrophotometer. The temperature was increased by 1 °C/min.
19F NMR Spectroscopy
An appropriate amount of oligonucleotides with sodium cations was reconstituted in a Tris-HCl buffer (10 mM, pH 7.5, 0.5 mL in deuterium oxide) containing appropriate concentration of NaCl (0.02–4 M) and 0.01 μM of sodium trifluoroacetate (as an internal standard for 19F NMR). 19F NMR spectra were processed by applying the linear backward prediction algorithm in order to eliminate background signals from NMR tubes.19
Acknowledgments
The authors thank Razvan Simionescu for assistance with NMR spectroscopy.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b02461.
Copies of NMR spectra of compounds (PDF)
This work was funded by the Natural Science and Engineering Research Council of Canada.
The authors declare no competing financial interest.
Supplementary Material
References
- Muller K.; Faeh C.; Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–1886. 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- Richardson P. Fluorination methods for drug discovery and development. Expert Opin. Drug Discov. 2016, 11, 983–999. 10.1080/17460441.2016.1223037. [DOI] [PubMed] [Google Scholar]
- Yerien D. E.; Bonesi S.; Postigo A. Fluorination methods in drug discovery. Org. Biomol. Chem. 2016, 14, 8398–8427. 10.1039/c6ob00764c. [DOI] [PubMed] [Google Scholar]
- Guo F.; Li Q.; Zhou C. Synthesis and biological applications of fluoro-modified nucleic acids. Org. Biomol. Chem. 2017, 15, 9552–9565. 10.1039/c7ob02094e. [DOI] [PubMed] [Google Scholar]
- Chen H.; Viel S.; Ziarelli F.; Peng L. 19F NMR: a valuable tool for studying biological events. Chem. Soc. Rev. 2013, 42, 7971–7982. 10.1039/c3cs60129c. [DOI] [PubMed] [Google Scholar]
- Nakamura S.; Yang H.; Hirata C.; Kersaudy F.; Fujimoto K. Development of 19F-NMR chemical shift detection of DNA B-Z equilibrium using 19F-NMR. Org. Biomol. Chem. 2017, 15, 5109–5111. 10.1039/c7ob00706j. [DOI] [PubMed] [Google Scholar]
- Schmidt S.; Pein C.-D.; Fritz H.-J.; Cech D. Chemical synthesis of 2’-deoxyoligonucleotides containing 5-fluoro-2’-deoxycytidine. Nucleic Acids Res. 1992, 20, 2421–2426. 10.1093/nar/20.10.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W. L. Chemical conversion of thymidine into 5-methyl-2’-deoxycytidine. J. Chem. Soc., Chem. Commun 1981, 1089. 10.1039/C3981001089A. [DOI] [Google Scholar]
- Reese C. B.; Skone P. A. The protection of thymine and guanine residues in oligodeoxyribonucleotide synthesis. J. Chem. Soc., Perkin Trans. 1 1984, 1263–1271. 10.1039/p19840001263. [DOI] [Google Scholar]
- Miah A.; Reese C. B.; Song Q. Convenient intermediates for the preparation of C-4 modified derivatives of pyrimidine nucleosides. Nucleosides Nucleotides 1997, 16, 53–65. 10.1080/07328319708002521. [DOI] [Google Scholar]
- Tram K.; Sanghvi Y. S.; Yan H. Further optimization on the detritylation in solid phase oligodeoxyribonucleotide synthesis. Nucleosides, Nucleotides Nucleic Acids 2011, 30, 12–19. 10.1080/15257770.2010.537291. [DOI] [PubMed] [Google Scholar]
- Kypr J.; Kejnovska I.; Renciuk D.; Vorlickova M. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res. 2009, 37, 1713–1725. 10.1093/nar/gkp026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H.; Powers R.; Gibbons A.; Joshi D.. Z-DNA: Chemistry and Biological Relevance. In Elsevier Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Reedijk J., Ed.; Elsevier: Waltham, MA, 2017. [Google Scholar]
- Jovin T.; Soumpasis D. M.; McIntosh L. P. The transition between B-DNA and Z-DNA. Annu. Rev. Phys. Chem. 1987, 38, 521–558. 10.1146/annurev.pc.38.100187.002513. [DOI] [Google Scholar]
- Rich A.; Zhang S. Z-DNA: the long road to biological function. Nat. Rev. Genet. 2003, 4, 566–572. 10.1038/nrg1115. [DOI] [PubMed] [Google Scholar]
- Fuertes M. A.; Cepeda V.; Alonso C.; Pérez J. M. Molecular mechanisms for the B-Z transition in the example of poly[d(G-C)×(G-C)] polymers. A critical review. Chem. Rev. 2006, 106, 2045–2064. 10.1021/cr050243f. [DOI] [PubMed] [Google Scholar]
- Unpublished results.
- Led J. J.; Gesmar H. Application of the linear prediction method to NMR-spectroscopy. Chem. Rev. 1991, 91, 1413–1426. 10.1021/cr00007a007. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.