

Abstract
Bacterial infections and sepsis are leading causes of morbidity and mortality in critically ill patients. Currently, there are no effective treatments available to improve clinical outcome in sepsis. Here, we elucidated a mechanism by which E. coli bacteria impair neutrophil (PMN) chemotaxis and we studied whether this mechanism can be therapeutically targeted to improve chemotaxis and antimicrobial host defense. PMNs detect bacteria with formyl peptide receptors (FPR). FPR stimulation triggers mitochondrial ATP production and release. Autocrine stimulation of purinergic receptors exerts excitatory and inhibitory downstream signals that induce cell polarization and cell shape changes needed for chemotaxis. Here we show that the bacterial cell wall product lipopolysaccharide (LPS) dose-dependently impairs PMN chemotaxis. Exposure of human PMNs to LPS triggered excessive mitochondrial ATP production and disorganized intracellular trafficking of mitochondria, resulting in global ATP release that disrupted purinergic signaling, cell polarization, and chemotaxis. In mice infected intraperitoneally with E. coli, LPS treatment increased the spread of bacteria at the infection site and throughout the systemic circulation. Removal of excessive systemic ATP with apyrase improved chemotaxis of LPS-treated human PMNs in vitro and enhanced the clearance of E. coli in infected and LPS-treated mice. We conclude that systemic ATP accumulation in response to LPS is a potential therapeutic target to restore PMN chemotaxis and to boost the antimicrobial host immune defense in sepsis.
Summary sentence: LPS causes dysregulated ATP release, which interferes with autocrine purinergic signaling mechanisms needed for neutrophil chemotaxis and antimicrobial host defense.
Keywords: purinergic signaling, ATP release, endotoxin, bacterial clearance, apyrase
Graphical Abstract
INTRODUCTION
Adenosine triphosphate (ATP) is an energy carrier that fuels virtually all cellular processes. Damaged and dying cells release ATP into the extracellular space, where ATP acts as a “danger signal” that promotes inflammation and regulates immune responses.1,2 Elevated plasma ATP levels in the peripheral circulation of trauma and critical care patients may contribute to the systemic inflammatory response and multiple-organ dysfunction syndrome that are the hallmarks of sepsis.3–5
However, ATP can also be released from intact cells. Under these circumstances, ATP acts as a messenger molecule that regulates cell functions and facilitates intercellular communications by binding to specific purinergic receptors.6 The nineteen known purinergic receptor members comprise the P2X, P2Y, and P1 (adenosine) receptor families. The seven known P2X receptors act as ATP-gated cation channels, while the eight different P2Y receptors are G protein-coupled receptors (GPCRs) that respond to ATP, ADP, UTP, UDP, or UDP-glucose. All four known P1 receptors (A1, A2a, A2b, A3) recognize adenosine and are also members of the GPCRs.6,7
Purinergic receptors have important roles in the regulation of immune cell responses.6,8–10 Our previous work has shown that cellular ATP release fuels autocrine feedback mechanisms that involve P2Y2 and A2a receptors, which are the most abundant purinergic receptor subtypes expressed by human polymorphonuclear neutrophils (PMNs). We found that these purinergic receptors regulate various processes involved in PMN chemotaxis.11 Chemoattractant receptors such as the formyl peptide receptors (FPRs) induce rapid ATP release from PMNs that triggers autocrine stimulation of adjacent P2Y2 receptors on the cell surface.12 This autocrine purinergic feedback mechanism amplifies FPR signals, which increases the sensitivity of PMNs to formylated bacterial peptides that are the primary ligands of FPRs. FPRs and P2Y2 receptors activate mitochondria to produce ATP that is released through pannexin-1 channels, which colocalize with FPRs and P2Y2 receptors on the cell surface of PMNs.12,13 We found that localized mitochondrial activation, ATP release, and P2Y2 receptor stimulation induces cell polarization and initiates and directs PMN migration towards chemoattractant sources, that is bacteria that release formylated peptides.11–13 While the excitatory autocrine signaling mechanisms that involve P2Y2 receptors promote pseudopod protrusions at the front, we found that conversion of ATP to adenosine and stimulation of inhibitory A2a adenosine receptors promotes uropod retraction at the back of migrating cells.8,14,15
PMNs rely on these opposing purinergic signaling mechanisms to detect, pursue, and kill invading bacteria. However, because purinergic signaling takes place on the cell surface, these signaling events are vulnerable to interference by external factors such as the systemic ATP that accumulates in the plasma of patients as a result of inflammation, trauma, and tissue damage.16 In the current study, we show that E. coli bacteria exploit this vulnerability by deploying lipopolysaccharide (LPS) that elicits excessive ATP release from neutrophils and thereby disables PMN chemotaxis. We propose that Gram-negative bacteria use this deceptive strategy to defeat antimicrobial host defenses and to facilitate the unhindered spread of bacterial infections.
MATERIALS AND METHODS
Materials
Lipopolysaccharide (LPS) from Escherichia coli (E. coli) O111:B4 was used for all experiments (Sigma-Aldrich, St. Louis, MO). A highly purified (by gel-filtration chromatography; Sigma product number L3012) was used for in vitro experiments. For in vivo experiments, we used endotoxin from E. coli O111:B4 that was purified by phenol extraction (Sigma product number L2630). Apyrase was obtained from Sigma-Aldrich as a lyophilized powder, which we reconstituted in Hanks’ balanced salt solution (HBSS; HyClone, Thermo Fisher Scientific, Waltham, MA). E. coli (strain DH5-α), Rhod-2 AM and Fluo-4 AM were purchased from Thermo Fisher Scientific (Waltham, MA). NF023 was from Tocris Bioscience (R&D Systems, Minneapolis, MI); N-formylmethionine-leucyl-phenylalanine (fMLF; also known as fMLP) and all other reagents were obtained from Sigma-Aldrich and of tissue culture grade unless otherwise stated.
Isolation of human PMNs
The Institutional Review Board of the Beth Israel Deaconess Medical Center approved all studies involving human subjects. PMNs were isolated from the peripheral blood of healthy volunteers using Percoll (GE Healthcare, Boston, MA) density gradient centrifugation as previously described.15 Purified cells were washed twice and resuspended in HBSS containing Ca2+ and Mg2+ and 1% autologous heparinized human plasma. The purity of PMN preparations obtained with this method was >93% as determined by flow cytometry. PMNs were incubated at 37°C for a minimum of 30 min before addition of inhibitors or other treatments. All reagents and supplies were pyrogen-free and osmotic or excessive mechanical stimulation was avoided during the isolation procedure.
Mice
All animal experiments were approved by the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center and performed according to the guidelines of the National Institutes of Health. Male and female C57BL/6J mice (Charles River Laboratories, Wilmington, MA), 8–12 weeks old and weighing 20–25 g, were used for all experiments. Male and female mice did not differ in their responses to the experimental treatments.
Mouse model of E. coli-induced peritonitis
E. coli cultures were grown in Luria-Bertani (LB) liquid media (MP Biomedicals, Santa Ana, CA) until they reached the exponential growth phase (0.5 OD600). Bacteria were washed three times with sterile normal saline to reduce free LPS. Then mice were injected intraperitoneally (i.p.) with 1×106 E. coli in a total volume of 500 μl saline. Unless otherwise indicated, mice were treated with LPS (0.5 mg/mouse, i.p.) and/or apyrase (0.6 IU/g bodyweight, i.p.), or with an equal volume (500 μl) of saline prior (2 h) to injection of bacteria. The concentration of apyrase was based on previously published reports and our own experience with similar experiments.3,17 One hour after bacterial infection, mice were anesthetized with isoflurane and sacrificed by cardiac puncture and exsanguination. The blood was collected in heparinized tubes and chilled in an ice bath. Prior to blood collection, peritoneal lavage was performed using 3 ml of normal saline that contained 0.1% bovine serum albumin.
Bacterial and cell counts
Serial dilutions of peritoneal lavage fluid and blood samples were plated on LB agar plates (MP Biomedicals, Solon, OH). Colony-forming units (CFU) were counted after 18-h incubations at 37°C. Total leukocyte numbers in blood samples and peritoneal lavage fluids were determined with a hemocytometer. Differential staining with Hema 3 staining solution (Thermo Fisher Scientific) was performed to determine the percentage of PMNs in these samples.
ATP measurements
ATP, ADP, AMP, and adenosine concentrations in plasma samples were determined with high performance liquid chromatography (HPLC) as previously described.16 ATP breakdown by apyrase was verified in HBSS as well as in LB medium using a luciferase ATP bioluminescence kit (Thermo Fisher Scientific). The cell-surface targeting fluorescent ATP probe 2–2Zn (kind gift from Itaru Hamachi, Kyoto University) and flow cytometry were used for real-time monitoring of ATP release from individual cells.18,19 PMNs were suspended in HBSS containing 1% heparinized human plasma and stained with 2–2Zn (500 nM) for 5 min. ATP release in response to stimulation with LPS or fMLF was measured with a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA).
Imaging of calcium signaling and ATP release
Freshly isolated human PMNs were allowed to attach to fibronectin-coated coverslip dishes, resuspended in HBSS containing 1% heparinized human plasma, and loaded with the mitochondrial Ca2+ indicator Rhod-2 AM (1 μM) for 10 min or with the cytosolic Ca2+ probe Fluo-4 AM (4 μM) for 30 min. For imaging of ATP release, cells were stained with the ATP probe 2–2Zn (500 nM) for 5 min. Changes in mitochondrial Ca2+ uptake, cytosolic Ca2+ levels or ATP release after addition of LPS and/or fMLF were recorded by live-cell imaging with an inverted Leica DMI6000B microscope (Leica Microsystems, Wetzlar, Germany) equipped with a temperature controlled (37°C) stage incubator (Ibidi, Fitchburg, WI) and a Leica DFC365 FX camera. Fluorescence images were acquired at a frame rate of 1 frame per second using a 100x oil objective with a numerical aperture (NA) of 1.4, TRITC (Rhod-2) or FITC (Fluo-4, 2–2Zn) filter sets, and LAS X microscope imaging software (Leica Microsystems). Images were analyzed with ImageJ software (National Institutes of Health).
PMN chemotaxis
Under agarose chemotaxis assays were performed with minor modifications as previously described.20 In brief, a total of 3 ml of low gelling agarose (Sigma-Aldrich; 1.2% in HBSS supplemented with 10% heat-inactivated fetal bovine serum; Gibco, Thermo Fisher Scientific) was poured into 35 mm x 10 mm culture dishes (BD Biosciences, Billerica, MA). Then, wells (3.5 mm diameters) were cut out at a distance of 2.4 mm between two wells. Dishes were incubated at 37°C for 1 h in a sealed, humidified chamber. Residual moisture was aspirated and 20 μl of cell solution (1×106 human PMNs in HBBS containing 1% heparinized human plasma) was added to one set of wells. After a 10-min incubation period, 20 μl fMLF (50 nM) was loaded into opposing target wells. Then, cells were incubated at 37°C for 3 h to allow PMN migration towards the fMLF-containing wells. Gels were cooled on ice and kept at 4°C to stop cell movement. Images of PMNs were acquired using a 2.5x objective (NA 0.07) and a Leica DMIRB microscope (Leica Microsystems, Wetzlar, Germany) equipped with a Spot Boost EMCCD camera (Diagnostic Instruments Inc.; Sterling Heights, MI). Image analysis was done with ImageJ.
Statistical analyses
Unless otherwise stated, all values are expressed as means ± standard deviations (SD). Unpaired two-tailed Student’s t tests were used to compare groups. The Bonferroni method was used to adjust significance levels if more than one t test was performed on the same sample set. Multiple group comparisons were performed with one-way ANOVA or with the Kruskal-Wallis test followed by the Tukey test, depending on whether the data were normally distributed or not. Differences between groups were considered statistically significant at p<0.05.
RESULTS
LPS impairs host immune defenses in a mouse model of peritonitis
Danger molecules such as LPS induce inflammasome activation, which initiates host immune defenses.21,22 LPS, however, is also associated with the pathogenesis of sepsis.23 We used a mouse model to study how LPS affects the antimicrobial resistance in bacterial infection. Treating mice with an intraperitoneal injection of LPS two hours prior to injection of live E. coli significantly increased the bacterial load in the peritoneal cavity and in the circulating blood when compared to mice that had been injected with vehicle control instead of LPS (Fig. 1A–B). These data suggest that treatment with LPS compromises host immune defenses and the ability of animals to contain bacterial infections.
Figure 1. Removal of extracellular ATP improves bacterial clearance.

(A-B) Mice were treated with LPS (0.5 mg/mouse, i.p.), apyrase (0.6 IU/g bodyweight, i.p.), or normal saline. Two hours later, they received a bolus injection (i.p.) of 1×106 live E. coli. Peritoneal lavages (A) and blood samples (B) were collected after one hour and bacteria were counted (n = 6–7; *p<0.05 vs. LPS, one-way ANOVA; #p<0.05, t test). CFU, colony forming units.
Removal of extracellular ATP with apyrase improves bacterial clearance
LPS induces the release of cellular ATP from monocytes and macrophages.24–27 We hypothesized that LPS-induced accumulation of systemic ATP may weaken the antimicrobial resistance in our mouse model. In support of this hypothesis, we found that treatment of mice with apyrase, an enzyme that hydrolyzes extracellular ATP, decreased the spread of bacteria. Apyrase significantly reduced the number of bacteria at the site of infection, i.e., the peritoneal cavity of mice that received LPS and E. coli injections (Fig. 1A). Importantly, apyrase also reduced the numbers of bacteria in the peritoneal cavity and the systemic circulation of infected mice in the absence of LPS, indicating that E. coli infection itself induces systemic ATP release that impairs bacterial clearance (Fig. 1). These results were not caused by direct effects of apyrase on the viability of bacteria per se. Apyrase rapidly hydrolyzed extracellular ATP but failed to reduce the growth of bacteria in in vitro cultures of E. coli in LB medium (Fig. S1). Apyrase treatment also did not simply enhance the recruitment of PMNs to the site of infection in the peritoneal cavity or increase PMN numbers in the systemic circulation as demonstrated in Fig. 2. Instead, our findings suggest that removal of extracellular ATP improves the ability of PMNs to find and kill bacterial invaders at the infection site and that fewer PMNs are sufficient to carry out these tasks when the concentration of extracellular ATP is reduced with apyrase.
Figure 2. Apyrase prevents the systemic and local increase in leukocytes in infected mice.

Mice were intraperitoneally injected with LPS (0.5 mg/mouse), apyrase (0.6 IU/g bodyweight), or saline. Two hours later, a bolus injection (i.p.) of 1×106 live E. coli was administered. After another hour, peritoneal lavages and blood samples were collected and PMNs (A and B) and white blood cell counts (C and D) were determined (n= 3–10; #p<0.05 vs. E. coli, one-way ANOVA; *p<0.05 vs. LPS + E. coli, one-way ANOVA).
Apyrase treatment converts ATP to AMP and adenosine
The findings shown above suggest that apyrase improves host immune defenses by removing extracellular ATP, which impairs bacterial clearance in infected mice. In order to confirm this concept, we assessed the levels of ATP and its breakdown products ADP, AMP, and adenosine in the systemic circulation of our mice. Animals challenged with LPS and/or E. coli were treated with apyrase as described above and plasma levels of ATP and its breakdown products were measured with HPLC (Fig. 3). In most mice that were challenged with E. coli, LPS, or both, plasma ATP levels were higher than in untreated control animals. Apyrase treatment lowered these levels, but the differences among the treatment groups were not statistically significant. However, apyrase treatment significantly increased plasma AMP and adenosine levels in mice infected with E. coli and challenged with LPS (Fig. 3C–D). Taken together with our previous finding that apyrase reduces peritoneal ATP levels in a cecal ligation and puncture model of sepsis,17 we conclude that apyrase converts systemic ATP to AMP and adenosine and that this effect is responsible for improved antimicrobial resistance of apyrase-treated mice.
Figure 3. Apyrase treatment converts ATP to AMP and adenosine.

Mice were treated with LPS (0.5 mg/mouse i.p.), apyrase (0.6 IU/g bodyweight i.p.), or saline as indicated and received a bolus injection (i.p.) of 1×106 live E. coli two hours later. After one more hour, plasma ATP (A), ADP (B), AMP (C), and adenosine (ADO; D) concentrations were measured by HPLC (n= 3–8; *p<0.05 vs. LPS + E. coli, one-way ANOVA). Solid and dotted lines in the box plots depict median and mean values, respectively.
LPS interferes with the purinergic signaling mechanisms that regulate PMN chemotaxis
PMN chemotaxis is essential for antimicrobial host defense. We have previously demonstrated that PMN chemotaxis depends on the regulated release of cellular ATP and autocrine stimulation of P2Y2 receptors on the cell surface.11 PMNs detect bacteria with FPRs that bind bacterial peptides such as fMLF. FPR stimulation triggers the release of cellular ATP that amplifies chemotactic signals and guides PMNs to bacteria releasing formylated peptides.12 In monocytes and macrophages, stimulation of TLR4 receptors by LPS has been shown to trigger rapid ATP release.25,26 Neutrophils also express TLR4 receptors, suggesting that LPS-induced ATP release may interfere with the purinergic signaling events that regulate PMN chemotaxis. We tested this possibility using a fluorescent ATP probe (2–2Zn) that attaches to the cell surface where it allows real-time monitoring of the spatiotemporal patterns of ATP release from living cells.18 Cellular ATP release from human PMNs was assessed using live-cell imaging and flow cytometry. LPS caused rapid and dose-dependent ATP release that was more robust than the ATP release induced by fMLF (Fig. 4A–C). In addition, we found that LPS induced global ATP release that occurred uniformly across the cell surface, whereas fMLF-stimulated PMNs released ATP from distinct foci that tended to coincide with pseudopod protrusions and other membrane deformations involved in PMN chemotaxis (Fig. 4A–B; Video 1).11,12 Furthermore, priming of PMNs with LPS significantly increased ATP release in response to subsequent fMLF stimulation (Fig. 4D). These findings suggest that LPS interferes with the ATP release patterns and the purinergic signaling mechanisms that are known to regulate FPR-induced cell polarization and PMN chemotaxis.11,12
Figure 4. LPS-induced ATP release from PMNs overwhelms chemotactic signals.

(A-B) ATP release from human PMNs stimulated with fMLF (10 nM) or LPS (100 ng/ml) was analyzed using the cell surface targeting fluorescent ATP probe 2–2Zn and fluorescence microscopy (see also Video 1). (A) Representative images of at least 6 experiments with cells from different donors demonstrating ATP release patterns 30 s after cell stimulation. The areas within rectangles were magnified in the images below. Scale bars: 10 μm. (B) Fluorescence profile of back-to-front line scans as indicated in (A) (dotted lines) are shown as means ± SD of different cells (n=7). (C) Human PMNs were stained with the ATP probe 2–2Zn and ATP release after addition of LPS (100 ng/ml), fMLF (10 nM), or vehicle control was analyzed by flow cytometry. Representative results of 5 independent experiments with similar results are shown. (D) Human PMNs stained with the ATP probe 2–2Zn were treated for 15 min with the indicated concentrations of LPS. Then, cells were stimulated with fMLF (1 nM) or with vehicle control for an additional 5 min and ATP release was analyzed by flow cytometry (means ± SD, n=3–5 independent experiments; *p<0.05 vs. no LPS, one-way ANOVA; #p<0.05; fMLF vs. no fMLF; t test and Bonferroni correction).
LPS disrupts the intracellular trafficking and activation of mitochondria that regulate PMN chemotaxis
Although PMNs use glycolytic metabolism to cover much of their cellular ATP demand, they also possess mitochondria that produce ATP that is needed to recognize chemoattractants and initiate chemotaxis.13 Differential activation and trafficking of mitochondria within the cell are required to deliver localized ATP that regulates pseudopod protrusion at the front of polarized cells. Activated mitochondria traffic to the front of migrating PMNs where they fuel purinergic signaling mechanisms that orchestrate the directional movement and chemotaxis of PMNs.14 Because LPS induced global ATP release across the surface of PMNs, we hypothesized that LPS may interfere with the differential activation and/or trafficking of mitochondria within the cell. In order to test this hypothesis, we labeled PMNs with the mitochondrial Ca2+ probe Rhod-2-AM that allows real-time monitoring of mitochondrial Ca2+ uptake, which is a prerequisite for mitochondrial ATP production.28 PMNs were stimulated with LPS and mitochondrial Ca2+ uptake was recorded using live cell imaging. Stimulation with fMLF immediately increased mitochondrial Ca2+ levels, followed by the redistribution of activated mitochondria to the leading edge (Fig. 5A).13 LPS also caused rapid and dose-dependent Ca2+ uptake by mitochondria (Fig. 5A–B). However, in the absence of chemotactic cues, PMNs failed to polarize and their mitochondria remained evenly distributed throughout the cell (Fig. 5A; Video 2). PMNs that were primed with LPS, however, showed markedly enhanced mitochondrial Ca2+ uptake in response to subsequent fMLF-stimulation (Fig. 5C). Similarly, LPS prolonged the cytosolic Ca2+ response triggered by fMLF, even though LPS alone caused only a modest increase in cytosolic Ca2+ levels (Fig. S2A–B). In LPS-primed cells, fMLF stimulation led to the uniform dispersion of mitochondria towards the cell periphery. Moreover, in contrast to intact PMNs, LPS-primed cells failed to form a single leading edge but formed multiple leading edges in response to fMLF stimulation. As a result of this, primed cells spread in all directions and assumed cell shapes that were significantly less polarized than fMLF-stimulated PMNs (Fig. S2C, Fig. 5A; Video 2). These findings demonstrate that LPS disrupts the spatiotemporal patterns of mitochondrial activation and trafficking that are required for PMN polarization and chemotaxis.
Figure 5. LPS disrupts mitochondrial trafficking and activation in stimulated PMNs.

(A) Human PMNs labeled with the mitochondrial Ca2+ indicator Rhod-2-AM were stimulated with LPS (100 ng/ml), fMLF (10 nM), or with LPS followed by fMLF 10 min later. Mitochondrial Ca2+ uptake was monitored using time-lapse fluorescence microscopy and mitochondrial movement was tracked. Representative images of 3 separate experiments taken before and 60 s after stimulation are shown. Arrows indicate the change in mitochondrial position during the 60 s observation period (see also Video 2). (B) Human PMNs were treated as described in (A) and mitochondrial Ca2+ uptake in response to LPS stimulation was recorded. Averaged results (mean + SEM) of different cells (n=55–83) analyzed in 3 separate experiments are shown. (C) PMNs were stimulated with LPS (100 ng/ml) or vehicle control for 2 min, followed by stimulation with fMLF (10 nM), and mitochondrial Ca2+ uptake was recorded. Data are expressed as mean + SEM of 85 (fMLF) or 173 (LPS → fMLF) cells using PMN preparations from 3 separate donors.
Removal of excessive ATP improves PMN chemotaxis
Chemotaxis is a key host defense function that enables PMNs to track and kill invading microbes.29 LPS was reported to enhance the expression of FPRs.30 However, using an under-agarose chemotaxis assay, we found that LPS dose-dependently inhibited chemotaxis in response to fMLF (Fig. 6A–B). PMN chemotaxis depends on localized purinergic signaling mechanisms that may be impaired by excessive ATP accumulation in the extracellular space.11,17 Therefore, we hypothesized that removal of accumulated ATP with apyrase may restore PMN chemotaxis in the presence of LPS. In order to test this possibility, we exposed PMNs simultaneously to LPS and low concentrations of apyrase. Treatment of cells with apyrase, but not heat-inactivated apyrase restored chemotaxis in the presence of LPS (Fig. 6A, C). These findings demonstrate that removal of excessive extracellular ATP that accumulates in response to LPS stimulation can indeed restore PMN chemotaxis.
Figure 6. Apyrase recovers PMN chemotaxis in the presence of LPS.

(A) Healthy human PMNs were treated with LPS (1 ng/ml) and apyrase (0.1 mU/ml), loaded into wells in an agarose gel, and incubated at 37°C for 3 h. Migration was determined by measuring the distance that the cell front traveled toward the wells containing fMLF. Representative images of independent experiments (n=3) are shown (scale bar: 1 mm). (B) PMNs were treated with the indicated concentrations of LPS and chemotaxis was analyzed as described in (A). Data are means ± SD of 3 independent experiments; *p<0.05 vs. no LPS (one-way ANOVA). (C) PMNs were treated with LPS (1 ng/ml) in the presence or absence of apyrase or heat-inactivated apyrase and chemotaxis was analyzed as described in (A). Data shown are means ± SD of 12–16 experiments performed with cells from 4 different donors. The distance control cells migrated in the absence of LPS ranged from 1307 to 2003 μm; *p < 0.05 vs. LPS without apyrase (Kruskal-Wallis test).
DISCUSSION
Sepsis remains a major cause of death among critical care patients with an estimated mortality of 20 to 30%.31 Intraabdominal infections are the second most common cause of sepsis and E. coli is one of the most common Gram-negative pathogens that are involved.32 Gram-negative bacteria are capable of shedding LPS from their outer cell wall, which stimulates TLR4 receptor signaling in monocytes and macrophages and induces an inflammatory host immune response.33,34 While TLR4 signaling can elicit antimicrobial host defense, uncontrolled TLR4 signaling may also provoke excessive inflammation that causes endotoxic shock and host tissue damage.35 Our current findings demonstrate that E. coli use LPS to trigger the inflammatory cascade and to disorient PMNs. Through this strategy of deceit, Gram-negative bacteria evade PMN-mediated host defenses and gain an advantage that allows them to spread and cause severe infections and sepsis.
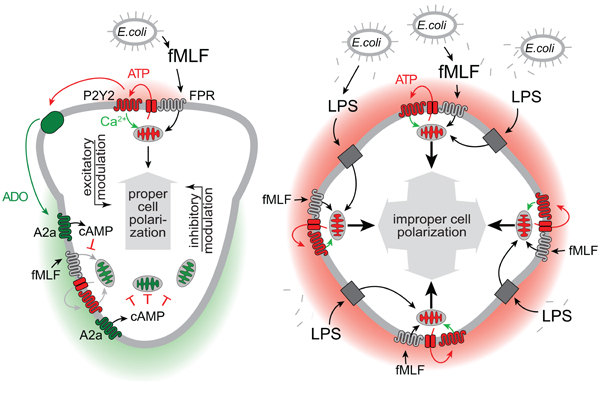
PMNs use dedicated receptors (FPRs) to recognize formylated peptides that are shed from bacteria. These receptors are highly sensitive and allow PMNs to detect and kill bacteria before they can spread to cause disseminated infections. We have previously identified a sophisticated feedback signaling system that PMNs employ to amplify and interpret chemotactic signals they receive.11–12 At the core of these signaling mechanisms are mitochondria that produce the ATP that fuels purinergic signaling (Fig. 7A).13 Pannexin-1 channels deliver ATP and colocalize with FPRs and P2Y2 receptors to form an excitatory purinergic signaling loop that amplifies chemotactic cues and regulates cell polarization.12 In addition to the excitatory purinergic signaling loop at the leading edge of cells, PMNs also need A2a adenosine receptors that translocate to the back during cell polarization where they exert an inhibitory cAMP-dependent purinergic signaling loop that promotes uropod retraction and the shutdown of mitochondrial ATP production.11,14
Figure 7. Proposed mechanism by which LPS impairs PMN chemotaxis.

(A) Detection of bacteria by formyl peptide receptors (FPRs) triggers mitochondrial ATP production and localized release of ATP through pannexin-1 channels. ATP stimulates adjacent P2Y2 receptors that induce an excitatory purinergic feed-forward loop and upregulate mitochondrial ATP production. This process amplifies FPR signaling, which results in cell polarization towards the chemoattractant source. Breakdown of ATP by CD39 and other ectonucleotidases generates adenosine (ADO) that stimulates inhibitory A2a receptors at the back of polarized cells. The combined actions of these excitatory and inhibitory purinergic signaling pathways via P2Y2 and A2a receptors, respectively, regulate PMN chemotaxis. (B) LPS liberated from the cell wall of Gram-negative bacteria triggers TLR4/CD14 signaling that induces global ATP release and P2Y2 receptor stimulation, resulting in mitochondrial activation throughout the cell. (C) In the presence of fMLF, LPS-primed PMNs release excessive amounts of ATP that overpower the differential purinergic signal mechanisms needed for cell polarization and PMN chemotaxis. As a result of this interference, LPS-stimulated PMNs are unable to properly polarize, to undergo chemotaxis, and to eliminate invading bacteria from the infected host.
Our current study suggests that bacterial LPS can impair PMN functions by interfering with the purinergic signaling system that regulates chemotaxis. We found that LPS stimulates mitochondrial Ca2+ uptake, consistent with increased mitochondrial respiration.28 The rise in mitochondrial Ca2+ was accompanied by only a modest rise in cytosolic Ca2+ levels. PMNs seem to differ in this respect from macrophages where LPS was recently reported to induce Ca2+ influx through TRPM7 channels.36 The activation of mitochondria by LPS led to uniform ATP release across the cell surface. When LPS-primed PMNs were exposed to chemotactic cues, FPR stimulation triggered excessive mitochondrial activation and ATP release. We therefore propose that LPS priming promotes excitatory purinergic signal amplification across the entire cell surface and thereby overpowers the locally defined purinergic signaling events that are necessary for PMN chemotaxis (Fig. 7A–C).11–13 As a result, LPS renders PMNs unable to clear bacteria from sites of infection such as the peritoneal cavity in mice after E. coli infection. Recently, we reported that LPS suppresses T cell functions in cocultures of monocytes and T cells by a mechanism that involves increased levels of extracellular ATP and P2Y11 receptor stimulation.37 Like A2a receptors that are highly expressed in PMNs, P2Y11 receptors are GPCRs that couple to Gαs and increase intracellular cAMP in T cells. We found that apyrase could restore PMN chemotaxis, indicating that ATP released in response to LPS directly impairs cell migration. Nevertheless, it is possible that enhanced breakdown of ATP to adenosine further disrupts chemotaxis in the presence of LPS by global stimulation of A2a receptors.15
In contrast to our finding that LPS impairs PMN chemotaxis, others have shown that LPS can also contribute to the recruitment of PMNs, for example in transwell assays using culture medium from LPS-stimulated monocytes or in air pouch models.38,39 In these models, LPS was shown to act indirectly on PMN chemotaxis, namely by promoting the production of chemotactic cytokines such as interleukin-8 (IL-8) that attracts PMNs to sites of inflammation. In this setting, apyrase has been shown to block IL-8 production and PMN recruitment while ATP promoted it.38,40 Differences in the experimental models and the chemoattractants involved are likely responsible for the different results of those studies and our current work. Further studies are needed to test whether and how LPS suppresses PMN chemotaxis in response to IL-8 in our in vitro assays and how other cells such as monocytes contribute to PMN dysfunction by inflammasome-induced ATP release.24–26
Our findings show that removal of the excessive ATP that is released in response to LPS can restore PMN chemotaxis in vitro and improve the clearance of bacteria in vivo. In addition to LPS, other pathological processes such as inflammation, hypoxia, tissue damage, and necrosis can also increase extracellular ATP concentrations.3 Like with LPS, excessive accumulation of systemic ATP in response to those insults will likely interfere with PMN chemotaxis and impair host immune defenses. In support of this notion, several animal studies have shown that removal of systemic ATP with apyrase has therapeutic potential by improving clinical outcome in mouse models of sepsis and inflammation.4,17,41 Another possible approach to mitigate the disruptive effects of systemic ATP on immune functions is to block the mechanisms involved in LPS-induced ATP release. In a recent study, LPS-induced ATP release from PMNs was reported to involve connexin-43 (Cx43) hemichannels and autocrine feedback via P2X1 receptors.42 The authors of this interesting report have shown that inhibition of P2X1 receptors with NF279 can recover PMN chemotaxis in the presence of LPS. We were unsuccessful to replicate these findings using another P2X1 receptor antagonist, namely NF023 (Fig. S3A–B). In addition, NF023 treatment did not significantly improve bacterial clearance in our mouse model of bacterial infection (Fig. S3C–D). The reasons for these dissimilar results are not clear and more work is needed to further elucidate the underlying mechanisms of this treatment approach compared to apyrase and other soluble ATPases.
Defective host immune function is increasingly recognized as a possible therapeutic target for the treatment of sepsis.43 Dozens of previous trials focusing on anti-inflammatory drugs to block the pro-inflammatory episode of sepsis have yielded largely disappointing results.44 This humbling experience has made it abundantly clear that the pathogenesis of sepsis is complex and comprises not only pro-inflammatory components that promote host tissue destruction but also an anti-inflammatory component that causes immunosuppression and hampers both innate and adaptive immune responses.43,45 Future research must therefore focus on a wide-reaching treatment approach that corrects both the inflammatory and suppressive aspects of immune dysfunction in sepsis.
Our current study shows that PMNs respond to LPS with inappropriate ATP release that disrupts their protective function and that may also contribute to indiscriminate PMN activation that causes host tissue damage and organ failure in sepsis.46 We show that these pathological processes can be addressed with apyrase which targets the root causes of immune dysfunction, namely defects in ATP signaling that impair the complex functions of PMN in host defense.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported in part by grants from the National Institutes of Health, GM-51477, GM-60475, GM-116162, AI-080582, and T32 GM-103702
Abbreviations
- ADP
adenosine diphosphate
- AMP
adenosine monophosphate
- ATP
adenosine triphosphate
- CFU
colony-forming units
- E. coli
Escherichia coli
- fMLF
N-formylmethionine-leucyl-phenylalanine
- FPR
formyl peptide receptor
- GPCR
G protein coupled receptor
- HBSS
Hanks’ balanced salt solution
- HPLC
high performance liquid chromatography
- LB
Luria-Bertani
- LPS
lipopolysaccharide
- NA
numerical aperture
- PMN
polymorphonuclear leukocyte
- TLR4
Toll-like receptor 4
- UDP
uridine diphosphate
- UTP
uridine triphosphate
Footnotes
DISCLOSURES
The authors declare no conflict of interest.
REFERENCES
- 1.Trautmann A Extracellular ATP in the immune system: more than just a “danger signal”. Sci Signal. 2009;2:pe6. [DOI] [PubMed] [Google Scholar]
- 2.Chekeni FB, Elliott MR, Sandilos JK, Walk SF, Kinchen JM, Lazarowski ER, Armstrong AJ, Penuela S, Laird DW, Salvesen GS, Isakson BE, Bayliss DA, Ravichandran KS. Pannexin 1 channels mediate “find-me” signal release and membrane permeability during apoptosis. Nature. 2010;467:863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cauwels A, Rogge E, Vandendriessche B, Shiva S, Brouckaert P. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014;5:e1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. 2014;509:310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ledderose C, Bao Y, Kondo Y, Fakhari M, Slubowski C, Zhang J, Junger WG. Purinergic signaling and the immune response in sepsis: A Review. Clin Ther. 2016;38:1054–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnstock G, Boeynaems JM. Purinergic signalling and immune cells. Purinergic Signal. 2014;10:529–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- 8.Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. 2011;11:201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Virgilio F, Vuerich M. Purinergic signaling in the immune system. Auton Neurosci. 2015;191:117–123. [DOI] [PubMed] [Google Scholar]
- 10.Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16:177–192. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Yao Y, Sumi Y, Li A, To UK, Elkhal A, Inoue Y, Woehrle T, Zhang Q, Hauser C, Junger WG. Purinergic signaling: a fundamental mechanism in neutrophil activation. Sci Signal. 2010;3:ra45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao Y, Ledderose C, Seier T, Graf AF, Brix B, Chong E, Junger WG. Mitochondria regulate neutrophil activation by generating ATP for autocrine purinergic signaling. J Biol Chem. 2014;289:26794–26803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bao Y, Ledderose C, Graf AF, Brix B, Birsak T, Lee A, Zhang J, Junger WG. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol. 2015;210:1153–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bao Y, Chen Y, Ledderose C, Li L, Junger WG. Pannexin 1 channels link chemoattractant receptor signaling to local excitation and global inhibition responses at the front and back of polarized neutrophils. J Biol Chem. 2013;288:22650–22657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sumi Y, Woehrle T, Chen Y, Bao Y, Li X, Yao Y, Inoue Y, Tanaka H, Junger WG. Plasma ATP is required for neutrophil activation in a mouse sepsis model. Shock. 2014;42:142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Kondo Y, Bao Y, Staudenmaier L, Lee A, Zhang J, Ledderose C, Junger WG. Systemic adenosine triphosphate im pairs neutrophil chemotaxis and host defense in sepsis. Crit Care Med. 2017;45:e97–e104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurishita Y, Kohira T, Ojida A, Hamachi I. Organelle-localizable fluorescent chemosensors for site-specific multicolor imaging of nucleoside polyphosphate dynamics in living cells. J Am Chem Soc. 2012;134:18779–18789. [DOI] [PubMed] [Google Scholar]
- 19.Sueyoshi K, Sumi Y, Inoue Y, Kuroda Y, Ishii K, Nakayama H, Iwabuchi K, Kurishita Y, Shigemitsu H, Hamachi I, Tanaka H. Fluorescence imaging of ATP in neutrophils from patients with sepsis using organelle-localizable fluorescent chemosensors. Ann Intensive Care. 2016;6:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heit B, Kubes P. Measuring chemotaxis and chemokinesis: the under-agarose cell migration assay. Sci STKE. 2003;2003(170):PL5. [DOI] [PubMed] [Google Scholar]
- 21.Cullen SP, Kearney CJ, Clancy DM, Martin SJ. Diverse activators of the NLRP3 inflammasome promote IL-1β secretion by triggering necrosis. Cell Rep. 2015;16;11:1535–1548. [DOI] [PubMed] [Google Scholar]
- 22.He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41:1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Opal SM, Scannon PJ, Vincent JL. Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J Infect Dis. 1999;180:1584–1589. [DOI] [PubMed] [Google Scholar]
- 24.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, Di Virgilio F. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol. 2006;176:3877–3883. [DOI] [PubMed] [Google Scholar]
- 25.Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A. 2008;105:8067–8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee AH, Ledderose C, Li X, Slubowski CJ, Sueyoshi K, Staudenmaier L, Bao Y, Zhang J, Junger WG. Adenosine triphosphate release is tequired for toll-like receptor-induced monocyte / macrophage activation, inflammasome signaling, interleukin-1β production, and the host immune response to infection. Crit Care Med. 2018;46:e1183–e1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP release by inflammatory cells. Int J Mol Sci. 2018;19(4).pii:E1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. [DOI] [PubMed] [Google Scholar]
- 30.Mandal P, Novotny M, Hamilton TA. Lipopolysaccharide induces formyl peptide receptor 1 gene expression in macrophages and neutrophils via transcriptional and posttranscriptional mechanisms. J Immunol. 2005;175:6085–6091. [DOI] [PubMed] [Google Scholar]
- 31.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med. 2013;369:840–851. [DOI] [PubMed] [Google Scholar]
- 32.Vincent JL, Rello J, Marshall J, Silva E, Anzueto A, Martin CD, Moreno R, Lipman J, Gomersall C, Sakr Y, Reinhart K. EPIC II Group of Investigators. International study of the prevalence and outcomes of infection in intensive care units. JAMA. 2009;302:2323–2329. [DOI] [PubMed] [Google Scholar]
- 33.Raetz CRH, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3:36–46. [DOI] [PubMed] [Google Scholar]
- 35.Tsujimoto H, Ono S, Efron PA, Scumpia PO, Moldawer LL, Mochizuki H. Role of Toll-like receptors in the development of sepsis. Shock. 2008;29:315–321. [DOI] [PubMed] [Google Scholar]
- 36.Schappe MS, Szteyn K, Stremska ME, Mendu SK, Downs TK, Seegren PV, Mahoney MA, Dixit S, Krupa JK, Stipes EJ, Rogers JS, Adamson SE, Leitinger N, Desai BN. Chanzyme TRPM7 mediates the Ca(2+) influx essential for lipopolysaccharide-iInduced Toll-like receptor 4 endocytosis and macrophage activation. Immunity. 2018;48:59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sueyoshi K, Ledderose C, Shen Y, Lee AH, Shapiro NI, Junger WG. Lipopolysaccharide suppresses T cells by generating extracellular ATP that impairs their mitochondrial function via P2Y11 receptors. J Biol Chem. 2019;294:6283–6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kukulski F, Ben Yebdri F, Lefebvre J, Warny M, Tessier PA, Sévigny J. Extracellular nucleotides mediate LPS-induced neutrophil migration in vitro and in vivo. J Leukoc Biol. 2007;81:1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kukulski F, Ben Yebdri F, Bahrami F, Lévesque SA, Martín-Satué M, Sévigny J. The P2 receptor antagonist PPADS abrogates LPS-induced neutrophil migration in the murine air pouch via inhibition of MIP-2 and KC production. Mol Immunol. 2010;47:833–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kukulski F, Ben Yebdri F, Lecka J, Kauffenstein G, Lévesque SA, Martín-Satué M, Sévigny J. Extracellular ATP and P2 receptors are required for IL-8 to induce neutrophil migration. Cytokine. 2009;46:166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Csóka B, Németh ZH, Törő G, Koscsó B, Kókai E, Robson SC, Enjyoji K, Rolandelli RH, Erdélyi K, Pacher P, Haskó G. CD39 improves survival in microbial sepsis by attenuating systemic inflammation. FASEB J. 2015;29:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X, Qin W, Xu X, Xiong Y, Zhang Y, Zhang H, Sun B. Endotoxin-induced autocrine ATP signaling inhibits neutrophil chemotaxis through enhancing myosin light chain phosphorylation. Proc Natl Acad SciU S A 2017;114:4483–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol. 2018;14:121–137. [DOI] [PubMed] [Google Scholar]
- 44.Cohen J, Opal S, Calandra T. Sepsis studies need new direction. Lancet Infect Dis. 2012;12:503–505. [DOI] [PubMed] [Google Scholar]
- 45.Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013;13:260–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sônego F, Castanheira FV, Ferreira RG, Kanashiro A, Leite CA, Nascimento DC, Colón DF, Borges V de F, Alves-Filho JC, Cunha FQ. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol. 2016;7:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.