

Abstract
Representing the most fundamental lipid class, fatty acids (FA) play vital biological roles serving as energy sources, cellular signaling molecules, and key architectural components of complex lipids. Direct infusion electrospray ionization spectrometry, also known as shotgun lipidomics, has emerged as a rapid and powerful toolbox for lipid analysis. While shotgun lipidomics can be a sensitive approach to FA detection, the diverse molecular structure of FA presents challenges for unambiguous identification and the relative quantification of isomeric contributors. In particular, pinpointing double bond position(s) in unsaturated FA and determining the relative contribution of double bond isomers has limited the application of the shotgun approach. Recently, we reported the use of gas-phase ion/ion reactions to facilitate the identification of FA. Briefly, singly deprotonated FA anions undergo charge inversion when reacted in the gas phase with tris-phenanthroline magnesium dications by forming [FA – H + MgPhen]+ complex ions. These charge-inverted FA complex cations fragment upon ion-trap collision-induced dissociation (CID) to generate product ion spectra unique to individual FA isomers. Herein, we report the development of a mass spectral library comprised of [FA – H + MgPhen]+ product ion spectra. The developed FA library permits confident FA identification, including polyunsaturated FA isomers. Furthermore, we demonstrate the ability to determine relative contributions of isomeric FA using multiple linear regression analysis paired with gas-phase ion/ion reactions. We successfully applied the presented method to generate a FA profile for bovine liver phospholipidome based entirely on gas-phase chemistries.
Graphical Abstract
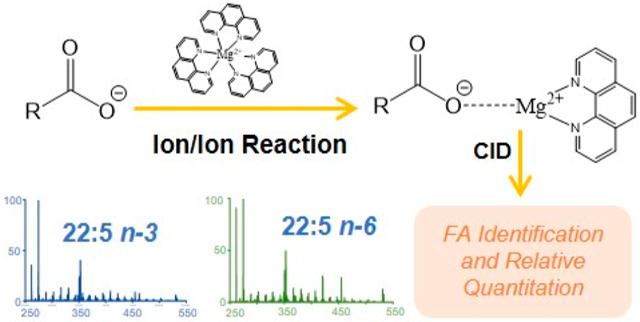
Fatty acids (FAs) constitute a class of ubiquitous lipids, present in all living organisms. FAs are known to serve vital roles in cellular energy storage, cellular signaling, modification of proteins, and as key architectural components of complex lipids such as glycerophospholipids, sphingolipids, and sterol esters.1-5 FA are comprised of at least one carboxylic acid moiety and an aliphatic chain. Despite their apparently simple composition, FA display dramatic structural diversity. Specifically, variations in aliphatic chain length, degree of unsaturation, site(s) of unsaturation, and modifications such as hydroxylation,6 nitrosylation,7 and methyl chain branching8 are observed. Recently, alterations to FA structure and composition have been linked with the progression of numerous chronic diseases such as heart disease, diabetes, several types of cancer, and neurodevelopmental disorders like attention-deficit/hyperactivity disorder.9-14 Therefore, efforts to both characterize and quantify FA are a principle focus of many recent research endeavors, including FA profiling for biomarker discovery.14-17 Moreover, in a recent investigation profiling human plasma samples from patients with breast cancer and type 2 diabetes, Zhang et al. report that the ratio of lipid isomers differing on sites of unsaturation (so-called double bond positional isomers) could be of particular importance in lipid biomarker discovery.14
Mass spectrometry (MS) coupled with some form of chromatography represents the most widely adopted analytical platform for routine FA identification and quantitation. Gas chromatography (GC) and liquid chromatography (LC) are mainstays of FA analysis yet are not without their respective challenges. For instance, the analysis of FA via GC–MS first requires conversion of FA to their respective methyl esters. A significant disadvantage to GC–MS analysis of FA methyl esters is the inability to distinguish the resulting electron ionization mass spectra of lipid isomers, particularly highly unsaturated FA isomers.18 Benefits of FA analysis with LC–MS/MS include no requirement of FA derivatization prior to analysis and validated methods for FA quantitation.19 Once again, however, the resulting mass spectra (in this instance, collision-induced dissociation mass spectra) do not readily distinguish FA double bond positional isomers. This restricts methods to those including LC workflows, but even the separation of some double bond positional FA isomers can only be achieved using silver-ion chromatography.18
Direct infusion electrospray ionization (ESI), commonly referred to as shotgun lipidomics, has gained popularity due to ease of use, reduced analysis time, and minimal sample volume requirements.3,20 Briefly, shotgun lipidomics involves the direct infusion of crude lipid extract into the mass spectrometer without prior fractionation. Direct infusion under negative ESI conditions produces abundant [M – H]− anions of FA due to facile deprotonation of the carboxylic acid moiety. From accurate mass measurements of the [M – H]− ion mass-to-charge (m/z) ratio, FA sum composition information (i.e., aliphatic chain length, degree of unsaturation) can be determined. However, accurate mass measurements alone do not provide insight into FA structural characteristics, such as double bond position(s). Furthermore, reliance on tandem MS (MS/MS) methods for FA analysis, specifically low-energy collision induced dissociation (CID), has been minimally effective. In negative ion mode, activation of deprotonated FA anions results in CID spectra dominated by small molecule losses (i.e., carbon dioxide and water neutral losses) that do not provide insight into structural characteristics important in biochemical function such as double bond position(s).21 Notably, relative abundances of water and carbon dioxide neutral losses are sensitive to structural features, which can further be exploited for FA identification and quantitation.22
To enhance the number of structurally informative product ions, two distinct strategies regarding FA derivatization are employed. The first approach involves derivatization of the double bond, while the second approach involves derivatization of the carboxylic acid moiety. Reactions used to derivatize the double bond include ozonolysis18,23-28 and the Paternò–Büchi (PB) reaction.29-31 Both strategies generate diagnostic product ions indicative of double bond position. Employing the PB reaction, Ma et al. demonstrated both relative and absolute quantitation of unsaturated lipids in rat brain tissue, reporting nearly one-half of the identified lipid species existing as mixtures of double bond positional isomers.30 While absolute quantitation has yet to be explored with ozonolysis, relative changes in isomeric lipid concentration can be achieved.32
Charge-switching strategies represent the second approach to FA derivatization. Charge-switching derivatives directly target modification of the FA carboxyl group with a fixed, positively charged reagent. Wang et al. describe covalent modification of the carboxyl moiety via amidation of the carboxyl moiety with N-[4-(aminomethyl)phenyl]pyridinium (AMPP) ion in solution.33 ESI–MS/MS analysis of the AMPP-derivatized FA ion provided informative CID spectra that could be utilized for both structural elucidation and quantitation of FA. Specifically, Han and co-workers have shown that wet-chemical derivatization of fatty acids to AMPP-conjugates followed by CID of the FA-AMPP ion can be used to undertake relative quantification of binary and ternary mixtures of fatty acid isomers utilizing a fitting routine similar to that outlined here.33 Extending this approach, Yang et al. successfully quantified double bond positional isomers of AMPP-derivatized linolenic acid (i.e., 18:3(9,12,15), 18:3-(6,9,12)) originating from neutral lipids found human serum.34 While effective, analysis of AMPP-derivatized FA relies on wet-chemical modification of free (i.e., nonesterified) FA. Thus, crude samples first require solution-based lipid extraction and hydrolysis to generate free FA prior to derivatization with AMPP and subsequent analysis. In contrast to covalent modification of the carboxyl moiety, numerous investigators have exploited metal cation adducted FA.35-44 Metal adducted FAs are typically generated prior to ionization via doping metal salts into ESI solution containing FA. It is well-known that charge-remote fragmentation (CRF) of alkali and alkaline earth metal cationized FA using high-energy CID is an effective method for structural elucidation of FA.35-37 As high-energy CID operates in the keV kinetic regime, workflows to access CRF are uncommon, mostly due to the replacement of multisector instruments with triple quadrupole (QqQ) and quadrupole time-of-flight (Q-TOF) mass spectrometers that operate under low-energy CID conditions. More recently, product ion spectra generated via low-energy CID of dilithiated40,41 (i.e., [M – H + 2Li]+), copper(II) adducted42 (i.e., [M – H + CuII]+), and barium cationized lipids43,44 (i.e., [M – H + Ba]+, [M + Ba]2+) have been advantageous in lipid structural analysis. However, the analysis of metalated lipid ions has been exploited solely for FA identification, and thus relative quantitation has remained unaddressed. Like AMPP-based methods, the construction of FA profiles utilizing metal adducted FA first requires the generation of free FA prior to metal cation adduction. Therefore, FA profiles developed from crude lipid extracts utilizing solution-based methods such as metal cation adduction or AMPP derivatization reflect the FA composition of all ester-linked lipids, unless lipid classes are first separated using additional fractionation steps prior to hydrolysis, derivatization, and analysis.
Recently, we reported the gas-phase charge inversion of FA anions via ion/ion reaction with tris-phenanthroline alkaline earth metal complex cations to identify the C═C double bond position(s).45 Gas-phase charge inversion of FA via ion/ion reactions offers the primary advantage of ionizing FA in the most efficient modality (i.e., negative ion mode), while characterizing the FA in the positive ion mode where structurally informative product ions are more readily observed. By conducting FA derivatization in the gas phase, electrospray and solution conditions can be optimized for each reactant.46 Furthermore, when compared to FA derivatization methods reliant on wet chemistry (i.e., metal cation adduction, AMPP derivatization, etc.), gas-phase ion/ion reactions provide enhanced control of reaction outcomes including product generation and reaction efficiency. Singly deprotonated [FA – H]− anions and tris-phenanthroline magnesium complex dications react in the gas phase to form the long-lived electrostatic complex comprised of the deprotonated fatty acid, the magnesium dication, and a phenanthroline ligand. Collisional activation of the [FA – H + MgPhen]+ ion generated reproducible spectral patterns that provided double bond position(s) for monounsaturated and diunsaturated FA. Double bond localization in highly unsaturated FA was hindered due to rearrangements and congested CID spectra; yet discrimination between isomeric polyunsaturated fatty acids (PUFA) was achieved.
Herein, we report the development of a mass spectral library, comprised of [FA – H + MgPhen]+ product ion spectra. The FA library permits the confident identification of FA, including PUFA. Furthermore, in this study, we demonstrate the ability to sensitively determine relative abundances of isomeric FA using multiple linear regression analysis in conjunction with ion/ion chemistry. In addition, we applied the mass spectral library, relative quantitation algorithm, and an MSn platform reliant entirely on gas-phase chemistries to develop a FA profile selectively for phospholipids present in bovine liver extract. Contrary to solution-based approaches, gas-phase FA profiling offers the ability to selectively release FA from the lipids ionized in negative ion mode permitting a quantitative profile of the fatty acid isomers carried by phospholipids. Within the literature, there is a growing awareness of the distinct profile of fatty acid isomers between lipid classes based on both traditional multistage fractionation47 and next generation methods.14 However, the method provided here is unique in that it provides a snapshot of the FA composition of phospholipids where changes will be related to membrane synthesis and function in isolation from changes to storage lipids such as triacylglycerols and cholesterol esters, which, in some contexts (i.e., plasma), will dominate total FA pool. To gain the same insights as provided by our approach utilizing wet-chemical methods, the lipid extract would need to be split in two, with one fraction subjected to intact phospholipid analysis while the other being hydrolyzed, derivatized, and analyzed. Therefore, gas-phase FA profiling in conjunction with ion/ion chemistry as presented herein provides a detailed, isomer-specific profile of all FA released from the phospholipid fraction in the same infusion experiment. In turn, gas-phase FA profiling offers a rapid, sensitive, and selective approach to quantitative lipid profiling.
EXPERIMENTAL SECTION
Materials.
HPLC-grade methanol and water were purchased from Fisher Scientific (Pittsburgh, PA). All FA standards were purchased from Cayman Chemical (Ann Arbor, MI) and used without further purification. Magnesium chloride and 1,10-phenanthroline (Phen) were purchased from Sigma-Aldrich (St. Louis, MO). Polar liver extract (bovine) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL).
Preparation of nESI Solutions.
Solutions of FA standards were prepared in methanol to a final concentration of 10 μM (m/v). Similarly, solutions of bovine liver extract (polar) were prepared to a final concentration of 50 μM (m/v) in methanol with 10 mM ammonium acetate, assuming an average lipid molecular mass of 760 g/mol. Mixtures of FA isomers were prepared from individual isomer stock solutions in methanol. For relative quantitation, the total FA concentration was held constant (10 μM), while the molar ratio of FA isomers was varied. Tris-phenanthroline magnesium complexes were prepared by mixing the 1:1 (mol/mol) metal salt:Phen in methanol to a final concentration of 50 μM (m/v).
Nomenclature.
The shorthand notation reported by Liebisch et al. is adopted to describe FA.48 Before the colon, the number of carbon atoms is first indicated. After the colon, the number of carbon–carbon double bonds is designated with definitive double bond position(s), with respect to the carboxylic acid moiety, indicated within parentheses. If known, carbon–carbon double bond stereochemistry is further indicated as Z (cis) and E (trans). If unknown, only the double bond position is indicated. In some instances, the n-x terminology is also employed to classify FA isomers. The n-x terminology describes the location of the first carbon–carbon double bond from the methyl end of the aliphatic chain.
Mass Spectrometry.
All data were collected on a Sciex QTRAP 4000 hybrid triple quadrupole/linear ion trap mass spectrometer (SCIEX, Concord, ON, Canada) with modifications analogous to those previously described.49 Alternately pulsed nanoelectrospray ionization (nESI) allows for sequential injection of the tris-phenanthroline magnesium dications, [Mg(Phen)3]2+, followed by singly deprotonated FA ions.50 First, [Mg(Phen)3]2+ was generated in the positive ion mode, then mass-selected in Q1 and transferred to the high-pressure collision cell, q2, for storage. Next, [FA – H]− anions were generated in the negative ion mode, isolated in Q1 during transit, and transferred to q2 for storage. In q2, the [FA – H]− and [Mg(Phen)3]2+ ion were simultaneously stored for 300 ms (ms). The mutual storage ion/ion reaction produced [FA – H + Mg(Phen)2]+. The [FA – H + Mg(Phen)2]+ ion was then subjected to 18 V of dipolar direct current (DDC)-CID for 25 ms, generating the [FA – H + MgPhen]+ ion.51 The resulting [FA – H + MgPhen]+ ion was then transferred to the low-pressure linear ion trap (LIT), Q3, for monoisotopic isolation. Monoisotopically isolated [FA – H + MgPhen]+ was then collisionally activated via single frequency resonance excitation (q = 0.383) for 150 ms. Product ions generated from CID were analyzed via mass-selective axial ejection (MSAE).52
As described in our original description of the charge inversion of FA with [Mg(Phen)3]2+,45 both singly and doubly hydrated FA complex cations can be observed resulting from ion/molecule reactions in the collision cell with adventitious water. This occurs most readily with saturated FA. This phenomenon can lead to more complicated spectra and isobaric interferences. In our experience, this can be an issue after the instrument has been vented and shortly after pumping down.
Development of Fatty Acid Mass Spectral Library.
A library of CID spectra of [FA – H + MgPhen]+ ions of FA standards was constructed to aid in the identification of unknown FA. Automated spectral matching was conducted in the MATLAB numerical computing environment (Mathworks, Natick, MA). After normalization of product ion intensities relative to the base peak, for both the library and the unknown [FA – H + MgPhen]+ CID spectra obtained under identical conditions, the residuals were calculated between the normalized product ion intensities of unknown FA and each individually detected FA standard. Calculated residuals were then summed, and the minimum summed residual was returned. The FA standard from the developed library that generated the minimum summed residual when compared to the unknown FA represents the identity of the unknown FA.
Relative Quantitation of FA Isomers.
The MATLAB numerical computing environment (Mathworks, Natick, MA) was used to perform a multiple linear regression analysis modeled after that reported by Wang et al.33 To generate relative quantities of isomeric components in a mixture, the library spectra of the standards, as described above, served as the predictors, while the normalized CID spectra of the [FA – H + MgPhen]+ ions served as the responses. Herein, it is assumed that product ions observed in an isomeric mixture can be equated to a linear combination of individually detected isomer product ions. Under that assumption, the least-squares coefficients represent the relative composition of each individual isomer. Mathematically, this concept is represented by eq 1, where xisomerj signifies the composition of isomer j present in an isomeric mixture, Iioniisomerj represents the normalized product ion intensity of the specified product ion i from an individually detected isomer j sample, and Iionimixture indicates the normalized product ion intensity of the specified product ion i in an isomeric mixture. Note that the number of specified product ions i is strictly greater than the number of individual isomers j present in the isomeric mixture (i.e., i = 1, 2, 3, …n and j = 1, 2, 3, …m such that n > m). The least-squares coefficients were constrained such that the sum of all coefficients was one and all coefficients were greater than or equal to zero, as modeled by eqs 2 and 3, respectively.
| (1) |
| (2) |
| (3) |
Data are presented as the mean ± standard deviation from at least three individually prepared samples (n = 3). Calculated standard deviations are reported to one significant figure.
RESULTS AND DISCUSSION
Development of CID Mass Spectral Library.
Singly deprotonated FA anions undergo charge inversion when reacted in the gas phase with tris-phenanthroline magnesium dications via a mutual storage ion/ion reaction (Scheme 1).45 The ion/ion reaction generates the [FA – H + Mg(Phen)2]+ complex ion. DDC-CID of [FA – H + Mg(Phen)2]+ results in the neutral loss of one phenanthroline ligand, producing [FA – H + MgPhen]+, as shown in Scheme 1. The [FA – H + MgPhen]+ fragments upon ion-trap collision-induced dissociation (CID) to generate product ion spectra indicative of double bond position(s). A priori double bond identification via direct interpretation of the [FA – H + MgPhen]+ CID spectrum is achieved only for monounsaturated and diunsaturated FA. However, distinct spectral differences are observed for polyunsaturated [FA – H + MgPhen]+ isomeric ions, as previously reported.45
Scheme 1.

Gas-Phase Charge Inversion Ion/Ion Reaction between [FA – H]− and [Mg(Phen)3]2+ for the Generation of the [FA – H + MgPhen]+ Ion
To aid in the identification of FA, we constructed a library comprised of ion trap CID product ion spectra resulting from collisional activation of the [FA – H + MgPhen]+ cations derived from the ion/ion charge inversion of the [FA – H]− anions of 40 unsaturated FA standards (Scheme 1). Ion-trap CID of the [FA – H + MgPhen]+ precursor ion generated CID spectra unique to individual FA isomers. Raw data for each of the FA standards of the library are shown in Tables S1-S40.
A notable advantage of the library matching approach is its ability to distinguish highly unsaturated FA isomers due to the fact that the CID spectra of the [FA – H + MgPhen]+ ions are isomer-specific. As an illustration, two double bond positional isomers of FA 22:5 were examined using the described ion/ion chemistry. The reaction between [Mg(Phen)3]2+ dications and [22:5 – H]− anions generated the [22:5 – H + MgPhen]+ complex cations (m/z 533.3). Product ion spectra for the 22:5 n-3 (i.e., 22:5(7Z, 10Z, 13Z, 16Z, 19Z)) and 22:5 n-6 (i.e., 22:5(4Z, 7Z, 10Z, 13Z, 16Z)) complex cations are shown in Figure 1a and b, respectively. To better visualize distinguishing product ions for 22:5 isomer differentiation, a difference plot was generated using Figure 1a and b, as illustrated with Figure 1c. To generate Figure 1c, the relative abundances of product ions produced via CID [22:5 – H + MgPhen]+ for the n-3 isomer (Figure 1a) were subtracted from those obtained from the n-6 isomer (Figure 1b). Therefore, product ions with a higher relative abundance in, or unique to, the charge inverted 22:5 n-6 isomer fragmentation pattern are depicted with positive relative abundances (shown in blue font) in Figure 1c, while those with a higher relative abundance, or unique to, the fragmentation of the n-3 isomer are presented with negative relative abundances (shown in red font). Ultimately, distinct differences in the relative abundances of fragment ions as well as product ions unique to individual isomers were observed between the CID spectra of the 22:5 FA ions. For example, the product ion generated via C2–C3 cleavage (m/z 262.1) is observed at a significantly higher relative abundance in the spectrum of the 22:5 n-6 isomer (Figure 1b) when compared to that of the 22:5 n-3 isomer (Figure 1a), as also highlighted with the difference plot (Figure 1c). This difference in m/z 262.1 fragment ion abundance is related to the proximity of the first double bond position relative to the carboxylate moiety; FA 22:5 n-6 contains a C4═C5, whereas the closest double bond to the carboxylate moiety in 22:5 n-3 is located at C7═C8. As the double bond moves closer to the carboxylate moiety, the relative abundance of the product ion m/z 262.1 increases. From Figure 1c, another example can be illustrated with the product ion at m/z 421.1, which is formed via C14–C15 cleavage. The product ion at m/z 421.1 is significantly more abundant in the 22:5 n-6 isomer. Carbon–carbon bond cleavages are correlated to double bond position(s), as the proximity to the charge site likely affects CRF. Numerous other reproducible differences in product ion relative abundances between the two isomeric ions are observed, many of which are highlighted in Figure 1c. Other notable differences include product ions at m/z 367.1, 393.2, 407.2, 433.2, 447.2, 455.3, and 461.2, which are significantly more abundant in the [22:5(4Z, 7Z, 10Z, 13Z, 16Z) – H + MgPhen]+ CID spectrum (Figure 1b) than in the [22:5(7Z, 10Z, 13Z, 16Z, 19Z) – H + MgPhen]+ CID spectrum (Figure 1a), as highlighted with the difference plot (Figure 1c). Conversely, product ions such as those observed at m/z 317.1, 331.1, 397.2, 409.2, 423.2, and 463.2 are indicative of the 22:5 n-3 isomer. By analogy with the comparison of Figure 1, all CID spectra of isomeric [FA – H + MgPhen]+ ions in the library were sufficiently unique to allow for unambiguous identification and thus isomeric discrimination.
Figure 1.

CID spectra resulting from the activation of (a) [22:5(7Z, 10Z, 13Z, 16Z, 19Z) – H + MgPhen]+ and (b) [22:5(4Z, 7Z, 10Z, 13Z, 16Z) – H + MgPhen]+. (c) Difference plot ((n-6) – (n-3)) highlighting the distinguishing product ions for 22:5 isomer differentiation.
Relative Quantitation of Isomeric FA.
Qualitative and quantitative analysis remains a challenge with shotgun lipidomics, as the innate complexity of the cellular lipidome gives rise to a multitude of both isomeric and isobaric lipid species.14,53,54 Therefore, methods to quantify relative compositions of FA isomers are of interest. We approach this issue using multiple linear regression analysis with the library data in conjunction with data derived from mixtures. It is assumed that the product ions generated from fragmentation of the [FA – H + MgPhen]+ precursor ion population containing multiple FA isomers can be represented by a linear combination of spectra from individual components.
Relative quantitation is first illustrated with octadecenoic (18:1) isomers. Oleic (18:1 n-9) and vaccenic (18:1 n-7) acids were dissolved in methanol at varying molar ratios, while the total FA concentration was held constant (10 μM). The following theoretical molar ratios of the n-9 to n-7 18:1 FA (denoted 18:1 n-9/n-7) were used: 5/95, 15/85, 50/50, 85/15, and 95/5. Isomeric FA mixtures of 18:1 n-9/n-7 were ionized in the negative ion mode and transformed in the gas phase to [18:1 – H + MgPhen]+ complex cations (m/z 485.2) as detailed above. CID spectra of [18:1 – H + MgPhen]+ generated from 18:1 FA isomer mixtures and individual isomer standards were acquired under identical CID conditions, and product ion abundances were normalized to the base peak in all cases. Normalized product ion abundances from the 18:1 FA isomer mixture were used as responses, while the normalized product ion abundances from individually analyzed 18:1 isomer standards were used as predictors for the multiple linear regression analysis described by eqs 1, 2, and 3. Specific diagnostic product ions, referred to as the ion set, were chosen as the basis of quantitation. The ion set utilized represents product ions indicative of double bond position for each 18:1 isomer. The ion set used for 18:1 n-9/n-7 relative quantitation includes the following product ions (depicted in red font in Figure 2a and b): m/z 345.1, 357.1, 359.2, 371.2, 373.2, 385.2, 387.2, 399.2, 413.3, and 415.3. Other ion sets were explored, including utilization of all product ions; however, the reported ion set yielded the most accurate results. Figures S1 and S2 provide structures for the [18:1 – H + MgPhen]+ ions, along with product ion m/z ratios from fragmentation of the complex cation for the n-9 and n-7 isomers, respectively.
Figure 2.

CID spectra representative of isomeric FA mixtures analyzed as the charge-inverted FA complex cation, [FA – H + MgPhen]+. CID spectra of [18:1 – H + MgPhen]+ for the isomeric mixture of 18:1 n-9/n-7 at the molar ratios of (a) 5/95 and (b) 95/5. CID spectra of [18:3 – H + MgPhen]+ for the isomeric mixture of 18:3 n-3/n-6 at the molar ratios of (c) 15/85 and (d) 95/5. Product ions used for relative quantitation are shown in red. Calculated isomer compositions (mean ± standard deviation, n = 3) are shown in green. The lightning bolt signifies the precursor ion subjected to ion-trap CID.
The multiple linear regression approach yielded 18:1 n-9/n-7 isomer ratios of (mean ± standard deviation, n = 3) 5.0 ± 0.2/95.0 ± 0.2 (Figure 2a), 14 ± 2/86 ± 2 (Figure S3), 46 ± 1/54 ± 1 (Figure S4), 86.3 ± 0.9/13.7 ± 0.9 (Figure S5), and 95.0 ± 0.2/5.0 ± 0.2 (Figure 2b), for the expected 18:1 n-9/n-7 isomeric ratios of 5/95, 15/85, 50/50, 85/15, and 95/5, respectively. The results show relative errors <9% and relative standard deviations <2% over a relatively wide dynamic range of molar ratios (i.e., 5/95 to 95/5). Ultimately, for isomeric monounsaturated FA, we are confident in the ability of the multiple linear regressions approach coupled with gas-phase ion/ion chemistry to both reliably and accurately calculate relative isomeric composition over a broad range of molar ratios for binary mixtures.
FA relative quantitation using multiple linear regression analysis was extended to the analysis of isomeric polyunsaturated fatty acids (PUFA). To illustrate, the n-3 and n-6 isomers of linolenic acid (18:3) were examined. Again, ion/ion reactions of [18:3 – H]− anions and [Mg(Phen)3]2+ dications were used to generate [18:3 – H + MgPhen]+ (m/z 481.2) from both isomeric mixtures and individual authentic isomer standards. The isomer ratios investigated for the relative quantitation of 18:3 n-3/n-6 isomers were identical to those utilized in the 18:1 experiments described above (i.e., 5/95, 15/85, 50/50, 85/15, and 95/5). For relative quantitation of 18:3 n-3/n-6 isomeric mixtures, the ion set that provided the best results includes product ions at m/z 317.1, 329.1, 331.1, 343.1, 345.2, 367.1, 371.2, 381.2, 383.2, 395.2, 397.2, 409.2, 423.2, and 425.2. [18:3 – H + MgPhen]+ CID spectra for the 95/5 and 15/85 isomer ratios of 18:3 n-3/n-6 are shown in Figure 2c and d, respectively. Representative [FA – H + MgPhen]+ structures and fragmentation patterns for the n-3 and n-6 isomers of FA 18:3 are shown in Figures S6 and S7, respectively. The results obtained from the analysis of the 15/85, 50/50, 85/15, and 95/5 isomeric ratios of FA 18:3 n-3/n-6 were (mean ± standard deviation, n = 3) 14 ± 2/86 ± 2 (Figure 2c), 48 ± 3/52 ± 3 (Figure S8), 86 ± 2/14 ± 2 (Figure S9), and 96.3 ± 0.4/3.7 ± 0.4 (Figure 2d). Again, our results show reasonable accuracy with most relative errors <5% and relative standard deviation <3% for these given isomeric molar ratios. Unfortunately, we were unable to reliably calculate the isomer ratio for the theoretical 5/95 n-3/n-6 18:3 isomer mixture, as the calculated isomer composition was 1 ± 1/99 ± 1. Thus, the observed limit of relative quantitation was 10% (mol) for the n-3 isomer as the minor component. However, this limit was not observed when the n-6 isomer was the minor component. The observed limit of relative quantitation for low molar ratios of the n-3 isomer relative to the n-6 isomer is most likely due the ratio of diagnostic ion abundance to the structurally uninformative product ion abundances. For the 18:3 n-3 FA, fewer diagnostic ions are generated relative to structurally uninformative product ions as compared to the relative abundance of diagnostic product ions for the 18:3 n-6 isomer.
We attempted to extend the approach to the relative quantitation of a three-component 18:1 isomer mixture. We used the n-7, n-9, and n-12 18:1 FA isomers for these experiments. However, using our approach, we were only able to achieve relative quantitation for isomeric mixtures with high concentrations of the n-9 isomer relative to that of the n-7 and n-12 isomers. A similar difference in diagnostic product ion generation relative to structurally uninformative product ion generation was also observed among the 18:1 isomers. Of the three 18:1 isomers, the n-9 isomer had the lowest ratio of diagnostic ions generated in relation to structurally uninformative product ions. Comparatively, the n-7 and n-12 isomers demonstrated increased ratios for the generation of diagnostic ions relative to structurally uninformative product ions. This decrease in the ratio of diagnostic ions to structurally uninformative product ions for the n-9 isomer equates to reliable relative quantitation only at high concentrations of the n-9 isomer relative to the other two 18:1 FA isomers. Furthermore, we also attempted to extend the approach to the relative quantitation of highly unsaturated FA isomers; however, due to extensive fragmentation, these efforts were also unsuccessful.
FA Profiling of Bovine Liver Extract.
To demonstrate the capabilities of the presented method, we applied ion/ion charge inversion chemistry, the FA library, and the relative quantitation algorithm for the profiling of FA in bovine liver extract (BLE). FA profiles reported are representative of the fraction of lipids ionizable in the negative ion mode (i.e., phosphatidylethanolamine, phosphatidylinositol, phosphatidylserine, etc.). In this polarity, it is important to recognize that phosphatidylcholine (PC) lipids ionize as chloride or acetate adducts; therefore, PC ionization is likely to be less efficient in the negative ion mode relative to some of the other lipid classes. For comparison, our approach is most closely analogous to multistage chromatography for lipid class separation (i.e., TLC) with subsequent hydrolysis and fatty acid analysis (i.e., GC). However, as described above, the entirely gas-phase profiling experiment presented herein is unique in that information regarding the intact phospholipidome and a quantitative, isomer-specific FA profile of the phospholipidome can be obtained from a single experiment. Specifically, we first achieve fractionation of the lipidome via direct ionization of the lipid extract in the negative ion mode, as nonpolar lipids are not readily ionized in this modality. FA identification and relative quantitation are then achieved via an MSn platform paired with ion/ion charge inversion chemistry. Although not demonstrated here, it is plausible that gas-phase FA profiling via precursor ion scanning could be used to identify lipid precursors for subsequent targeted analysis by ion/ion charge inversion.
To generate the FA profile for BLE, total lipid extract is first ionized in the negative ion mode, as illustrated with Figure 3a. To obtain [FA – H]− anions from complex lipid anions, we employ ion-trap CID over a broad range of m/z ratios. To do so, the excitation frequency is fixed (114.06 kHz) while the RF amplitude in q2 is scanned, producing what is referred to as a ramped ion-trap CID experiment. Over the phospholipid m/z range (m/z 650–900), ramp CID was used to generate fatty acyl anions directly from the ionized lipid precursor. A second ramp CID was applied over the range of m/z 400–650 to produce [FA – H]− from remaining product ions generated via the first ramp CID experiment. The result of both ramp CID experiments for BLE is shown in Figure 3b. The resulting product ion spectrum is representative of the FA profile of all precursor lipids ionizable in negative ion mode and is enlarged in Figure 3c over the m/z range of 200–400 (i.e., the FA anion range).
Figure 3.

Demonstration of gas-phase generation of fatty acyl anions from bovine liver extract. (a) Nano-ESI mass spectrum of 50 μM BLE obtained via direct negative ion mode ionization. (b) Fatty acid profile CID spectrum resulting from collisional activation of BLE precursors in the phospholipid range (m/z 650–900), followed by a subsequent collisional activation of remaining anions from m/z 400 to 650. (c) Enlargements of the fatty acid profile shown in panel (b) over the mass-to-charge range of 200–400.
Following generation of FA anions from ionized lipids, positive ion nESI directly generated [Mg(Phen)3]2+ dications. In q2, a mutual storage reaction between the [FA – H]− anions and the [Mg(Phen)3]2+ dications followed by q2 DDC-CID of the resulting mutual storage product ion generated the complexes of interest, [FA – H + MgPhen]+ (Figure 4). Note that the m/z range in Figure 4 is equivalent to that shown in Figure 3c. Unidentified product ions were also generated following the ion/ion reaction, as marked in Figure 4 with blue circles. Mass-selection and collisional activation of each unidentified product ion (blue circles) indicated that these species were not FA because the fragmentation patterns were inconsistent with those observed via CID of a charge-inverted FA. Additionally, both singly and doubly hydrated product ions are observed following the mutual storage ion/ion reaction and DDC-CID. This observation is discussed in more detail below. To identify FA present in BLE, [FA – H + MgPhen]+ ions were transferred to Q3. Once in Q3, [FA – H + MgPhen]+ complex cations were monoisotopically isolated and subsequently activated via ion-trap CID (q = 0.383). The resulting CID spectra were used to identify FA in BLE via automated spectral matching with the FA library as described above. If applicable, relative compositions of FA isomers were determined using multiple linear regression analysis, also as described above.
Figure 4.

Mass spectrum resulting from the gas-phase ion/ion reaction of [Mg(Phen)3]2+ dications with fatty acyl anions derived from bovine liver extract via the described ion/ion chemistry. Peaks labeled with the blue circle are not fatty acids, as confirmed by CID of the precursor ion.
The FA profile of BLE is summarized in Table 1. To generate the FA profile of BLE, each charge-inverted FA complex cation with a relative abundance >1% (Figure 4) was mass-selected and subjected to ion-trap CID. Saturated FAs in BLE include 16:0, 17:0, and 18:0, as confirmed via CID of [FA – H + MgPhen]+. The following polyunsaturated FAs (PUFA) were identified in BLE in the pure n-6 form: 18:2(9,12), 20:3(8,11,14), 20:4(5,8,11,14), and 22:4(7,10,13,16). FA 22:5(7,10,13,16,19) existed in the pure n-3 form. Thus, all PUFA present in BLE were found to be “pure” (i.e., no double bond positional isomers detectable). The 18:1 FA in BLE existed as a mixture of 18:1(9) and 18:1(11) isomers (i.e., n-9 and n-7 isomers). Employing our relative quantitation approach, the relative composition of 18:1 isomers was calculated to be 88.4 ± 0.7 and 11.6 ± 0.7 for the 18:1(9) and 18:1(11) isomers, respectively. Our results are in good agreement with a previously developed lipid profile for BLE.55
Table 1.
Fatty Acid Profile of Bovine Liver Phospholipidome (Polar Bovine Liver Extract)
| m/z | fatty acid composition |
double bond position(s) |
relative abundance (mean ± SD) |
|---|---|---|---|
| 459.3 | 16:0 | N/A | N/A |
| 473.3 | 17:0 | N/A | N/A |
| 487.3 | 18:0 | N/A | N/A |
| 485.3 | 18:1 | 9 and 11 | 88.4 ± 0.7 and 11.6 ± 0.7 |
| 483.3 | 18:2 | 9, 12 | N/A |
| 509.3 | 20:3 | 8, 11, 14 | N/A |
| 507.3 | 20:4 | 5, 8, 11, 14 | N/A |
| 535.3 | 22:4 | 7, 10, 13, 16 | N/A |
| 533.3 | 22:5 | 7, 10, 13, 16, 19 | N/A |
A challenge for any shotgun lipidomics approach arises from the presence of isobaric FA species. In the present case, the ramped CID approach generates FA from all phospholipids in the BLE. We found that using a relatively wide mass isolation for the [Mg(Phen)3]2+ reagent led to isobaric overlap between [18:1 – H + 24MgPhen]+ and [18:2 – H + 26MgPhen]+ at m/z 485.3, for example (see Figures S10-S12). Fortunately, this did not lead to isobaric product ions and did not affect the ability to quantitate 18:1 isomers in BLE, as relative quantitation is based on specific product ions unique to the [18:1 – H + 24MgPhen]+ precursor ion. Nevertheless, this was confirmed by repeating the experiment using isolated [24Mg(Phen)3]2+ reagent ions (Figures S13 and S14). While is it straightforward to isolate [24Mg(Phen)3]2+ reagent ions prior to reaction, other isobaric interferences can arise when all phospholipids undergo ramped CID with collection of all FA. An alternate approach would be a top-down shotgun lipidomics workflow that would entail the direct negative mode ionization of a complex lipid (or lipid extract), followed by mass-selection of a specific lipid precursor anion. Using traditional MS/MS, class information (phospholipid head-group) and fatty acyl anions could be obtained from the mass-selected phospholipid precursor ion. Subsequent ion/ion reaction of [FA – H]− derived from a mass-selected lipid precursor with the [Mg(Phen)3]2+ dications permits identification of FA double bond position(s). This workflow is currently under investigation and is the subject of a future report.
CONCLUSIONS
In this work, we have demonstrated a unique shotgun approach for selective FA profiling of the that provides composition, double bond, and relative quantity (for binary isomeric mixtures) information regarding the phospholipidome. The workflow begins with the generation of phospholipids in the negative ion mode, thereby selecting for polar lipids, followed by ramped CID (i.e., a broad-band activation approach) of all of the lipid anions to release and capture the FA anions from the phospholipids in the precursor ion mixture. All FA anions are then simultaneously subjected to reactions with [Mg(Phen)3]2+ to generate [FA – H + MgPhen]+ cations. Ion trap CID spectra generated from the mixture are then compared to library CID spectra derived from the [FA – H + MgPhen]+ cations of 40 standards. Confident identification of even highly unsaturated FA could be made via the library comparison approach. Furthermore, we demonstrated a method to determine relative compositions of FA double bond positional isomers in binary mixtures via a multiple linear regression approach applied to [FA – H + MgPhen]+ cations. In this case, library CID spectra serve as predictors, while the spectrum of a mixture serves as the response. A significant advantage of a multiple linear regressions approach is rapid analysis time, as external calibration curve construction is not required; thus, both sample volume requirements and analysis times are greatly reduced. Collectively, the approach provides a relatively rapid and sensitive approach for lipid profiling that provides composition, double bond location, and relative abundance information for binary isomeric mixtures. Variations of this workflow, such as the addition of a precursor ion scan or the use of ion/ion reactions after lipid anion isolation and activation could also, in principle, provide lipid class information.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (NIH) under grants GM R37-45372 and GM R01-118484. S.J.B. acknowledged project funding through the Discovery Program (DP150101715 and DP190101486) Australian Research Council (ARC).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.9b01333.
Additional information discussed in the text that supports the presentation of the work (PDF)
FA mass spectral library discussed in the text (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Murphy RC Tandem Mass Spectrometry of Lipids: Molecular Analysis of Complex Lipids; RSC Publishing: UK, 2015; pp 1–39. [Google Scholar]
- (2).Murphy RC; Axelsen PH Mass Spectrom. Rev 2011, 30, 579–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Han XL; Gross RW Mass Spectrom. Rev 2005, 24, 367–412. [DOI] [PubMed] [Google Scholar]
- (4).Melo T; Montero-Bullon J-F; Domingues P; Domingues MR Redox Biol. 2019, 101106–101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Batthyany C; Schopfer FJ; Baker PRS; Duran R; Baker LMS; Huang Y; Cervenansky C; Branchaud BP; Freeman BA J. Biol. Chem 2006, 281, 20450–20463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Oh HJ; Kim SU; Song JW; Lee JH; Kang WR; Jo YS; Kim KR; Bornscheuer UT; Oh DK; Park JB Adv. Synth. Catal 2015, 357, 408–416. [Google Scholar]
- (7).Iles KE; Wright MM; Cole MP; Welty NE; Ware LB; Matthay MA; Schopfer FJ; Baker PRS; Agarwal A; Freeman BA Free Radical Biol. Med 2009, 46, 866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Vlaeminck B; Fievez V; Cabrita ARJ; Fonseca AJM; Dewhurst RJ Anim. Feed Sci. Technol 2006, 131, 389–417. [Google Scholar]
- (9).Cascio G; Schiera G; Di Liegro I Curr. Diabetes Rev 2012, 8, 2–17. [DOI] [PubMed] [Google Scholar]
- (10).Simopoulos AP Nutrients 2016, 8, 17. [Google Scholar]
- (11).Kuhajda FP Nutrition 2000, 16, 202–208. [DOI] [PubMed] [Google Scholar]
- (12).Antalis CJ; Stevens LJ; Campbell M; Pazdro R; Ericson K; Burgess JR Prostaglandins, Leukotrienes Essent. Fatty Acids 2006, 75, 299–308. [DOI] [PubMed] [Google Scholar]
- (13).Richardson AJ Int. Rev. Psychiatry 2006, 18, 155–172. [DOI] [PubMed] [Google Scholar]
- (14).Zhang WP; Zhang DH; Chen QH; Wu JH; Ouyang Z; Xia Y Nat. Commun 2019, 10, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hu CX; van der Heijden R; Wang M; van der Greef J; Hankemeier T; Xua GW J. Chromatogr. B-Anal. Technol. Biomed. Life Sci 2009, 877, 2836–2846. [DOI] [PubMed] [Google Scholar]
- (16).Li F; Qin XZ; Chen HQ; Qiu L; Guo YM; Liu H; Chen GQ; Song GG; Wang XD; Li FJ; Guo S; Wang BH; Li ZL Rapid Commun. Mass Spectrom 2013, 27, 24–34. [DOI] [PubMed] [Google Scholar]
- (17).Min HK; Lim S; Chung BC; Moon MH Anal. Bioanal. Chem 2011, 399, 823–830. [DOI] [PubMed] [Google Scholar]
- (18).Mitchell TW; Pham H; Thomas MC; Blanksby SJ J. Chromatogr. B: Anal. Technol. Biomed. Life Sci 2009, 877, 2722–2735. [DOI] [PubMed] [Google Scholar]
- (19).Hewawasam E; Liu G; Jeffery DW; Muhlhausler BS; Gibson RA Prostaglandins, Leukotrienes Essent. Fatty Acids 2017, 125, 1–7. [DOI] [PubMed] [Google Scholar]
- (20).Han XL; Yang K; Gross RW Mass Spectrom. Rev 2012, 31, 134–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kerwin JL; Wiens AM; Ericsson LH J. Mass Spectrom 1996, 31, 184–192. [DOI] [PubMed] [Google Scholar]
- (22).Yang K; Zhao Z; Gross RW; Han X Anal. Chem 2011, 83, 4243–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Brown SHJ; Mitchell TW; Blanksby SJ Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2011, 1811, 807–817. [DOI] [PubMed] [Google Scholar]
- (24).Poad BLJ; Pham HT; Thomas MC; Nealon JR; Campbell JL; Mitchell TW; Blanksby SJ J. Am. Soc. Mass Spectrom 2010, 21, 1989–1999. [DOI] [PubMed] [Google Scholar]
- (25).Thomas MC; Mitchell TW; Blanksby SJ J. Am. Chem. Soc 2006, 128, 58–59. [DOI] [PubMed] [Google Scholar]
- (26).Thomas MC; Mitchell TW; Harman DG; Deeley JM; Murphy RC; Blanksby SJ Anal. Chem 2007, 79, 5013–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Thomas MC; Mitchell TW; Harman DG; Deeley JM; Nealon JR; Blanksby SJ Anal. Chem 2008, 80, 303–311. [DOI] [PubMed] [Google Scholar]
- (28).Thomas MC; Mitchell TW; Blanksby SJ Methods Mol. Biol 2009, 579, 413–441. [DOI] [PubMed] [Google Scholar]
- (29).Ma XX; Xia Y Angew. Chem., Int. Ed 2014, 53, 2592–2596. [DOI] [PubMed] [Google Scholar]
- (30).Ma XX; Chong L; Tian R; Shi RY; Hu TY; Ouyang Z; Xia Y Proc. Natl. Acad. Sci. U. S. A 2016, 113, 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Stinson CA; Xia Y Analyst 2016, 141, 3696–3704. [DOI] [PubMed] [Google Scholar]
- (32).Paine MRL; Poad BLJ; Eijkel GB; Marshall DL; Blanksby SJ; Heeren RMA; Ellis SR Angew. Chem., Int. Ed 2018, 57, 10530–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Wang M; Han RH; Han XL Anal. Chem 2013, 85, 9312–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yang K; Dilthey BG; Gross RW Anal. Chem 2013, 85, 9742–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Adams J; Gross ML Anal. Chem 1987, 59, 1576–1582. [Google Scholar]
- (36).Davoli E; Gross ML J. Am. Soc. Mass Spectrom 1990, 1, 320–324. [DOI] [PubMed] [Google Scholar]
- (37).Gross ML Int. J. Mass Spectrom. Ion Processes 1992, 118, 137–165. [Google Scholar]
- (38).Hsu FF; Turk J J. Am. Soc. Mass Spectrom 2010, 21, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Trimpin S; Clemmer DE; McEwen CN J. Am. Soc. Mass Spectrom 2007, 18, 1967–1972. [DOI] [PubMed] [Google Scholar]
- (40).Hsu FF; Turk J J. Am. Soc. Mass Spectrom 1999, 10, 600–612. [DOI] [PubMed] [Google Scholar]
- (41).Hsu FF; Turk J J. Am. Soc. Mass Spectrom 2008, 19, 1673–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Afonso C; Riu A; Xu Y; Fournier F; Tabet JC J. Mass Spectrom 2005, 40, 342–349. [DOI] [PubMed] [Google Scholar]
- (43).Duncan KD; Volmer DA; Gill CG; Krogh ET J. Am. Soc. Mass Spectrom 2016, 27, 443–450. [DOI] [PubMed] [Google Scholar]
- (44).Hale OJ; Cramer R Anal. Bioanal. Chem 2018, 410, 1435–1444. [DOI] [PubMed] [Google Scholar]
- (45).Randolph CE; Foreman DJ; Betancourt SK; Blanksby SJ; McLuckey SA Anal. Chem 2018, 90, 12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Newton KA; Amunugama R; McLuckey SA J. Phys. Chem. A 2005, 109, 3608–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Astudillo AM; Meana C; Guijas C; Pereira L; Lebrero P; Balboa MA; Balsinde J J. Lipid Res 2018, 59, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Liebisch G; Vizcaino JA; Kofeler H; Trotzmuller M; Griffiths WJ; Schmitz G; Spener F; Wakelam MJO J. Lipid Res 2013, 54, 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Yu X; Jin W; McLuckey SA; Londry FA; Hager JW J. Am. Soc. Mass Spectrom 2005, 16, 71–81. [DOI] [PubMed] [Google Scholar]
- (50).Xia Y; Liang XR; McLuckey SA J. Am. Soc. Mass Spectrom 2005, 16, 1750–1756. [DOI] [PubMed] [Google Scholar]
- (51).Webb IK; Londry FA; McLuckey SA Rapid Commun. Mass Spectrom 2011, 25, 2500–2510. [DOI] [PubMed] [Google Scholar]
- (52).Londry FA; Hager JW J. Am. Soc. Mass Spectrom 2003, 14, 1130–1147. [DOI] [PubMed] [Google Scholar]
- (53).Sansone A; Tolika E; Louka M; Sunda V; Deplano S; Melchiorre M; Anagnostopoulos D; Chatgilialoglu C; Formisano C; Di Micco R; Mennella MRF; Ferreri C Plos One. 2016, 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Shevchenko A; Simons K Nat. Rev. Mol. Cell Biol 2010, 11, 593–598. [DOI] [PubMed] [Google Scholar]
- (55).Franklin ET; Betancourt SK; Randolph CE; McLuckey SA; Xia Y Anal. Bioanal. Chem 2019, 1 DOI: 10.1007/s00216-018-1537-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.