

Abstract
Background
Baitouweng is a traditional Chinese medicine with a long history of different applications. Although referred to as a single medicine, Baitouweng is actually comprised of many closely related species. It is therefore critically important to identify the different species that are utilized in these medicinal applications. Knowledge about their phylogenetic relationships can be derived from their chloroplast genomes and may provide additional insights into development of molecular markers.
Methods
Genomic DNA was extracted from six species of Pulsatilla and then sequenced on an Illumina HiSeq 4000. Sequences were assembled into contigs by SOAPdenovo 2.04, aligned to the reference genome using BLAST, and then manually corrected. Genome annotation was performed by the online DOGMA tool. General characteristics of the cp genomes of the six species were analyzed and compared with closely related species. Additionally, phylogenetic trees were constructed, based on single nucleotide polymorphisms (SNPs) and 51 shared protein-coding gene sequences in the cp genome among all 31 species via maximum likelihood.
Results
The size of cp genomes of P. chinensis (Bge.) Regel, P. chinensis (Bge.) Regel var. kissii (Mandl) S. H. Li et Y. H. Huang, P. cernua (Thunb.) Bercht. et Opiz f. plumbea J. X. Ji et Y. T. zhao, P. dahurica (Fisch.) Spreng, P. turczaninovii Kryl. et Serg, and P. cernua (Thunb.) Bercht. et Opiz. were 163,851 bp, 163,756 bp, 162,481 bp, 162,450 bp, 162,795 bp, and 162,924 bp, respectively. Each species included two inverted repeat regions, a small single-copy region, and a large single-copy region. A total of 134 genes were annotated, including 90 protein-coding genes, 36 tRNAs, and eight rRNAs across all species. In simple sequence repeat analysis, only P. dahurica was found to contain hexanucleotide repeats. A total of 26, 39, 32, 37, 32 and 43 large repeat sequences were identified in the genic regions of the six Pulsatilla species. Nucleotide diversity analysis revealed that the rpl36 gene and ccsA-ndhD region have the highest Pi value. In addition, two phylogenetic trees of the cp genomes were constructed, which laced all Pulsatilla species into one branch within Ranunculaceae.
Conclusions
We identified and analyzed the cp genome features of six species of P. Miller, with implications for species identification and phylogenetic analysis.
Keywords: Pulsatilla chinensis, Pulsatilla Miller, Chloroplast genome, Phylogeny
Background
Baitouweng is the dry root of Pulsatilla chinensis, Ranunculaceae. It is a traditional Chinese medicine that has been used to alleviate fever and treat dysentery [1]. A total of 43 species have been identified in Europe and Asia, with 11 found in China [2]. Triterpenoid saponins are thought to be one of the main active components in Baitouweng [3]. It is included in the Chinese Pharmacopoeia as a genuine medicinal material, but not all species are included. Our previous investigation into the market of Chinese medicine found that there are many counterfeits, partially due to mistaking closely related species. This seriously affects the quality of medicinal materials and clinical efficacy. Previously, our team has studied DNA barcodes of Pulsatilla [4]. However, there are limited studies on the phylogenetic position and species diversity of these species, which could be improved by focusing on their chloroplast (cp) genomes.
The chloroplast is an important organelle in plants, which provides energy through photosynthesis and plays an important role in carbon uptake. Additionally, it contains its own genome, which takes the form of a cyclic double-stranded DNA molecule, with a maternal inheritance pattern [5–8]. Typical cp genome structure consists of four stable parts—two inverted repeats (IRs), a large single copy (LSC) region, and a small single copy (SSC) region [7]. In general, the cp genome contains an average of about 120 kb of unique sequences. In addition to the rRNA and tRNA genes, the number of protein-coding genes in the cp genome is about 100 [9]. In recent years, molecular identification has been widely used to discern true Chinese medicines from their counterfeits [10, 11]. With the rapid development of next generation sequencing, the acquisition of genomes is faster and cheaper than traditional Sanger sequencing [12]. Compared with nuclear genome DNA, cp genome DNA has a low molecular weight, multiple copies, and a simple structure, which are conducive to cp genome analysis [13]. Simple sequence repetition (SSR) has high mutation rate and multiple copies, and SSR markers have been widely used in genetic diversity and evolutionary research [14, 15].
In this study, the cp genomes of P. chinensis, P. chinensis var. kissii, P. cernua f. plumbea, P. dahurica, P. turczaninovii, and P. cernua were sequenced to analyze their structures and explore differences at molecular level. Meanwhile, compared with the cp genomic characteristics of Aconitum carmichaelii (NC_030761.1) and Coptis chinensis (NC_036485.1), whether there was a characteristic variation. We also analyzed SSRs, large sequence repeats, IR boundaries, and nucleotide diversity in an attempt to identify differences. Phylogenetic analysis was carried out to determine the evolutionary relationship and phylogenetic positions of six Pulsatilla species.
Methods
DNA extraction and sequencing
Fresh leaves of six species of Pulsatilla were collected from Liaoning Provincial Preservation Nursery of Key Species of Chinese Medicinal Plants in Dalian Campus of Liaoning University of Traditional Chinese Medicine (N 39°06′, E 121°87′, Dalian, Liaoning Province, China). P. chinensis was introduced in Dalian, Liaoning Province. P. chinensis var. kissii was introduced in Anshan, Liaoning Province. P. cernua f. plumbea was introduced in Jiaohe, Jilin Province. P. dahurica was introduced in Yichun, Heilongjiang Province. P. turczaninovii was introduced in Tongliao, Inner Mongolia Autonomous Region. P. cernua was introduced in Dandong, Liaoning Province. All the species were introduced by Xu Liang. Professor Kang Tingguo at Liaoning University of Traditional Chinese Medicine, identified the certificate specimens (P. chinensis 10162180425513LY, P. chinensis var. kissii 10162180429514LY, P. cernua f. plumbea 10162180503515LY, P. dahurica 10162180503516LY, P. turczaninovii 10162180504517LY, P. cernua 10162180504518LY) and deposited them in the Herbarium of Liaoning University of Traditional Chinese Medicine. Approximately 5 g of fresh leaves was harvested for cp DNA isolation using a modified cetyl trimethylammonium bromide method [16]. After DNA isolation, 1 μg of purified DNA was fragmented and used to construct short-insert libraries (insert size 430 bp) according to the manufacturer’s instructions (Illumina), then sequenced on the Illumina Hiseq 4000 [17].
Genome assembly and annotation
Prior to assembly, raw reads were filtered to remove reads with adaptor contamination, low quality (Q < 20), or a high percentage of uncalled bases (> 10%). The cp genome was reconstructed using a combination of de novo and reference-guided assemblies [18]. First, the filtered reads were assembled into contigs using SOAP denovo 2.04 [19]. Contigs were then aligned to the reference genome using BLAST and aligned contigs (≥ 80% similarity and query coverage) were ordered according to the reference genome. Finally, the clean reads were mapped to the assembled draft cp genome for base correction, and most gaps were filled through local assembly. The cp genes were annotated using an online DOGMA tool [20] with default parameters to predict protein-coding genes, transfer RNA (tRNA) genes, and ribosome RNA (rRNA) genes. A whole cp genome BLAST [21] search was performed against five databases, with cutoffs of < 1e−5 E-value and minimum alignment length percentage of > 40%. Searched databases included KEGG (Kyoto Encyclopedia of Genes and Genomes) [22–24], COG (Clusters of Orthologous Groups) [25, 26], NR (Non-Redundant Protein Database databases), Swiss-Prot [27], and GO (Gene Ontology) [28]. The sequencing data and gene annotations were then submitted to GenBank and assigned accession numbers (P. chinensis: MK860682, P. chinensis var. kissii: MK860683, P. cernua f. plumbea: MK860684, P. dahurica: MK860685, P. turczaninovii: MK860686, P. cernua: MK860687).
The cp genomes were then mapped using Organellar Genome Draw (OGDRAW) (Max Planck Institute of Molecular Plant Physiology, Am Mühlenberg, Potsdam, Germany) (http://ogdraw.mpimp-golm.mpg.de/index.Shtml) [29].
Comparative analysis of the cp genomes
The SSR software MicroSAtellite (MISA) (http://pgrc.ipk-gatersleben.de/misa/) was used to identify SSR sequences and tandem repeats of 1–6 nucleotides were considered microsatellites. The minimum numbers of repeats were set to 10, 6, 5, 5, 5, and 5 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively. The maximum number of bases interrupting two SSRs in a compound microsatellite was set to 100. The data were then compared with A. carmichaelii (NC_030761.1) and C. chinensis (NC_036485.1), with an emphasis on perfect repeat sequences [30]. Web-based REPuter (http://bibiserv.techfak.uni-bielefeld.de/reputer/) was used to analyze the long repeat sequences, which included forward, reverse, complement, and palindromic repeats with minimum sequence length of 30 bp and edit distances of 3 bp [31]. DnaSP v5.10 was utilized to determine the average number of nucleotide differences between the six genomes [32].
Phylogenetic analysis
In order to analyze the relationship between the phylogenetic position of Pulsatilla and other genera in Ranunculaceae, phylogenetic trees were constructed by aligning cp genome sequences from 31 species, 25 of which were obtained from GenBank. Among the 31 species, there were two outgroups: Arabidopsis thaliana (NC_003076.8) and Panax ginseng (NC_006290.1). Phylogenetic analysis included 29 species in Ranunculaceae, one species of Cruciferae, and one species of Araliaceae. Single nucleotide polymorphisms (SNPs) and 51 shared protein-coding genes of the cp genome for all 31 species were analyzed. The PhyML V3.0 software was used to construct a phylogenetic tree by maximum likelihood method (ML), and a model GTR+I+G was selected for ML analyses with 1000 bootstrap replicates to calculate bootstrap values [33].
Results and discussion
General characteristics
The cp genome sizes of the six species were 163,851, 163,756, 162,481, 162,450, 162,795 and 162,924 bp for P. chinensis, P. chinensis var. kissii, P. cernua f. plumbea, P. dahurica, P. turczaninovii, and P. cernua, respectively. The similar sizes of these genomes indicate that they were highly conserved during the course of evolution. The overall GC content of the P. chinensis, P. chinensis var. kissii, P. cernua f. plumbea, P. dahurica, P. turczaninovii, and P. cernua cp genomes was 37.14%, 37.16%, 37.42%, 37.42%, 37.40%, and 37.35%, respectively. The minor differences in cp genome size is mainly caused by changes in the LSC region. Most variations occur in the non-coding regions, which has been reported in other systems previously [34]. It is reflected in the species of Cerasus humilis [35], Talinum paniculatum [36] and Heimia myrtifolia [37]. The largest gene-coding region was the LSC, which was 82,432 bp, 82,294 bp, 81,923 bp, 81,894 bp, 82,177 bp and 82,427 bp, respectively. The SSC regions were 19,273 bp, 19,225 bp, 17,872 bp, 17,844 bp, 18,244 bp and 17,677 bp in size, respectively. The IR (IRa, IRb) regions were 31,118 bp, 31,119 bp, 31,343 bp, 31,356 bp, 31,187 bp and 31,410 bp in size, respectively. We also compared the cp genomes of A. carmichaelii and C. chinensis with those of the six sequenced species, and found that their sizes were highly divergent (Table 1). In addition, we found that the total cp genome size of P. chinensis var. kissii was only 203 bp different from that of Amomum compactum in different families [38].
Table 1.
Comparison of general characteristics of the cp genomes of the eight Ranunculaceae species
| Type | P. chinensis | P. chinensis var. kissii | P. cernua f. plumbea | P. dahurica | P. turczaninovii | P. cernua | A. carmichaelii | C. chinensis |
|---|---|---|---|---|---|---|---|---|
| Size (bp) | 163,851 | 163,756 | 162,481 | 162,450 | 162,795 | 162,924 | 155,737 | 155,484 |
| GC content (%) | 37.14 | 37.16 | 37.42 | 37.42 | 37.40 | 37.35 | 38.10 | 38.17 |
| LSC length (bp) | 82,342 | 82,294 | 81,923 | 81,894 | 82,177 | 82,427 | 86,330 | 84,585 |
| SSC length (bp) | 19,272 | 19,224 | 17,871 | 17,843 | 18,243 | 17,676 | 17,021 | 17,383 |
| IR length (bp) | 31,118 | 31,119 | 31,343 | 31,356 | 31,187 | 31,410 | 26,193 | 26,758 |
| Gene number | 134 | 134 | 134 | 134 | 134 | 134 | 112 | 128 |
| Gene number in IR regions | 46 | 46 | 46 | 46 | 46 | 46 | 36 | 36 |
| Protein-coding gene number | 90 | 90 | 90 | 90 | 90 | 90 | 78 | 92 |
| rRNA gene number | 8 | 8 | 8 | 8 | 8 | 8 | 4 | 8 |
| tRNA gene number | 36 | 36 | 36 | 36 | 36 | 36 | 30 | 28 |
A total of 134 genes were observed in each Pulsatilla cp genome, which was the same as that observed in the earliest differentiated group of flowering plants, Amborella trichopoda [39]. All genes, including 36 tRNAs, eight rRNAs and 90 protein-coding genes, were consistent in number. The six species of Pulsatilla all had 14 tRNAs and all eight rRNAs located in the IR region. Based on short read sequencing, we found that the LSC regions of the six Pulsatilla cp genomes were very similar in size, indicating that the evolution of the cp genes from the six Pulsatilla species was highly conserved (Table 1).
The relative locations and sizes of different genes were mostly similar in the six species of Pulsatilla. For example, the matK genes of the six species were all located in the trnK-UUU gene region. The rpl16 gene of P. chinensis in the IRb region was always larger than that in the IRa region. On the other hand, the rpl16 gene of P. chinensis var. kissii in the IRa region was larger than of the one in the IRb region. The rpl16 genes in the IRa and IRb regions of P. cernua were larger than the other five Pulsatilla species, which can be used as a species identification characteristic. In the rpl16 gene characteristic, P. dahurica and P. cernua were identical, although the total genome size of the latter was 31 bp longer than that of the former. The only difference between P. dahurica and P. turczaninovii is was the rps12 gene in the IRa region, with the latter having a much smaller size. This was a unique characteristic of P. turczaninovii, compared with all five other Pulsatilla species. Although the cp genes of P. dahurica and P. cernua were mostly similar. The cp genome of the former was 474 bp longer that the latter. The ycf15 gene, which is present in angiosperms, was missing in the cp genomes of Pulsatilla and Illicium (Schisandraceae) [40]. Due to these various differences, genes such as rpl16 and rps12 can be used to develop molecular markers for the differentiation of these species (Fig. 1, Additional file 1: Figure S1, Additional file 2: Figure S2, Additional file 3: Figure S3, Additional file 4: Figure S4 and Additional file 5: Figure S5).
Fig. 1.

Circular gene map of P. chinensis. Genes on the outside circle are transcribed counterclockwise, while genes on the inside circle are transcribed clockwise. LSC large single copy, SSC small single copy, IRa inverted repeat a, IRb inverted repeat b
Among the cp genes of the six Pulsatilla species, the two largest gene families were involved in photosynthesis and self-replication (Table 2). There were six genes coding for subunits of the ATP synthase (atpA, atpB, atpE, atpF, atpH and atpI) and 12 genes coding for subunits of NADH dehydrogenase (ndhA, ndhB, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ and ndhK), which contained two ndhB genes. Five (psaA, psaB, psaC, psaI and psaJ) and 15 genes (psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT and psbZ) coded for the subunits of photosystem I and photosystem II, respectively. This was consistent with the number of genes identified in Acer miaotaiense [34]. The function of five of the 134 genes, including ycf1, ycf3, ycf4, and two copies of ycf2, were not known.
Table 2.
List of the genes in the cp genomes of six species of Pulsatilla
| Gene category | Gene group | Gene name |
|---|---|---|
| Photosynthesis | Subunits of ATP synthase (6) | atpA, atpB, atpE, atpFa, atpH, atpI |
| Subunits of NADH dehydrogenase (12) | ndhAa, ndhBa (x2), ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome (6) | petA, petBa, petDa, petG, petL, petN | |
| Subunits of photosystem I (5) | psaA, psaB, psaC, psaI, psaJ | |
| Subunits of photosystem II (15) | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Other genes | Subunit of rubisco (1) | rbcL |
| Subunit of Acetyl-CoA-carboxylase (1) | accD | |
| c-type cytochrome synthesis gene (1) | ccsA | |
| Envelop membrane protein (1) | cemA | |
| Protease (1) | clpPb | |
| Maturase (1) | matK | |
| Self-replication | Large subunit of ribosome (13) | rpl2a (x2), rpl14 (x2), rpl16a (x2), rpl20, rpl22 (x2), rpl23 (x2) rpl33, rpl36 |
| DNA dependent RNA polymerase (4) | rpoA, rpoB, rpoC1a, rpoC2 | |
| Small subunit of ribosome (18) | rps2, rps3 (x2), rps4, rps7 (x2), rps8 (x2), rps11, rps12a (x3), rps14, rps15, rps16a, rps18, rps19 (x2) | |
| rRNA Genes (8) | rrn4.5 (x2), rrn5 (x2), rrn16 (x2), rrn23 (x2) | |
| tRNA Genes (36) | trnA-UGC (x2), trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnG-GCC, trnG-UCC, trnH-GUG, trnI-CAU (x2), trnI-GAU (x2), trnK-UUU, trnL-CAA (x2), trnL-UAA, trnL-UAG, trnfM-CAU, trnM-CAU, trnN-GUU (x2), trnP-UGG, trnQ-UUG, trnR-ACG (x2), trnR-UCU, trnS-GCU, trnS-GGA, trnS-UGA, trnT-GGU, trnV-GAC (x2), trnV-UAC, trnW-CCA, trnY-GUA | |
| Unknown function | Conserved open reading frames (5) | ycf 1, ycf 2 (x2), ycf 3b, ycf4 |
Numbers in brackets behind the name of gene group refer to the number of repetitive genes
aContains one intron
bContains two introns
Introns play an important role in regulating gene expression. Recent studies have found that many introns can enhance the expression of exogenous genes at specific times and locations, with implications for agronomic trait improvement [41]. The protein coding genes of all six Pulsatilla species contained the same number of introns. rps16, rpoC1, atpF, petB, petD, rpl16, rpl2, ndhB, rps12, and ndhA all display one intron, while ycf3 and clpP have two introns (Additional file 6: Table S1).
Repeat sequence analysis
SSR analysis
Microsatellite markers, also known as SSR markers, are PCR-based DNA molecular markers [42]. Because of the characteristics of neutral markers, the highly variable numbers of repeats and the relative conservation of flanking sequences of SSRs, they have been widely utilized in genotyping. SSR marker are also easy to design and have high repeatability and codominant inheritance among alleles, making them the best choice for evaluating the genetic diversity of crop species [43–45].
The cp genomes of P. chinensis, P. chinensis var. kissii, P. cernua f. plumbea, P. dahurica, P. turczaninovii, and P. cernua, contained 196, 197, 207, 208, 196, and 196 SSRs, respectively (Table 3). This is significantly different from that observed with Arctium, which had far less SSRs [46]. The length of SSRs were 116 bp, 116 bp, 114 bp, 114 bp, 126 bp and 176 bp, respectively. The number of SSRs in the formation of compound microsatellites was 48, 48, 55, 57, 46 and 47, respectively. There were six types of SSRs in P. dahurica, including mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide, other five species of Pulsatilla contained five types of SSRs other than hexanucleotide (Fig. 2). The six species of Pulsatilla contained mostly A/T SSRs, with 149, 150, 157, 156, 150 and 147, respectively. They accounted for 76.00%, 76.14%, 75.85%, 75.00%, 76.53%, 75.00% of all SSR nucleotides, respectively (Additional file 7: Table S2). The AT content was relatively high, which may be related to the relative stability of AT and GC base pairs [47]. In SSRs, only P. turczaninovii contained seven C/G repeats, while other Pulsatilla contained eight C/G, which can be used as a distinguishing characteristic. In addition, the AT content of P. cernua f. plumbea and P. dahurica was very high (6.76% and 6.73%, respectively), which was significantly different from those of other Pulsatilla. The SSRs of the six species of Pulsatilla were also compared with those of A. carmichaelii and C. chinensis. The SSRs of C. chinensis also contained hexanucleotide SSRs, which were found in P. dahurica. However, A. carmichaelii did not contain pentanucleotide or hexanucleotide SSRs. Compared with other types of nucleotide repeats, the six species of Pulsatilla contained more mononucleotides repeats (Additional file 7: Table S2). The SSRs of several species in Ranunculaceae were similar, making SSR markers a tenable approach.
Table 3.
SSRs identified in the cp genomes of the six Pulsatilla species
| Unit size | P. chinensis | P. chinensis var. kissii | P. cernua f. plumbea | P. dahurica | P. turczaninovii | P. cernua |
|---|---|---|---|---|---|---|
| Mononucleotide | 157 | 158 | 165 | 164 | 157 | 155 |
| Dinucleotide | 9 | 9 | 14 | 14 | 8 | 7 |
| Trinucleotide | 10 | 10 | 10 | 10 | 11 | 14 |
| Tetranucleotide | 14 | 14 | 16 | 16 | 17 | 17 |
| Pentanucleotide | 6 | 6 | 2 | 3 | 3 | 3 |
| Hexanucleotide | 0 | 0 | 0 | 1 | 0 | 0 |
Fig. 2.

SSRs in the cp genomes of eight species in Ranunculaceae. MonoNucl represents mononucleotide repeats, DiNucl represents dinucleotide repeats, TriNucl represents trinucleotide repeats, TetraNucl represents tetranucleotide repeats, PentaNucl represents pentanucleotide repeats, and HexaNucl represents hexalnucleotide repeats
Taking P. chinensis as the representative species, the distribution of palindromic-type SSR (p-type SSR) was analyzed (Additional file 8: Table S3). The repeat sequences were mainly distributed in the non-coding sequences (CNS), intergenic spacers, and intron regions. Some were found in the coding regions of certain genes, including matK, psbC, rpoB, rpoC2, rps2, atpA, ndhJ, atpB, accD, and others. The other five Pulsatilla species were similar to P. chinensis in terms of p-type SSR (Additional file 9: Table S4, Additional file 10: Table S5, Additional file 11: Table S6, Additional file 12: Table S7 and Additional file 13: Table S8).
Large repeat analysis
Many repeats are present in gene deserts, although whole-genome sequencing has shown that they can occur in functional regions as well [48]. Repeat of 30 bases or more are typically considered large repeats. Forty-nine (P. chinensis), 49 (P. chinensis var. kissii), 38 (P. cernua f. plumbea), 38 (P. dahurica), 49 (P. turczaninovii) and 49 (P. cernua) pairs of large repeat sequences were found in the six Pulsatilla cp genomes, with sequence identity exceeding 90%. The repeats from P. chinensis and P. chinensis var. kissii ranged from 30 to 59 bp in length, and in P. cernua f. plumbea, P. dahurica, P. turczaninovii and P. cernua, the repeats ranged from 30 to 52 bp in length. A total of 26, 39, 32, 37, 32 and 43 large repeats were located in the genic regions of the six Pulsatilla species, respectively (Additional file 14: Table S9, Additional file 15: Table S10, Additional file 16: Table S11, Additional file 17: Table S12, Additional file 18: Table S13 and Additional file 19: Table S14).
Analysis of the LSC, SSC, and IR border regions
Contraction and expansion of the IR regions may have caused the differences in cp genome size in plant groups [49]. By comparing the IR boundary characteristics of P. chinensis, P. chinensis var. kissii, P. cernua f. plumbea, P. dahurica, P. turczaninovii, P. cernua, A. carmichaelii and C. chinensis, we found that the length of IR region of the eight species ranged from 26,193 to 31,410 bp, while that of the six Pulsatilla species had a relatively narrower range between 31,118 and 31,410 bp (Fig. 3). Among the six species of Pulsatilla, the ndhF gene was located downstream of the IRa/LSC border, which was the same as Arctium lappa and Magnolia grandiflora [46, 50] but different from most angiosperms [51]. The ycf1 gene of the six Pulsatilla species was located across the IRb and SSC region boarder, which was the same as that observed in A. carmichaelii. The IRa/SSC and IRa/LSC boarders in A. carmichaelii and C. chinensis were located inside the ycf1 and rps19 genes. The rps19 genes of both species were 278 bp in length, whereas the length of ycf1 varied, which was not observed in the six Pulsatilla species. The trnN-GUU genes were in the IRa region in the six Pulsatilla species and the distance between trnN-GUU genes and the IRa/SSC border were not all the same in the six Pulsatilla species. Among six Pulsatilla species, the distances in P. chinensis and P. chinensis var. kissii were the smallest (1983 bp), while that in P. cernua was the largest (2212 bp). However, the trnN-GUU gene of C. chinensis was in the IRb region. These results showed that the IR regions of six Pulsatilla species were highly conserved, but were quite different from those of A. carmichaelii and C. chinensis, which are from different genera of the same family. The length of IR region is not constant during the evolution of cp genomes. Some studies have shown that the amplification of reverse repeats of the buckwheat cp genome may be a result of reverse transcription [52].
Fig. 3.

Comparison of the LSC, SSC, and IR border regions among the eight Ranunculaceae cp genomes. The number above gene features represents the distance between gene ends and the borders sites
Nucleotide diversity analysis
Both the coding and non-coding regions of cp genome sequences are highly variable, these sequences can thus be utilized to determine the phylogenetic relationship between species and genera. In total, 108 coding genes (Fig. 4a) and 105 non-coding genes (Fig. 4b) were found in the six Pulsatilla cp genomes. The Pi values of coding genes ranged between 0 and 0.0140351, while those of the non-coding regions were between 0 and 0.0575316. In both cases, most Pi values were zero (Additional file 20: Table S15). Overall, the non-coding regions were more conserved than the coding regions. The two most variable loci were rpl36 (Pi = 0.0140351) and ccsA-ndhD (Pi = 0.0575316), which are located in the LSC and SSC region, respectively. This is consistent with earlier results that the IR region is generally more conserved than the LSC and SSC regions [53, 54].
Fig. 4.

Comparative analysis of the nucleotide variability by Pi values of the six Pulsatilla species (a coding region, b non-coding region)
Phylogenetic analysis
The variation of angiosperm cp genomes is much higher than can be monitored with a single DNA barcode sequence. This sequence variability can be utilized for studying the phylogenetic relationships among closely related species, and is also valuable for species identification. The cp genome has also been suggested as a super barcode [55]. In our previous study, DNA barcodes were used to identify eight P. Miller species present in different medicinal materials. These earlier results showed that ITS2 and psbA-trnH sequences can identify these species accurately, while rbcL and matK sequences cannot [4, 56]. To determine the phylogenetic position of P. Miller in Ranunculaceae, a phylogenetic tree was constructed based on SNPs of the whole cp genomes of the six Pulsatilla species, 23 species from the other genera in Ranunculaceae, and two species of the outgroups (Fig. 5). In the resulting phylogenetic tree, 13 of 28 nodes exhibited bootstrap values of 100% and seven had bootstrap values of 90%. The six Pulsatilla species clustered closely together into one branch. This result is consistent with earlier phylogenetic analyses based on DNA molecular identification of several mixed Pulsatilla samples [57].
Fig. 5.
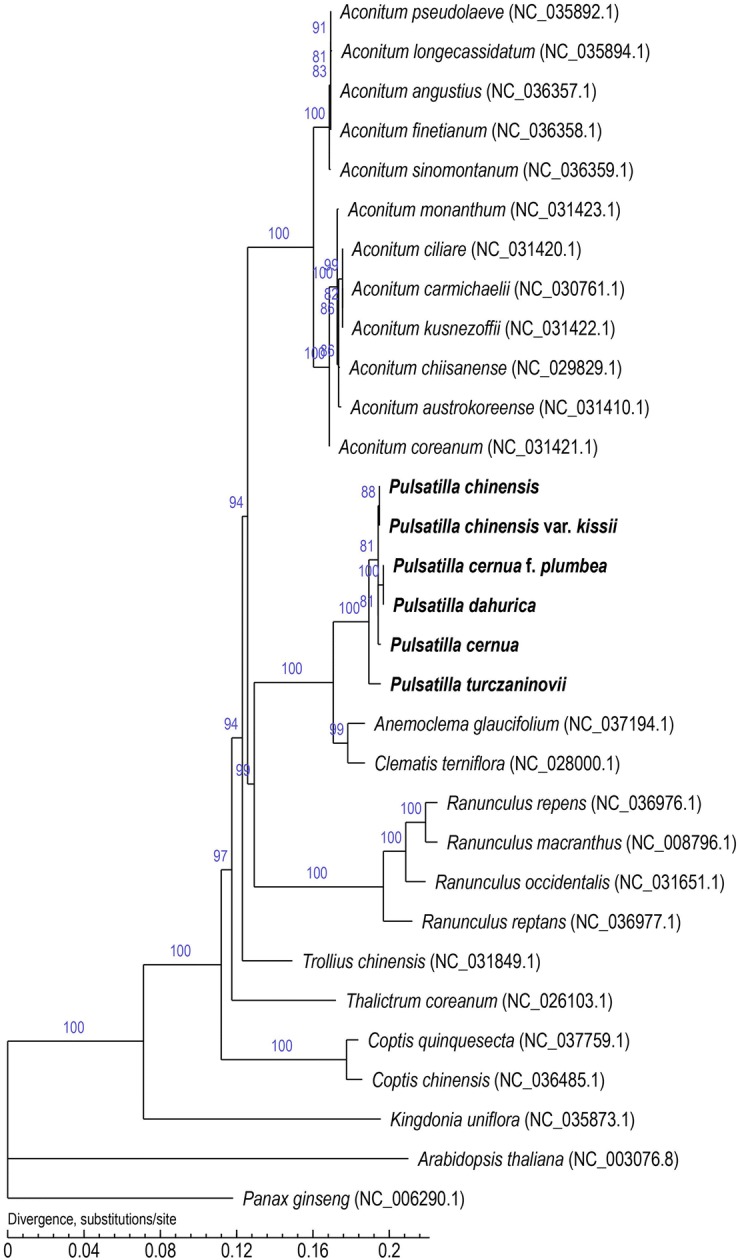
Molecular phylogenetic tree of 31 species based on whole cp genome SNPs. The tree was constructed using the maximum likelihood method in PhyML v3.0 with 1000 bootstrap replications
Pulsatilla chinensis and P. chinensis var. kissii clustered into one branch, implying that they are very closely related. P. chinensis var. kissii is also recorded as a variety of P. chinensis in both Flora Reipubicae Popularis Sinicae and Herbaceous Flora of Northeast China [2, 58], which was further validated by our clustering results. P. cernua f. plumbea and P. dahurica were clustered into one branch. In fact, P. cernua f. plumbea is considered a forma of P. cernua [59], but our results showed that they are relatively distant within the Pulsatilla branch. P. turczaninovii was relatively far from other Pulsatilla species. This indicates that P. turczaninovii and other Pulsatilla species were relatively distantly related. In reality, P. turczaninovii is also recorded as a separate species in some literature [2, 58, 60]. Among these six species, only P. chinensis is included in the Chinese Pharmacopoeia as a original plant, but other Pulsatilla members also clustered with P. chinensis in our tree. Whether other Pulsatilla species can replace P. chinensis requires further verification.
In our phylogenetic tree, Anemoclema glaucifolium and Clematis terniflora were clustered in one clade, and they both were closely related. At the same time, they both were clustered in one large clade with six species of Pulsatilla. This indicates that Pulsatilla species were more closely related to both than was previously thought. Ranunculus repens, R. macranthus, R. occidentalis and R. reptans gathered in one clade, with R. repens and R. macranthus clustered into one small branch. Trollius chinensis and Thalictrum coreanum were also closely related. C. quinquesecta and C. chinensis were gathered in one clade. In addition, Kingdonia uniflora was clustered in a single branch, which indicated that it was distantly related to other Ranunculaceae species. Aconitum L. were clustered into one branch, with A. pseudolaeve, A. longecassidatum, A. angustius, A. finetianum and A. sinomontanum gathered in one small clade. Previous studies on A. longecassidatum and A. pseudolaeve have shown that they have highly conserved cp genome structure [61], which fits with our results. A. monanthum, A. ciliare, A. carmichaelii, A. kusnezoffii, A. chiisanense, A. austrokoreense and A. coreanum gathered in one small clade, which was also consistent with previous research results [62]. Arabidopsis thaliana and Panax ginseng were located at the bottom of the phylogenetic tree, and clustered into one branch. The phylogenetic relationship of cp genomes in Ranunculaceae was analyzed by SNPs sequence, which indicated the cp genomes in Ranunculaceae was relatively conserved, and the six species of Pulsatilla were closely related.
Another phylogenetic tree of 31 species was constructed based on 51 shared protein-coding sequences, with protein similarity threshold set at > 40% (Fig. 6). Twelve of the 28 nodes had bootstrap values of 100% and nine nodes had bootstrap values ≥ 90%. The six Pulsatilla species formed one closely related clade. Overall, the results of the two methods of building trees were similar, with P. chinensis, P. chinensis var. kissii, P. cernua f. plumbea, and P. dahurica clustering into one small clade. P. turczaninovii was once again far away from other Pulsatilla. Twelve species of Aconitum were grouped into one branch. Two species of Coptis and four species of Ranunculus were also clustered into one branch. C. terniflora and A. glaucifolium composed one closely related clade. T. chinensis, T. coreanum and K. uniflora were clustered into different branches. P. ginseng and A. thaliana were also located at the bottom of the phylogenetic tree, and clustered into one branch. Ranunculaceae diverged early during eudicot evolution and is increasingly being used as a model for the study of plant evolution. Previous studies have shown that two radiation waves occurred early on in Ranunculaceae evolution, generating most extant tribes and genera [63]. Through the construction of two phylogenetic trees, the position of Pulsatilla in Ranunculaceae was elucidated, with implications for future evolutionary analysis.
Fig. 6.
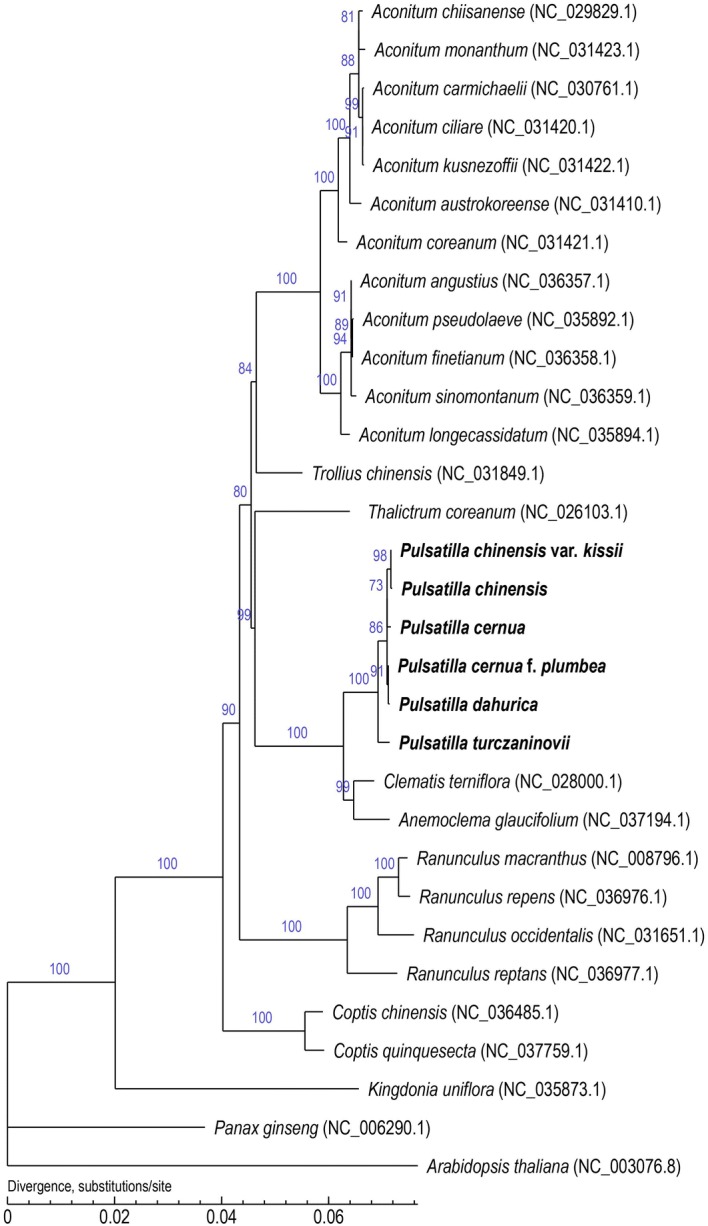
Molecular phylogenetic tree of 31 species based on 51 shared chloroplastic protein-coding. The tree was constructed via a maximum likelihood analysis using PhyML v3.0 with 1000 bootstrap replications
Conclusion
Analysis of the cp genome sequences of six Pulsatilla species showed that they had very similar genome sizes. Comparison to A. carmichaelii and C. chinensis in Ranunculaceae revealed differences in genome sizes. For example, the size of annotated cp genes in the six Pulsatilla was different, particularly rpl16 and rps12. These differences may be useful for marker development and phylogenetic analysis. In addition, P. dahurica contained six types of SSRs, while the other five Pulsatilla species had only five SSR types (with no hexanucleotides identified). Thirty-eight to 49 pairs of large repeats were found in the six Pulsatilla cp genomes, which are valuable for marker development and phylogenetic analysis. In addition, the size of IR regions was conserved among Pulsatilla species but different from that of A. carmichaelii and C. chinensis in Ranunculaceae. We also analyzed the nucleotide diversity of 108 genes and 105 non-coding regions, and rpl36 and ccsA-ndhD were the most variable, which are potentially suitable for marker design. The phylogenetic analysis was conducted based on SNPs of the whole cp genome and 51 shared chloroplastic protein-coding, and the position of six Pulsatilla species in Ranunculaceae was determined. These two phylogenetic trees will provide a reference for studying the evolutionary history of Ranunculaceae.
Supplementary information
Additional file 1: Figure S1. Circular gene map of P. chinensis var. kissii.
Additional file 2: Figure S2. Circular gene map of P. cernua f. plumbea.
Additional file 3: Figure S3. Circular gene map of P. dahurica.
Additional file 4: Figure S4. Circular gene map of P. turczaninovii.
Additional file 5: Figure S5. Circular gene map of P. cernua.
Additional file 6: Table S1. Location and length of intron-containing cp genes within the six Pulsatilla species.
Additional file 7: Table S2. Type and abundance of different SSRs in six species of Pulsatilla.
Additional file 8: Table S3. SSRs distribution of the P. chinensis cp genome.
Additional file 9: Table S4. SSRs distribution of the P. chinensis var. kissii cp genome.
Additional file 10: Table S5. SSRs distribution of the P. cernua f. plumbea cp genome.
Additional file 11: Table S6. SSRs distribution of the P. dahurica cp genome.
Additional file 12: Table S7. SSRs distribution of the P. turczaninovii cp genome.
Additional file 13: Table S8. SSRs distribution of the P. cernua cp genome.
Additional file 14: Table S9. Large repeats identified in the P. chinensis cp genome.
Additional file 15: Table S10. Large repeats identified in the P. chinensis var. kissii cp genome.
Additional file 16: Table S11. Large repeats identified in the P. cernua f. plumbea cp genome.
Additional file 17: Table S12. Large repeats identified in the P. dahurica cp genome.
Additional file 18: Table S13. Large repeats identified in the P. turczaninovii cp genome.
Additional file 19: Table S14. Large repeats identified in the P. cernua cp genome.
Additional file 20: Table S15. Pi values of the coding and no-coding regions in the six Pulsatilla cp genomes.
Acknowledgements
We thank the Shanghai BIOZERON Biotechnology Co., Ltd. for processing the raw sequencing data.
Abbreviations
- cp
chloroplast
- LSC
large single copy
- SSC
small single copy
- IR
inverted repeat
- tRNA
transfer RNA
- rRNA
ribosomal RNA
Authors’ contributions
Conceptualization, TZ; Methodology, TZ; Software, JW; Validation, LX and TK; Formal analysis, DZ; Investigation, SL; Resources, GB; Data curation, YX; Writing—original draft, TZ; Writing—review and editing, TZ; Visualization, YY; Supervision, ZZ; Project administration, LX; Funding acquisition, LX and TK. All authors read and approved the final manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (General Program, Grant Numbers 81874338, 81773852), the Major Expenditure Increase and Reduction Project at the Central Level “Capacity Building for Sustainable Utilization of Precious Traditional Chinese Medicine Resources” (Grant Number 2060302) and the Liaoning Province Education Department (Liaoning Higher School Outstanding Young Scholar Growth Plan, Grant Number LJQ2014101).
Availability of data and materials
All data generated or analyzed during the course of this study are included in this document or obtained from the appropriate author(s) at reasonable request.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Liang Xu, Email: 861364054@qq.com.
Tingguo Kang, Email: kangtingguo@163.com.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13020-019-0274-5.
References
- 1.Chinese Pharmacopoeia Commission . Chinese Pharmacopoeia. 342. Beijing: China Medical Science and Technology Press; 2015. p. 104. [Google Scholar]
- 2.Editorial Board of Flora Reipubicae Popularis Sinicae . Flora Reipubicae Popularis Sinicae. Beijing: Science Press; 1980. p. 62. [Google Scholar]
- 3.Ye WC, Ji NN, Zhao SX, Liu JL, Ye T, Makervey MA. Triterpenoids from Pulsatilla chinensis. Phytochemistry. 1996;42:799–802. doi: 10.1016/0031-9422(96)00043-X. [DOI] [PubMed] [Google Scholar]
- 4.Liang YM, Chen SY, Xu L, Wang B, Kang TG. Identification of plants and herbs of Pulsatilla genus based on ITS2 barcode. J Chin Med Mater. 2017;40:1547–1551. [Google Scholar]
- 5.Douglas SE. Plastid evolution: origins, diversity, trends. Curr Opin Genet Dev. 1998;8:655–661. doi: 10.1016/S0959-437X(98)80033-6. [DOI] [PubMed] [Google Scholar]
- 6.McFadden GI. Primary and secondary endosymbiosis and theorigin of plastids. J Phycol. 2001;37:951–959. doi: 10.1046/j.1529-8817.2001.01126.x. [DOI] [Google Scholar]
- 7.Palmer DJ. Comparative organization of chloroplast genomes. Annu Rev Genet. 1985;19:325–354. doi: 10.1146/annurev.ge.19.120185.001545. [DOI] [PubMed] [Google Scholar]
- 8.Wicke S, Schneeweiss GM, dePamphilis CW, Müller KF, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76:273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dyer TA. chloroplast genome: its nature and role in development. Top Photosynth. 1984;5:23–69. [Google Scholar]
- 10.Xin TY, Yao H, Luo K, Xiang L, Ma XC, Han JP, et al. Stability and accuracy of the identification of Notopterygii Rhizoma et Radix using the ITS/ITS2 barcodes. Acta Pharm Sin. 2012;47:1098–1105. [PubMed] [Google Scholar]
- 11.Luo K, Ma P, Yao H, Xin TY, Hu Y, Zheng SH, et al. Identification of gentianae macrophyllae radix using the ITS2 barcodes. Acta Pharm Sin. 2012;47:1710–1717. [PubMed] [Google Scholar]
- 12.Li YG, Xu WQ, Zou WT, Jiang DY, Liu XH. Complete chloroplast genome sequences of two endangered Phoebe (Lauraceae) species. Bot Stud. 2017;58:37. doi: 10.1186/s40529-017-0192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang Y, Li CL, Ma C, Wu NH. Chloroplast DNA and its application to plant systematic studies. Chin Bull Bot. 1994;11:11–25. [Google Scholar]
- 14.Huang H, Shi C, Liu Y, Mao SY, Gao LZ. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol Biol. 2014;14:151. doi: 10.1186/1471-2148-14-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao YB, Yin JL, Guo HY, Zhang YY, Xiao W, Sun C, et al. The complete chloroplast genome provides insight into the evolution and polymorphism of Panax ginseng. Front Plant Sci. 2015;5:696. doi: 10.3389/fpls.2014.00696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mcpherson H, Van der Merwe M, Delaney SK, Edwards MA, Henry RJ, Malntosh E, et al. Capturing chloroplast variation for molecular ecology studies: a simple next generation sequencing approach applied to a rainforest tree. BMC Ecol. 2013;13:8. doi: 10.1186/1472-6785-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.BorgstrÖm E, Lundin S, Lundeberg J. Large scale library generation for high throughput sequencing. PLoS ONE. 2011;6:e19119. doi: 10.1371/journal.pone.0019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cronn R, Liston A, Parks M, Gernandt DS, Shen RK, Mockler T. Multiplex sequencing of plant chloroplast genomes using Solexa sequencing-by-synthesis technology. Nucleic Acids Res. 2008;36:e122. doi: 10.1093/nar/gkn502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo RB, Liu BH, Xie YL, Li ZY, Huang WH, Yuan ZY, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1:18. doi: 10.1186/2047-217X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20:3252–3255. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- 21.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 22.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M. A database for post-genome analysis. Trends Genet. 1997;13:375. doi: 10.1016/S0168-9525(97)01223-7. [DOI] [PubMed] [Google Scholar]
- 24.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- 26.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, et al. The COG database: an updated version includes eukaryotes. BMC Bioinf. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Magrane M, Consortium U UniProt Knowledgebase: a hub of integrated protein data. Databases (Oxford). 2011;2011:bar009. doi: 10.1093/database/bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lohse M, Drechsel O, Bock R. Organellar genome DRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52:267–274. doi: 10.1007/s00294-007-0161-y. [DOI] [PubMed] [Google Scholar]
- 30.Mayer C, Leese F, Tollrian R. Genome-wide analysis of tandem repeats in Daphnia pulex-a comparative approach. BMC Genome. 2010;11:277. doi: 10.1186/1471-2164-11-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. RePuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29:4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, et al. VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000;16:1046–1047. doi: 10.1093/bioinformatics/16.11.1046. [DOI] [PubMed] [Google Scholar]
- 33.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 34.Zhao JT, Xu Y, Xi LJ, Yang JW, Chen HW, Zhang J. Characterization of the chloroplast genome sequence of Acer miaotaiense: comparative and phylogenetic analyses. Molecules. 2018;23:1740. doi: 10.3390/molecules23071740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mu XP, Wang PF, Du JJ, Gao YG, Zhang JC. The chloroplast genome of Cerasus humilis: genomic characterization and phylogenetic analysis. PLoS ONE. 2018;13:e0196473. doi: 10.1371/journal.pone.0196473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Li Y, Yang HY, Zhou BY. Chloroplast genome of the folk medicine and vegetable plant Talinum paniculatum (Jacq.) Gaertn.: gene organization, comparative and phylogenetic analysis. Molecules. 2018;23:857. doi: 10.3390/molecules23040857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gu CH, Dong B, Xu L, Tembrock LR, Zheng SY, Wu ZQ. The complete chloroplast genome of Heimia myrtifolia and comparative analysis within myrtales. Molecules. 2018;23:846. doi: 10.3390/molecules23040846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu ML, Li Q, Xu J, Li XW. Complete chloroplast genome of the medicinal plant Amomum compactum: gene organization, comparative analysis, and phylogenetic relationships within Zingiberales. Chin Med. 2018;13:10. doi: 10.1186/s13020-018-0164-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cui L, Veeraraghavan N, Richter A, Wall K, Jansen RK, Leebens-Mack J, et al. ChloroplastDB: the chloroplast genome database. Nucleic Acids Res. 2006;34:D692–D696. doi: 10.1093/nar/gkj055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hansen DR, Dastidar SG, Cai ZQ, Penafior C, Kuehl JV, Boore JL, et al. Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early diverging angiosperms: buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae) Mol Phylogenet Evol. 2007;45:547–563. doi: 10.1016/j.ympev.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 41.Jiao Y, Jia HM, Li XW, Chai ML, Jia HJ, Chen Z, et al. Development of simple sequence repeat (SSR) markers from a genome survey of Chinese bayberry (Myrica rubra) BMC Genomics. 2012;13:201. doi: 10.1186/1471-2164-13-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akkaya MS, Shoemaker RC, Specht JE, Bhagwat AA, Cregan PB. Integration of simple sequence repeat DNA markers into a soybean linkage map. Crop Sci. 1995;35:1439–1445. doi: 10.2135/cropsci1995.0011183X003500050030x. [DOI] [Google Scholar]
- 43.Bindler G, van der Hoeven R, Gunduz I, Plieske J, Ganal M, Rossi L, et al. A microsatellite marker based linkage map of tobacco. Theor Appl Genet. 2007;114:341–349. doi: 10.1007/s00122-006-0437-5. [DOI] [PubMed] [Google Scholar]
- 44.Rajput SG, Santra DK. Evaluation of genetic diversity of proso millet germplasm available in the United States using simple-sequence repeat markers. Crop Sci. 2016;56:2401–2409. doi: 10.2135/cropsci2015.10.0644. [DOI] [Google Scholar]
- 45.Li LJ, Yang KC, Pan GT, Rong TZ. Genetic diversity of maize populations developed by two kinds of recurrent selection methods investigated with SSR markers. Agr Sci China. 2008;7:1037–1045. doi: 10.1016/S1671-2927(08)60144-3. [DOI] [Google Scholar]
- 46.Xing YP, Xu L, Chen SY, Liang YM, Wang JH, Liu CS, et al. Comparative analysis of complete chloroplast genomes sequences of Arctium lappa and A. tomentosum. Biol Plantarum. 2019;63:565–574. [Google Scholar]
- 47.Yang Y, Dang YY, Li Q, Lu JJ, Li XW, Wang YT. Complete chloroplast genome sequence of poisonous and medicinal plant Datura stramonium: organizations and implications for genetic engineering. PLoS ONE. 2014;9:e110656. doi: 10.1371/journal.pone.0110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duitama J, Zablotskaya A, Gemayel R, Jansen A, Belet S, Vermeesch JR, et al. Large-scale analysis of tandem repeat variability in the human genome. Nucleic Acids Res. 2014;42:5728–5741. doi: 10.1093/nar/gku212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim KJ, Lee HL. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004;11:247–261. doi: 10.1093/dnares/11.4.247. [DOI] [PubMed] [Google Scholar]
- 50.Li XW, Gao HH, Wang YT, Song JY, Henry R, Wu HZ, et al. Complete chloroplast genome sequence of Magnolia grandiflora and comparative analysis with related species. Sci China life Sci. 2013;56:189–198. doi: 10.1007/s11427-012-4430-8. [DOI] [PubMed] [Google Scholar]
- 51.Raubeson LA, Peery R, Chumley TW, Dziubek C, Foursade HM, Boore JL, et al. ComParative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics. 2007;8:174. doi: 10.1186/1471-2164-8-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aii J, Kishima Y, Mikami T, Adachi T. Expansion of the IR in the chloroplast genomes of buckwheat species is due to incorporation of an SSC sequence that could be mediated by an inversion. Curr Genet. 1997;31:276–279. doi: 10.1007/s002940050206. [DOI] [PubMed] [Google Scholar]
- 53.Hong SY, Cheon KS, Yoo KO, Lee HO, Cho KS, Suh JT, et al. Complete chloroplast genome sequences and comparative analysis of Chenopodium quinoa and C. album. Front Plant Sci. 2017;8:1696. doi: 10.3389/fpls.2017.01696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu LX, Wang YW, He PZ, Li P, Lee J, Soltis DE, et al. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genomics. 2018;19:235. doi: 10.1186/s12864-018-4633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li XW, Yang Y, Henry RJ, Rossetto M, Wang YT, Chen SL. Plant DNA barcoding: from gene to genome. Biol Rev. 2015;90:157–166. doi: 10.1111/brv.12104. [DOI] [PubMed] [Google Scholar]
- 56.Liang YM, Xu L, Chen SY, Wang JH, Wang B, Kang TG. Classification of Pulsatilla Adans and molecular identification of DNA barcodes based on ITS2 sequence in Liaoning Province. Chin J ETMF. 2018;24:36–42. [Google Scholar]
- 57.Zhang TT, Liang YM, Xu L, Yang YY, Xing YP, Liu T, et al. Study on DNA molecular identification of mix samples of five species of Baitouweng medicinal materials based on high-throughput sequencing technology. Acta Pharm Sin. 2018;53:1918–1923. [Google Scholar]
- 58.Liaoning Forestry Soil Research Institute . Herbaceous flora of Northeast China. Beijing: Science Press; 1975. p. 162. [Google Scholar]
- 59.Jin JX, Zhao YT. A new form of genus pulsatilla from northeast China. Bull Bot Res. 1989;9:69–70. [Google Scholar]
- 60.Editorial Board of Flora of China . Flora of China. Beijing: Science Press; 2001. p. 332. [Google Scholar]
- 61.Park I, Yang SY, Choi G, Kim WJ, Moon BC. The complete chloroplast genome sequences of Aconitum pseudolaeve and Aconitum longecassidatum, and development of molecular markers for distinguishing species in the Aconitum Subgenus Lycoctonum. Molecules. 2017;22:2012. doi: 10.3390/molecules22112012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Choi JE, Kim JB, Lim CE, Yu HJ, Mun JH. The complete chloroplast genome of Aconitum austrokoreense Koidz. (Ranunculaceae), an endangered endemic species in Korea. Mitochondrial DNA Part B. 2016;1:688–689. doi: 10.1080/23802359.2016.1219644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhai W, Duan XS, Zhang R, Guo CC, Li L, Xu GX, et al. Chloroplast genomic data provide new and robust insights into the phylogeny and evolution of the Ranunculaceae. Mol Phylogenet Evol. 2019;135:12–21. doi: 10.1016/j.ympev.2019.02.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. Circular gene map of P. chinensis var. kissii.
Additional file 2: Figure S2. Circular gene map of P. cernua f. plumbea.
Additional file 3: Figure S3. Circular gene map of P. dahurica.
Additional file 4: Figure S4. Circular gene map of P. turczaninovii.
Additional file 5: Figure S5. Circular gene map of P. cernua.
Additional file 6: Table S1. Location and length of intron-containing cp genes within the six Pulsatilla species.
Additional file 7: Table S2. Type and abundance of different SSRs in six species of Pulsatilla.
Additional file 8: Table S3. SSRs distribution of the P. chinensis cp genome.
Additional file 9: Table S4. SSRs distribution of the P. chinensis var. kissii cp genome.
Additional file 10: Table S5. SSRs distribution of the P. cernua f. plumbea cp genome.
Additional file 11: Table S6. SSRs distribution of the P. dahurica cp genome.
Additional file 12: Table S7. SSRs distribution of the P. turczaninovii cp genome.
Additional file 13: Table S8. SSRs distribution of the P. cernua cp genome.
Additional file 14: Table S9. Large repeats identified in the P. chinensis cp genome.
Additional file 15: Table S10. Large repeats identified in the P. chinensis var. kissii cp genome.
Additional file 16: Table S11. Large repeats identified in the P. cernua f. plumbea cp genome.
Additional file 17: Table S12. Large repeats identified in the P. dahurica cp genome.
Additional file 18: Table S13. Large repeats identified in the P. turczaninovii cp genome.
Additional file 19: Table S14. Large repeats identified in the P. cernua cp genome.
Additional file 20: Table S15. Pi values of the coding and no-coding regions in the six Pulsatilla cp genomes.
Data Availability Statement
All data generated or analyzed during the course of this study are included in this document or obtained from the appropriate author(s) at reasonable request.