

Abstract
A novel series of thieno[3,2-d]pyrimidine derivatives were synthesised and their inhibitory effects against diacylglycerol acyltransferase 1 (DGAT-1) were assessed. cis-Isomer 17a showed potent and selective inhibitory activity against DGAT-1 in SF9 cells. In addition, 17a had an acceptable pharmacokinetic profile and accumulated mainly in the small intestine and liver. Oral administration of 17a led to a significant reduction in plasma triacylglycerol level during an oral lipid tolerance test (OLTT) in murine and canine models. Taken together, 17a is a high-quality candidate that deserves further investigation.
Keywords: DGAT-1, obesity, small molecules, thienopyrimidine, type 2 diabetes
Introduction
Obesity is characterised by abnormal fat accumulation due to a systemic dysfunction of energy homeostasis. It is a significant risk factor for diabetes, hypertension, cardiovascular disease, and non-alcoholic fatty liver disease1. Excessive accumulation of triglycerides (or triacylglycerol, TAG) in adipocytes and non-adipocytes, including in the liver, skeletal muscle, myocardium, and pancreas, is a crucial feature of obesity2. In particular, abnormal TAG levels increases the risk of metabolic syndrome, characterised by as insulin resistance, dyslipidemia and cardiomyopathy3.
Therefore, preventing excessive accumulation of TAG could be beneficial in the treatment of metabolic diseases such as obesity and type 2 diabetes. Two routes of TAG biosynthesis have been identified: the glycerol phosphate pathway, and the monoacylglycerol pathway4. Both routes have a common intermediate, diacylglycerol (DAG), which is then converted to TAG by acyl CoA: diacylglycerol acyltransferase (DGAT)5. DGAT exists as two isoforms, DGAT-1 and DGAT-2, which share minimal sequence homology6. These enzymes catalyse the final dedicated step in TAG synthesis: i.e. the esterification of a fatty acid moiety to DAG to generate TAG. DGAT-1 is highly expressed in the small intestine, adipose tissue, and liver6. DGAT-1 deficient mice showed a significant reduction in the postprandial increase of plasma TAG and were resistant to diet-induced obesity due to increased energy expenditure. Moreover, the DGAT1 knockout mice had enhanced sensitivity to insulin and leptin. Indeed, DGAT-1 inhibitors had significant pharmacologic effects, including decreased TAG, which were consistent with the DGAT1 knockout mice phenotypes7. However, DGAT2 knockout mice had extremely low levels of TAG, and their skin could not protect against moisture, so these mice died after birth8. Therefore, selective DGAT-1 inhibitors have been developed to manage metabolic diseases such as obesity and type 2 diabetes9–11. Pharmaceutical companies, including AstraZeneca (1)12, Novartis (2)13, Pfizer (3)14, and Abbott (4)15, and Hoffmann-La Roche (5)16 are developing selective DGAT-1 inhibitors, whose structures are described in Figure 1. Other novel DGAT1 inhibitors have also been reported17–19.
Figure 1.

(a) Structures of reported DGAT1 inhibitors; (b) diagram showing structural features of DGAT-1 inhibitors.
Herein, we described the discovery of novel thieno[3,2-d]pyrimidine derivatives as DGAT-1 inhibitors. Target molecules were designed based on a bioisosteric replacement strategy (Figure 2). Introduction of a heterobicylcle instead of a phenyl linker was attempted. Thienopyrimidines showed various pharmacologic activities as a privileged scaffold20, and the introduction of thieno[3,2-d]pyrimidine was the first approach to designing novel inhibitors based on the structural features of reported DGAT-1 inhibitors.
Figure 2.

A schematic strategy for the design of target molecules.
Materials and methods
Chemistry
All commercial chemicals and solvents were reagent grade and were used without further purification. Completion of the reactions was monitored by analytical thin layer chromatography (TLC) using precoated glass-backed plates (E-Merck, silica gel 60 F254 0.25 mm). For normal pressure and flash column chromatography purifications, Merck silica gel 60 (size 70–230 and 230–400 mesh, respectively) was used. 1H and 13C NMR spectra were recorded with an Avance DPX-300 NMR spectrometer (Bruker, Germany) and Jeol 600 MHz spectrometer (Jeol Resonance ECZ 600 R, USA). All the 1H and 13C NMR spectra were recorded in deuterated chloroform (CDCl3) with tetramethylsilane (TMS) as an internal standard or deuterated dimethyl sulfoxide (DMSO)-d6 as solvents; chemical shifts are reported in δ values (ppm) relative to the residual solvent peak. Multiplicity was indicated as follows: s (singlet); d (doublet); t (triplet); m (multiplet); dd (doublet of doublet); brs (broad singlet). Mass spectra were obtained on Waters Aquity UPLC/QTOF (Waters Corporation, USA). HPLC was used Agilent, 1200 series using capcellpak MGII (4.6 × 150 mm, 5 μm) eluted with a 30 min gradient from 20–70% acetonitrile in water.
Synthesis of 17a and 17b
Tert-butyl (4–(7-bromothieno[3,2-d]pyrimidin-4-yl)phenyl)carbamate (8)
A mixture of 7-bromo-4-chlorothieno[3,2-d]pyrimidine 6 (13 g, 52.10 mmol, CY C37043, KindChem Corporation, China), t-butyl 4–(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) phenylcarbamate 7 (20 g, 62.70 mmol), tetrakis(triphenylphosphine)palladium (3.16 g, 2.73 mmol) and sodium carbonate (8.28 g. 78.12 mmol) were added to 100 mL of 1,4-dioxane/water (4:1), and then stirred at 80 °C for 18 h under argon atmosphere. The organic layer was extracted with 300 mL of ethyl acetate and 300 mL of water, dried over anhydrous magnesium sulphate, and then concentrated in vacuo. Subsequently, 30 mL of methanol was added, and the resulting mixture was stirred to precipitate out solids, and then filtered to obtain 12.5 g of the yellow title compound.
1H-NMR (300 MHz, CDCl3): δ 9.80(s, 1H), 8.18(d, J = 6.9 Hz, 2H), 8.04(s, 1H), 7.62(d, J = 6.9 Hz, 2H), 6.76(s, 1H), 1.58(s, 9H).
Methyl 2–(4–(4-(4-((tert-butoxycarbonyl)amino)phenyl)thieno[3,2-d]pyrimidin-7-yl)cyclohex-3-en-1-yl)acetate (10)
A mixture of 8 (0.56 g, 1.38 mmol), 2–(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)cyclohex-3-en-1-yl)acetate 9 (0.5 g, 1.78 mmol), which was prepared according to the reported method21, and tetrakis(triphenylphosphine)palladium (94.9 mg, 0.08 mmol) was added to a solution of 2.06 mL of 2 N aqueous sodium carbonate and 8.2 mL of 1,4-dioxane, and then stirred at 100 °C for 12 h under argon. The reaction mixture was extracted with 300 mL of ethyl acetate and 300 mL of water. The organic layer was dried with anhydrous magnesium sulphate, filtered and then concentrated. Subsequently, methanol was added to the resulting solution, stirred to precipitate out solids, and then filtered to obtain 315 mg of the yellow title compound.
1H-NMR (300 MHz, CDCl3): δ 9.30(s, 1H), 8.17(d, J = 8.7 Hz, 2H), 7.43(s, 1H), 7.59(d, J = 8.7 Hz, 2H), 7.16(bs, 1H), 6.69(s, 1H), 3.71(s, 3H), 2.64 ∼ 2.38(m, 3H), 2.2 6 ∼ 2.02(m, 6H), 1.56(s, 9H). LCMS (ESI) m/z 480.2 [M + H]+; HRMS calcd for C26H29N3O4S [M + H]+ 480.1957, found 480.1986
Methyl 2–(4–(4-(4-((t-butoxycarbonyl)amino)phenyl)thieno[3,2-d]pyrimidin-7-yl)cyclohexyl) acetate (11)
To a solution of 10 (315 mg, 0.66 mmol) in ethanol (50 mL) and 1,4-dioxane (20 mL) was added 20% charcoal-shaped palladium hydroxide (158 mg, 50%w/w). The mixture was stirred at room temperature under H2 overnight. The reaction mixture was filtered through Celite, and then the filtrate was concentrated to obtain the title compound 11 (190 mg), which was used in the following step without further purification.
1H-NMR (300 MHz, CDCl3): δ 9.26(s, 2H), 8.17(d, J = 8.5 Hz, 4H), 7.69(s, 1H), 7.63(s, 1H), 7.62(d, J = 8.2 Hz, 4H), 6.71(s, 2H), 3.69(s, 6H), 3.3 2 ∼ 3.17(m, 2H), 2.50(d, J = 7.5 Hz, 2H), 2.4 3 ∼ 2.35(m, 1H), 2.33(d, J = 6.6 Hz, 2H), 2.2 8 ∼ 2.18(m, 2H), 2.0 7 ∼ 1.19(m, 9H), 1.7 3 ∼ 1.60(m, 4H), 1.57(s, 18H), 1.4 0 ∼ 1.25(m, 2H). LCMS (ESI) m/z 482.2 [M + H]+; HRMS calcd for C26H31N3O4S [M + H]+ 482.2114, found 482.2258
Methyl 2–(4–(4-(4-aminophenyl)thieno[3,2-d]pyrimidin-7-yl)cyclohexyl)acetate (12)
To a solution of 11 (280 mg, 0.58 mmol) in dichloromethane (5 mL) was added 0.6 mL of trifluoroacetic acid. The mixture was stirred at room temperature for 12 h. Then the reaction mixture was concentrated to a residue that was partitioned between dichloromethane and saturated aqueous sodium bicarbonate. The organic layer was separated from the aqueous layer, washed with brine, dried over anhydrous magnesium sulphate, and then concentrated to afford 190 mg of the title compound.
1H-NMR (300 MHz, DMSO-d6): δ 9.08(s, 2H), 8.13(s, 1H), 8.04(s, 1H), 8.01(d, J = 8.6 Hz, 4H), 6.76(d, J = 8.6 Hz, 4H), 5.90(s, 4H), 3.60(s, 6H), 3.3 0 ∼ 3.10(m, 2H), 2.48(d, 2H), 2.28(d, J = 6.5 Hz, 2H), 2.2 5 ∼ 2.13(m, 1H), 2.2 8 ∼ 2.16(m, 3H), 2.1 0 ∼ 1.98(m, 2H), 1.9 0 ∼ 1.46(m, 14H), 1.3 0 ∼ 1.10(m, 2H).
Methyl 2–(4–(4-(4-(3–(3-chlorophenyl)ureido)phenyl)thieno[3,2-d]pyrimidin-7-yl)cyclohexyl)acetate (13)
To a solution of 12 (3.1 g, 8.13 mmol) in anhydrous tetrahydrofuran (60 mL) was added 3-chlorophenyl isocyanate (1.37 g, 8.92 mmol). The mixture was stirred at room temperature for 18 h. The mixture was concentrated in part and diluted with diethyl ether to give a precipitate, which was filtered to afford 4.06 g of a light-yellow title compound.
1H-NMR (300 MHz, DMSO-d6): δ 9.21(s, 2H), 9.16(s, 2H), 9.03(s, 2H), 8.24(s, 1H), 8.2 1 ∼ 8.15(m, 5H), 7.7 4 ∼ 7.71(m, 6H), 7.4 0 ∼ 7.29(m, 4H), 7.1 0 ∼ 7.00(m, 2H), 3.61(s, 6H), 3.3 2 ∼ 3.04(m, 2H), 2.46(d, 2H), 2.29(d, J = 6.5 Hz, 2H), 2.2 2 ∼ 2.14(m, 1H), 2.2 1 ∼ 2.01(m, 2H), 1.8 9 ∼ 1.49(m, 14H), 1.3 0 ∼ 1.11(m, 2H). LCMS (ESI) m/z 535.2 [M + H]+; HRMS calcd for C28H27ClN4O3S [M + H]+ 535.1571, found 535.1793
Methyl cis-2–(4–(4-(4-(3–(3-chlorophenyl)ureido)phenyl)thieno[3,2-d]pyrimidin-7-yl)cyclohexyl)acetate (15a)
13 (150 mg, 0.28 mmol) was added to ethyl acetate (2 mL) and stirred at 70 °C for 12 h, and then cooled to room temperature to give a solid. The solid (15b, 72 mg) was filtered, and then the filtrate was concentrated and the residue was purified by silica gel column chromatography to give 76 mg of the yellow title compound 15a.
1H-NMR (300 MHz, DMSO-d6): δ 9.23(s, 1H), 9.17(s, 1H), 9.04(s, 1H), 8.25(s, 1H), 8.19(d, J = 8.8 Hz, 2H), 7.7 5 ∼ 7.72(m, 3H), 7.3 3 ∼ 7.31(m, 2H), 7.0 7 ∼ 7.03(m, 1H), 3.61(s, 3H), 3.3 1 ∼ 3.18(m, 1H), 2.52(d, 2H), 2.2 8 ∼ 2.14(m, 1H), 1.8 9 ∼ 1.49(m, 8H). LCMS (ESI) m/z 535.2 [M + H]+; HRMS calcd for C28H27ClN4O3S [M + H]+ 535.1571, found 535.1793
Methyl trans-2–(4–(4-(4-(3–(3-chlorophenyl)ureido)phenyl)thieno[3,2-d]pyrimidin-7-yl)cyclohexyl)acetate (15b)
1H-NMR (300 MHz, DMSO-d6): δ 9.23(s, 1H), 9.23(s, 1H), 9.16(s, 1H), 9.03(s, 1H), 8.19(d, J = 8.8 Hz, 2H), 8.16(s, 1H), 7.7 5 ∼ 7.72(m, 3H), 7.3 6 ∼ 7.29(m, 2H), 7.0 7 ∼ 7.03(m, 1H), 3.61(s, 3H), 3.11(t, J = 11.9 Hz, 1H), 2.30(d, J = 6.5 Hz, 2H), 2.1 3 ∼ 2.01(m, 2H), 1.9 0 ∼ 1.72(m, 3H), 1.6 8 ∼ 1.50(m, 2H), 1.2 9 ∼ 1.11(m, 2H). LCMS (ESI) m/z 535.2 [M + H]+; HRMS calcd for C28H27ClN4O3S [M + H]+ 535.1571, found 535.1793
cis-2–(4–(4-(4-(3–(3-chlorophenyl)ureido)phenyl)thieno[3,2-d]pyrimidin-7-yl)cyclohexyl)acetic acid (16a)
To a solution of 15a (7.8 g, 14.58 mmol) in tetrahydrofuran/methanol/water (120 mL, 1:1:1) was added 1.17 g of sodium hydroxide (29.25 mmol), and the mixture was stirred. The reaction mixture was acidified with 1 N HCl (pH was adjusted to between 5 and 6) to give a solid. The solid was filtered, and washed with water to afford 16a quantitatively.
1H-NMR (300 MHz, DMSO-d6): δ 9.28(s, 1H), 9.23(s, 1H), 9.16(s, 1H), 8.24(s, 1H), 8.19(d, J = 8.8 Hz, 2H), 7.7 5 ∼ 7.72(m, 3H), 7.3 3 ∼ 7.31(m, 2H), 7.1 0 ∼ 7.00(m, 1H), 3.3 2 ∼ 3.20(m, 1H), 2.39(d, J = 7.5 Hz, 2H), 2.2 8 ∼ 2.14(m, 1H), 1.9 0 ∼ 1.50(m, 8H). 13C -NMR (150 MHz, DMSO-d6): δ 174.08, 160.01, 158.32, 154.04, 152.20, 142.37, 142.10, 141.00, 133.20, 131.36, 130.43, 129.97, 129.20, 127.20, 121.70, 118.21, 117.66, 116.75, 37.03, 35.51, 30.03, 29.31, 27.18. LCMS (ESI) m/z 521.1 [M + H]+; HRMS calcd for C27H25ClN4O3S [M + H]+ 521.1414, found 521.1441
trans-2–(4–(4-(4-(3–(3-chlorophenyl)ureido)phenyl)thieno[3,2-d]pyrimidin-7-yl)cyclohexyl)acetic acid (16b)
16b was obtained from the procedure described for 16a
1H-NMR (300 MHz, DMSO-d6): δ 12.04(s, 1H), 9.29(s, 1H), 9.23(s, 1H), 9.16(s, 1H), 8.18(d, J = 9.0 Hz, 2H), 8.15(s, 1H), 7.7 5 ∼ 7.72(m, 3H), 7.3 6 ∼ 7.29(m, 2H), 7.0 7 ∼ 7.02(m, 1H), 3.11(t, J = 12.1, 1H), 2.19(d, J = 6.7 Hz, 2H), 2.1 3 ∼ 2.01(m, 2H), 1.9 0 ∼ 1.72(m, 3H), 1.6 8 ∼ 1.50(m, 2H), 1.2 9 ∼ 1.11(m, 2H). 13C -NMR (150 MHz, DMSO-d6): δ 173.90, 159.93, 158.35, 154.10, 152.22, 142.36, 141.05, 133.19, 130.81, 130.41, 129.97, 129.17, 127.27, 121.68, 118.25, 117.70, 116.79, 41.60, 36.15, 34.13, 32.42, 32.05. LCMS (ESI) m/z 521.1 [M + H]+; HRMS calcd for C27H25ClN4O3S [M + H]+ 521.1414, found 521.1441
Sodium cis-2–(4–(4-(4-(3–(3-chlorophenyl)ureido)phenyl)thieno[3,2-d]pyrimidin-7-yl)cyclohexyl)acetate (17a)
To a solution of 16a (7.6 g, 14.59 mmol) in 100 mL of methanol was added 14 mL of a 1 N sodium hydroxide. The resulting mixture was stirred at room temperature for 2 h. The solvent was removed to give 8.3 g of the yellow title compound 17a.
1H-NMR (300 MHz, DMSO-d6): δ 12.50(s, 1H), 12.37(s, 1H), 9.21(s, 1H), 8.23(s, 1H), 8.17(d, J = 8.8 Hz, 2H), 7.7 5 ∼ 7.72(m, 3H), 7.56 (d, J = 9.0 Hz, 1H), 7.27 (t, J = 8.1 Hz, 1H), 6.96 (d, J = 7.9 Hz, 1H), 3.3 2 ∼ 3.18(m, 1H), 2.4 0 ∼ 2.20(m, 3H), 2.0 0 ∼ 1.60(m, 8H). 13C -NMR (150 MHz, DMSO-d6): δ 178.28, 160.00, 158.50, 154.04, 153.53, 144.38, 142.88, 142.70, 132.90, 130.91, 130.04, 128.93, 128.77, 126.97, 120.44, 117.95, 117.40, 116.55, 41.40, 39.08, 30.92, 30.03, 27.70. LCMS (ESI) m/z 521.1 [M + H]+; HRMS calcd for C27H25ClN4O3S [M + H]+ 521.1414, found 521.1441 (as free base)
Sodium trans-2–(4–(4-(4-(3–(3-chlorophenyl)ureido)phenyl)thieno[3,2-d]pyrimidin-7-yl)cyclohexyl)acetate (17b)
1H-NMR (300 MHz, DMSO-d6): δ 12.27(s, 1H), 12.25(s, 1H), 9.18(s, 1H), 8.17(d, J = 8.9 Hz, 2H), 7.99(s, 1H), 7.89(d, J = 8.9 Hz, 2H), 7.83(s, 1H), 7.56(d, J = 9.3 Hz, 1H), 7.27(t, J = 8.1 Hz, 1H), 6.96(d, J = 8.0 Hz, 1H), 3.11(t, J = 11.6, 1H), 2.1 0 ∼ 1.83(m, 2H), 1.6 8 ∼ 1.46(m, 2H), 1.2 9 ∼ 1.11(m, 2H). 13C -NMR (150 MHz, DMSO-d6): δ 178.21, 159.84, 158.38, 154.07, 153.42, 144.40, 142.93, 142.83, 132.90, 130.28, 130.05, 128.94, 128.60, 126.87, 120.48, 117.80, 117.32, 116.49, 46.16, 36.07, 35.92, 33.08, 32.68. LCMS (ESI) m/z 521.1 [M + H]+; HRMS calcd for C27H25ClN4O3S [M + H]+ 521.1414, found 521.1441 (as free base)
DGAT-1 inhibition assay (IC50)
The activity of DGAT-1 inhibitors in vitro was evaluated by using a human recombinant DGAT1 enzyme expressed in insect cells (SF9 cells). SF9 cells were homogenised by washing them with DPBS (Dulbecco’s phosphate-buffered saline) and then suspending cell pellets with a tris buffer (250 mM sucrose; 10 mM Tris-HCl [pH 7.4]; proteinase inhibitor). The resulting mixture was centrifugally separated at 10,000 × g for 30 min to remove the cell debris remaining in the lower layer thereof, and was centrifugally separated at 100,000 × g for 60 min to obtain a microsomal membrane. Further, membrane fractions were resuspended by the tris buffer, and then stored at -80 °C.
The activity of DGAT1 was measured according to the reported method20. Specifically, 0.0001 – 10 μΜ (final concentration, FC) of the test compounds were cultured at room temperature (25 °C) for 15 min with a 10 μΜ of SF9 microsomal protein solution and 100 mM of MgCl2 solution, and were then further cultured at room temperature (25 °C) for 30 min after the addition of 100 μΜ (FC in 12.5% EtOH) of 1,2-dioleyl-sn-glycerol and 30 μΜ (final conc.) of 14C-oleyl coenzyme A. The reaction was completed by the addition of 300 μL of a mixed solution of 2-propanol/heptane (7:1), and radioactive triglyceride was separated from an organic solvent layer by using 200 μL of heptane and 200 μL of a 0.1 M carbonate buffer (pH 9.5). The amount of triglyceride was measured by liquid scintillography (Perkin Elmer, USA) after mixing with an organic solvent and an equivalent amount of scintillation solvent (MicroScint-O, Perkin Elmer, USA). The effects of inhibiting DGAT1 were calculated as percent in comparison with a control material.
Oral lipid tolerance test (OLTT)
Mouse
Male, Institute of Cancer Research (ICR) mice were obtained from OrientBio Inc. (Republic of Korea). ICR mice (8-weeks old, n = 5/group) fasted 16 h, received 5 mL/kg corn oil via oral administration (p.o.) 30 min after compound administration. Plasma was collected at different time points (0, 1, 2, and 4 h).
Dog
Beagle dogs (9 months old, n = 2/group) were obtained from Woojung Bio Inc. (Republic of Korea) were fasted for 16 h and received heavy cream (2.7 mL/kg), glucose (1 g/kg), water (5.3 mL/kg), and acetaminophen (20 mg/kg p.o.) 1 h after compound administration. Blood samples were collected at –0.5, 0, 1, 2, and 4 h.
Statistical analysis
Statistical analysis was performed using one-way ANOVA, followed by Dunnett’s multiple comparison test using Prism 6.0 (GraphPad, USA). The Kruskal-Wallis test was applied when data did not pass the normality test.
Results and discussion
We found that thieno[3,2-d]pyrimidine could be a core scaffold connecting an aryl moiety at one end to a cyclo-aliphatic carboxylic acid group at the other. Synthetic schemes for a series of thieno[3,2-d]pyrimidine derivatives are outlined in Schemes 1 and 2.
Scheme 1.

Synthesis of thienopyrimidine derivatives. Reagents and conditions: (a) Pd(PPh3)4, 2 N Na2CO3, 1,4-Dioxane; (b) H2, 20% PdOH, EtOH/1,4-Dioxane; (c) TFA, DCM, rt; (d) aryl chloride, pyridine, DCM 0 °C to rt or aryl isocyanate, THF, 12 h, rt; (e) NaOH or LiOH, THF/MeOH/H2O (1:1:1), rt.
Scheme 2.

Reagents and conditions: (a) THF, rt; (b) separation by using ethyl acetate at 60–70 °C; filtrate for 15a and solid for 15b; (c) NaOH, THF/MeOH/H2O; (d) 1 M NaOH, MeOH, rt.
Desired compounds 14a–14l were synthesised from commercially available 7-bromo-4-chlorothieno[3,2-d]pyrimidine (6), as shown in Scheme 1. Consecutive Suzuki coupling reactions gave 8 and 10. Compound 9 was prepared using the reported method19. Hydrogenation of 10, followed by de-protection of the Boc group with trifluoroacetic acid afforded the key intermediate 12. The aniline 12 was converted to amides 14a–14h or ureas 14i–14l by coupling with acid chlorides or aryl isocyanates, respectively, followed by hydrolysis with sodium hydroxide or lithium hydroxide.
The activities of synthesised compounds were evaluated using SF9 insect cells expressing human DGAT-122. The IC50 values are summarised in Table 1. A potent DGAT-1 inhibitor, LCQ-908 (2 or pradigastat) was used as a positive control. To optimise the alkyl chain length of the acid moiety, compound 14b (n = 1) was more potent than short chain 14a (n = 0) and long chain 14c (n = 2). The calculated lowest energy conformation model of 14c provided that the distance between the terminal carboxylic acid and NH of urea was over 16 Å, which may result in reduced activity. 14a exhibited a loss of activity, which suggested that the terminal carboxylic acid should be placed one-carbon away from cyclohexane. Next, structure-activity relationships for the aryl moiety on the left side were investigated, with derivatives bearing chain lengths the same as that of 14b. 5-chloro-2-nitro phenyl (14d), 2-trifluoromethylpyridinyl (14f), 4-chloropyridinyl (14k), and 5-bromopyridinyl (14l) derivatives had no activity (IC50 >1 μM), while 14b, 14c, 14e, 14h, and 14j had good activity (IC50 values of 0.3 μM–0.4 μM). Compounds 14g and 14i were equipotent to the known compound 2. The most potent compound, 14i, was subjected to further investigation. Cis and trans isomers of 14i were separated to determine the activity and properties of each isomer. As shown in Scheme 2, the racemic mixture 13 was conveniently separated into cis-isomer 15a and trans-isomers 15b by the treatment with ethyl acetate. 15a and 15b exhibited greatly different solubility for ethyl acetate, and were soluble and insoluble for ethyl acetate, respectively. Hydrolysis of 15a and 15b afforded cis isomer 17a and trans isomer 17b, respectively.
Table 1.
DGAT-1 inhibitors and their in vitro data.
 |
Data present mean values.
Interestingly, the isomers 17a and 17b were more potent than mixture 14i, and even compound 2, in SF9 cells (Table 2). However, pharmacokinetic studies demonstrated that cis-isomer 17a had a much better profile than trans-isomer 17b (Table 3). 17a had a shorter half-life (1.2 h), but had much better bioavailability, compared to 17b. In enzymatic assays, 17a had an IC50 of 61 nM for DGAT-1 and displayed high off-target selectivity against DGAT-2, acyl-coenzyme A (CoA):cholesterol acyltransferases (ACAT1 and ACAT2) (Table 2). ACATs have sequence homology to DGAT-1 and play essential roles in cholesterol homeostasis23. 17b was not explored in the enzymatic assays due to its poor pharmacokinetic profile.
Table 2.
Enzyme and cellular inhibitory potencies for selected compounds.
| IC50 (nM) | |||||
|---|---|---|---|---|---|
|
Compound |
DGAT1(SF9) | hDGAT1 | hDGAT2 | hACAT1 | hACAT2 |
| 2 | 157 | 57 | >10,000 | >10,000 | >10,000 |
| 17a | 74 | 61 | >10,000 | >10,000 | >10,000 |
| 17b | 89 | – | – | – | – |
Table 3.
Pharmacokinetic profiles of compounds 17a, 17b in ICR mice.a
| Compounds | 17a | 17b |
|---|---|---|
| AUC 0∼24 (ng·h/mL) | 861.2 | 233.7 |
| Cmax (ng/mL) | 479.3 | 74.8 |
| T1/2 (h) | 1.2 | 5.2 |
| Bioavailability (%) | 19 | 0.7 |
All parameters were determined after oral administration (10 mg/kg, n = 3) and intravenous (iv) injection (3 mg/kg, n = 2) in ICR mice; both compounds were dissolved in a vehicle solution of DMSO/Tween80/saline (1:6:23) for iv., and 0.5% methylcellulose/0.5% Tween80 in distilled water for oral administration. Data represent mean values.
Considering potency, selectivity, and pharmacokinetic profiles, 17a was investigated for further pharmacokinetic properties in four species, and results are summarised in Tables 4 and 5. 17a exhibited low to moderate clearance and acceptable oral bioavailability in rat and dog.
Table 4.
Oral pharmacokinetic profile of 17a in rat and dog.
| Parameters | AUC 0 ∼ 24 (ng·h/mL) | T 1/2 (h) | Cl (L/hr/kg)) | Bioavailability (%) |
|---|---|---|---|---|
| Rata | 18,594.5 ± 6764.2 | 3.0 ± 0.4 | 0.5 ± 0.2 | 30 |
| Dogb | 8243.0 ± 2296.3 | 3.6 ± 0.7 | 0.4 ± 0.1 | 53 |
Dose; 3 mg/kg for iv, 10 mg/kg for po.; Vehicle: DMSO/Tween80/saline (1:6:23) for iv.; 0.5% methylcellulose/0.5% Twee80 in distilled water for po.
Dose: 1 mg/kg for iv, 3 mg/kg for po.; Vehicle: DMSO/Tween80/saline (1:3:26) for iv; 0.5% methylcellulose/0.5% Tween80 in distilled water for po.
Table 5.
In vitro microsomal stability and cytochrome P450 (CYP) inhibition assay of compound 17a.
| Microsomal stability, (%)a |
CYPs enzyme inhibition, IC50 (μM)b |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| No | Human | Dog | Rat | Mouse | CYP3A4 | CYP1A2 | CYP2C9 | CYP2C19 | CYP2D6 |
| 17a | 94 | 82 | 87 | 82 | 16.2 | 10.6 | 2.9 | >20 | 8.7 |
Remaining percent of metabolism by incubation of the parent molecule (5 μM) with liver microsomes of mouse, rat, dog, and human for 60 min (duplicate).
Data were analysed by a fluorescence detection method.
17a exhibited durable metabolic stabilities, as shown in Table 5. In particular, 17a was metabolically stable in four species: human, dog, rat, and mouse. Further evaluation of 17a was carried out for inhibition of clinically relevant cytochrome 450 (CYP) isoforms (3A4, 1A2, 2C9, 2C19, 2D6). No significant inhibition by 17a was observed (Table 5). Thus, 17a would be very unlikely to affect the pharmacokinetics of other drugs.
To determine in vivo efficacy, we evaluated the activity of 17a against serum TAG level induced by the oral administration of corn oil (oral lipid tolerance test, OLTT; Figures 3 and 4), since intestinal targeted DGAT-1 inhibition results in a reduced serum TAG level14. 17a was orally administered to ICR mice at 3 mg/kg or to dogs at 1 mg/kg before administration of corn oil. As shown in Figure 3, 17a showed significant activity, as potent as compound 2, in reducing plasma TAG level.
Figure 3.
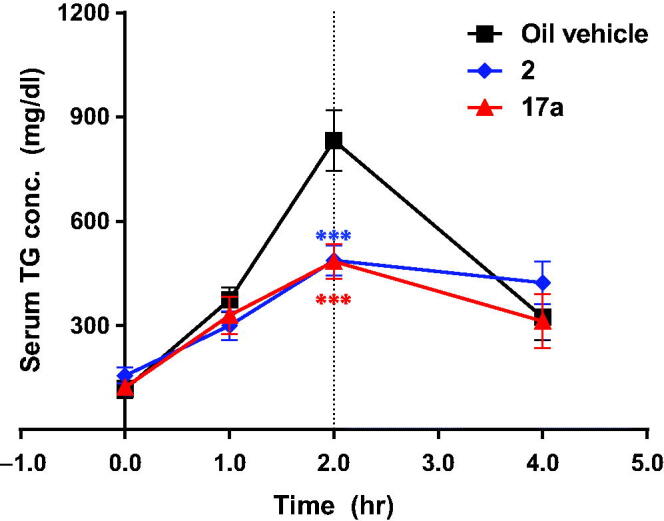
Effect of 17a in the oral lipid tolerance test (n = 5, ***p < .001). Serum TG (or TAG) concentration was measured before and 1, 2, and 4 h after oral administration of corn oil in ICR mice treated with 17a (3 mg/kg) or 2 (1 mg/kg). Vehicle for 2: 0.5% methylcellulose/0.5% Tween80 in distilled water, vehicle for 17a: 0.5% Tween80 in distilled water.
Figure 4.
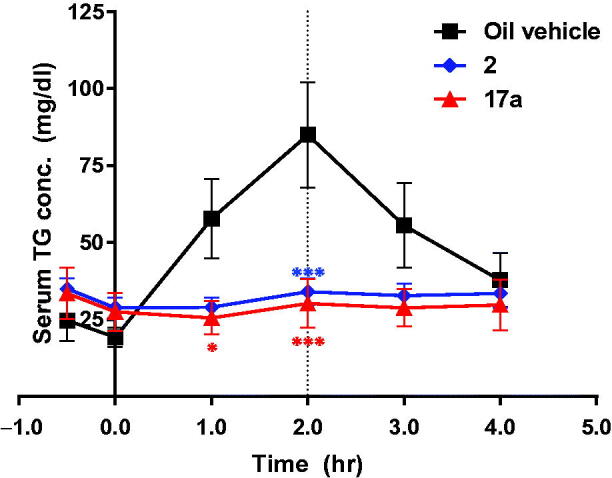
Effect of 17a in a canine oral lipid tolerance test (n = 2, *p < .05; ***p < .001). The dog was treated with 17a (po. 1 mg/kg) or 2 (po. 1 mg/kg) before oral administration of corn oil. Vehicle for 2: 0.5% methylcellulose/0.5% Tween80 in distilled water, vehicle for 17a: 0.5% Tween80 in distilled water.
In addition, tissue distribution of 17a was investigated. 17a was orally administered to mice at 30 mg/kg, and 2 was administrated as a positive control (Figure 5). The concentration levels of 17a in the liver and small intestine were much higher than in other tissues, indicating that liver and small intestine are the main target organs of 17a, while 2 showed much high concentrations in the liver and large intestine. Analysis of tissue-to-plasma ratios indicated that 17a had favourable distributions in the liver and small intestine. Significant activity of 17a in the OLTT appeared to result from high concentration in the small intestine, even though bioavailability of 17a was not great in mice. This result could be proof of a pharmacokinetic–pharmacodynamic correlation.
Figure 5.

Tissue distribution of 17a in mice. 17a and 2 were orally administered to ICR mice at 30 mg/kg (n = 3). (a) AUC values of 17a and 2 in indicated tissues. Each bar shows AUC obtained from concentrations measured at 0.5 h, 1 h, 2 h, 4 h, and 24 h after dosing. Data represent mean value. (b) Tissue-to-plasma (T/P) ratio of 17a and 2. S.I.: small intestine, L.I.: large intestine.
Conclusion
We found that a novel series of thieno[3,2-d]pyrimidine derivatives had potent DGAT-1 inhibitory activity. A representative compound, cis-isomer 17a showed potent DGAT-1 inhibitory activity, metabolic stability in four species, an acceptable pharmacokinetic profile in rodents and dogs, and a significant reduction of TAG in the OLTT in mice and dogs. In a tissue-distribution study, 17a was mainly distributed to the liver and small intestine. Therefore, 17a could be a candidate with considerable potential and deserves for further investigation.
Funding Statement
This research was supported by the Advanced Research Centre Programme (2015R1A5 A1008958) funded by the Korean Government.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Kusminski CM, Bickel PE, Scherer PE.. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov 2016;15:639–60. [DOI] [PubMed] [Google Scholar]
- 2.Subauste A, Burant CF.. Dgat: novel therapeutic target for obesity and type 2 diabetes mellitus. Curr Drug Targets Immune Endocr Metabol Disord 2003;3:263–70. [DOI] [PubMed] [Google Scholar]
- 3.Capeau J. Insulin resistance and steatosis in humans. Diabetes Metab 2008;34:649–57. [DOI] [PubMed] [Google Scholar]
- 4.Kennedy EP. Metabolism of lipides. Annu Rev Biochem 1957;26:119–48. [DOI] [PubMed] [Google Scholar]
- 5.Yen CL, Stone SJ, Koliwad S, et al. . Thematic review series: glycerolipids. Dgat enzymes and triacylglycerol biosynthesis. J Lipid Res 2008;49:2283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatt-Wessel B, Jordan TW, Miller JH, Peng L.. Role of dgat enzymes in triacylglycerol metabolism. Arch Biochem Biophys 2018;655:1–11. [DOI] [PubMed] [Google Scholar]
- 7.Smith SJ, Cases S, Jensen DR, et al. . Obesity resistance and multiple mechanisms of triglyceride synthesis in mice lacking dgat. Nat Genet 2000;25:87–90. [DOI] [PubMed] [Google Scholar]
- 8.Stone SJ, Myers HM, Watkins SM, et al. . Lipopenia and skin barrier abnormalities in dgat2-deficient mice. J Biol Chem 2004;279:11767–76. [DOI] [PubMed] [Google Scholar]
- 9.Birch AM, Buckett LK, Turnbull AV.. Dgat1 inhibitors as anti-obesity and anti-diabetic agents. Curr Opin Drug Discov Devel 2010;13:489–96. [PubMed] [Google Scholar]
- 10.King AJ, Judd AS, Souers AJ.. Inhibitors of diacylglycerol acyltransferase: a review of 2008 patents. Expert Opin Ther Pat 2010;20:19–29. [DOI] [PubMed] [Google Scholar]
- 11.Motiwala H, Kandre S, Birar V, et al. . Exploration of pyridine containing heteroaryl analogs of biaryl ureas as dgat1 inhibitors. Bioorg Med Chem Lett 2011;21:5812–7. [DOI] [PubMed] [Google Scholar]
- 12.McCoull W, Addie MS, Birch AM, et al. . Identification, optimisation and in vivo evaluation of oxadiazole dgat-1 inhibitors for the treatment of obesity and diabetes. Bioorg Med Chem Lett 2012;22:3873–8. [DOI] [PubMed] [Google Scholar]
- 13.Meyers C, Gaudet D, Tremblay K, et al. . The dgat1 inhibitor lcq908 decreases triglyceride levels in patients with the familial chylomicronemia syndrome. J Clin Lipidol 2012;6:266–7. [Google Scholar]
- 14.King AJ, Segreti JA, Larson KJ, et al. . Diacylglycerol acyltransferase 1 inhibition lowers serum triglycerides in the zucker fatty rat and the hyperlipidemic hamster. J Pharmacol Exp Ther 2009;330:526–31. [DOI] [PubMed] [Google Scholar]
- 15.Zhao G, Souers AJ, Voorbach M, et al. . Validation of diacyl glycerolacyltransferase i as a novel target for the treatment of obesity and dyslipidemia using a potent and selective small molecule inhibitor. J Med Chem 2008;51:380–3. [DOI] [PubMed] [Google Scholar]
- 16.Qian Y, Wertheimer SJ, Ahmad M, et al. . Discovery of orally active carboxylic acid derivatives of 2-phenyl-5-trifluoromethyloxazole-4-carboxamide as potent diacylglycerol acyltransferase-1 inhibitors for the potential treatment of obesity and diabetes. J Med Chem 2011;54:2433–46. [DOI] [PubMed] [Google Scholar]
- 17.Harrison TJ, Bauer D, Berdichevsky A, et al. . Successful strategies for mitigation of a preclinical signal for phototoxicity in a dgat1 inhibitor. ACS Med Chem Lett 2019;10:1128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu Y, Wu Z, Shi Z-C, et al. . Accelerating the discovery of dgat1 inhibitors through the application of parallel medicinal chemistry (pmc). Bioorg Med Chem Lett 2019;29:1380–5. [DOI] [PubMed] [Google Scholar]
- 19.Nakajima K, Chatelain R, Clairmont KB, et al. . Discovery of an orally bioavailable benzimidazole diacylglycerol acyltransferase 1 (dgat1) inhibitor that suppresses body weight gain in diet-induced obese dogs and postprandial triglycerides in humans. J Med Chem 2017;60:4657–64. [DOI] [PubMed] [Google Scholar]
- 20.Devani MB, Shishoo CJ, Pathak US, et al. . Synthesis of 3-substituted thieno [2, 3-d] pyrimidin-4(3h)-one-2-mercaptoacetic acids and their ethyl esters for pharmacological screening. J Pharm Sci 1976;65:660–4. [DOI] [PubMed] [Google Scholar]
- 21.Dow RL, Munchhof MJ, Preparation of substituted dihydropyrimidooxazepine derivatives for use as antiobesity agents. In: Preparation of substituted dihydropyrimidooxazepine derivatives for use as antiobesity agents; 20080722; WO: Pfizer Products Inc.; 2009. [Google Scholar]
- 22.Birch AM, Birtles S, Buckett LK, et al. . Discovery of a potent, selective, and orally efficacious pyrimidinooxazinyl bicyclooctaneacetic acid diacylglycerol acyltransferase-1 inhibitor. J Med Chem 2009;52:1558–68. [DOI] [PubMed] [Google Scholar]
- 23.Pramfalk C, Eriksson M, Parini P.. Cholesteryl esters and acat. Eur J Lipid Sci Technol 2012;114:624–33. [Google Scholar]