

Abstract
Accumulated evidence suggests that sporadic cases of Alzheimer's disease (AD) make up more than 95% of total AD patients, and diabetes has been implicated as a strong risk factor for the development of AD. Diabetes shares pathological features of AD, such as impaired insulin signaling, increased oxidative stress, increased amyloid-beta (Aβ) production, tauopathy and cerebrovascular complication. Due to shared pathologies between the two diseases, anti-diabetic drugs may be a suitable therapeutic option for AD treatment. In this article, we will discuss the well-known pathologies of AD, including Aβ plaques and tau tangles, as well as other mechanisms shared in AD and diabetes including reactive glia and the breakdown of blood brain barrier in order to evaluate the presence of any potential, indirect or direct links of pre-diabetic conditions to AD pathology. In addition, clinical evidence of high incidence of diabetic patients to the development of AD are described together with application of anti-diabetic medications to AD patients.
Keywords: Diabetes, Alzheimer's Disease, Insulin Signaling, Oxidative Stress, Blood Brain Barrier, Brain Inflammation
Graphical Abstract
INTRODUCTION OF ALZHEIMER'S DISEASE (AD)
AD is a devastating brain disorder which gradually develops over an extended period of time, causing loss of memory and cognition accompanied by neuronal death in certain regions including the entorhinal cortex, hippocampus, and basal forebrain.1,2,3 AD affects about 12%–13% of people aged over 65, and nearly 50% of people aged 85 and older. Considering that humanity's average life span is continually increasing in the modern era, AD is noted to be one of the most problematic health issues of our time.4 AD can be categorized into familial Alzheimer's disease (FAD) or sporadic Alzheimer's disease (SAD), the latter accounting for more than 95% of all cases.5 SAD is largely misunderstood, due to components involving both genetic and environmental influences. Age-related risk factors associated with SAD include cardiovascular disease, cancer, stroke, diabetes mellitus (DM), and impaired glucose tolerance.6,7 AD pathology is also associated with increased oxidative stress in early stages of the disease process,8,9 and this oxidative stress may be the driving force behind impaired insulin signaling in AD affected brains. Cellular damage from oxidative stress can initiate the interruption of synthesis and/or function of lipids and proteins, leading to inactivation of enzymes, changes in receptor activity, and ultimately cell death.10 Synaptic damage, impaired neurogenesis, mitochondrial dysfunction, and lack of growth factors such as nerve growth factor and brain derived neurotrophic factor, are also implicated in AD.11,12,13 With progression, the symptoms of AD include irritability, aggression, depression, confusion, and decline of language abilities. Although AD progression has been studied extensively, there is still a paucity of evidence regarding the causes and mechanisms involved. Moreover, the initial symptoms are often mistaken as responses to stress or considered as normal ‘age-related’ changes. Behavioral tests and brain scans can aid in the diagnosis, but a final definitive diagnosis requires postmortem analysis of the brain. Symptomatic relief is attainable through means of active brain stimulation, physical activities, pharmacotherapy, and incorporating a balanced diet into one's lifestyle. Nonetheless, disease progression will inevitably continue.14
Two features in AD brains are extracellular amyloid-beta (Aβ) containing neuritic plaques, which are generated by overproduction of Aβ peptides from the amyloid precursor protein (APP), and intracellular accumulation of phosphor tau-positive neurofibrillary tangles from precipitated paired helical filaments (PHFs).1,2,3 These two abnormal structures are known to contribute to AD progression and have been argued for as either a cause or a result. Other pathology includes dysfunction of energy metabolism and neuronal death in selective areas of the brain including the hippocampus and cortex.14 Studies indicate that AD may be triggered by a multitude of factors including age, genetic background, and/or prolonged inflammation by means of physical injury (brain trauma or stroke leading to overproduction of reactive oxygen species [ROS]). Environmental factors also appear to contribute to AD pathogenesis.15,16
There are three major gene mutations associated with early onset forms of FAD: those for APP, presenilin 1 (PSEN1) and presenilin 2 (PSEN2).16 However, FADs represent less than 5% of all AD cases, suggesting many other factors that may be involved in disease progression. One such genetic risk factor is the possession of one or two copies of the ε4 alleles of the gene for apolipoprotein E (ApoE), which are linked to a late onset of AD cases with a buildup of amyloid plaques in the brain before AD symptoms arise.17 All these genetic mutations are linked to the progression of Aβ plaque formation in defined brain regions, which have been implicated as one of the initial symptoms of AD. Aβ peptides are generated from a transmembrane APP by β- and γ-secretases via consecutive endomembrane-proteolytic cleavage. Early studies have suggested that fibrillar Aβ aggregates, the main constituents of senile plaques, might have a key role in initiating neurodegenerative progression in AD brains.18 However, later studies have reported that oligomeric Aβ is more toxic than insoluble fibrillary Aβ, and an increase of oligomeric Aβ is known to be strongly correlated with the degree of cognitive dysfunction in AD,19,20 and associated with synaptic deficiency.21 Research has shown that both insoluble and soluble (oligomeric) species of Aβ exist in brains of AD patients and of a transgenic mouse model of AD.22,23,24
Aβ AND τ PATHOLOGIES IN AD
Although the exact pathological mechanisms in AD remain unclear, evidence indicates that accumulated Aβ peptides may initiate the process of neurodegeneration in AD brains.25,26 Aβ is known to induce its toxicity through direct interaction with an receptor for advanced glycation end product (RAGE), p75NTR, α-7 nicotinic AChR or an amylin receptor, or by indirectly mediating through glutamate excitotoxicity.27,28,29,30,31 The mechanisms underlying Aβ toxicity are not clear, but likely involve alteration of intracellular calcium signaling, generation of free radicals, phosphorylated tau, and a caspase-mediated apoptosis.31,32,33 Although evidence of a direct interaction of Aβ with receptors is still controversial, many studies have shown that Aβ peptides are internalized by interacting with RAGE, scavenging receptor, low-density lipoprotein receptor related protein-1 (LRP-1), NMDA glutamate receptors or α-7 nicotinic AChR receptors.34,35 Glutamate excitotoxicity has been implicated in neuronal death by Aβ toxicity: first, Aβ increases glutamate release and inhibits uptake; second, Aβ induces glutamate-mediated neurotoxicity that is reversed by antagonists of the glutamatergic NMDA receptor; third, the NMDA receptor has been found to be involved in Aβ neurotoxicity in rat brain, and Aβ toxicity increases susceptibility to glutamate toxicity in Aβ-generating transgenic mice.36 Not only neurons, but also astrocytes are a major source of glutamate from the glutamate-glutamine cycle occurring between neurons and astrocytes37,38; impaired glutamate uptake function by stressed neurons and reactive astrocytes can contribute to neurodegeneration by enhancing glutamate excitotoxicity in neurons. It is of interest that Aβ toxicity is more prominent in diabetic rats associated with an oxidative stress condition.39
AD is the best known tauopathy, and several mutations in the tau gene have been implicated in dementia and neurodegeneration.33,40 Tau is normally phosphorylated and dephosphorylated to function in axonal integrity and transport. The sustained phosphorylation of tau causes an impaired ability for tau binding to microtubules, leading to impaired axonal transport, increased toxicity and subsequent formation of PHFs. Although there are inconsistencies in reports of tau pathology in AD brains,41 many studies have proposed a close relationship between tau pathology and Aβ toxicity such that tau abnormalities induce accumulation of Aβ in AD mice42,43,44 and Aβ treatment causes tau phosphorylation through activation of multiple kinases.2,31,41 In addition, inhibition of tau phosphorylation is known to reduce Aβ-induced neurotoxicity45,46,47,48 and tau knockout neurons show resistance to Aβ-induced toxicity.49
LINKS BETWEEN AD AND DIABETES
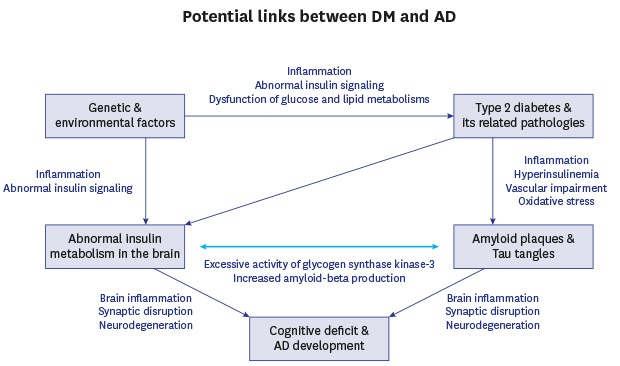
As mentioned above, causes of SAD are multifactorial, involving several environmental and genetic factors, in particular, insulin resistance, an acquired condition in type 2 diabetic mellitus (T2DM). T2DM is associated with metabolic disorders such as obesity and high blood pressure. Whereas type 1 diabetes mellitus (T1DM) is linked more closely with familial inheritance, both are known to contribute to the incidence of SAD.50 It is estimated that the number of diabetic patients will increase dramatically by the end of 2030, with most of these patients over 64 years of age.51 Considering the fact that aging is a major risk factor in AD, many studies have indicated increasing evidence of links between AD and insulin dysfunction.52,53,54 This combination of aging and insulin dysfunction has already emerged as a major risk factor in AD development. Population studies have reported a close relationship between AD and insulin dysfunction such as abnormal insulin signaling further leading to glucose intolerance, impaired insulin secretion and insulin resistance.6,55,56 Higher incidences of AD have been observed in elderly diabetic patients55,57 and decreased mental ability has been reported in diabetic children.58 Other clinical studies have also shown the significant increase of the risk of AD in diabetic patients,59,60,61,62 suggesting a potential association between diabetes and AD (Fig. 1).
Fig. 1. Potential links between diabetes mellitus and AD.

AD = Alzheimer's disease.
Insulin, a polypeptide hormone produced by β-cells in pancreatic islets of Langerhans, consists of a 21-amino acid chain linked to a 30 amino acid chain by two disulfide bonds.63 It regulates blood glucose levels by converting glucose to glycogen in the cell and functions in lipid and protein metabolism. De novo synthesis of insulin in the brain occurs in certain brain areas including hippocampus and prefrontal cortex64; however, insulin can also be actively transported from the periphery to the brain via the blood-brain barrier (BBB).65 This transport can be modulated by multiple factors such as hormones, fasting, obesity or some other conditions like aging and diabetes.66 The insulin receptor (IR) is a tetrameric transmembrane receptor composed of two α-subunits (extracellular) with an insulin binding site, and two β-subunits that have tyrosine kinase activity. IRs are expressed in the hippocampal and medial temporal cortical areas of the brain, suggesting their potential roles in memory processes. In fact, a study done by Zhao et al.67 has reported that mRNA and protein levels of IRs are upregulated following a spatial memory task, implicating insulin modulation in memory and cognition. Insulin binding to IR activates two signaling pathways, the phosphoinositide-3 kinase (PI3K)/AKT pathway and the mitogen-activated protein kinase (MAPK) pathway. The PI3K/AKT pathway is important in cellular differentiation, proliferation and survival of neurons, and the MAPK pathway is mainly involved in cell growth and protein synthesis.68,69 Activation of PI3K leads to stimulation of the downstream kinase, 3-phosphoinositide-dependent protein kinase-1 (PDK1). Activated PDK1 then activates protein kinase B, also known as AKT, which inhibits glycogen synthase kinase-3 (GSK3-β), a major tau kinase. Therefore, a downregulation of insulin signaling may lead to a decrease in glucose metabolism and an increase in tau phosphorylation and neurofibrillary tangles through the activation of GSK3-β.70,71 Increased activity of GSK3-α is also associated with the processing of APP and generation of Aβ.72 Liu et al.71 found downregulation of signaling molecules in the insulin-PI3K–AKT pathway in both AD and T2DM cases, and the effect was even more severe in individuals with both T2DM and AD (T2DM–AD).
Another important downstream signaling partner is mitogen activated protein kinase (MAPK).73 In particular, C-Jun N-terminal kinase (JNK), known as a stress- activated protein kinase has been detected in AD brain.74 JNK activation is also associated with Aβ deposition, tau phosphorylation, and a decrease of synaptophysin75 and also participates in amyloidogenesis through β-secretase-1.76 Insulin degrading enzyme (IDE) is another downstream target of insulin signaling that can be negatively regulated by the insulin-PI3K-AKT pathway via a negative feedback mechanism.77 Therefore, impaired insulin signaling may result in decreased IDE levels due to a reduction of AKT activation. Since IDE plays a significant role in enzymatic degradation of Aβ,78,79 a decrease in IDE levels would reduce Aβ clearance leading to an increase of Aβ accumulation in the brain. Thus, the insulin signaling pathway seems to be important in two major hallmarks of AD pathology, namely plaques and tangles.
APOEε4, one of major risk factors for AD development, modulates insulin activity and exhibits effects on memory of those afflicted with AD.77 Chan et al.80 have further reported that hippocampus-dependent memory deficit observed in APP mice was accelerated when APP was co-expressed with APOEε4, with a mechanism via impaired insulin signaling. Alternatively, the insulin resistance observed in APOEε4 carriers is not associated with a change on Aβ levels in plasma but linked to the abnormal hyperphosphorylation of tau recognized in cerebrospinal fluid.81 These studies generally suggest APOEε4 as a possible connection between diabetes and AD.
Insulin-like growth factor-1 (IGF-1) is important in neuronal survival and neurogenesis in hippocampus, and alteration of IGF-1 is implicated in early stages of diabetic conditions.82 IGF-1 treatment also inhibits abnormal tauopathy and Aβ deposition in cell culture and AD mouse models,82 and a recent report from a human study shows that low levels of serum IGF-1 are closely related with incidence of AD in older and middle-aged individuals83 which supports the role of IGF-1 in diabetes and AD.84 Although the data seem complicated and somewhat controversial, alteration of insulin and IGF-1 signaling is likely to be involved in the development of diabetic conditions linked to AD pathologies.
OXIDATIVE STRESS, INFLAMMATION AND CEREBROVASCULAR COMPLICATIONS IN DIABETES AND AD
Inflammation, an important feature of neurodegenerative and metabolic disorders has been suggested to play a critical role in the pathogenesis of many diseases.85,86,87 Inflammation is a vital biological response for homeostasis of our body to re-establish normal physiology from stress stimuli, such as injury or infection. Various studies have indicated inflammatory responses in the brain, such as upregulation of inflammatory factors and activation of glia cells.88 The increase of inflammatory factors, including tumor necrosis factor-α (TNF-α), interleukin-6, and interleukin-1β, is evident in blood samples from AD patients.89 In particular, overexpression of TNF-α by prolonged inflammation may cause peripheral insulin resistance.90 In addition, activation of astrocytes and microglia, a brain inflammatory response, is also observed in diabetes,91 suggesting that increased levels of inflammation in the CNS and PNS may be a trigger in diabetes subsequent to neurodegeneration. The AD features, i.e., formation of plaques and tangles, may be linked to impaired insulin signaling in the brain; nonetheless, the underlying mechanism is unclear. Oxidative stress seems to be a common factor in many neurodegenerative disorders including AD,8,9 thus being a potential link between diabetes and AD. In fact, the onset of diabetic complications such as altered insulin sensitivity and neuropathy is closely associated with increased oxidative stress due to lack of removal of ROS92; thus oxidative stress could be a driving force behind insulin signaling impairments in AD brains. Increases of ROS inhibit IR activity through prolonged inhibition of phosphotyrosine phosphatase, the enzyme responsible for dephosphorylation of IR,93,94 suggesting that disrupting IR activity by oxidative stress may result in impairment of insulin signaling seen in SAD.
The effect of insulin on inflammation is controversial. Low doses of insulin exert anti-inflammatory effects; conversely, high levels of insulin during chronic hyperinsulinemia may exacerbate inflammatory responses and increase oxidative stress.95 Hyperinsulinemia induces a dramatic increase of inflammatory factors including TNF-α, IL-1β and IL-6 and a lipid peroxidation marker, F2-lsoprostane, which are potentiated by obesity, given that insulin elevates TNF-α and free forms of fatty acids released from adipocytes.52 Insulin may enhance inflammatory responses in the brain through upregulated levels of Aβ, resulting in increases of the inflammatory factors mentioned above.96 The pro- or anti-inflammatory factors require only tiny quantities to exert multiple physiological effects in the brain, mainly for homeostasis and also functioning as growth or trophic factors; thus chronic imbalance of these factors could be a direct link between diabetes and AD.97
Although pathologies of Aβ and tau are features commonly focused on AD, it is evident that many other causes may contribute to disease pathogenesis. One possible mechanism could be a compromised cerebrovascular system, a common pathology in diabetes and AD.98 The underlying mechanism of cerebrovascular disruption could be a loss of vascular pericytes and astrocytes that can occur by mitochondrial oxidative stress during hyperinsulinemia in a diabetic animal model.99 Thus, aside from impaired insulin signaling, diabetes may affect AD pathology via other mechanisms such as cerebrovascular impairment and oxidative stress-induced inflammation. These multiple factors may have synergistic effects on Aβ pathologies by interrupting metabolism and clearance of Aβ via degradation enzymes or Aβ transport across the BBB.100
Low-density LRP-1, a 600 kDa type-1 trans-membrane receptor, recognizes at least 20 structurally diverse ligands including cholesterol, and transports them across the BBB.101,102 LRP-1 provides a homeostatic control mechanism for Aβ clearance at the BBB and for cerebrovascular cells mediating brain-to-systemic clearance of Aβ.103 Soluble LRP-1 in circulating blood stream acts as a peripheral “sink” for Aβ by restricting access of free Aβ into the brain, and LRP-1 in the liver also involves systemic clearance of Aβ.102,103,104 RAGE is another system for Aβ clearance; circulating Aβ is transported through RAGE in the luminal surface of brain vessels.28 The expression of RAGE is upregulated in cerebral vessels, neurons and microglia when Aβ species accumulate in AD brains. RAGE interaction with Aβ at the BBB has been implicated in the development of cerebrovascular impairment through transcytosis of circulating Aβ across the BBB, inflammation of the endothelium and suppression of cerebral blood flow.28,105
Takeda et al.98 generated an animal model that reflected pathologies for both AD and diabetes by crossing APP transgenic mice with leptin-deficient ob/ob mice. Their findings indicate that diabetes exacerbates memory and cognitive dysfunction of AD, even without an increase of Aβ. They also showed cerebral vascular inflammation and severe Aβ angiopathy in these mice, with upregulation of RAGE and inflammatory changes at the BBB prior to the angiopathy. Similar data from Liu et al.106 have shown up-regulation of RAGE in streptozotocin-induced diabetic mice such that hyperinsulinemia-induced stress in these mice serve as a trigger for Aβ transcytosis from the bloodstream to the brain that may eventually contribute to an interruption of the BBB.
ACTIVATION OF ASTROCYTES IN BRAIN INFLAMMATION AND DIABETES
Astrocytes comprise about 50% of brain cells and support neurons structurally, metabolically, and trophically. Activation of astrocytes is a typical brain response to stress stimuli that is evident in changes in cellular morphology and function as well as the upregulation of glial fibrillary acidic protein (GFAP). Reactive astrocytes result in decreased glutamate uptake, subsequent to an increase in extracellular glutamate levels, thereby contributing toward excitotoxicity.107 A recent study by our group shows that at the initiation stage of inflammation, astrocytes become active to make the stress conditions return to homeostasis, but chronic activation of astrocytes eventually causes astrocytic death by losing their own neuroprotective properties, although astrocytes are less vulnerable than neurons to brain stress stimuli.108
Astrocytes are important players in the brain immune response against infection, trauma, ischemia and neurodegenerative diseases, such that in response to stress stimuli they secrete inflammatory/anti-inflammatory factors and neurotoxic factors.109 Astrocytes also undergo structural and functional changes, called astrogliosis that is evident by increased GFAP expression110,111 often leading to scar formation, an indicator of many brain injuries.
Upregulation of GFAP expression may induce secretion of some factors from astrocytes that could be beneficial or harmful depending on the degree and period of the pathological conditions of the disease or injury112,113,114 Among the molecules released from reactive astrocytes, nitric oxide and prostaglandins modulate blood flow, thus affecting BBB permeability.115 As mentioned earlier, the BBB is a highly specialized structure in cerebrovascular system to restrict molecular movement from systemic blood circulation to the brain. This dedicated structure is formed by vascular endothelial cells covered by astrocytic end-feet processes that form a continuous barrier for brain homeostasis.116,117 The BBB is tightly regulated in young and healthy subjects, but gradually becomes permeable with age resulting in more invasions of peripheral microbes into the brain.118 In addition, LRP-1 expressed on astrocytes functions to clear out brain-derived Aβ across the BBB was decreased in a diabetic animal model,119 and thus possibly causes the accumulation of Aβ peptides in the brain when BBB is impaired by the activation of astrocytes.102,120 Thus, prolonged activation of astrocytes in a diabetic condition could be an initial mechanism of destruction of brain homeostasis via BBB dysfunction, further affecting the onset and development of AD- associated pathologies.121
TREATMENTS RELATED TO DIABETES AND AD
Unfortunately, there is currently no cure for AD. Only two kinds of symptomatic medications are currently available for AD patients. Based on the close relationship between diabetes and AD, and decrease of insulin signaling in AD, many clinical trials have been done in the past using insulin to slow AD progression. In fact, intranasal insulin has been proven beneficial as a result of decrease of Aβ42 in cerebrospinal fluid,122 but with some side effects such as nasal mucosa damage, irritation or induction of high blood pressure. Thus, improved delivery into the brain is required for insulin therapy.
Many studies with other antidiabetic medications such as Metformin and Liraglutide that can cross the BBB, have been further tested in AD symptoms. In fact, Liraglutide, a GLP-1 analog, has proven beneficial for memory and synaptic plasticity by reducing neurotoxic oligomeric Aβ and decreasing Aβ plaques as well as increasing neurogenesis in a APP/PS1 mouse model of AD.123 Interestingly, this drug works similarly to glucagon-like peptide 1 (GLP-1), a 30-amino acid incretin hormone produced in the gut as well as in the brainstem and hypothalamus.124,125,126 GLP-1 receptors are expressed in the temporal cortex and hippocampus, areas of the brain that are affected in AD.125,126 GLP-1 has neuroprotective effects on AD-associated pathologies such as Aβ plaque accumulation and oxidative stress and decreases in synaptic plasticity,127 suggesting GLP-1 to be a possible treatment for AD.
Two pilot studies have shown a clinical trial with Liraglutide, GLP-1 analog tested on 200 AD patients conducted by the Imperial College London (Clinical Trials identification: NCT01255163), and the effect of Exenatide, another GLP-1 analog, has been tested on 230 AD patients for three years conducted by National Institute on Aging (Clinical Trials identifier: NCT01255163). In accordance with these clinical trials, recent studies have reported that Liraglutide has beneficial effects in maintaining glucose level in the brain by preventing the decline of glucose metabolism128 and restores glucose transport at the BBB.129
Metformin is another anti-diabetic medication that has shown glucose lowering effect, recovery of insulin sensitivity, increases of glucose uptake, decreases of hepatic glucose syntheses, and activation of protein kinase pathways required for glucose metabolism.130 Metformin also has beneficial effects on reduction of Aβ production by inhibiting β-secretase and tau phosphorylation, suggesting metformin as a suitable drug in AD treatment. A clinical trial conducted by a Taiwanese group has reported that metformin significantly decreases the risk of dementia.131 On the other hand, long term use of Metformin, as studied by a UK group, may increase the risk of AD development.132 Similar results by an Australian group have been reported that Metformin decreased cognitive performance.133 This discrepancy may be due to difficulty finding optimal doses and the limited size of the patient population, which requires larger scale studies.
GSK-3β inhibitors have long been tested as AD therapy and recently studied for the treatments of both diabetes and AD as GSK-3β plays an important role in insulin signaling as mentioned earlier. GSK-3β inhibitors might be beneficial because insulin promotes activation of glycogen synthase by suppressing GSK-3β activity.134 Overexpression of GSK-3β attenuates insulin signaling by phosphorylating and downregulating insulin receptor substrates,135 and therapeutic doses of lithium chloride, an inhibitor for GSK-3β, show reduction of Aβ peptide expression in the brain72 and tau phosphorylation in both neuron and glia,82,136,137 suggesting that inhibition of GSK-3β activity may be a promising form of therapy for both diabetes and AD.
The nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ) is a transcription factor that regulates glucose and lipid metabolism and suppresses gene expression in inflammation.138 PPARγ is particularly important as this nuclear receptor regulates the metabolism of lipids and carbohydrates, glucose levels in serum, and insulin sensitivity.139 However, the effects of PPARγ agonists from a study using Tg2576 mice are conflicting, such that Pioglitazone, a PPARγ agonist, exhibits no changes in Aβ pathology and no changes in reactive glia, an inflammatory response,140 while rosiglitazone, an insulin sensitizer acting on PPARγ has effects on recovery of insulin sensitivity and improvements in behavioral deficits.141 Pioglitazone also shows an anti-oxidant effect in an experiment using human serum.142 A clinical study with Pioglitazone shows that a low dose of the drug has a better effect than placebo in AD patients, suggesting Pioglitazone as a potential treatment in AD.143 A large scale of studies requires to validate and replicate the beneficial effects of drugs in the future.
CONCLUSION
Although various mechanisms and markers have been suggested in establishing the idea that T2DM and AD might be linked, there is still a need for further elaboration due to several conflicting findings in the scientific literature, particularly in epidemiology studies. Even with such conflicts, altered insulin or insulin-related signaling seems to be involved in many of pathologies of AD brains. Regulation of diabetic complications helps to alleviate these pathologies, as evidenced by prolonged lifespan, reduction of Aβ plaques, and improvement of cognitive function. By evaluating the given evidence along with conflicting data, it appears that T2DM may serve as a factor in accelerating pathologies in AD development. T2DM should be considered as part of a larger complication accompanying pathologies such as inflammation, oxidative stress, DNA damage, and mitochondrial dysfunction which may contribute to a degenerative domino effect. With studies on diabetic drugs as a useful tool in ameliorating AD symptoms, AD can be considered as a form of type 3 diabetes.53 Nevertheless, memory and cognitive functions are not only observed in AD patients but in other degenerative diseases. Hence with this observation in mind, it is vital that the individuals involved in any future advancements of AD treatment prioritize the distinguishing routes or etiologies of disease causality and should investigate further to clarify underlying mechanisms of pathologies in diabetes and AD.
Footnotes
Funding: This research was supported by Leading Foreign Research Institute Recruitment Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT) (NRF-2018K1A4A3A02060572).
Disclosure: The authors have no potential conflicts of interest to disclose.
- Conceptualization: Song MS.
- Formal analysis: Chung PS, Kim HG.
- Investigation: Learman CR, Weaver CL.
- Validation: Song MS.
- Writing - original draft: Ahn KC, Song MS.
- Writing - review & editing: Baker GB, Ahn KC, Song MS.
References
- 1.Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43(1):545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 2.Brion JP, Anderton BH, Authelet M, Dayanandan R, Leroy K, Lovestone S, et al. Neurofibrillary tangles and tau phosphorylation. Biochem Soc Symp. 2001;67(67):81–88. doi: 10.1042/bss0670081. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20(4):154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- 4.Alzheimer's Association. 2012 Alzheimer's disease facts and figures. Alzheimers Dement. 2012;8(2):131–168. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 5.Piaceri I, Nacmias B, Sorbi S. Genetics of familial and sporadic Alzheimer's disease. Front Biosci (Elite Ed) 2013;5(1):167–177. doi: 10.2741/e605. [DOI] [PubMed] [Google Scholar]
- 6.Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology. 2004;63(7):1187–1192. doi: 10.1212/01.wnl.0000140292.04932.87. [DOI] [PubMed] [Google Scholar]
- 7.Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430(7000):631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Di Domenico F, Perluigi M, Butterfield DA, Cornelius C, Calabrese V. Oxidative damage in rat brain during aging: interplay between energy and metabolic key target proteins. Neurochem Res. 2010;35(12):2184–2192. doi: 10.1007/s11064-010-0295-z. [DOI] [PubMed] [Google Scholar]
- 9.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7(12):548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 10.Unsal C, Oran M, Albayrak Y, Aktas C, Erboga M, Topcu B, et al. Neuroprotective effect of ebselen against intracerebroventricular streptozotocin-induced neuronal apoptosis and oxidative stress in rats. Toxicol Ind Health. 2016;32(4):730–740. doi: 10.1177/0748233713509429. [DOI] [PubMed] [Google Scholar]
- 11.Scheff SW, Sparks L, Price DA. Quantitative assessment of synaptic density in the entorhinal cortex in Alzheimer's disease. Ann Neurol. 1993;34(3):356–361. doi: 10.1002/ana.410340309. [DOI] [PubMed] [Google Scholar]
- 12.Wollen KA. Alzheimer's disease: the pros and cons of pharmaceutical, nutritional, botanical, and stimulatory therapies, with a discussion of treatment strategies from the perspective of patients and practitioners. Altern Med Rev. 2010;15(3):223–244. [PubMed] [Google Scholar]
- 13.Rodríguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM, et al. Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer's disease. PLoS One. 2008;3(8):e2935. doi: 10.1371/journal.pone.0002935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB. Diabetes mellitus and Alzheimer's disease: shared pathology and treatment? Br J Clin Pharmacol. 2011;71(3):365–376. doi: 10.1111/j.1365-2125.2010.03830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.George-Hyslop PS, Rossor M. Alzheimer's disease. Unravelling the disease process. Lancet. 2001;358(Suppl):S1. doi: 10.1016/s0140-6736(01)07014-3. [DOI] [PubMed] [Google Scholar]
- 16.Guerreiro RJ, Gustafson DR, Hardy J. The genetic architecture of Alzheimer's disease: beyond APP, PSENs and APOE. Neurobiol Aging. 2012;33(3):437–456. doi: 10.1016/j.neurobiolaging.2010.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polvikoski T, Sulkava R, Haltia M, Kainulainen K, Vuorio A, Verkkoniemi A, et al. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med. 1995;333(19):1242–1247. doi: 10.1056/NEJM199511093331902. [DOI] [PubMed] [Google Scholar]
- 18.Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci U S A. 1994;91(25):12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, et al. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8(1):79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 20.Song MS, Saavedra L, de Chaves EI. Apoptosis is secondary to non-apoptotic axonal degeneration in neurons exposed to Abeta in distal axons. Neurobiol Aging. 2006;27(9):1224–1238. doi: 10.1016/j.neurobiolaging.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 21.Selkoe DJ. Resolving controversies on the path to Alzheimer's therapeutics. Nat Med. 2011;17(9):1060–1065. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 22.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46(6):860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 23.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, et al. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100(18):10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lei M, Xu H, Li Z, Wang Z, O'hMalley TT, Zhang D, et al. Soluble Aβ oligomers impair hippocampal LTP by disrupting glutamatergic/GABAergic balance. Neurobiol Dis. 2016;85:111–121. doi: 10.1016/j.nbd.2015.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith WW, Gorospe M, Kusiak JW. Signaling mechanisms underlying Abeta toxicity: potential therapeutic targets for Alzheimer's disease. CNS Neurol Disord Drug Targets. 2006;5(3):355–361. doi: 10.2174/187152706784111515. [DOI] [PubMed] [Google Scholar]
- 27.Kuner P, Schubenel R, Hertel C. Beta-amyloid binds to p57NTR and activates NFkappaB in human neuroblastoma cells. J Neurosci Res. 1998;54(6):798–804. doi: 10.1002/(SICI)1097-4547(19981215)54:6<798::AID-JNR7>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 28.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382(6593):685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 29.Wang HY, Lee DH, Davis CB, Shank RP. Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J Neurochem. 2000;75(3):1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- 30.Jhamandas JH, MacTavish D. Antagonist of the amylin receptor blocks beta-amyloid toxicity in rat cholinergic basal forebrain neurons. J Neurosci. 2004;24(24):5579–5584. doi: 10.1523/JNEUROSCI.1051-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song MS, Rauw G, Baker GB, Kar S. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur J Neurosci. 2008;28(10):1989–2002. doi: 10.1111/j.1460-9568.2008.06498.x. [DOI] [PubMed] [Google Scholar]
- 32.Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer's disease. Cell Calcium. 2003;34(4-5):385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- 33.Blurton-Jones M, Laferla FM. Pathways by which Abeta facilitates tau pathology. Curr Alzheimer Res. 2006;3(5):437–448. doi: 10.2174/156720506779025242. [DOI] [PubMed] [Google Scholar]
- 34.Bi X, Gall CM, Zhou J, Lynch G. Uptake and pathogenic effects of amyloid beta peptide 1-42 are enhanced by integrin antagonists and blocked by NMDA receptor antagonists. Neuroscience. 2002;112(4):827–840. doi: 10.1016/s0306-4522(02)00132-x. [DOI] [PubMed] [Google Scholar]
- 35.Nagele RG, D'Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer's disease. Neuroscience. 2002;110(2):199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- 36.Ong WY, Tanaka K, Dawe GS, Ittner LM, Farooqui AA. Slow excitotoxicity in Alzheimer's disease. J Alzheimers Dis. 2013;35(4):643–668. doi: 10.3233/JAD-121990. [DOI] [PubMed] [Google Scholar]
- 37.Schousboe A, Waagepetersen HS. Role of astrocytes in glutamate homeostasis: implications for excitotoxicity. Neurotox Res. 2005;8(3-4):221–225. doi: 10.1007/BF03033975. [DOI] [PubMed] [Google Scholar]
- 38.Song MS, Baker GB, Dursun SM, Todd KG. The antidepressant phenelzine protects neurons and astrocytes against formaldehyde-induced toxicity. J Neurochem. 2010;114(5):1405–1413. doi: 10.1111/j.1471-4159.2010.06857.x. [DOI] [PubMed] [Google Scholar]
- 39.Pereira C, Moreira P, Seiça R, Santos MS, Oliveira CR. Susceptibility to beta-amyloid-induced toxicity is decreased in goto-kakizaki diabetic rats: involvement of oxidative stress. Exp Neurol. 2000;161(1):383–391. doi: 10.1006/exnr.1999.7270. [DOI] [PubMed] [Google Scholar]
- 40.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95(13):7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43(3):321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 42.Busciglio J, Lorenzo A, Yeh J, Yankner BA. Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14(4):879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 43.Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 44.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 45.Alvarez G, Muñoz-Montaño JR, Satrústegui J, Avila J, Bogónez E, Díaz-Nido J. Regulation of tau phosphorylation and protection against beta-amyloid-induced neurodegeneration by lithium. Possible implications for Alzheimer's disease. Bipolar Disord. 2002;4(3):153–165. doi: 10.1034/j.1399-5618.2002.01150.x. [DOI] [PubMed] [Google Scholar]
- 46.Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribé E, Dalfó E, et al. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer's disease and tauopathies. Curr Alzheimer Res. 2005;2(1):3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- 47.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, et al. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 48.Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142(3):387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 49.Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A. Tau is essential to beta -amyloid-induced neurotoxicity. Proc Natl Acad Sci U S A. 2002;99(9):6364–6369. doi: 10.1073/pnas.092136199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.El Khoury NB, Gratuze M, Papon MA, Bretteville A, Planel E. Insulin dysfunction and Tau pathology. Front Cell Neurosci. 2014;8:22. doi: 10.3389/fncel.2014.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 52.Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004;3(3):169–178. doi: 10.1016/S1474-4422(04)00681-7. [DOI] [PubMed] [Google Scholar]
- 53.de la Monte SM, Wands JR. Alzheimer's disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2(6):1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sims-Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol. 2010;6(10):551–559. doi: 10.1038/nrneurol.2010.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rönnemaa E, Zethelius B, Sundelöf J, Sundström J, Degerman-Gunnarsson M, Berne C, et al. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology. 2008;71(14):1065–1071. doi: 10.1212/01.wnl.0000310646.32212.3a. [DOI] [PubMed] [Google Scholar]
- 56.Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, et al. Glucose levels and risk of dementia. N Engl J Med. 2013;369(6):540–548. doi: 10.1056/NEJMoa1215740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu WL, Qiu CX, Wahlin A, Winblad B, Fratiglioni L. Diabetes mellitus and risk of dementia in the Kungsholmen project: a 6-year follow-up study. Neurology. 2004;63(7):1181–1186. doi: 10.1212/01.wnl.0000140291.86406.d1. [DOI] [PubMed] [Google Scholar]
- 58.S Roriz-Filho J, Sá-Roriz TM, Rosset I, Camozzato AL, Santos AC, Chaves ML, et al. (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta. 2009;1792(5):432–443. doi: 10.1016/j.bbadis.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 59.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O'Brien PC, et al. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997;145(4):301–308. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- 60.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology. 1999;53(9):1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 61.Arvanitakis Z, Wilson RS, Bienias JL, Evans DA, Bennett DA. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 2004;61(5):661–666. doi: 10.1001/archneur.61.5.661. [DOI] [PubMed] [Google Scholar]
- 62.Peila R, Rodriguez BL, Launer LJ Honolulu-Asia Aging Study. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the Honolulu-Asia Aging Study. Diabetes. 2002;51(4):1256–1262. doi: 10.2337/diabetes.51.4.1256. [DOI] [PubMed] [Google Scholar]
- 63.Nelson TJ, Alkon DL. Insulin and cholesterol pathways in neuronal function, memory and neurodegeneration. Biochem Soc Trans. 2005;33(Pt 5):1033–1036. doi: 10.1042/BST20051033. [DOI] [PubMed] [Google Scholar]
- 64.Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature. 1978;272(5656):827–829. doi: 10.1038/272827a0. [DOI] [PubMed] [Google Scholar]
- 65.Margolis RU, Altszuler N. Insulin in the cerebrospinal fluid. Nature. 1967;215(5108):1375–1376. doi: 10.1038/2151375a0. [DOI] [PubMed] [Google Scholar]
- 66.Blázquez E, Velázquez E, Hurtado-Carneiro V, Ruiz-Albusac JM. Insulin in the brain: its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer's disease. Front Endocrinol (Lausanne) 2014;5(5108):5161. doi: 10.3389/fendo.2014.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, et al. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem. 1999;274(49):34893–34902. doi: 10.1074/jbc.274.49.34893. [DOI] [PubMed] [Google Scholar]
- 68.Le Roith D, Zick Y. Recent advances in our understanding of insulin action and insulin resistance. Diabetes Care. 2001;24(3):588–597. doi: 10.2337/diacare.24.3.588. [DOI] [PubMed] [Google Scholar]
- 69.Tremblay ML, Giguère V. Phosphatases at the heart of FoxO metabolic control. Cell Metab. 2008;7(2):101–103. doi: 10.1016/j.cmet.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 70.Rankin CA, Sun Q, Gamblin TC. Tau phosphorylation by GSK-3beta promotes tangle-like filament morphology. Mol Neurodegener. 2007;2(1):12. doi: 10.1186/1750-1326-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Deficient brain insulin signalling pathway in Alzheimer's disease and diabetes. J Pathol. 2011;225(1):54–62. doi: 10.1002/path.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature. 2003;423(6938):435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- 73.Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology. 2010;58(3):561–568. doi: 10.1016/j.neuropharm.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 74.Otth C, Mendoza-Naranjo A, Mujica L, Zambrano A, Concha II, Maccioni RB. Modulation of the JNK and p38 pathways by cdk5 protein kinase in a transgenic mouse model of Alzheimer's disease. Neuroreport. 2003;14(18):2403–2409. doi: 10.1097/00001756-200312190-00023. [DOI] [PubMed] [Google Scholar]
- 75.Puig B, Gómez-Isla T, Ribé E, Cuadrado M, Torrejón-Escribano B, Dalfó E, et al. Expression of stress-activated kinases c-Jun N-terminal kinase (SAPK/JNK-P) and p38 kinase (p38-P), and tau hyperphosphorylation in neurites surrounding betaA plaques in APP Tg2576 mice. Neuropathol Appl Neurobiol. 2004;30(5):491–502. doi: 10.1111/j.1365-2990.2004.00569.x. [DOI] [PubMed] [Google Scholar]
- 76.Quiroz-Baez R, Rojas E, Arias C. Oxidative stress promotes JNK-dependent amyloidogenic processing of normally expressed human APP by differential modification of alpha-, beta- and gamma-secretase expression. Neurochem Int. 2009;55(7):662–670. doi: 10.1016/j.neuint.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 77.Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci. 2004;24(49):11120–11126. doi: 10.1523/JNEUROSCI.2860-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chesneau V, Vekrellis K, Rosner MR, Selkoe DJ. Purified recombinant insulin-degrading enzyme degrades amyloid beta-protein but does not promote its oligomerization. Biochem J. 2000;351(Pt 2):509–516. [PMC free article] [PubMed] [Google Scholar]
- 79.Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100(7):4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chan ES, Shetty MS, Sajikumar S, Chen C, Soong TW, Wong BS. ApoE4 expression accelerates hippocampus-dependent cognitive deficits by enhancing Aβ impairment of insulin signaling in an Alzheimer's disease mouse model. Sci Rep. 2016;6(1):26119. doi: 10.1038/srep26119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Starks EJ, Patrick O'Grady J, Hoscheidt SM, Racine AM, Carlsson CM, Zetterberg H, et al. Insulin resistance is associated with higher cerebrospinal fluid tau levels in asymptomatic APOEɛ4 carriers. J Alzheimers Dis. 2015;46(2):525–533. doi: 10.3233/JAD-150072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem. 1997;272(40):25326–25332. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- 83.Westwood AJ, Beiser A, Decarli C, Harris TB, Chen TC, He XM, et al. Insulin-like growth factor-1 and risk of Alzheimer dementia and brain atrophy. Neurology. 2014;82(18):1613–1619. doi: 10.1212/WNL.0000000000000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Teppala S, Shankar A. Association between serum IGF-1 and diabetes among U.S. adults. Diabetes Care. 2010;33(10):2257–2259. doi: 10.2337/dc10-0770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gregor MF, Hotamisligil GS. Inflammatory mechanisms in obesity. Annu Rev Immunol. 2011;29(1):415–445. doi: 10.1146/annurev-immunol-031210-101322. [DOI] [PubMed] [Google Scholar]
- 86.Clark I, Atwood C, Bowen R, Paz-Filho G, Vissel B. Tumor necrosis factor-induced cerebral insulin resistance in Alzheimer's disease links numerous treatment rationales. Pharmacol Rev. 2012;64(4):1004–1026. doi: 10.1124/pr.112.005850. [DOI] [PubMed] [Google Scholar]
- 87.Ferreira ST, Clarke JR, Bomfim TR, De Felice FG. Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer's disease. Alzheimers Dement. 2014;10(1) Suppl:S76–S83. doi: 10.1016/j.jalz.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 88.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6(4):193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 89.Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta-analysis of cytokines in Alzheimer's disease. Biol Psychiatry. 2010;68(10):930–941. doi: 10.1016/j.biopsych.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 90.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science. 1996;271(5249):665–668. doi: 10.1126/science.271.5249.665. [DOI] [PubMed] [Google Scholar]
- 91.De Felice FG, Ferreira ST. Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer disease. Diabetes. 2014;63(7):2262–2272. doi: 10.2337/db13-1954. [DOI] [PubMed] [Google Scholar]
- 92.Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med. 2011;50(5):567–575. doi: 10.1016/j.freeradbiomed.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mahadev K, Zilbering A, Zhu L, Goldstein BJ. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J Biol Chem. 2001;276(24):21938–21942. doi: 10.1074/jbc.C100109200. [DOI] [PubMed] [Google Scholar]
- 94.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9(2):387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 95.Fishel MA, Watson GS, Montine TJ, Wang Q, Green PS, Kulstad JJ, et al. Hyperinsulinemia provokes synchronous increases in central inflammation and β-amyloid in normal adults. Arch Neurol. 2005;62(10):1539–1544. doi: 10.1001/archneur.62.10.noc50112. [DOI] [PubMed] [Google Scholar]
- 96.Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer's disease. Neurobiol Aging. 2001;22(6):903–908. doi: 10.1016/s0197-4580(01)00287-1. [DOI] [PubMed] [Google Scholar]
- 97.Plata-Salamán CR, ffrench-Mullen JM. Interleukin-1 beta inhibits Ca2+ channel currents in hippocampal neurons through protein kinase C. Eur J Pharmacol. 1994;266(1):1–10. doi: 10.1016/0922-4106(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 98.Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107(15):7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shah GN, Morofuji Y, Banks WA, Price TO. High glucose-induced mitochondrial respiration and reactive oxygen species in mouse cerebral pericytes is reversed by pharmacological inhibition of mitochondrial carbonic anhydrases: Implications for cerebral microvascular disease in diabetes. Biochem Biophys Res Commun. 2013;440(2):354–358. doi: 10.1016/j.bbrc.2013.09.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Erickson MA, Banks WA. Blood-brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J Cereb Blood Flow Metab. 2013;33(10):1500–1513. doi: 10.1038/jcbfm.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. 2001;108(6):779–784. doi: 10.1172/JCI13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jaeger S, Pietrzik CU. Functional role of lipoprotein receptors in Alzheimer's disease. Curr Alzheimer Res. 2008;5(1):15–25. doi: 10.2174/156720508783884675. [DOI] [PubMed] [Google Scholar]
- 103.Deane R, Bell RD, Sagare A, Zlokovic BV. Clearance of amyloid-beta peptide across the blood-brain barrier: implication for therapies in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2009;8(1):16–30. doi: 10.2174/187152709787601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yamada K, Hashimoto T, Yabuki C, Nagae Y, Tachikawa M, Strickland DK, et al. The low density lipoprotein receptor-related protein 1 mediates uptake of amyloid beta peptides in an in vitro model of the blood-brain barrier cells. J Biol Chem. 2008;283(50):34554–34562. doi: 10.1074/jbc.M801487200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zlokovic BV, Deane R, Sagare AP, Bell RD, Winkler EA. Low-density lipoprotein receptor-related protein-1: a serial clearance homeostatic mechanism controlling Alzheimer's amyloid β-peptide elimination from the brain. J Neurochem. 2010;115(5):1077–1089. doi: 10.1111/j.1471-4159.2010.07002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu LP, Hong H, Liao JM, Wang TS, Wu J, Chen SS, et al. Upregulation of RAGE at the blood-brain barrier in streptozotocin-induced diabetic mice. Synapse. 2009;63(8):636–642. doi: 10.1002/syn.20644. [DOI] [PubMed] [Google Scholar]
- 107.Maragakis NJ, Rothstein JD. Mechanisms of Disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006;2(12):679–689. doi: 10.1038/ncpneuro0355. [DOI] [PubMed] [Google Scholar]
- 108.Ahn KC, MacKenzie EM, Learman CR, Hall TC, Weaver CL, Dunbar GL, et al. Inhibition of p53 attenuates ischemic stress-induced activation of astrocytes. Neuroreport. 2015;26(14):862–869. doi: 10.1097/WNR.0000000000000439. [DOI] [PubMed] [Google Scholar]
- 109.Neal M, Richardson JR. Epigenetic regulation of astrocyte function in neuroinflammation and neurodegeneration. Biochim Biophys Acta Mol Basis Dis. 2018;1864(2):432–443. doi: 10.1016/j.bbadis.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000) Neurochem Res. 2000;25(9-10):1439–1451. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- 111.Song MS, Learman CR, Ahn KC, Baker GB, Kippe J, Field EM, et al. In vitro validation of effects of BDNF-expressing mesenchymal stem cells on neurodegeneration in primary cultured neurons of APP/PS1 mice. Neuroscience. 2015;307:37–50. doi: 10.1016/j.neuroscience.2015.08.011. [DOI] [PubMed] [Google Scholar]
- 112.Pekny M, Wilhelmsson U, Pekna M. The dual role of astrocyte activation and reactive gliosis. Neurosci Lett. 2014;565:30–38. doi: 10.1016/j.neulet.2013.12.071. [DOI] [PubMed] [Google Scholar]
- 113.Wang DD, Bordey A. The astrocyte odyssey. Prog Neurobiol. 2008;86(4):342–367. doi: 10.1016/j.pneurobio.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14(4):388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Newman EA. Glial cell regulation of neuronal activity and blood flow in the retina by release of gliotransmitters. Philos Trans R Soc Lond B Biol Sci. 2015;370(1672):20140195. doi: 10.1098/rstb.2014.0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 117.Abbott NJ. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J Inherit Metab Dis. 2013;36(3):437–449. doi: 10.1007/s10545-013-9608-0. [DOI] [PubMed] [Google Scholar]
- 118.Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 2003;10(6):463–470. doi: 10.1038/sj.mn.7800212. [DOI] [PubMed] [Google Scholar]
- 119.Ma LY, Fei YL, Wang XY, Wu SD, Du JH, Zhu M, et al. The research on the relationship of RAGE, LRP-1, and Aβ Accumulation in the hippocampus, prefrontal lobe, and amygdala of STZ-induced diabetic rats. J Mol Neurosci. 2017;62(1):1–10. doi: 10.1007/s12031-017-0892-2. [DOI] [PubMed] [Google Scholar]
- 120.Herz J. Apolipoprotein E receptors in the nervous system. Curr Opin Lipidol. 2009;20(3):190–196. doi: 10.1097/MOL.0b013e32832d3a10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- 122.Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol. 2012;69(1):29–38. doi: 10.1001/archneurol.2011.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McClean PL, Parthsarathy V, Faivre E, Hölscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. J Neurosci. 2011;31(17):6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cabou C, Burcelin R. GLP-1, the gut-brain, and brain-periphery axes. Rev Diabet Stud. 2011;8(3):418–431. doi: 10.1900/RDS.2011.8.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.During MJ, Cao L, Zuzga DS, Francis JS, Fitzsimons HL, Jiao X, et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med. 2003;9(9):1173–1179. doi: 10.1038/nm919. [DOI] [PubMed] [Google Scholar]
- 126.McIntyre RS, Powell AM, Kaidanovich-Beilin O, Soczynska JK, Alsuwaidan M, Woldeyohannes HO, et al. The neuroprotective effects of GLP-1: possible treatments for cognitive deficits in individuals with mood disorders. Behav Brain Res. 2013;237:164–171. doi: 10.1016/j.bbr.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 127.Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer's disease. J Alzheimers Dis. 2010;19(4):1205–1219. doi: 10.3233/JAD-2010-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gejl M, Gjedde A, Egefjord L, Møller A, Hansen SB, Vang K, et al. In Alzheimer's disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front Aging Neurosci. 2016;8(8):108. doi: 10.3389/fnagi.2016.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gejl M, Brock B, Egefjord L, Vang K, Rungby J, Gjedde A. Blood-brain glucose transfer in Alzheimer's disease: effect of GLP-1 analog treatment. Sci Rep. 2017;7(1):17490. doi: 10.1038/s41598-017-17718-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li J, Deng J, Sheng W, Zuo Z. Metformin attenuates Alzheimer's disease-like neuropathology in obese, leptin-resistant mice. Pharmacol Biochem Behav. 2012;101(4):564–574. doi: 10.1016/j.pbb.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hsu CC, Wahlqvist ML, Lee MS, Tsai HN. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimers Dis. 2011;24(3):485–493. doi: 10.3233/JAD-2011-101524. [DOI] [PubMed] [Google Scholar]
- 132.Imfeld P, Bodmer M, Jick SS, Meier CR. Metformin, other antidiabetic drugs, and risk of Alzheimer's disease: a population-based case-control study. J Am Geriatr Soc. 2012;60(5):916–921. doi: 10.1111/j.1532-5415.2012.03916.x. [DOI] [PubMed] [Google Scholar]
- 133.Moore EM, Mander AG, Ames D, Kotowicz MA, Carne RP, Brodaty H, et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care. 2013;36(10):2981–2987. doi: 10.2337/dc13-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov. 2004;3(6):479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 135.Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci U S A. 1997;94(18):9660–9664. doi: 10.1073/pnas.94.18.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6(12):1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- 137.Muñoz-Montaño JR, Moreno FJ, Avila J, Diaz-Nido J. Lithium inhibits Alzheimer's disease-like tau protein phosphorylation in neurons. FEBS Lett. 1997;411(2-3):183–188. doi: 10.1016/s0014-5793(97)00688-1. [DOI] [PubMed] [Google Scholar]
- 138.Landreth G, Jiang Q, Mandrekar S, Heneka M. PPARgamma agonists as therapeutics for the treatment of Alzheimer's disease. Neurotherapeutics. 2008;5(3):481–489. doi: 10.1016/j.nurt.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126(4):789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, et al. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Neurosci. 2003;23(20):7504–7509. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pedersen WA, Flynn ER. Insulin resistance contributes to aberrant stress responses in the Tg2576 mouse model of Alzheimer's disease. Neurobiol Dis. 2004;17(3):500–506. doi: 10.1016/j.nbd.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 142.Ceconi C, Francolini G, Bastianon D, Gitti GL, Comini L, Ferrari R. Differences in the effect of angiotensin-converting enzyme inhibitors on the rate of endothelial cell apoptosis: in vitro and in vivo studies. Cardiovasc Drugs Ther. 2007;21(6):423–429. doi: 10.1007/s10557-007-6068-5. [DOI] [PubMed] [Google Scholar]
- 143.Umegaki H. Therapeutic potential of antidiabetic medications in the treatment of cognitive dysfunction and dementia. Drugs Aging. 2016;33(6):399–409. doi: 10.1007/s40266-016-0375-0. [DOI] [PubMed] [Google Scholar]