

Abstract
Background
Sirtuin1 (SIRT1) participates in a wide variety of cellular processes, but the molecular mechanism remains largely unknown. miR-155 is an element of the inflammatory signaling pathway in atherosclerosis. Therefore, we tested the hypothesis that TNF-α stimulates miR-155 to target SIRT1 and thereby regulates endothelial senescence, and we also explored the function of miR-155 as a regulator of cardiovascular diseases.
Material/Methods
TNF-α was used to stimulate human umbilical vein endothelial cells (HUVECs), after which protein and gene expression were assessed via Western blotting and RT-qPCR. miR-155 targeting of SIRT1 was confirmed via luciferase reporter assays, while MTT and senescence-associated β-galactosidase (SA-β-gal) assays were used for quantifying cellular proliferation and senescence.
Results
We found that miR-155 was upregulated in response to TNF-α treatment, in addition to inducing marked changes in SIRT1/FoxO-1/p21 pathway protein level. When we overexpressed miR-155 mimics, SIRT1 was markedly reduced, whereas miR-155 inhibition had the opposite effect in TNF-α-treated cells. We additionally confirmed that miR-155 was able to directly bind to SIRT1 3′-UTR, and that inhibition of miR-155 reduced the ability of TNF-α to induce senescence in HUVECs, thereby leading to their enhanced proliferation.
Simvastatin was associated with suppression of miR-155 expression in HUVECs following TNF-α treatment, and with a corresponding reduction in TNF-α-induced senescence, whereas miR-155 overexpression had the opposite effect.
Conclusions
Our findings suggest that TNF-α upregulates miR-155, which then targets SIRT1, suppressing its expression and driving HUVEC apoptosis. Simvastatin disrupted this senescence mechanism via the miR-155/SIRT1/FoxO-1/p21 pathway signaling. Hence, miR-155 is a possible therapeutic approach to endothelial senescence in the development of cardiovascular diseases.
MeSH Keywords: Atherosclerosis, Endothelial Cells, MicroRNAs, Sirtuin 1, Tumor Necrosis Factor-alpha
Background
Atherosclerosis is a highly prevalent chronic inflammatory disorder that causes extensive mortality [1], but the details of the underlying mechanism are unknown. In recent decades, accumulating evidence has revealed the essential regulatory role played by inflammation throughout atherosclerotic progression [2]. Moreover, proinflammatory tumor necrosis factor-α (TNF-α) promotes atherosclerosis progression and subsequent cardiovascular events via induction of inflammatory responses [3]. TNF-α is a typical member of the cytokine family, which promotes inflammation, differentiation, and apoptosis of endothelial cells (ECs). Atherosclerosis is increasingly thought to be an age-associated chronic disorder, and endothelial dysfunction always precedes the development of atherosclerosis. Endothelial senescence is an important mediator of vascular inflammation and dysfunction [4,5].
Sirtuins, a group of NAD+-dependent class III histone/protein deacetylases (HDACs), are involved in essential processes such as proliferation, apoptosis, senescence, and differentiation [6,7]. Sirtuin1 (SIRT1) is the most extensively characterized and best-studied member of the sirtuin family. SIRT1 acts as a regulatory switch in vascular endothelial cells homoeostasis [8,9] and is critically involved in regulating inflammation-related disorders via inhibition of proinflammatory cytokine secretion [10–12]. Previous studies have demonstrated that hypoxia, lipopolysaccharide (LPS), TNF-α, and high glucose decreased SIRT1 expression in ECs [9,10,13]. SIRT1 expression is modulated at diverse levels, and previous studies have shown that post-transcriptional regulation is important in modifying SIRT1 expression. Previous reports have shown that the SIRT1 3′-untranslated region (3′ UTR) is critical for its mRNA stability. Additionally, a series of microRNAs (miRNAs), such as miR-34a, miR-23b-3p, and miR-132-3p, modulate SIRT1 expression [9,14,15].
miRNAs are small noncoding RNAs (containing approximately 22 nucleotides) responsible for post-transcriptional modulation of gene expression and suppression of mRNA translation and stability, typically through binding to the 3′ UTR of target mRNAs [16]. Several miRNAs have been found to exert a vital role in cardiac and endothelial function, which is associated with cardiovascular disease progression [17,18]. miR-155, a typical miRNA with multiple functions, was recently identified as an element of the inflammatory signaling pathway in atherosclerosis [19,20]. miR-155 regulates gene expression associated with inflammation in various cell types in vitro and affects atherogenesis in vivo [21–26]. miR-155 can enhance or suppress lesion formation at a range of atherosclerotic stages. Therefore, miR-155 may be linked to the pathogenesis of atherosclerosis [27].
Recent evidence has demonstrated that some clinical drugs can modulate miRNA expression, suggesting that miRNAs may be therapeutic targets of these agents [9]. Statins and sartan are widely applied to treat dyslipidemia and related vascular disorders. However, whether valsartan and simvastatin modulate the effects of proinflammatory cytokines on endothelial function is largely unknown.
Despite the acknowledged importance of SIRT1 and miR-155 in the modulation of endothelial function, the relationship between them and the regulatory effects of inflammation on endothelial function during atherosclerosis remain unclear. Therefore, this study was designed to assess the possible association between SIRT1 and miR-155 and to determine how miR-155 regulates cardiovascular homeostasis. We found that simvastatin or valsartan can ameliorate TNF-α-induced HUVEC senescence through inhibition of miR-155.
Material and Methods
Endothelial cell culture
Human umbilical vein endothelial cells (HUVECs) (ScienCell, CA, USA) were cultured in endothelial cell medium (ECM) (ScienCell) containing 5% FBS, 1% endothelial cell growth supplement (ECGS) (ScienCell), and penicillin/streptomycin and were cultured at 37˚C with 5% CO2 and 21% O2. ECs were seeded at a proper density in accordance with each experimental procedure. HUVECs were treated with ±TNF-α (20 ng/ml) (PeproTech, USA).
miR-155 inhibition and overexpression
To inhibit miR-155 expression, HUVECs at 50%–70% confluency were transfected using miR-155 inhibitor (GenePharma, Shanghai, China) or miRNA inhibitor negative control (NC) (50 nmol/l) for 36 h using Lipofectamine 3000 (Invitrogen, Life Technologies, NY, USA) in Opti-MEM, following the manufacturer’s instructions. After transfection, Opti-MEM was exchanged for medium containing TNF-α, and cells were cultured for another 12 h. HUVECs were transfected using miR-155 mimic (GenePharma) to overexpress miR-155, or with NC mimic (50 nmol/l) for 48 h prior to additional analysis.
Western blotting
After washing, cold RIPA with protease inhibitors (Roche) was used to lyse cells for 15 min. Then, after centrifugation, the cell supernatant was kept frozen at −80°C until use. Bradford assays (Bio-Rad Laboratories; CA, USA) were employed to assess protein concentrations. Samples (20 μg) were separated via SDS-PAGE, after which dry transfer to nitrocellulose membranes was conducted. After being blocked in 5% BSA for 1 h, primary antibodies were used to probe the membranes overnight at 4°C, including antibodies targeting human SIRT1, acetylated-p53 at K382 (Ac-p53), p21, forkhead box O1 (FoxO-1) (Cell Signaling Technology; USA), Ac-FoxO-1, total p53 (1: 2000 dilution), and senescence marker protein-30 (SMP-30) (all from Santa Cruz Biotechnology). β-actin served as an internal control and was purchased from Sigma Aldrich. After incubation with the appropriate secondary antibodies (1: 10 000 dilution; GE Healthcare, Buckinghamshire, UK), the immunostained protein bands were visualized with an ECL system (ProteinSimple; Santa Clara, USA), followed by quantification using ProteinSimple image software. All samples had 3 biological replicates, and each biological replicate was assayed in duplicate; therefore, the data for each time point are an average of 6 individual replicate runs. Representative images of the immunostained bands are presented in the figures.
qPCR
TRIzol (Invitrogen) was used for EC total RNA extraction in accordance with standard protocols. A Nanodrop 2000 spectrophotometer (Invitrogen) was employed to determine RNA concentration and purity. cDNA was produced from 500 ng of total RNA utilizing a qScript microRNA Quantification System kit (microRNAs; Quanta Biosciences) or PrimeScript RT Master Mix (mRNAs; TaKaRa) in line with standard protocols. qPCR was conducted utilizing PerfeCTa SYBR Green SuperMix (microRNAs; Quanta Biosciences) and Power SYBR® Green PCR Master Mix (mRNAs; Applied Biosystems; USA) using an ABI 7500 machine (Applied Biosystems). The primer sequences were commercially obtained from Invitrogen and were as follows (forward and reverse, respectively):
SIRT1, 5′-GCT GTG AAA TTA CTG CAA GAG TG-3′ and
5′-AAT ACC ATC CCT TGA CCT GAA G -3′;
CYPA, 5′-TTG CTG TTC CTT AGA ATT TTG CC-3′ and
5′-ACC CTG ACA CAT AAA CCC TG -3′;
miR-155, 5′-TTA ATG CTA ATC GTG ATA GGG GT-3′;
U6, 5′-GAT GAC ACG CAA ATT CGT G-3′.
mRNA and miRNA expression were normalized to CYPA and U6, respectively. The ΔΔCt method was utilized to calculate fold changes in expression. All mRNA and miRNA samples had 3 biological replicates, and each biological replicate was also assayed in triplicate; therefore, the data for each time point are an average of 9 individual replicate runs.
Luciferase reporter assay
Firefly luciferase cDNA fused with the amplified 3′ UTR of human SIRT1 from a genomic DNA sample was cloned into psiCHECK-2 (Promega, USA). Luciferase in this vector was replaced by the produced wild-type (WT) and mutant 3′ UTR of SIRT1. HUVECs were added to 24-well plates, grown to 70–80% confluency, and then transfected using psiCHECK-2 that had been cloned so as to contain the WT or mutant SIRT1 3′ UTR (300 ng/well) using Lipofectamine 3000 in line with standard protocols. Following co-transfection using miR-155 mimic or NC mimic (50 nmol/l) for 48 h, a dual-luciferase assay system (Promega) was utilized, with firefly luciferase normalized to activity from Renilla luciferase for a control to determine transfection efficiency.
Senescence assessment
Senescence-associated β-galactosidase (SA β-Gal) activities were quantified in cells that had been fixed using formaldehyde following standard protocols (Sigma Aldrich). In brief, after PBS washing, cells underwent a 6-min fixation step followed by washing again in PBS. Next, staining buffer was added to wells overnight at 37°C. Stained cells were then imaged via microscopy (DM IL LED, Leica). Blue (positive) cells versus total cells in every field were determined by ImageJ software (NIH). The data are fold changes in activity relative to controls.
Cell proliferation assay (BrdU assay)
A Cell Proliferation ELISA kit (Roche) was used to facilitate BrdU incorporation during DNA synthesis to measure proliferation. To do so, we added 10 μl of BrdU labeling solution per sample in 100 μl of culture medium in 96-well plates. After 2 h at 37°C, FixDenat (200 uL) was administered per well and then discarded after 30 min at 25°C. Next, 100 μl per well of anti-BrdU-POD was added for 90 min at 25°C. Following 3 washes and the addition of 100 μl of substrate solution, absorbance was assessed at 370 nm (reference: 490 nm) after 20 min at 25°C. The values are presented as the fold absorbance difference relative to that of the control group.
Cell viability assay
We used CellTiter 96 AQueous One Solution (Promega) to assess the number of viable cells. This solution consists of MTT together with a reagent for electron coupling (PES). In brief, 20 μl of this compound was added per sample in 100 μl of media in 96-well plates for 1 h at 37°C. Absorbance was then determined at 490 nm (reference wavelength, 630 nm). The results are presented as the fold absorbance difference relative to that of the control group.
Statistical analysis
GraphPad Prism6 (CA, USA) was used for statistical testing. Data are means±SEM. Comparison of multiple groups or 2 groups was performed via one-way ANOVA and t tests, respectively. P<0.05 was the significance threshold. All experimental procedures were conducted in triplicate or more.
Results
TNF-α increased miR-155 expression and reduced SIRT1 expression in HUVECs
miR-155 is a frequently analyzed miRNA in the progression of atherosclerosis. To assess whether miR-155 participates in the effects of specific inflammatory stimuli, HUVECs were exposed to TNF-α (0, 10, 20, or 40 ng/ml) for 8 h, followed by qPCR to measure miRNA expression. Compared with NG, treatment with TNF-α from 10 to 40 ng/ml mediated dose-dependent increases in miR-155 expression by more than 2-fold (Figure 1A). We further performed a time-response assay of miR-155 expression in TNF-α-treated HUVECs. Specifically, miR-155 expression in HUVECs was determined after administration with TNF-α (20 ng/mL) for 0, 1, 2, 4, 6, and 8 h, and the results revealed that TNF-α (20 ng/ml) significantly enhanced miR-155 expression. This was apparent at 2 h, and expression peaked at 6 h (Figure 1B). Moreover, miR-155 expression was also triggered by LPS, an inflammatory cytokine (Figure 1C).
Figure 1.

TNF-α regulated miR-155 and SIRT1 expression in human umbilical vein endothelial cells (HUVECs). (A) Concentration-dependent effect of TNF-α on miR155 expression. (n=3, ** p<0.01 vs. control). (B) Time-dependent effect of TNF-α on miR-155 expression. (n=3, * p<0.05, ** p<0.01 vs. control). (C) LPS induced miR-155 expression in HUVECs. (n=3, * p<0.05 vs. control). (D) TNF-α reduced SIRT1 expression in HUVECs (n=3, * p<0.05 vs. control).
Following the above-described results, we further determined the effects of TNF-α on SIRT1 expression. HUVECs were administered TNF-α (20 ng/ml) for 8 h, followed by WB analysis and qPCR to assess the SIRT1 protein and mRNA levels, respectively. SIRT1 protein expression in HUVECs was reduced by TNF-α at 20 ng/ml (Figure 1D).
miR-155 regulated TNF-α-mediated SIRT1 expression in HUVECs
TNF-α bolstered miR-155 expression in addition to having reduced SIRT1 expression, which suggested that miR-155 was likely to contribute to TNF-α-mediated SIRT1 downregulation. To verify this hypothesis, we examined whether miR-155 could decrease SIRT1 expression. Following HUVEC miR-155 mimic transfection for 48 h, the SIRT1 mRNA and protein levels were assessed by qPCR and WB, respectively. miR-155 mimic transfection significantly enhanced miR-155 expression (Figure 2A), demonstrating that upregulation of miR-155 in HUVECs reduced the SIRT1 protein level by nearly 50% (Figure 2B). Therefore, miR-155 inhibited SIRT1 expression in HUVECs.
Figure 2.

Modulation of miR-155 regulated TNF-α-induced SIRT1 expression in HUVECs. (A, B) miR-155 mimic treatment impact on SIRT1 expression in HUVECs (n=3, ** P<0.01 vs. mimics NC). (C, D) Effect of miR-155 inhibitor on SIRT1 expression in HUVECs treated with or without TNFa. HUVECs were transfected with miR-155 inhibitor for 36 h followed by TNF-α stimulation for 12 h (n=3, * p<0.05, ** P<0.01 vs. inhibitor NC).
To confirm whether miR-155 inhibition attenuated TNF-α-mediated downregulation of SIRT1 expression, we transfected HUVECs with miR-155 inhibitor for 36 h, after which stimulation with TNF-α for 12 h was performed. Then, qPCR and WB were utilized to determine SIRT1 mRNA and protein levels, respectively. Transfection with miR-155 inhibitor induced a significantly decrease in miR-155 expression, showing that inhibition of miR-155 in HUVECs significantly upregulated SIRT1 protein expression by more than 30% (Figure 2C). Moreover, we transfected cells with miR-155 inhibitor under basal conditions and observed a significant increase in SIRT1 expression (Figure 2D), indicating that miR-155 modulated SIRT1 expression under physiological and pathological conditions. These results confirmed that upregulation of miR-155 in HUVECs suppressed SIRT1 expression, while downregulation of miR-155 enhanced SIRT1 expression, suggesting that SIRT1 may be a target of miR-155.
miR-155 directly targets SIRT1
Our present results provided a strong rationale to test the hypothesis that miR-155 directly targets SIRT1. miRNAs inhibit the translation initiation step via binding to target mRNA 3′ UTR. Therefore, the online prediction tool TargetScan was employed to recognize putative miR-155 targets. Bioinformatics analysis showed that SIRT1 was a miR-155 target, suggesting this sequence (perfect match) might be a molecular target of miR-155 (Figure 3A). According to the bioinformatics analysis, 2 PCR product fragments (F1-R1 and mut F1-R1 T1) were cloned into a psiCHECK-2 vector (Figure 3A). Unfortunately, based on the results of luciferase reporter assays, co-transfection with miR-155 mimic decreased the luciferase activity in both the FI-R1 and mut F1-R1 T1 groups (Figure 3B, 3C). Thus, we failed to confirm our hypothesis. These findings indicated that this sequence (perfect match) is not a molecular target of miR-155 (Figure 3A).
Figure 3.

SIRT1 may be a miR-155 target. Alignment between the miR-155 seed sequence and putative targeting sites in the SIRT1 3′UTR represented in red and green letters, respectively. (A) Sequence of human miR-155 and the predicted binding sequence with miR-155 within SIRT1 3′ untranslated regions (3′ UTRs) depend on the online prediction tool Target Scan are shown. (B, C, F) Luciferase reporter constructs containing F1-R1, mut F1-R1 T1, and F5-R2 sites of SIRT1 gene co-transfected with miR-155 mimics and the luciferase activities were assayed (n=5, ** P<0.01 vs. control). (D) All potential target sides within SIRT1 3′ UTRs we predicated are shown. (E) Sequence of human miR-155 and potential target binding sequences with a mismatch base pairing range from T1 to T4 are shown.
Previous work has demonstrated that if miRNA and target mRNA are an exact or nearly exact complementary match, target mRNA can be degraded. However, translational repression occurs when there is partial (imperfectly matched) complementarity between miRNA and target mRNA [20]. Therefore, new potential target sequences were predicted that had a mismatch base pairing in the seed sequence (Figure 3D). In addition, we identified 3 other possible sites and named these sites with or without a mismatch target sequence 1 to 4 (T1–T4, where T1 is the perfect match mentioned above). Subsequently, we evaluated the sequences T1 to T4 (Figure 3E). Based on these potential target sequences, we cloned the F5-R2 PCR product into the psiCHECK-2 vector and detected no difference between the miR-155 mimic and NC group, as indicated by luciferase reporter assays (Figure 3F). These findings suggested that the target sequence may be in the F1-R1 sites. Thus, we designed different forward primers, and the amplified sequences (F2-R1, F3-R1, F4-R1, and F5-R1) were cloned into the psiCHECK-2 vector. As shown in Figure 4A–4D, as the cloning sequence become closer to R1, the results of the luciferase reporter assay changed from positive to negative, suggesting that the real target sequence was between the F3-R1 and F4-R1 fragment (the T2 sequence). Therefore, we generated a T2 sequence mutant (Figure 4E). According to the result of luciferase reporter assays (Figure 4F), co-transfection of cells the WT SIRT1 3′ UTR T2 plasmid with the miR-155 mimic decreased relative luciferase activity. Furthermore, miR-155 did not repress the activity of the luciferase reporter with a mutant SIRT1 3′ UTR T2 plasmid, and these differences were all significant (P<0.05). Overall, these data provided experimental evidence that SIRT1 is directly targeted by miR-155.
Figure 4.

SIRT1 is a, miR-155 target. (A–D) Luciferase reporter constructs containing F2-R1, F3-R1, F4-R1, and F5-R1 sites of SIRT1 gene were co-transfected with miR-155 mimics and the luciferase activities were assayed (n=5, ** P<0.01 vs. control). (E) Sequence of human miR-155 and the predicted binding sequence within SIRT1 3′-UTRs T2 are shown, as is the MUT SIRT1 3′-UTR T2 reporter construct. (F) Cells were transfected with psiCHECK-2 vector containing the WT of MUT SIRT1 3′UTR T2. Cells were co-transfected with miR-155 mimics or mimics NC, respectively. After 48 h, luciferase activity was quantified. (n=5, ** P<0.01 vs. control).
Effects of miR-155 on HUVEC proliferation and senescence
BrdU and MTT assays were used to evaluate proliferation of HUVECs, which demonstrated that proliferation of HUVECs was significantly inhibited by miR155 expression (Figure 5A, 5B). Then, HUVECs underwent transfection with miR-155 mimic to observe whether upregulation of miR-155 could reduce cell proliferation. A significant effect on cell proliferation was observed in transfected HUVECs compared to the effect on the control cells after incubation for 48 h (Figure 5C, 5D). We confirmed that miR-155 downregulation increased proliferation of HUVECs. As expected, the results showed that suppression of miR-155 significantly promoted TNF-α-induced HUVEC proliferation (Figure 5E, 5F).
Figure 5.

Effects of TNF-α on HUVECs proliferation. (A, B) Effects of miR-155 on HUVECs proliferation. Cells were measured by BrdU assay (A), MTT assay (B). (n=3, * p<0.05 vs. control). (C, D) Overexpression of miR-155 reduced proliferation in HUVECs. HUVECs were measured by BrdU assay (C) and MTT assay (D) (n=3, * p<0.05 vs. mimics NC). (E, F) Inhibition of miR-155 increased proliferation in HUVECs. HUVECs were measured by BrdU assay (E), MTT assay (F) (n=3, * p<0.05 vs. inhibitor NC).
SIRT1 is an anti-aging gene that is has a vital role in preventing endothelial senescence. The results revealed that SIRT1 was directly targeted by miR-155, and TNF-α could upregulate miR-155 expression, implying that miR-155 might participate in senescence promotion. Thus, determination of cellular senescence of HUVECs was conducted using 2 indicators – anti-SMP-30 and SA β-Gal staining – and TNF-α significantly reduced SMP-30 protein expression by almost 50% and enhanced SA β-Gal staining by nearly 2-fold (Figure 6A). Then, the role of miR-155 in senescence of HUVECs was assessed, and the results showed that miR-155 mimic transfection increased SA β-Gal staining and downregulated SMP-30 (Figure 6B). Moreover, miR-155 was suppressed to verify reversal of the increased senescence. Briefly, after transfection of HUVECs with miR-155 inhibitor, the senescence of TNF-α-stimulated cells was examined as described above. Addition of the miR-155 inhibitor decreased SA β-Gal staining and increased SMP-30 protein levels (Figure 6C). Thus, we concluded that miR-155 promoted senescence in TNF-α-induced HUVECs.
Figure 6.

Effects of TNF-α on HUVECs senescence. (A) Impact of miR-155 on HUVECs senescence. Cells were measured by SA β-Gal activity and SMP-30. (n=3, * p<0.05 vs. control). (B) miR-155 overexpression promoted senescence in HUVECs. HUVECs were measured by SA β-Gal activity and SMP-30. (n=3, * p<0.05 vs. mimics NC). (C) Inhibition of miR-155 reversed the increase senescence in HUVECs. HUVECs were measured by SA β-Gal activity and SMP-30 (n=3, * p<0.05 vs. inhibitor NC).
miR-155 regulated HUVECs proliferation and senescence through SIRT1/FoxO-1/p53/p21 signaling
We next investigated the signaling pathway by which miR-155 regulated HUVECs proliferation and senescence. SIRT1 siRNA transfection decreased the protein level of SIRT1, confirming the silencing effect of SIRT1 siRNA on SIRT1 (Figure 7A). SIRT1, a deacetylase for multiple transcription factors, including FoxO-1 and p53, has been demonstrated to modulate critical cellular processes, including proliferation, senescence, differentiation, and apoptosis. Thus, the downstream signaling targets of SIRT1, including the protein levels of acetylated FoxO-1, acetylated p53, and p21, were also determined. Knockdown of miR-155 decreased p21, Ac-p53, and Ac-FoxO-1 protein expression, and SIRT1 silencing reversed this effect (Figure 7B).
Figure 7.

The role of miR-155 in the SIRT1/FoxO-1/P53/P21 pathway. (A) SIRT1 siRNA transfection reduced the expression of SIRT1. (B) Acetylated FoxO-1, FoxO-1, acetylated p53, p53, and p21 in HUVECs of each group were measured by Western blotting. β-actin was used as an internal control (n=3, * p<0.05 vs. untreated, # p<0.05 vs. NC+TNF-α, & p<0.05 vs. miR-155 inhibitor+TNF-α, @ p<0.05 vs. simock+TNF-α).
Then, we assessed whether HUVEC proliferation and senescence were influenced by siSIRT1, and the results showed that siSIRT1 prevented the decrease in TNF-α-induced cell proliferation triggered by miR-155 (Figure 8A). Similarly, the effect of miR-155 on the upregulation of senescence and SMP-30 expression were substantially decreased by SIRT1 knockdown (Figure 8B). These outcomes indicated that miR-155 regulated HUVEC proliferation and senescence through the SIRT1/FoxO-1/p53/p21 pathway.
Figure 8.

miR-155 regulated proliferation and senescence through the SIRT1/FoxO-1/P53/P21-dependent pathway. (A) The proliferation effects of miR-155 downregulation were mostly abolished after SIRT1 knockdown. (B) SA β-Gal activity and Western blot analysis of SMP-30 protein levels in HUVECs after miR-155 knockdown. (n=3, * p<0.05 vs. untreated, # p<0.05 vs. NC+TNF-α, & p<0.05 vs. miR-155 inhibitor+TNF-α, @ p<0.05 vs. simock+TNF-α)
Valsartan and simvastatin ameliorated TNF-α-induced HUVEC senescence through inhibition of miR-155
HUVECs were pretreated with or without valsartan (40 μmol/L) or simvastatin (1 μmol/L) for 3 h, followed by subsequent stimulation with TNF-α for 8 h. WB analysis and qPCR were employed to determine expression changes in miR-155 and SIRT1. Our results demonstrated significantly enhanced miR-155 expression in HUVECs exposed to TNF-α, accompanied by a parallel reduction in SIRT1 levels, as previously shown. When HUVECs were treated with TNF-α together with valsartan or simvastatin, miR-155 expression was significantly reduced, accompanied by a parallel upregulation in SIRT1 expression (Figure 9A, 9B) and a decrease in TNF-α-induced HUVEC senescence (Figure 9C). In addition, we examined the effect of valsartan or simvastatin in HUVECs overexpressing miR-155 to confirm whether the endothelial-protective function of valsartan or simvastatin depended on miR-155. Transfection of HUVECs with miR-155 mimic and exposure to TNF-α with valsartan or simvastatin significantly increased miR-155 expression. However, transfection with miR-155 mimic negated the protective effects of valsartan or simvastatin on SIRT1 and senescence in TNF-α-induced HUVECs (Figure 10A–10C). Overall, downregulation of miR-155 expression mediated the protective effects of valsartan and simvastatin on endothelial dysfunction.
Figure 9.

Effect of valsartan and simvastatin on senescence and miR-155 and SIRT1 expression in HUVECs treated with TNF-α. (A–C) HUVECs were pretreated with or without valsartan (40 umol/L) or simvastatin (1μmol/L) for 3 h and subsequently stimulated with TNF-α for 8 h, and then changes in the expression of miR-155 (A) and SIRT1 (B) were assessed by WB and qPCR. Cells senescence was measured by SA β-Gal activity and SMP-30 (C). (n=3, ** p<0.01, * p<0.05 vs. TNF-α only group).
Figure 10.

Valsartan and simvastatin ameliorated TNF-α-induced HUVECs senescence through inhibition of miR-155 expression. HUVECs were transfected using miR-155 inhibitor for 36 h followed by valsartan (40 umol/L) or simvastatin (1 μmol/L) stimulation for 3 h, and subsequently stimulated with TNF-α for 8 h, then changes in the expression of SIRT1 were assessed by WB (A). Cells senescence was measured by SA β-Gal activity and SMP-30 (B) and (C). (n=3, ** P<0.01, * p<0.05 vs. mimics NC).
Discussion
Here, we found TNF-α significantly enhanced miR-155 expression and decreased SIRT1 expression. Moreover, SIRT1 was directly targeted by miR-155. In addition, miR-155 regulated endothelial cell senescence and proliferation via the SIRT1/FoxO-1/p53/p21 pathway. Overexpression of miR-155 contributed to the TNF-α-triggered reduction in SIRT1 expression and impairment of endothelial senescence. Moreover, anti-miR-155 inhibitor partially reduced TNF-α-induced SIRT1 suppression. Functional j ju showed that miR-155 inhibitor reduced TNF-α-mediated endothelial senescence and bolstered cellular proliferation. It was also verified that miR-155 inhibitor enhanced SIRT1 expression under basal conditions, suggesting that miR-155 can control SIRT1 expression under physiological conditions. Moreover, the presence of simvastatin or valsartan restored SIRT1 expression and inhibited senescence via suppression of miR-155 in TNF-α-treated HUVECs. However, miR-155 mimic prevented simvastatin- or valsartan-mediated protection. These outcomes suggested that miR-155 might be a valuable intervention target to prevent cardiovascular disease (Figure 11).
Figure 11.
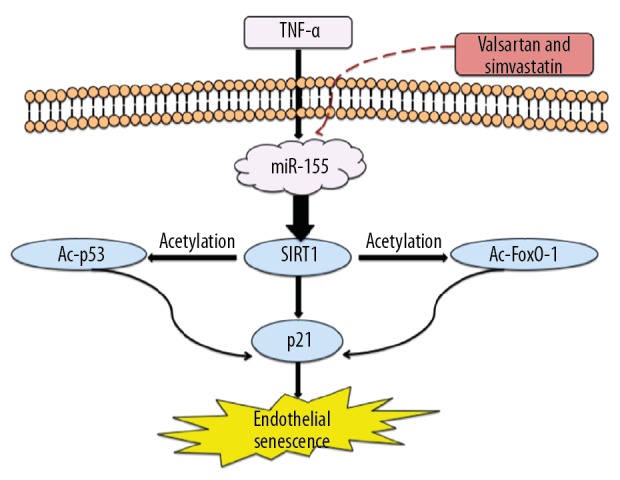
Schematic overview of the mechanisms of valsartan and simvastatin modulated TNF-α-induced endothelial senescence via miR-155/SIRT1 signaling. TNF-α enhanced intracellular miR-155 production, which in turn downregulated SIRT1 and thereby modulated FoxO-1/p53/p21-mediated endothelial senescence. Furthermore, valsartan and simvastatin decreased miR155, upregulated SIRT1, and attenuated TNF-α-induced endothelial dysfunction.
miR-155 directly targets SIRT1
This study is the first to identify that SIRT1 is a miR-155 target, which promotes TNF-α-induced senescence via the SIRT1/FoxO-1/p53/p21 pathway in HUVECs, and is also the first study to demonstrate that valsartan modulates the effects of proinflammatory cytokines on endothelial function.
Accumulating evidence has suggested a vital role of miR-155 in regulating a variety of cellular functions, including inflammation, hematopoiesis, immunological response, and viral infection [22,23,25]. A recent report showed significant downregulation of the circulating level of miR-155 in people with coronary artery disease [28]. Previous studies demonstrated that miR-155 can enhance or suppress lesion formation, based on atherosclerosis stage [27]. Furthermore, previous data have indicated that miR-155 is critically involved in inflammation and is potentially correlated with atherosclerosis pathogenesis in numerous cell types [2,27]. Together, these studies demonstrated that miR-155 might be a vital modulator of cardiovascular disease processes.
miR-155, a typical miRNA with multiple functions, simultaneously modulates numerous biological process by targeting diverse genes [20]. Thus, luciferase reporter assays have identified several direct miR-155 targets, such as helper T 1 (Th1), Sma- and Mad-related protein 2 (SMAD2), secretogranin II (SCG2), SH2-containing inositol 5′-phosphatase 1 (SHIP1), and SOCS1 [2,29]. Moreover, other research groups have found that eNOS and NF-κB are direct miR-155 targets in ECs [30]. Taken together, the results show that miR-155 might exert a conflicting function in endothelial cells.
This is the first study showing that miR-155 targets SIRT1. miR-155 downregulated SIRT1 primarily via SIRT1 3′ UTR binding. First, the online prediction tool TargetScan indicated that miR-155 may target SIRT1. However, the luciferase reporter assays indicated that this sequence, which was a perfect match, was not a molecular target of miR-155. Previous studies have shown that when a miRNA and target mRNA are exactly or almost exactly complementary to one another, degradation of target mRNA will occur. However, translation repression occurs only in the case of partial (imperfectly matched) complementarity between miRNA and target mRNA [20]. Therefore, we predicted new potential target sequences that had a mismatch base pairing in the seed sequence. To determine the general position of the sequence, these mismatch sequences were cloned into cells. The luciferase reporter assays indicated that one of these mismatch sequences is the real target binding sequence of miR-155. Although a previous [31] study reported that SIRT1 may be a direct target of miR-155, the authors revealed that miR-155 downregulated SIRT1 expression by binding to 3 sites on the SIRT1 3′ UTR. The sequences in the previous study and our study were different. Moreover, our study was performed in different cells.
SIRT1, referred to as an anti-aging gene, is critical for preventing endothelial senescence. Accumulating evidence indicates that senescence of the endothelium plays an important role in promoting vascular inflammation and dysfunction. Further evidence has suggested that the SIRT1 3′ UTR is critical for SIRT1 mRNA stability. Additionally, a series of miRNAs, including miR-132-3p, miR-23b-3p, and miR-34a, were found to modulate SIRT1 expression [9,14,15]. Our results demonstrated that miR-155 upregulation inhibited SIRT1 expression and HUVEC proliferation and promoted TNF-α-induced senescence in HUVECs. The data showed that miR-155 reduced the protein level of SIRT1 but had no effect on the mRNA level. These findings indicate that SIRT1 expression is reduced by miR-155 via translational repression, not degradation, of the target mRNA. Furthermore, inhibition of miR-155 attenuated TNF-α-triggered downregulation of SIRT1 expression and decreased TNF-α-induced senescence in HUVECs. Interestingly, we also found that transfection with a miR-155 inhibitor enhanced SIRT1 expression under basal conditions, indicating that miR-155 regulates SIRT1 expression under physiological and pathological conditions. Moreover, the data revealed that miR-155 regulates HUVEC proliferation and senescence through the SIRT1/FoxO-1/p53/p21 pathway.
This indicates that miR-155 plays a key role in cardiovascular homeostasis regulation via the inhibition of SIRT1 expression.
Valsartan and simvastatin ameliorated TNF-α-induced HUVEC senescence through inhibition of miR-155 expression
Recent growing evidence has demonstrated that some clinical drugs can modulate miRNA expression, suggesting that miRNAs may be therapeutic targets of these agents [9]. Statins are widely applied to treat dyslipidemia and relevant vascular disorders. In vitro and in vivo studies has revealed statins to have pleiotropic actions, including maintenance of plaque stability, induction of increased NO availability, anti-platelet effects, reduction of inflammatory effects, and immune regulation, in addition to their anti-fibrotic and anti-oxidant properties and lipid-lowering effects [32,33]. Previous studies have reported that simvastatin ameliorated TNF-α-induced endothelial vasorelaxation through inhibition of miR-155 expression via mechanisms involving upregulation of eNOS and its downstream signaling pathway in HUVECs [29]. Another previous study reported that SIRT1 expression was reduced by TNF-α and enhanced by simvastatin [34]. Nevertheless, whether miR-155 modulates the protective roles of simvastatin on SIRT1 expression and endothelial cell function following treatment with TNF-α remains unknown. Sartan, an orally administered selective antagonist of angiotensin II (Ang II) receptor blockers (ARBs), is approved for treating hypertension. Previous experimental studies have demonstrated that telmisartan, an ARB, modulates endothelial inflammation induced by inflammatory stimuli in HUVECs [35]. Moreover, effects similar to those of telmisartan were achieved with 2 lipophilic ARBs – olmesartan and candesartan. However, whether valsartan modulates the effects of proinflammatory cytokines on endothelial function is largely unknown.
To investigate this potential relationship, we pretreated HUVECs with valsartan or simvastatin. To the best of our knowledge, the present study is the first to demonstrate that valsartan attenuates the inflammatory process induced by TNF-α. Here, our results showed that simvastatin or valsartan decreased miR-155 expression, accompanied by a parallel upregulation in SIRT1 expression and a decrease in TNF-α-induced HUVEC senescence. After transfection of miR-155 mimics, simvastatin or valsartan led to a negation of the protective roles of SIRT1 expression, as well as senescence, in TNF-α-induced HUVECs. The above findings indicate that downregulation of miR-155 expression mediated the protective effects of simvastatin or valsartan on endothelial dysfunction. Thus, simvastatin or valsartan promoted SIRT1 expression via the downregulation of miR-155 expression. Hence, these results might supply novel insights into the potential mechanism of the anti-senescence effect of simvastatin or valsartan in ECs.
Conclusions
We showed that miR-155 mediates TNF-α-induced endothelial senescence via suppressing SIRT1. Simvastatin disrupted this through altering miR-155/SIRT1/FoxO-1/p21 pathway signaling. As such, miR-155 may be a viable therapeutic approach for endothelial senescence during the development of cardiovascular diseases.
Footnotes
Source of support: This study was supported by grants from the National Natural Science Foundation of China (NSFC) 81700405 and 51890894 to E. Zhang, and 81970326 and 51890891 to Y. Wu. Q. Guo was supported by Chinese Scholarship Council (CSC) scholarship 201706210415 and China Postdoctoral Science Foundation 2019M650032
References
- 1.Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Ma X, Ma C, Zheng X. MicroRNA-155 in the pathogenesis of atherosclerosis: Aa conflicting role? Heart Lung Circ. 2013;22:811–18. doi: 10.1016/j.hlc.2013.05.651. [DOI] [PubMed] [Google Scholar]
- 3.Skoog T, Dichtl W, Boquist S, et al. Plasma tumour necrosis factor-alpha and early carotid atherosclerosis in healthy middle-aged men. Eur Heart J. 2002;23:376–83. doi: 10.1053/euhj.2001.2805. [DOI] [PubMed] [Google Scholar]
- 4.Zhou Z, Chen Y, Ni W, Liu T. Upregulation of nuclear factor IA suppresses oxidized low-density lipoprotein-induced endoplasmic reticulum stress and apoptosis in human umbilical vein endothelial cells. Med Sci Monit. 2019;25:1009–16. doi: 10.12659/MSM.912132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minamino T, Komuro I. Vascular cell senescence: Contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- 6.Haigis MC, Guarente LP. Mammalian sirtuins – emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–21. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 7.Michan S, Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shah A, Gray K, Figg N, et al. Defective base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation. 2018;138(14):1446–62. doi: 10.1161/CIRCULATIONAHA.117.033249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arunachalam G, Lakshmanan AP, Samuel SM, et al. Molecular interplay between microRNA-34a and sirtuin1 in hyperglycemia-mediated impaired angiogenesis in endothelial cells: Effects of metformin. J Pharmacol Exp Ther. 2016;356:314–23. doi: 10.1124/jpet.115.226894. [DOI] [PubMed] [Google Scholar]
- 10.Yang L, Zhang J, Yan C, et al. SIRT1 regulates CD40 expression induced by TNF-alpha via NF-κB pathway in endothelial cells. Cell Physiol Biochem. 2012;30:1287–98. doi: 10.1159/000343318. [DOI] [PubMed] [Google Scholar]
- 11.Rajendrasozhan S, Yang SR, Kinnula VL, Rahman I. SIRT1, an anti-inflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;177:861–70. doi: 10.1164/rccm.200708-1269OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moon MH, Jeong JK, Lee YJ, et al. SIRT1, a class III histone deacetylase, regulates TNF-alpha-induced inflammation in human chondrocytes. Osteoarthritis Cartilage. 2013;21:470–80. doi: 10.1016/j.joca.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 13.Zhang E, Guo Q, Gao H, et al. Metformin and resveratrol inhibited high glucose-induced metabolic memory of endothelial senescence through SIRT1/p300/p53/p21 pathway. PLoS One. 2015;10:e0143814. doi: 10.1371/journal.pone.0143814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou W, Xu J, Wang C, et al. MiR-23b-3p regulates apoptosis and autophagy via suppressing SIRT1 in lens epithelial cells. J Cell Biochem. 2019;120(12):19635–46. doi: 10.1002/jcb.29270. [DOI] [PubMed] [Google Scholar]
- 15.Qu H, Li T, Jin H, et al. Silent mating type information regulation 2 homolog (SIRT1) influences osteogenic proliferation and differentiation of MC3T3-E1 cells via regulation of miR-132-3p. Med Sci Monit. 2019;25:2289–95. doi: 10.12659/MSM.912392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beermann J, Piccoli MT, Viereck J, Thum T. Non-coding RNAs in development and disease: Background, mechanisms, and therapeutic approaches. Physiol Rev. 2016;96:1297–325. doi: 10.1152/physrev.00041.2015. [DOI] [PubMed] [Google Scholar]
- 17.Duygu B, de Windt LJ, da Costa Martins PA. Targeting microRNAs in heart failure. Trends Cardiovasc Med. 2016;26:99–110. doi: 10.1016/j.tcm.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 18.Ye Y, Perez-Polo JR, Qian J, Birnbaum Y. The role of microRNA in modulating myocardial ischemia-reperfusion injury. Physiol Genomics. 2011;43:534–42. doi: 10.1152/physiolgenomics.00130.2010. [DOI] [PubMed] [Google Scholar]
- 19.Elton TS, Selemon H, Elton SM, Parinandi NL. Regulation of the MIR155 host gene in physiological and pathological processes. Gene. 2013;532:1–12. doi: 10.1016/j.gene.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 20.Faraoni I, Antonetti FR, Cardone J, Bonmassar E. miR-155 gene: A typical multifunctional microRNA. Biochim Biophys Acta. 2009;1792:497–505. doi: 10.1016/j.bbadis.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Duan Q, Mao X, Xiao Y, et al. Super enhancers at the miR-146a and miR-155 genes contribute to self-regulation of inflammation. Biochim Biophys Acta. 2016;1859:564–71. doi: 10.1016/j.bbagrm.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Pan Q, Zhao Y, et al. MicroRNA-155 regulates ROS production, NO generation, apoptosis and multiple functions of human brain microvessel endothelial cells under physiological and pathological conditions. J Cell Biochem. 2015;116:2870–81. doi: 10.1002/jcb.25234. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Zhao F, Yu X, et al. MicroRNA-155 modulates the proliferation of vascular smooth muscle cells by targeting endothelial nitric oxide synthase. Int J Mol Med. 2015;35:1708–14. doi: 10.3892/ijmm.2015.2181. [DOI] [PubMed] [Google Scholar]
- 24.Wu XY, Fan WD, Fang R, Wu GF. Regulation of microRNA-155 in endothelial inflammation by targeting nuclear factor (NF)-kappaB P65. J Cell Biochem. 2014;115:1928–36. doi: 10.1002/jcb.24864. [DOI] [PubMed] [Google Scholar]
- 25.Ye J, Guo R, Shi Y, et al. miR-155 regulated inflammation response by the SOCS1-STAT3-PDCD4 axis in atherogenesis. Mediators Inflamm. 2016;2016 doi: 10.1155/2016/8060182. 8060182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cerutti C, Soblechero-Martin P, Wu D, et al. MicroRNA-155 contributes to shear-resistant leukocyte adhesion to human brain endothelium in vitro. Fluids Barriers CNS. 2016;13(1):8. doi: 10.1186/s12987-016-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang E, Wu Y. Dual effects of miR-155 on macrophages at different stages of atherosclerosis: LDL is the key? Med Hypotheses. 2014;83:74–78. doi: 10.1016/j.mehy.2014.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Fichtlscherer S, De Rosa S, Fox H, et al. Circulating microRNAs in patients with coronary artery disease. Circ Res. 2010;107:677–84. doi: 10.1161/CIRCRESAHA.109.215566. [DOI] [PubMed] [Google Scholar]
- 29.Wei Y, Nazari-Jahantigh M, Neth P, et al. MicroRNA-126, -145, and -155: A therapeutic triad in atherosclerosis? Arterioscler Thromb Vasc Biol. 2013;l33:449–54. doi: 10.1161/ATVBAHA.112.300279. [DOI] [PubMed] [Google Scholar]
- 30.Sun HX, Zeng DY, Li RT, et al. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension. 2012;60:1407–14. doi: 10.1161/HYPERTENSIONAHA.112.197301. [DOI] [PubMed] [Google Scholar]
- 31.Jens H, Benjamin L, Ludwig C, et al. miR-124a and miR-155 enhance differentiation of regulatory T cells in patients with neuropathic pain. J Neuroinflammation. 2016;13:248. doi: 10.1186/s12974-016-0712-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mascitelli L, Goldstein MR. Changes in plaque composition and the pleiotropic effects of statins. J Am Coll Cardiol. 2016;68:1250–51. doi: 10.1016/j.jacc.2016.05.095. [DOI] [PubMed] [Google Scholar]
- 33.Margaritis M, Channon KM, Antoniades C. Statins as regulators of redox state in the vascular endothelium: Beyond lipid lowering. Antioxid Redox Signal. 2014;20:1198–215. doi: 10.1089/ars.2013.5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du G, Song Y, Zhang T, et al. Simvastatin attenuates TNFalphainduced apoptosis in endothelial progenitor cells via the upregulation of SIRT1. Int J Mol Med. 2014;34:177–82. doi: 10.3892/ijmm.2014.1740. [DOI] [PubMed] [Google Scholar]
- 35.Cicha I, Urschel K, Daniel WG, Garlichs CD. Telmisartan prevents VCAM-1 induction and monocytic cell adhesion to endothelium exposed to non-uniform shear stress and TNF-alpha. Clin Hemorheol Microcirc. 2011;48:65–73. doi: 10.3233/CH-2011-1394. [DOI] [PubMed] [Google Scholar]