

Abstract
Genetic variation influences how the genome is interpreted in individuals and in mouse strains used to model immune responses. We developed approaches to utilize next-generation sequencing datasets to identify sequence variation in genes and enhancer elements in congenic and backcross mouse models. We defined genetic variation in the widely used B6-CD45.2 and B6.SJL-CD45.1 congenic model, identifying substantial differences in SJL genetic content retained in B6.SJL-CD45.1 strains based on the vendor source of the mice. Genes encoding PD-1, CD62L, Bcl-2, cathepsin E and Cxcr4 were within SJL genetic content in at least one vendor source of B6.SJL-CD45.1 mice. SJL genetic content affected enhancer elements, gene regulation, protein expression, and amino acid content in CD4+ T helper 1 cells, and mice infected with influenza showed reduced expression of Cxcr4 on B6.SJL-CD45.1 T follicular helper cells. These findings provide information on experimental variables and aid in creating approaches that account for genetic variables.
Graphical Abstract
eTOC blurb
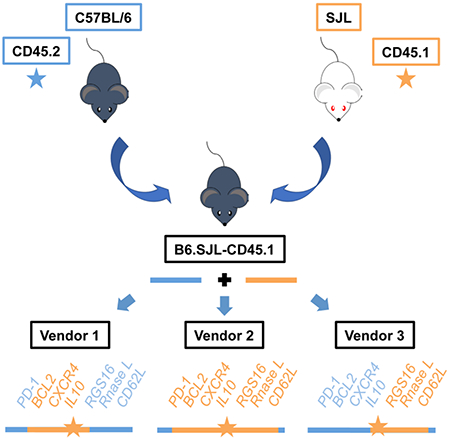
Congenic and backcrossed mouse models retain genetic content from their original parent strains, but this genetic variable often remains unknown. Chisolm et al. present alternative processing strategies for next-generation sequencing datasets to define genetic variation, and find substantial differences in SJL genetic content retained in B6.SJL-CD45.1 congenic mice by vendor, encompassing immune-related genes encoding BCL2, CD62L, CXCR4, RNaseL and PD-1.
Introduction
Genetic variation contributes to the regulation of gene expression, and a great deal of research has been performed to define genetic variation associated with diseases (Link et al., 2018; Timpson et al., 2018). In addition, inbred mouse strains have differences in immune responses and disease susceptibilities that are related to genetic variation between strains (Tacchini-Cottier et al., 2012). Researchers have also used genetic variation at specific loci in mice as an experimental tool to identify cells in adoptive transfer and mixed bone marrow chimera experiments. For example, genetic variation in the Ptprc locus (encodes CD45, also named Ly5) was identified between C57BL/6 and SJL mice (Shen et al., 1985). C57BL/6 mice have the Ptprcb allele (encodes CD45.2), while SJL mice have the Ptprca allele (encodes CD45.1). The encoded variation between CD45.1 and CD45.2 does not impact its function, but it does create unique epitopes that can be recognized by monoclonal antibodies. To use the variation in CD45 as an experimental tool, the Ptprca locus from SJL mice was backcrossed to C57BL/6 mice to create the B6.SJL-PtprcaPepcb/Boy strain (referred to here as B6.SJL-CD45.1)(Shen et al., 1985). C57BL/6 (referred to here as B6-CD45.2) and B6.SJL-CD45.1 mice are now one of the most widely used tools in adoptive transfer and mixed bone marrow chimera experiments because the transferred cells can be identified using monoclonal antibodies specific to CD45.2 or CD45.1 (Levine et al., 2017; Taylor et al., 2012; Xu et al., 2018).
The assumption in using this as an experimental tool is that B6-CD45.2 and B6.SJL-CD45.1 cells are genetically and phenotypically the same (“wild-type B6”), with the exception of CD45 epitope recognition. However, several studies have uncovered differences in the phenotype of immune cells from B6-CD45.2 and B6.SJL-CD45.1 mice. For example, researchers have observed that hematopoietic stem cell (HSC) reconstitution is less efficient with bone marrow isolated from B6.SJL-CD45.1 mice compared to B6-CD45.2 mice (Basu et al., 2013; Jafri et al., 2017; Mercier et al., 2016; Waterstrat et al., 2010). The HSC defect was absent in a gene targeted knock-in mouse created to genetically engineer the CD45.1 epitope (Mercier et al., 2016), indicating that genetic variation outside of the Ptprc locus between B6-CD45.2 and B6.SJL-CD45.1 mice impacts HSC reconstitution. Several studies have also reported differences in immune cell development or function, including in B cells as well as γδT cells, in comparisons between cells of B6-CD45.2 and B6.SJL-CD45.1 origin (Basu et al., 2013; Gray et al., 2013; Jang et al., 2018; Mercier et al., 2016). Although some differences might involve spontaneous mutations, together this highlights the undervalued importance of genetic variation in interpreting experiments comparing even highly related mouse strains.
The issue of defining genetic variation is not limited to B6-CD45 congenic mice because genomic intervals from the original parent strains are retained in all backcrossed mouse models. For instance, most transgenic and knockout mice have been created using embryonic stem cells originating from 129 mouse strains (Eberl et al., 2004; Rubtsov et al., 2008). This means 129 genetic content will remain in proximity to genetically manipulated regions, and at present, these genomic intervals are not typically identified. Thus, genetic variation might be an unrecognized variable in comparisons with the designated “wild-type” controls (Vanden Berghe et al., 2015). Indeed, independently generated knockout mice sometimes have yielded different experimental results, indicating the presence of an unidentified genetic variable (Mahajan et al., 2016; Purtha et al., 2012; Wong et al., 2018). This highlights the need to develop strategies to assess genetic variation.
In this study, we present strategies to analyze next-generation sequencing datasets to define genetic variation in B6 congenic and transgenic strains. Using these approaches, we identified differences in the extent of SJL genetic content in B6.SJL-CD45.1 strains from different vendor sources. This included regions encompassing genes of immunological importance in immune cell homing, T cell exhaustion, T and B cell differentiation, and innate immune responses. Our data also indicated that SJL genetic content retained in B6.SJL-CD45.1 congenic mice impacts enhancer elements, gene regulation, protein expression and amino acid content, highlighting the importance of defining genetic variation in mouse models.
Results
Microbiota-independent effects in T cells from conventional versus germ-free mice
Our initial goal was to identify whether the microbiota influences the enhancer landscape in T cells because the microbiota produces short chain fatty acids such as butyrate and acetate, which can play roles in modulating histone acetylation (Chisolm and Weinmann, 2018). To examine this question, we isolated CD4+ or CD8+ T cells from either conventional or germ-free mice. In all experiments described in this study, the cells were polarized in type 1 conditions and will be referred to as CD4+ T helper 1 (Th1) or CD8+ T cytotoxic 1 (Tc1) cells. We then performed histone 3 lysine 27 acetylation (H3K27Ac) chromatin immunoprecipitation sequencing (ChIP-seq) experiments to define the putative enhancer landscape in the cells that had developed in the presence or absence of a microbiota. We identified several H3K27Ac ChIP-seq peaks in CD4+ Th1 or CD8+ Tc1 cells that were differentially enriched in cells isolated from conventional as compared to germ-free mice (Fig. 1A). This included peaks in proximity to Sox13, Serpinb10, Atp2b4, and Kif14 (Figs. 1A, S1A). Based on these results, and a preliminary RNA-seq experiment, we then performed quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analyses on RNA from CD4+ Th1 or CD8+ Tc1 cells that were isolated from conventional or germ-free mice. We detected differential expression for several genes between cells isolated from conventional as compared to germ-free mice (Fig. S1B–C). However, we were unable to rescue the changes in gene expression when we performed co-housing experiments to reconstitute the microbiota in germ-free mice (Fig. S1D), suggesting the differences in CD4+ and CD8+ T cells isolated from conventional versus germ-free mice might not be related to the microbiota.
Figure 1. Genetic variation between B6-CD45.2 and B6.SJL-CD45.1 mice impacts H3K27Ac in T cells.

(A, C) H3K27Ac ChIP-seq analyses were performed on (A, C) CD4+ or (A) CD8+ T cells polarized in Th1 conditions. Cells were isolated from (A) conventional (B6-CD45.2) or germ-free (B6.SJL-CD45.1) mice, or (C) B6-CD45.2, B6.SJL-CD45.1, or SJL mice from Jax. (B) Schematic representation of chromosome 1, with the red box indicating a region identified to contain SJL genetic content (Waterstrat et al., 2010). (A, C) UCSC genome browser tracks with genes (below) and cell types or genetic backgrounds (left) indicated. (A, C) Representative of at least 2 independent biological replicates. See also Fig. S1.
Genetic variation between B6-CD45.2 and B6.SJL-CD45.1 mice impacts enhancers in T cells
We first reexamined the data from the conventional and germ-free experiments, and noticed that many changes in H3K27Ac and gene expression were on chromosome 1 in proximity to the Ptprc locus, which encodes CD45. In our experiments, the conventional mice were on the B6-CD45.2 background while the germ-free mice were on the B6.SJL-CD45.1 background. A previous publication using single nucleotide polymorphism (SNP) mapping indicated that SJL genetic content was retained in an approximately 40 Mb interval surrounding the Ptprc locus on chromosome 1 in B6.SJL-CD45.1 mice (Fig. 1B)(Waterstrat et al., 2010). Therefore, we next assessed whether the genetic background of the closely related B6 congenic mice impacted H3K27Ac enhancer peaks and gene expression. To test this, we first performed H3K27Ac ChIP-seq experiments on CD4+ Th1 cells isolated from conventionally housed mice that were on the B6-CD45.2, B6.SJL-CD45.1, or SJL genetic background. Similar to the data in Fig. 1A, we identified differences in H3K27Ac ChIP-seq peaks in cells isolated from B6-CD45.2 mice in comparison to B6.SJL-CD45.1 mice (Fig. 1C). In these regions, H3K27Ac peaks were similar between the SJL and B6.SJL-CD45.1 genetic backgrounds. Together, the data indicated there were differences in the H3K27Ac enhancer landscape in the genomic interval surrounding the Ptprc locus in CD4+ Th1 cells between B6-CD45 congenic mice, and this was likely related to SJL genetic content remaining in B6.SJL-CD45.1 mice.
Cathepsin E expression is impaired in T cells isolated from B6-CD45.2 mice
We next wanted to define the genetic elements that impact immunologically important genes in the chromosome 1 interval surrounding Ptprc. We first focused on the Ctse locus (encodes cathepsin E) because it was one of the most differentially expressed genes in RNA-seq experiments comparing CD4+ Th1 cells isolated from B6-CD45.2 and B6.SJL-CD45.1 mice. Consistent with this, cathepsin E protein was expressed in CD4+ Th1 cells isolated from B6.SJL-CD45.1 and SJL mice, but was not detected in cells isolated from B6-CD45.2 mice (Fig. 2A). We observed similar results in CD8+ Tc1 cells (Fig. 2B). These data suggested that either SJL genetic content in B6.SJL-CD45.1 mice promoted cathepsin E expression, or alternatively, a genetic element in B6-CD45.2 mice inhibited its expression.
Figure 2. Viral integration impacts cathepsin E expression.

(A, D) CD4+ or (B) CD8+ T cells were isolated from B6-CD45.2, B6.SJL-CD45.1, or SJL mice from Jax and cells were polarized in Th1 conditions. (A, B) Western blot analysis of cathepsin E protein expression with STAT4 as a control. (C) Schematic of Ctse locus displaying the location of an alternative Ctse annotation (Ctse-LTR) and a viral LTR integration. (D) qRT-PCR analysis of Ctse and Ctse-LTR transcripts. Transcripts were normalized to Rps18 as a control and then expression levels were compared relative to B6-CD45.2 genotype cells. (E) PCR analysis of DNA isolated from B6-CD45.2, B6.SJL-CD45.1, or SJL mice using a common forward primer, with either a reverse primer inside (blue arrow in (C); LTR present) or outside (purple arrow in (C); LTR absent) of the LTR. (F) Ctse expression in CD4+ T cells isolated from mouse strains indicated to the left of the graph as defined in ImmGen datasets (GSE60337). Asterisks indicate strains that have the viral integration in the first intron of Ctse. (A, B, D, E) Data are compiled from, or representative of, at least (E) 2 or (A, B, D) 3 independent biological replicates. (D, F) Error bars represent standard error of the mean (SEM) and (D) p-values were calculated by an unpaired Student’s t test (***≤ 0.001 and ** ≤ 0.01). See also Fig. S2.
A viral integration in the B6-CD45.2 genome disrupts Ctse transcript expression
We next used the UCSC genome browser to interrogate the genome annotations surrounding the Ctse locus in the mm10 genome, which is derived from C57BL/6 mice, to start to define genetic element(s) that impact cathepsin E expression. We noticed an alternative annotation for the Ctse transcript that contained a unique exon terminating it in the first intron of the typical Ctse gene structure (Fig. 2C). We designed qRT-PCR primers to this alternative transcript (referred to here as Ctse-LTR), and compared its expression to the typical Ctse transcript in CD4+ Th1 cells isolated from B6-CD45.2, B6.SJL-CD45.1, or SJL mice. The Ctse-LTR transcript was expressed in CD4+ Th1 cells isolated from B6-CD45.2 mice whereas it was not detected in cells isolated from B6.SJL-CD45.1 or SJL mice (Fig. 2D). This was in contrast to the expression pattern for the typical Ctse transcript, which was more highly expressed in CD4+ Th1 cells isolated from B6.SJL-CD45.1 and SJL mice in comparison to B6-CD45.2 mice (Fig. 2D; Ctse). We observed similar results in CD8+ Tc1 cells (Fig. S2A). The expression of the typical Ctse transcript corresponded with cathepsin E protein expression, confirming it was the natural transcript that encoded the known protein product (Fig. 2A and 2D).
Genome annotations indicate the unique exon in the Ctse-LTR transcript is not conserved between species, and instead is located within repetitive sequence. We examined the annotations associated with repetitive elements in the mm10 genome and found that the unique exon overlapped with the integration of a viral long terminal repeat (LTR) approximately 7.5 kb in length (Fig. 2C). We hypothesized that the LTR integration disrupts the natural Ctse transcript when present in the genome. To start to test this, we used a PCR-based method because the SJL genome has not been sequenced. We designed a common forward primer in the conserved 5’ DNA sequence outside of the LTR. We then designed one reverse primer inside the LTR that would only form a product if the LTR was present (Fig. 2C; blue arrow), and another reverse primer in the 3’ sequence outside of the LTR that would only form an easily amplified product if the LTR was absent (Fig. 2C; purple arrow). The PCR analysis indicated the viral integration was present in DNA from B6-CD45.2 mice and was absent in DNA from B6.SJL-CD45.1 and SJL mice (Fig. 2E).
We next examined the Ctse gene structure in the mouse genomes with available sequence annotations in the Wellcome Sanger Institute database (Yalcin et al., 2011; Yalcin et al., 2012) to define whether the viral LTR was present or absent in different mouse strains. We then compared this information to the expression profile of Ctse in CD4+ T cells from the same mouse strains as defined by ImmGen consortium datasets. In all cases, Ctse expression was robust in CD4+ T cells from mouse strains that did not contain the viral LTR integration, whereas it was severely diminished in CD4+ T cells from mouse strains containing the integration (Fig. 2F). These data provided correlative support for the hypothesis that the viral integration disrupts the natural Ctse exon structure when present, and this genetic variation impacts cathepsin E expression between mouse strains.
Genetic variation between B6-CD45.2 and B6.SJL-CD45.1 mice impacts Cxcr4 expression
We noticed several sites of differential H3K27Ac enrichment in proximity to Cxcr4 in comparisons between CD4+ Th1 cells isolated from B6-CD45.2 and B6.SJL-CD45.1 mice (data not shown). Therefore, we hypothesized that genetic variation might impact Cxcr4 expression. To address this, we first assessed Cxcr4 protein expression. In a western blot analysis, Cxcr4 expression was diminished in CD4+ Th1 cells isolated from B6.SJL-CD45.1 compared to B6-CD45.2 mice (Fig. 3A). There was also diminished expression of Cxcr4 on the surface of CD4+ Th1 cells from B6.SJL-CD45.1 mice in flow cytometry experiments (Fig. 3B). We observed similar results in CD8+ Tc1 cells (Fig. 3A, 3B). These data suggested there was a defect in Cxcr4 expression in T cells isolated from B6.SJL-CD45.1 compared to B6-CD45.2 mice.
Figure 3. Genetic variation occurs in the Cxcr4 locus in B6.SJL-CD45.1 compared to B6.CD45.2 mice.

(A-F) CD4+ or (A, B) CD8+ T cells were isolated from B6-CD45.2, B6.SJL-CD45.1, or SJL mice from Jax and polarized in Th1 conditions. (A) Western analysis monitoring Cxcr4 protein expression, with STAT4 shown as a control. (B) Flow cytometry analyzing Cxcr4 cell surface expression. (C) qRT-PCR analyses with primer sets monitoring different regions of the Cxcr4 transcript. Data were processed as in Fig. 2D, and p-values were calculated with an unpaired Student’s t test (*** ≤ 0.001 and ** ≤ 0.01). (D) Primer sequence locations (color arrows) used in (C) and Sanger sequencing for a partial Cxcr4 transcript. (E) Graphs of normalized counts for RNA-seq datasets from B6-CD45.2 (dark blue), B6.SJL-CD45.1 (light blue) and SJL (orange) mice processed with default (solid bars) or stringent (hatched bars) parameters. DESeq2 performs a Benjamini-Hochberg test to calculate adjusted p-values (*** ≤ 0.001 and ** ≤ 0.01). (F) IGV browser display of RNA-seq alignment files from default parameters with nucleotide changes from the mm10 genome shown by color lines (red (T), blue (C), green (A), orange (G)). (A-F) Data are compiled from, or representative of, at least (D) 2, (A, E, F) 3, (C) 4, or (B) 6 independent biological replicates. See also Fig. S2.
There is sequence variation in the Cxcr4 locus in B6.SJL-CD45.1 mice
We next wanted to address the genetic elements that contribute to the differential regulation of Cxcr4 in the congenic backgrounds. RNA-seq experiments with CD4+ Th1 cells isolated from B6-CD45.2 or B6.SJL-CD45.1 mice suggested there was a modest difference in Cxcr4 gene expression between the two genotypes (1.4 fold-change). However, there was a substantial difference in the detection of Cxcr4 transcripts in a qRT-PCR analysis performed with RNA from CD4+ Th1 cells isolated from B6-CD45.2 compared to B6.SJL-CD45.1 mice (Fig. 3C; Cxcr4). We designed additional qRT-PCR primers throughout the Cxcr4 transcript to better understand the discrepancy in the results between the two assays (Fig. 3D). The ability to detect Cxcr4 transcripts was impacted by the location of the primers, with some primers detecting modest to no change in Cxcr4 transcripts, while other primers detected substantial changes in Cxcr4 transcripts in comparisons between CD4+ Th1 cells isolated from B6-CD45.2 and B6.SJL-CD45.1 mice (Fig. 3C). Similar results were observed in CD8+ Tc1 cells (Fig. S2C).
One possibility for the discrepancy in the results between the primer pairs is if different splice products are expressed in the B6 congenic backgrounds. We detected both the known Cxcr4 spliced transcript (Fig. S2B, upper band), and a second alternatively spliced transcript that had not previously been defined (Fig. S2B, lower band). The alternatively spliced transcript was expressed at similar levels in both B6 congenics, which meant it did not explain the discrepancy in Cxcr4 transcript detection with the different primer pairs. Another possibility for discrepancies in the detection of a transcript could be caused by genetic variation in the underlying sequence the primers directly anneal with, or significant changes in the genomic content being amplified. We sequenced a partial Cxcr4 transcript (Fig. S2B upper band) and found 8 nucleotide substitutions in the coding sequence for B6.SJL-CD45.1 compared to B6-CD45.2 mice (Fig. 3D, S2D). One substitution was within the original Cxcr4 primers that detected substantial changes in the transcript, while another substitution caused a nonsynonymous amino acid change in the Cxcr4 protein (V to I at amino acid 216). In the 3’ UTR, there were several variant nucleotides and an inserted AT repeat. The AT repetitive insertion was located within one of the amplicons that was severely diminished in the qRT-PCR analyses (Fig. 3C, 3-Cxcr4). These data indicated there was variation in the Cxcr4 transcript sequence in B6.SJL-CD45.1 as compared to B6-CD45.2 mice.
Approach to define genetic variation with RNA-seq datasets
We next wanted to develop an approach to broadly define genetic variation using RNA-seq datasets instead of Sanger sequencing individual regions. We also wanted this approach to utilize available informatics tools so it would be accessible. We reasoned that the standard RNA-seq processing alignment tools use default alignment parameters that allow for mismatched base pairs, which increases the number of sequencing reads that align to the genome to improve coverage. Additionally, most mouse next-generation sequencing alignment programs use the mm9 or mm10 reference genomes, which as indicated earlier, are derived from the C57BL/6 genome (i.e. B6-CD45.2). We hypothesized that using stringent alignment parameters (0 mismatches) would exclude genetic content that diverges from the B6-CD45.2 genome because reads with sequence variation would not be mapped to a location in the genome. Data from the stringent alignment criteria could then be compared to data from typical default alignment parameters, using programs such as DESeq2, as a method to identify genes containing significant regions with genetic variation (Fig. S3A). Alternatively, visualizing the default alignment files in the integrative genomics viewer (IGV) could identify genes with modest numbers of variant nucleotides, and allow for the underlying sequence variation relative to the B6-CD45.2 genome to be defined.
We tested these approaches with RNA-seq datasets generated from CD4+ Th1 cells isolated from B6-CD45.2, B6.SJL-CD45.1 or SJL mice. We first analyzed Cxcr4 to determine whether the approach could identify the genetic variation defined by Sanger sequencing (Fig. S2D). As a control, RNA-seq datasets from B6-CD45.2 mice processed with the stringent alignment criteria did not impact the detection of Cxcr4 in comparison to the same datasets processed with default alignment parameters (Fig. 3E, S2E). In contrast, the detection of Cxcr4 transcripts was diminished when comparing the stringent and default alignment parameters for RNA-seq datasets from B6.SJL-CD45.1 or SJL mice (Fig. 3E, S2E). The regions with reduced alignments using the stringent criteria corresponded to the areas of Cxcr4 with genetic variation defined by Sanger sequencing.
We next visualized in IGV the RNA-seq alignment files created with default parameters to define the underlying sequence variation within the Cxcr4 transcript in the B6.SJL-CD45.1 and SJL backgrounds compared to B6-CD45.2. The nucleotide variants in the coding sequence correlated with the Sanger sequencing data (Fig. 3F and S2D). In the 5’UTR, the data indicated there were a nucleotide deletion and a change in the neighboring nucleotide. To confirm this, we designed qRT-PCR primers to this region, anchoring one primer within the variant nucleotide sequence. The ability of these primers to detect Cxcr4 transcripts was diminished in CD4+ Th1 cells isolated from B6.SJL-CD45.1 in comparison to B6-CD45.2 mice (Fig. S2F). This was similar to the qRT-PCR data in which we fortuitously designed primers in regions that contained sequence variation (Fig. 3C). These data provided proof-of-concept that regions with sequence variation can be detected with the alternative RNA-seq processing approaches.
SJL genomic interval in B6.SJL-CD45.1 mice varies substantially by vendor source
We next used these approaches to define regions with sequence variation in B6.SJL-CD45.1 mice. Several studies have identified subtle genetic variation in mouse strains obtained from different colonies or vendor sources, and this can impact gene expression and function (Gray et al., 2013; Purtha et al., 2012). Therefore, we performed experiments with mice obtained from the Jackson Laboratory (Jax), Charles River (CR), and Taconic (Tac). We also examined SJL mice to define whether any genetic variation in B6.SJL-CD45.1 mice was likely from retaining SJL genetic content or rather was due to spontaneous mutations. We generated RNA-seq datasets for CD4+ Th1 cells isolated from the different mice and vendor sources, and then processed the datasets with either the default or stringent alignment parameters, and compared the two parameters using DESeq2. We also visualized the default alignment files in IGV to precisely define regions of SJL genetic content.
The analysis comparing default and stringent alignment parameters detected an extended 37 Mb genomic interval with sequence variation on chromosome 1 in B6.SJL-CD45.1 mice from Jax (Fig. 4A, 4B, S3B, S4). This interval was similar in location to the previously defined interval with SJL SNPs (Waterstrat et al., 2010), and it included genes such as Cxcr4, Ctse, Ptprv, and Bcl2. In contrast, the analyses examining B6.SJL-CD45.1 mice from Taconic detected sequence variation starting at the Ptprc locus on the 5’ end of chromosome 1 and extending to the Tiprl locus on the 3’ end (Fig. 4B, S4, S5). This genomic interval was approximately 27 Mb in length, and it included genes such as Sell, Rgs16, Rnasel, and Glul. Finally, the analysis for B6.SJL-CD45.1 mice from CR detected three distinct genomic intervals with sequence variation on chromosomes 1, 7, and 19 (Fig. 4B, S4, S5). The intervals with SJL genetic content on chromosomes 1 and 19 spanned approximately 72 Mb and 38 Mb in length, respectively. On chromosome 1, the interval included the regions with sequence variation in B6.SJL-CD45.1 mice from Jax and Tac, and extended in the 5’ direction to include the Pdcd1 (encodes PD1) and Mterf4 loci. We also identified loci with genetic variation in B6-CD45.2 mice as compared to the mm10 reference genome, which were commonly found in both B6-CD45.2 and B6.SJL-CD45.1 mice from the different vendors (Fig. 4A, 4B).
Figure 4. Substantial differences in SJL genetic content are present in B6.SJL-CD45.1 mice from different vendors.

(A, B) CD4+ T cells were isolated from B6-CD45.2, B6.SJL-CD45.1, and SJL mice from Jax (blue highlight), B6N-CD45.2 and B6.SJL-CD45.1 mice from Charles River (purple highlight), or B6.SJL-CD45.1 mice from Taconic (green highlight) and cells were polarized in Th1 conditions. RNA-seq datasets were processed with default (mismatches; blue, purple, or green lettering) or stringent (0 mismatches; red lettering) parameters. Shown are heatmaps representing the z scores for the normalized counts from the DESeq2 analyses of the indicated samples from (A) Jax or (B) CR, Tac and Jax mice. See methods for heatmap gene selection. Datasets from at least three independent biological replicates are shown. (C) Schematic of chromosomes 1, 7, and 19. Blue (Jax), purple (CR), and green (Tac) boxes indicate the regions defined to contain SJL genetic content in B6.SJL-CD45.1 mice from each vendor. White dashed line represents a region detected to have B6 content in H3K27Ac analysis for genetic variation. See also Figs. S3–5.
We next analyzed RNA-seq datasets generated from SJL mice. The data indicated genes with genetic variation specifically in the B6.SJL-CD45.1 genomes from the different vendors also had genetic variation in SJL mice (4A, S3B, S4). We also visualized the alignment files for the B6-CD45.2, B6.SJL-CD45.1 and SJL datasets in the IGV browser (Fig. S4A, S5A). This analysis confirmed the sequences with variation unique to B6.SJL-CD45.1 mice were derived from sequence in the SJL genome indicating regions with sequence variation in B6.SJL-CD45.1 strains were caused by the retention of SJL genetic content. A summary of the regions retaining SJL genetic content in the B6.SJL-CD45.1 mice from different vendors is provided in Fig. 4C (Table S1, S2).
Genetic variation in B6.SJL-CD45.1 mice impacts protein content
We wanted to begin assessing the functional consequences for genetic variation in B6.SJL-CD45.1 mice from different vendors. As will be discussed, we found examples of differences in protein expression, amino acid content, gene regulation, and H3K27Ac enhancer elements between CD4+ Th1 cells isolated from B6-CD45.2 and B6.SJL-CD45.1 mice from the various vendors. In most cases, these changes correlated with the retention of SJL genetic content at the region of interest in each assay.
The Ctse locus was derived from SJL genetic content in B6.SJL-CD45.1 mice from Jax and CR, but was derived from B6 genetic content in Tac mice (Fig. 4C). Ctse transcript (Fig. S6C) and cathepsin E protein (Fig. 5A) expression corresponded with whether the locus was derived from SJL (no viral integration; Fig. S6A) or B6 (viral integration; Fig. S6B) genetic content in B6.SJL-CD45.1 mice from each vendor. There were also amino acid content changes caused by the sequence variation in B6.SJL-CD45.1 mice. For example, the Ptprv locus is derived from SJL genetic content in B6.SJL-CD45.1 mice from Jax and CR, whereas it is derived from B6 genetic content in B6.SJL-CD45.1 mice from Tac (Fig. S4A). There were premature stop codons at amino acid positions 66 and 962 in the B6 genome, whereas the SJL genome encoded through 1706 amino acids (Fig. S4A). RNAse L also had substantial amino acid content changes in the different B6 congenic backgrounds. The Rnasel locus was derived from SJL genetic content in B6.SJL-CD45.1 mice from CR and Tac, but was derived from B6 genetic content in B6.SJL-CD45.1 mice from Jax. SJL genetic content encoded 16 nonsynonymous amino acid changes in the RNAse L protein in comparison to the B6 genome (Fig. S4A).
Figure 5. SJL genetic content functionally impacts CD4+ Th1 cells from B6.SJL-CD45.1 mice.

(A-D) CD4+ T cells were isolated from B6-CD45.2 or B6.SJL-CD45.1 mice from Jax, CR, or Tac and polarized in Th1 conditions (vendor indicated in panels). (A) Western blot analysis of cathepsin E protein expression with STAT4 shown as a control. (B) IGV browser display of RNA-seq default alignment files as indicated. Partial Sell transcript is shown indicating nucleotide substitutions in the C-type lectin (red arrow) and Egf-like (blue arrow) domains. Color lines in transcript coverage denote nucleotide change from mm10 genome as indicated in Fig. 3F. (C) qRT-PCR of genes on chromosomes 1, 7, or 19 of CD4+ Th1 cells isolated from B6-CD45.2 or B6.SJL-CD45.1 mice from Jax (blue) or CR (purple) as indicated. Error bars represent SEM and p values were calculated with an unpaired Student’s t test (*** ≤ 0.001, ** ≤ 0.01, and * ≤ 0.05). (D) Heatmap of H3K27Ac ChIP-seq datasets from CD4+ Th1 cells isolated from B6-CD45.2, B6.SJL-CD45.1 or SJL mice from Jax or CR. H3K27Ac ChIP-seq data from B6.SJL-CD45.1 germ-free (GF) mice are also shown. See methods for heatmap peak selection. Data are compiled from, or representative of, at least (A, D) 2 or (B, C) 3 independent biological replicates. See also Fig. S6.
Sell (encodes CD62L) was derived from SJL genetic content in B6.SJL-CD45.1 mice from CR and Tac, but had B6 genetic content in Jax mice (Fig. 5B). There were several nucleotide substitutions in the SJL genetic content that encode for nonsynonymous amino acid changes in CD62L (I to T at amino acid 32 and A to E at amino acid 184) (Fig. 5B). Amino acid 184 is in the Egf-like domain of CD62L, which plays a role in the rolling speed of lymphocytes (Dwir et al., 2000; Phan et al., 2006). We performed Sanger sequencing to confirm B6.SJL-CD45.1 mice from CR contained the nucleotide from the SJL genome encoding a nonsynonymous amino acid substitution at position 32, whereas B6.SJL-CD45.1 mice from Jax contained the sequence found in the B6 genome (Fig. S6F). We also designed qRT-PCR primers to encompass two nucleotide substitutions between the SJL and B6 genomes, which included the sequence encoding the amino acid substitution at position 184. The primers that hybridize in this region were unable to detect the Sell transcript in CD4+ Th1 cells isolated from B6.SJL-CD45.1 mice from CR and Tac, whereas they efficiently detected Sell transcripts in cells isolated from B6-CD45.2 and B6.SJL-CD45.1 mice from Jax (Fig. S6G). Together these data confirmed the Sell locus was derived from SJL genetic content in B6.SJL-CD45.1 mice from CR and Tac, whereas Sell was derived from B6 genetic content in B6.SJL-CD45.1 mice from Jax.
Genetic variation in B6.SJL-CD45.1 mice influences gene regulation
We next wanted to determine if there were any effects on gene expression related to the differences in SJL and B6 genetic content in B6.SJL-CD45.1 mice from different vendors. Standard analyses of RNA-seq datasets identified 40 genes in the regions of chromosomes 1, 7, and 19 with SJL content that were differentially expressed in CD4+ Th1 cells from B6.SJL-CD45.1 mice from CR as compared to cells from B6-CD45.2 mice, with 22 displaying at least a 2-fold change. This included genes such as Ctse and Ptprv, which have SJL genetic content in Jax and CR mice, as well as genes specific to the SJL genomic interval in B6.SJL-CD45.1 mice from CR and Tac, such as Glu1 (encodes glutamate synthase 1) and Rgs16 (encodes regulator of G protein signaling 16). There were also examples of genes with expression changes on chromosomes 7 and 19 in B6.SJL-CD45.1 mice from CR, such as Atrnl1 (encodes attractin like 1), Mfsd13a (encodes major facilitator superfamily domain containing protein 13a), and Pop4 (encodes ribonuclease P/MRP subunit).
We next performed qRT-PCR experiments to confirm the RNA-seq data. Rgs16 and Glul were differentially expressed between CD4+ Th1 cells isolated from B6-CD45.2 as compared to B6.SJL-CD45.1 mice from CR or Tac (Fig. 5C, S6D). In contrast, Rgs16 and Glul were similarly expressed in cells isolated from B6-CD45.2 and B6.SJL-CD45.1 mice originating from Jax (Fig. 5C). On chromosome 19, Atrnl1, Tmem2 and Mfsd13a were differentially expressed in CD4+ Th1 cells isolated from B6.SJL-CD45.1 mice from CR in comparison to B6-CD45.2 mice, but were similarly expressed in B6-CD45.2 and B6.SJL-CD45.1 mice from Jax and Tac (Fig. 5C, S6D). These data suggested the SJL genetic content in B6.SJL-CD45.1 mice impacted the expression of a subset of genes in CD4+ Th1 cells.
SJL genetic content influences H3K27Ac elements in B6.SJL-CD45.1 mice
To determine whether SJL genetic content in B6.SJL-CD45.1 mice impacts H3K27Ac enhancer elements, we performed H3K27Ac ChIP-seq experiments with CD4+ Th1 cells isolated from B6-CD45.2 or B6.SJL-CD45.1 mice from Jax and CR. We first defined the H3K27Ac ChIP-seq peaks on chromosomes 1, 7, and 19 that were differential between CD4+ Th1 cells isolated from B6-CD45.2 mice as compared to B6.SJL-CD45.1 mice from CR. We then created a heatmap to compare the levels of H3K27Ac associated with those peaks in CD4+ Th1 cells isolated from B6-CD45.2 and B6.SJL-CD45.1 mice from the two vendors. We also included in the analysis H3K27Ac ChIP-seq data from SJL mice and germ-free mice on the B6.SJL-CD45.1 background.
The patterns and levels of H3K27Ac were similar between SJL mice and B6.SJL-CD45.1 mice from CR at these peaks (Fig. 5D). This suggested SJL genetic content influenced H3K27Ac levels in these genomic regions. In addition, H3K27Ac levels in CD4+ Th1 cells isolated from B6.SJL-CD45.1 mice from Jax were similar to SJL mice in some 5’ aspects of chromosome 1, but resembled B6-CD45.2 mice in the 3’ area of chromosome 1 and in the genomic regions on chromosomes 7 and 19 that were differential in B6.SJL-CD45.1 mice from CR (Fig. 5D). These data were consistent with the distribution of B6 and SJL genetic content in B6.SJL-CD45.1 mice from Jax in comparison to B6.SJL-CD45.1 mice from CR. H3K27Ac levels in CD4+ Th1 cells from germ-free B6.SJL-CD45.1 mice were similar to B6.SJL-CD45.1 mice from CR indicating that the genetics of these chromosomal regions dominantly influenced H3K27Ac states at this subset of peaks (Fig. 5D). Together, the data indicated sequence variation derived from SJL genetic content influenced a subset of H3K27Ac elements in B6.SJL-CD45.1 mice.
Cxcr4 expression is reduced on Tfh cells from B6.SJL-CD45.1 mice during an influenza infection
We next wanted to address whether genetic variation in B6.SJL-CD45.1 mice impacts CD4+ T cells differentiating in the context of an infection. We examined Cxcr4 expression on Tfh cells developing during an influenza infection because Cxcr4 can play a role in homing within the germinal center. We transferred T cells from B6-CD45.2 or B6.SJL-CD45.1 mice from Jax or CR into TCR−/− mice, and infected the mice with influenza. We then analyzed the expression of Cxcr4 on Tfh cells at 12 days postinfection. Cxcr4 expression was diminished on CD4+ Tfh cells from B6.SJL-CD45.1 mice from CR in comparison to CD4+ Tfh cells from B6-CD45.2 or B6.SJL-CD45.1 mice originating from Jax (Fig. S6H).
The Cxcr4 locus was derived from SJL genetic content in B6.SJL-CD45.1 mice from both Jax and CR, and this genetic content was sufficient to dampen the IL-2-induced expression of Cxcr4 in the context of Th1 polarization in vitro (Fig. 3A, 3B). In contrast, the influenza experiments suggested the genetics of the Cxcr4 locus was not sufficient to dampen Cxcr4 expression on Tfh cells in the context of an influenza infection, and rather a genetic element outside of the locus contributed to the inhibition of Cxcr4 in this setting. A candidate for the additional genetic element might be Rgs16, which was contained within SJL genetic content in B6.SJL-CD45.1 mice from CR, but was within B6 genetic content in Jax mice. Rgs16 is a negative regulator of Cxcr4 (Estes et al., 2004; Lippert et al., 2003), and notably, Rgs16 expression was upregulated in B6.SJL-CD45.1 mice from CR, but not Jax (Fig. 5C). Further experiments will be necessary to confirm the genetic element(s) outside of the Cxcr4 locus that contributed to the differences observed in the B6.SJL-CD45.1 mice from the two vendors, but together the data show the complexity of the genetic regulation of Cxcr4 expression in B6-CD45 congenic mice.
There is genetic variation in H3K27Ac elements in B6.SJL-CD45.1 mice
We next wanted to determine if there is genetic variation in potential regulatory elements located within the genomic intervals that contain SJL genetic content in B6.SJL-CD45.1 mice. H3K27Ac is a widely distributed histone modification that is often associated with enhancers, super-enhancers, as well as transcription initiation sites, which provides the opportunity to assess genetic variation in a range of regulatory elements (Vahedi et al., 2015). To define genetic variation in regions with H3K27Ac, we reanalyzed the H3K27Ac ChIP-seq datasets from CD4+ Th1 cells isolated from B6-CD45.2 and B6.SJL-CD45.1 mice from Jax and CR. We filtered the alignment files to retain only the sequence alignments with 0 mismatches, and then compared these to the alignment files that included the default mismatch parameters using DESeq2. Consistent with the RNA-seq analysis for genetic variation, there was a small subset of H3K27Ac ChIP-seq peaks in B6-CD45.2 mice with sequence variation from the mm10 genome (Fig. 6A). In contrast, numerous H3K27Ac peaks were diminished in comparisons between the stringent and default alignment parameters for datasets from B6.SJL-CD45.1 mice from CR (Fig. 6A). The H3K27Ac peaks with genetic variation were located in the genomic intervals on chromosomes 1, 7, and 19 that were defined in the RNA-seq analysis to retain SJL genetic content. This included H3K27Ac peaks in proximity to genes such as Bcl2, Cxcr4, Rgs16, Rnasel, Got1, Slc18a2, and Ccne1.
Figure 6. Genetic variation is found in H3K27Ac elements in CD4+ Th1 cells.

(A) Heatmap of H3K27Ac ChIP-seq datasets comparing default and stringent alignment parameters for CD4+ Th1 cells isolated from B6-CD45.2 and B6.SJL-CD45.1 mice from Jax or CR. See results and methods for heatmap peak selection. (B) UCSC genome browser display of H3K27Ac ChIP-seq tracks for CD4+ Th1 cells isolated from B6-CD45.2 or B6.SJL-CD45.1 mice from Jax. Viral LTR annotation is shown below the tracks. Primer locations for PCR in (C) are shown with blue arrows. (C) PCR analysis examining DNA from B6-CD45.2 or B6.SJL-CD45.1 mice from Jax. (A-C) Data are representative of at least 2 independent biological replicates. See also Fig. S5.
We also noticed a gap with no detectable sequence variation within the chromosome 19 interval with SJL genetic content in B6.SJL-CD45.1 mice from CR. To assess this, we visually examined the region more closely in the IGV browser, and compared default H3K27Ac ChIP-seq alignment files from CR B6.SJL-CD45.1, SJL and B6-CD45.2 mice. The analysis defined a region of B6 genetic content embedded within the SJL genetic interval on chromosome 19 in B6.SJL-CD45.1 mice from CR (Fig. S5B, 4C white dotted lines). Thus, defining sequence variation in H3K27Ac peaks further clarified the SJL genetic content on chromosome 19 in B6.SJL-CD45.1 mice from CR.
We next performed comparisons between default and stringent alignment parameters for H3K27Ac ChIP-seq datasets for B6.SJL-CD45.1 mice from Jax (Fig. 6A). Consistent with the defined regions of SJL genetic content in the RNA-seq analysis, the peaks with genetic variation in B6.SJL-CD45.1 mice from Jax were localized within an approximately 37 Mb interval on chromosome 1 (Fig. 6A). We confirmed the sequence variation in H3K27Ac peaks in proximity to genes such as Bcl2 and Cxcr4 was derived from SJL genetic content by viewing the alignment files with the IGV browser (data not shown). Collectively, the data indicated there was genetic variation in elements with H3K27Ac in the genomic intervals containing SJL content in B6.SJL-CD45.1 mice.
Viral integrations are a component of genetic variation in H3K27Ac elements
As discussed, one component of genetic variation between mouse strains is viral integrations. Viral integrations are enriched in transcription factor binding sites and are thought to serve as enhancers in some circumstances. We noticed an H3K27Ac peak in proximity to the Kif14 locus that appeared to be interrupted in CD4+ Th1 cells isolated from B6.SJL-CD45.1 and SJL mice (Fig. 6B, S1A). The mm10 genome annotations for this region indicated a viral integration directly overlapped with the area devoid of H3K27Ac in the CD4+ Th1 cells from B6.SJL-CD45.1 and SJL mice (Fig. 6B). We designed PCR primers to specifically detect the viral integration at this location to test the hypothesis that the lack of H3K27Ac in B6.SJL-CD45.1 mice was because the viral integration was absent in the SJL genetic content in this region (Fig. 6B). The viral integration was detected in DNA from B6-CD45.2 mice and B6.SJL-CD45.1 mice from Tac, but not in B6.SJL-CD45.1 mice from Jax and CR (Fig. 6C, S6I). This was consistent with the sequence variation data indicating that this genomic region was derived from SJL genetic content in B6.SJL-CD45.1 mice from both Jax and CR, whereas the region was derived from B6 genetic content in B6.SJL-CD45.1 mice from Tac. These data indicated that viral integrations were a component of genetic variation in potential enhancer elements.
Alternative RNA-seq processing approaches can detect genetic variation in backcross models
We next wanted to address if the alternative next-generation sequencing processing approaches could be utilized to identify genetic content that is retained from the originating parent strain in other backcross models. We also wanted to define the most efficient sequencing parameters for detecting genetic variation. Knockout or transgenic mice are often created on the 129 genetic background. These mice are typically backcrossed to another strain because 129 mice are not used in most immunological research studies. Therefore, we wanted to determine if we could identify 129 genetic content in a knockout mouse model. We examined Tbx21−/− mice that were made with 129 ES cells and were backcrossed to the B6-CD45.2 background (Szabo et al., 2002).
We performed RNA-seq experiments with CD4+ Th1 cells isolated from B6-CD45.2 or Tbx21−/− mice. One set of RNA-seq datasets was generated with single-end 50bp reads, while another set was generated with paired-end 150bp reads, which was the technology used in the B6.SJL-CD45.1 studies. We then performed DESeq2 comparing default and stringent alignment parameters for these datasets. The analyses using datasets with 150bp reads identified more of the genes with sequence variation in Tbx21−/− mice in comparison to the analyses performed with 50bp reads (Fig. 7A, 7B, S7). From a technical standpoint, the data indicated that longer sequencing reads more efficiently detected sequence variation compared to shorter sequencing reads.
Figure 7. 129 genetic content was detected in Tbx21−/− transcripts.

(A-C) CD4+ T cells were isolated from B6-CD45.2 or Tbx21−/− mice or (D-E) B6-CD45.2/CD45.1 heterozygous mice and polarized in Th1 conditions. RNA-seq was performed with (A, C) 50bp single-end or (B, D, E) 150bp paired-end reads on two independent biological replicates. Graphs of normalized counts for RNA-seq data from (A, B) B6-CD45.2 (dark blue) and Tbx2T−/− (pink) or (D) B6-CD45.2/CD45.1 (light blue) mice processed with default (solid bars) or stringent (hatched bars) parameters. P-values defined by DESeq2 analysis (***≤ 0.001, * ≤ 0.05). (C, E) IGV browser tracks displaying RNA-seq alignments from default processing parameters with nucleotide changes from the mm10 genome displayed as in Fig. 3F. See also Fig. S7.
Many next-generation sequencing datasets deposited in GEO were produced with 50bp reads because this has been a standard sequencing technology. Thus, we wanted to determine a feasible approach to define sequence variation if 50bp single-end reads are the only datasets available for a retrospective analysis. We hypothesized that using the IGV browser to view default alignment files might be a feasible approach because it allowed us to identify modest sequence variation in B6.SJL-CD45.1 mice that was not detected with the statistical thresholds in the DESeq2 analysis. We examined the default alignment files for the RNA-seq 50bp reads from Tbx21−/− mice in the IGV browser to define areas of genetic variation located in proximity to the Tbx21 locus on chromosome 11. This approach identified sequence variation in several transcripts, including Ccr7, Igfbp4, Nme2, Smarce1, Top2a, Ikzf3, Cdk12, and Med1 (Fig. 7C, Table S3). We then compared this sequence variation data to the sequencing data for three 129 mouse strains contained in the Wellcome Sanger Institute database. We found the sequence variation in proximity to the Tbx21 locus was identified as SNPs in the 129 genomes (Table S3). This included sequence variation that resulted in nonsynonymous amino acid changes in Cdk12, Ikzf3 (Aiolos), Med1, and Eme1.
To validate the sequence variation in Tbx21−/− mice, we designed qRT-PCR primers to the region with sequence variation in the Smarce1 transcript using a similar strategy to the Cxcr4 analysis performed with the B6 congenics. Similar levels of Smarce1 transcripts were detected in CD4+ Th1 cells isolated from Tbx21−/− and B6-CD45.2 mice with primers annealing to conserved sequences (Fig. S7D). In contrast, Smarce1 transcripts were not as efficiently detected in CD4+ Th1 cells isolated from Tbx21−/− mice in comparison to B6-CD45.2 mice when the amplification was performed with a primer that hybridized to a region containing sequence variation (Fig. S7D). Together, the data suggested that sequence variation can be detected using RNA-seq datasets in knockout mouse models, and the genetic variation in Tbx21−/− mice was likely related to the retention of 129 genetic content in proximity to the Tbx21 locus.
Sequence variation can be detected in heterozygous mice
Finally, we wanted to determine if the alternative RNA-seq processing approaches could detect sequence variation in a heterozygous genetic background. We generated RNA-seq datasets with 150bp reads for CD4+ Th1 cells that were isolated from mice heterozygous for the CD45.2 and CD45.1 alleles, and compared the datasets after processing them with either default or stringent alignment parameters. We also visualized the default alignment files in the IGV browser. Genes with a substantial number of nucleotides with sequence variation were detected in the DESeq2 analysis comparing the default and stringent parameters for B6-CD45.2/CD45.1 heterozygous mice (Fig. 7D, S7E). However, DESeq2 could not detect genes with modest numbers of nucleotides with sequence variation. The heterozygous content in both circumstances was detectable using the IGV browser, which visually showed two different nucleotides at locations with sequence variation (Fig. 7E, S7F). Therefore, heterozygous alleles can be detected using these approaches.
Discussion
In this study, we developed alternative processing approaches for next-generation sequencing datasets to identify genetic variation, and we applied these to define variation in the widely used B6 CD45.1 and CD45.2 congenic strains. We found substantial differences in the SJL genetic content in B6.SJL-CD45.1 mice purchased from different vendors, and we defined how this contributed to differences in amino acid content, H3K27Ac, gene regulation, and protein expression in CD4+ Th1 cells isolated from B6-CD45.2 or B6.SJL-CD45.1 mice. The results of our study highlight the importance of defining and controlling for genetic variables in congenic and backcross mouse models.
We identified substantial differences in SJL genetic content in B6.SJL-CD45.1 mice from different vendor sources. Predicting how SJL genetic content influences individual activities and cell types in each B6.SJL-CD45.1 strain is complicated. This concept was highlighted by the different impact that SJL genetic content had on Cxcr4 expression depending upon the experimental system and cell type examined. We defined sequence variation in B6.SJL-CD45.1 mice located in the exons, 3’ UTR, 5’ UTR, and H3K27Ac potential enhancer elements of the Cxcr4 locus, and variation in the locus of a negative regulator of Cxcr4 in some vendor sources of the mice. The experiments with CD4+ Th1 cells in comparison to CD4+ Tfh cells showed the complexity of predicting how the combination of genetic variables within regulatory elements and the gene body impact the expression and function of a gene product in different settings. It also highlighted how genetic variables at different loci can work in cooperation to influence activities. Sequence variation in potential enhancer elements might affect different cell types in similar or opposing manners. This is because any effects will depend upon if sequence variation impacts transcription factor binding motifs, and impacted motifs might be utilized in a context-dependent manner. The take-home message is that any individual component of sequence variation can have a positive, negative, or no effect on activity, and there is not a single method to define which of these options is the case in a given circumstance.
There were a large number of differences in H3K27Ac peaks in CD4+ Th1 cells isolated from B6-CD45.2 and B6.SJL-CD45.1 mice, and this number varied by the vendor source. This, along with genetic variation in the underlying sequence in these elements, might influence gene regulation in a context-dependent manner. From an experimental perspective, this is also an important consideration for ChIP-seq analyses that compare wild-type and knockout cells on different B6 congenic backgrounds. This is because a significant number of differential peaks might be related to genetic variation between congenic backgrounds rather than the transcription factor deficiency being studied. If congenic-specific peaks are included in downstream bioinformatics comparisons, this might dilute the analyses by including a variable that is independent of the knockout under investigation.
Our study found that viral integrations represent one source of genetic variation that impacts both gene expression and H3K27Ac enhancer elements. Viral integrations are a source of genetic material that is often highly variable between species, and in this case, variable in different mouse strains (Chuong et al., 2017; Rebollo et al., 2012). Viral elements can have inhibitory effects on gene regulation, such as the disruption in the gene structure observed at Ctse. Viral integrations can also serve as a source of transcription factor binding sites and play roles in promoting gene regulation, often in the context of cell-type specific gene expression (Rebollo et al., 2012). Our study highlights the importance of defining this source of genetic variation and deciphering its role in widely used mouse models.
Advances in technologies are facilitating the generation of increasingly complex mouse models, which often include backcrosses with multiple different strains or congenic backgrounds. Thus, it is possible that seemingly identical experiments performed in different laboratories might have an overlooked variable related to the genetic content of parent strains retained after backcrossing (Vanden Berghe et al., 2015). It is important for immunologists to have the ability to assess whether genetic variation might be a relevant variable, especially when discrepancies are identified between experimental results, and to have a way to retrospectively address this variable. All experiments are imperfect and have both known and unknown variables. Genetics is often an undefined variable in inbred mouse experiments because spontaneous mutations and the retention of genetic content from parent strains after backcrosses have been difficult to identify. In the approaches presented here, genetic variation can be assessed in genes that are expressed in RNA-seq datasets or if the content is located within elements that are present in DNA-seq-based assays (e.g. open chromatin in ATAC-seq, epigenetic modifications in ChIP-seq). This will provide useful information on experimental variables and aid in creating approaches that account for the variables.
STAR Methods
CONTACT FOR REAGENT AND REAGENT SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Amy Weinmann (weinmann@uab.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The mouse strains used in this study include C57BL/6/J (000664), SJL/J (000686), B6.SJL-PtprcaPepb/BoyJ (002014), and B6.129S6-Tbx21tm1Glm/J (004648) from Jackson Laboratory (Jax), C57BL/6NCrl (027) and B6.SJL-PtprcaPepb/BoyCrCrl (564) from Charles River (CR), and B6.SJL-Ptprca/BoyAiTac (4007) from Taconic (Tac). Both male and female mice at least six weeks of age were used. Mice were bred in the University of Alabama at Birmingham (UAB) animal facility or directly purchased from vendor sources. Germ-free mice were obtained from the Gnotobiotic Core Facility at UAB. To generate B6-CD45.2 and B6.SJL-CD45.1 heterozygous mice, a parent of each genotype (vendor source Jax) were bred together and the F1 generation was used for experiments. All experimental procedures involving animals were approved by the UAB Institutional Animal Care and Use Committee (IACUC) and were performed according to guidelines outlined by the National Research Council.
METHOD DETAILS
Primary T cell isolations
Primary CD4+ or CD8+ T cells were isolated from the lymph nodes or spleen of both male and female mice that were at least 6 weeks old. Negative selection kits (CD4+ T cell MAGM202; or CD8+ T cell MAGM203) from R&D systems were used for the T cell isolations. The cells were stimulated with aCD3 and aCD28, and polarized in type 1 conditions (IL-2, IL-12 and anti-IL-4)(Chisolm et al., 2017). The initial anti-CD3 (clone 145-2C11, 5mg/mL) and anti-CD28 (clone 37.51, 10μg/mL) activation in Th1 polarizing conditions (IL-12 (R&D catalog number 419ML-010, 5ng/mL) and anti-IL-4 (clone 11B11, 10μg/mL)) was for three days. The cells were then split and plated without TCR stimulation in Th1 polarizing conditions and IL-2 (100 units/mL) for an additional two days before harvest (Chisolm et al., 2017).
Western blot and flow cytometry
A western analysis was performed with 2×106 CD4+ Th1 or CD8+ Tc1 cells that were isolated from either B6-CD45.2 or B6.SJL-CD45.1 mice. Antibodies used in the western analysis were to Cxcr4 (Abcam, ab124824), cathepsin E (R&D, BAF1130), or as a control to STAT4 (Santa Cruz, C-20, sc-486X). For the flow cytometry analysis of Cxcr4 surface expression, 0.5 ×106 CD4+ Th1 or CD8+ Tc1 cells were stained with a biotinylated antibody to Cxcr4 (BD Bioscience; 551968), followed by a secondary antibody that was conjugated with streptavidin PE (BD Bioscience, 554061). Flow cytometry analyses were done with FlowJo v.10 software. Numbers in Fig. 3B represent the frequency of cells from each genotype and the standard deviation. P values were calculated with unpaired Student’s t tests and the differences between the genotypes were statistically significant (p value ≤ 0.05).
Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) and RT-PCR
RNA was isolated using the Nucleospin kit from Clontech, and cDNA was made using the PrimeScript first strand cDNA synthesis kit from Clontech. Transcript abundance was quantified with iQSybr Green Mix (BioRad) using gene specific primers, quantitated relative to the control gene Rps18 and relative expression was determined in comparison to the indicated samples. For the schematic representation of primer locations in Fig. 3D that were used to monitor the Cxcr4 transcript, the primers for Cxcr4 (green arrows), 2Cxcr4 (blue arrows), 3Cxcr4 (yellow arrows) and 4Cxcr4 (grey arrows) primer pairs are shown. An unpaired Student t-test was performed to determine the p values by using the GraphPad Prism online program. Anova tests for multiple sample comparisons were also performed. For the reverse transcriptase PCR reaction, cDNA was prepared from the RNA followed by a standard PCR reaction with primers specific to the Cxcr4 gene. The products of the PCR reaction were run on an agarose gel for visualization.
RNA-sequencing (RNA-seq) and analysis
RNA-seq was performed on at least 3 independent biological replicates for the CD4+ Th1 cells isolated from B6-CD45.2, B6.SJL-CD45.1, and SJL mice from Jax; CD4+ Th1 cells isolated from B6N-CD45.2 and B6.SJL-CD45.1 mice from Charles River; CD4+ Th1 cells isolated from B6.SJL-CD45.1 mice from Taconic Biosciences. RNA-seq was performed on 2 independent biological replicates for the CD4+ Th1 cells isolated Tbx21−/− B6-CD45.2, and B6-CD45.2/CD45.1 heterozygous mice. Library preparation and next-generation sequencing were performed by Genewiz. For the B6-CD45.2, B6.SJL-CD45.1, SJL, B6-CD45.2/CD45.1, and Tbx21−/− experiments, libraries were prepared to detect strand specific reads with rRNA depletion and subjected to paired-end sequencing 150bp reads. For the Tbx21−/− experiments, a second set of libraries were generated that did not denote strand and were based on polyA, and were subjected to single-end 50bp reads on the HiSeq2500 platform. For both Tbx21−/− RNA-seq experiments, the RNA was harvested from CD4+ Th1 cells at day 3.
The analyses of the RNA-seq datasets were performed using the Cheaha HPC cluster at UAB. FastQ files were trimmed to remove primer adapters using Trim Galore! version 0.4.1. The trimmed sequences were then aligned to the mouse reference genome (mm10) from UCSC using RNA STAR version 2.5.4b with default settings (mismatches allowed): ‘--alignEndsType EndtoEnd’, or stringent settings (no-mismatches): ‘--outFilterMismatchNmax 0’ ‘--alignEndsType EndtoEnd’. To output a sorted BAM file, ‘--outSAMtype BAM SortedByCoordinate’ was used. For visualization of the tracks in the UCSC Genome Browser, alignment files were normalized using deepTools version 3.1.0 bamCoverage using the option: ‘--normalizeUsing RPKM’ (Ramirez et al., 2016). Gene counts were generated using FeatureCounts from the Rsubread Package in R version 3.5.1 (Liao Y et al., 2013) followed by differential gene expression using the DESeq2 package (Love et al., 2014). The DESeq2 was performed to compare two conditions, and it also included all samples from an experimental batch to generate normalized counts for all samples simultaneously. Heatmaps were then generated using the ComplexHeatmap Package in R version 3.5.1. The genes selected for the RNA-seq heatmap analyses presented in Figure 4 are the genes that had an adjusted p value (Benjamini-Hochberg) of ≤ 0.05 in the DESeq2 analysis comparing the default and stringent alignment parameters for B6.SJL-CD45.1 mice from Charles River. It is important to note that using an adjusted p value of ≤ 0.05 did not identify all genes with sequence variation. For example, the known sequence variation in the Ptprc locus reached this threshold in some cases, but not in others. Relaxing the criteria for the adjusted p value, or using the non-adjusted p value in DESeq2, identified more genes with sequence variation. Relaxing statistical parameters also identified false positives. Therefore, depending upon the goals of the analyses (e.g. define a genomic interval from a parent strain in a backcross model, define specific loci with variation, etc.) a more or less stringent statistical cut-off can be used. If a less stringent statistical cut-off is used in the analysis, or the goal is to define all transcripts with sequence variation, it is important to assess the genes predicted to have sequence variation by viewing default alignment tracks in the IGV browser to define the sequence variation specifically in each gene. For this, sorted and indexed default alignment files are viewed with the IGV browser (version 2.4.11). Comparing normalized default and stringent alignment tracks in the UCSC genome browser is another way to visualize the data. In this case, one can look for regions with diminished alignments in the stringent parameters in comparison to the default parameters to localize regions with sequence variation. The same considerations apply to the other next-generation sequencing approaches (e.g. ChIP-seq) to define sequence variation as well. Finally, one way to identify the list of genes contained within a genetic interval is to use the gene list in DESeq2, which is ordered by chromosome location. This means that all genes between the first and last genes containing sequence variation (as confirmed in IGV) within a given chromosomal interval can be extracted from the list of genes in DESeq2.
Chromatin immunoprecipitation sequencing (ChIP-seq) and analysis
ChIP-seq was performed on CD4+ Th1 cells or CD8+ Tc1 cells. In short, 20 million cells were crosslinked with 1% formaldehyde for 10 minutes (Chisolm et al., 2017; Oestreich et al., 2014). Nuclei were isolated and chromatin was sheared by sonication with a Bioruptor (Diagenode). An antibody specific to H3K27Ac (Abcam, ab4729) was used to precipitate the chromatin. ChIP-seq was performed on at least 2 independent biological replicates. Libraries were prepared and sequenced with 150bp paired-end sequencing reads by Genewiz.
Sequencing reads were trimmed to remove primer adapter using Trim Galore! Version 0.4.1. They were then mapped to the mouse UCSC mm10 reference genome using Bowtie2 version 2.3.3. Aligned reads were then sorted, followed by removal of duplicates using Picard version 2.18.11 SortSam and MarkDuplicates. For the stringent parameters, “0-mismatches” were filtered using Bamtools version 2.4.0‘-tag XM:0’ parameter. Peaks were identified using the MACS peak caller (Zhang et al., 2008; MACS2) and normalized to sequencing depth with the --SPMR parameter. The bedgraph files were then converted to bigwig to view in the UCSC genome browser using bedGraphToBigWig, and annotations were generated by a Genomic Regions Enrichment Annotations Tool (GREAT) analysis (Bejerano lab, Stanford University (McLean et al., 2010)) using the basal plus extension default parameters (proximal:5.0 kb; 1.0 kb downstream; plus distal up to 1000 kb). Differential peak analysis was performed by using featureCounts from the Rsubread Package in R version 3.5.1 and then pairwise comparisons of conditions were calculated using DESeq2 version 1.22.2. The DESeq2 analyses were performed to compare two conditions, and included all the samples for the experiment to generate normalized counts. Heatmaps of H3K27Ac differential peaks were generated using the EnrichedHeatmap package in R 3.5.1. In Figure 5D, the peaks selected for the heatmap were from chromosomes 1, 7 and 19 and had at least a 2-fold change and a base mean of 50 in a DESeq2 comparison between B6N-CD45.2 and B6.SJL-CD45.1 mice from CR. A repetitive element that could not be accurately mapped from B6N-CD45.2 mice was excluded in this analysis. In Figure 6A, the peaks selected for the heatmap were those with an adjusted p value ≤ 0.05 in a DESeq2 comparison of the default and stringent alignment parameters for the H3K27Ac ChIP-seq datasets from B6.SJL-CD45.1 mice from CR.
Additional considerations
It should be noted that detailed bioinformatics analysis tools for quantitatively defining genetic variation have been developed if a more comprehensive approach is required (Gezsi et al., 2015; Hwang et al., 2015). There is also a tool available to estimate 129 genetic content in knockout mice based upon predictions of how much genetic content is likely to remain with the number of backcrosses performed (Vanden Berghe et al., 2015). However, the advantage of the strategies described in this study is that it can be added to next-generation sequencing processing pipelines and does not require extensive specialized bioinformatics skills. It also can identify the exact sequence content. Therefore, these approaches should be accessible to immunologists with basic bioinformatics knowledge to estimate regions that retain genetic variation during backcrosses. It is worth noting that publicly available platforms (e.g. Galaxy) have been developed to aid in basic next-generation sequencing analyses for researchers without extensive bioinformatics training.
UCSC genome browser and IGV browser
ChIP-seq and RNA-seq data were displayed using the University of California Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu/)(Kent et al., 2002) or the Integrative Genomics Viewer (IGV; Broad Institute; version 2.4.11) (Robinson et al., 2011; Thorvaldsdottir et al., 2013).
Sanger sequencing
Transcripts were amplified from 40ng of cDNA and gel purified using a Nucleospin Gel and PCR clean up kit (Clontech). Amplicons were cloned into the pcDNA3.1 vector using the TOPO TA Expression Kit (Invitrogen). Clones were then purified and subjected to Sanger sequencing at Eurofins. For visualization, sequences were uploaded into the BLAT function of the UCSC genome browser for alignment to the mm10 reference genome.
Adoptive transfer experiments
Splenocyte cell suspensions were prepared from the spleens of C57BL/6, B6.SJL-PtprcaPepb/BoyJ (Jax), and B6.SJL-PtprcaPepb/BoyCrCrl (Charles River) mice. CD4+ and CD8+ T cells were enriched using magnetic beads. A mix of CD4+ and CD8+ T cells from the same donor strain was prepared and adjusted to contain the same amount of CD4+ (2×106) and CD8+ (1×106) T cells among cell suspensions. Three groups of 5-6 T-cell deficient mice (B6.129P2-Tcrbtm1Mom Tcrdtm1Mom/J) were transferred i.v. with one of the three T cell suspensions. The following day the reconstituted TCR−/− mice were infected with a sublethal dose of influenza virus A/PR8/34. After 12 days of infection, the mice were euthanized and cells suspensions from the mediastinal lymph node were prepared for flow cytometry analysis. T follicular helper cells were gated as CD3+ CD4+ FoxP3− PD1+ CXCR5+.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details for the different analyses performed are described in the specific methods. In most cases, error bars represent standard error of the mean (SEM), and the p values were calculated with unpaired Student’s t tests. For experiments with multiple sample comparisons, we first analyzed them with an anova test, and then performed a Student’s t test to obtain p values for pairwise comparisons. The DESeq2 program uses a Wald test for calculating the p value, and a Benjamini-Hochberg test for calculating the adjusted p value.
DATA AND SOFTWARE AVAILABILITY
Accession Numbers
Next-generation sequencing data are publicly available on the NCBI GEO website with the accession code for the SuperSeries (GSE129577). The individual sequencing types are also available with the following accession codes: RNA-seq (GSE129575) and ChIP-seq (GSE129576). ImmGen Consortium datasets were from the GEO accession code (GSE60337).
All software used for data analysis is publicly available.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| αCD4 (Clone: GK1.5) APC | Biolegend | Cat# 100412 |
| αCxcr4 | Abcam | Cat# ab124824 |
| αCathepsin E | R&D Systems | Cat# BAF1130 |
| αCxcr4 Biotin | BD Biosciences | Cat# 551968 |
| αStat4 (C-20) | Santa Cruz | Cat# sc-486X |
| αStreptavidin PE | BD Biosciences | Cat# BDB554061 |
| αH3K27Ac | Abcam | Cat# ab4729 |
| αCD3 (Clone: 145-2C11) | BD Biosciences | Cat# 553057 |
| αCD28 (Clone: 37.51) | BD Biosciences | Cat# 553294 |
| αIL-4 (11B11) | Biolegend | Cat# 504115 |
| PD-1 FITC (Clone: J43) | eBioscience | Cat# 11-9985-85 |
| Bcl-6 PerCP-Cy5.5 (Clone: K112-91) | BD Bioscience | Cat# 562198 |
| FOXP3 PE-Cy7 (Clone: FJK-16s) | eBioscience | Cat# 25-5773-82 |
| CXCR4 PE (Clone: 2B11) | BD Bioscience | Cat# 551966 |
| CXCR5 Brilliant Violet 421 (Clone: 2G8) | BD Biosciences | Cat# 562889 |
| CD3 APC-eFlour780 (Clone: 17A2) | eBioscience | Cat# 47-0032-82 |
| CD4 Qdot - 605 (Clone: RM4-5) | Invitrogen | Cat# Q10092 |
| CD8a V500 (Clone: 53-6.7) | BD Biosciences | Cat# 560776 |
| Bacterial and Virus Strains | ||
| Influenza Virus A/PR8/34 | generated in house | N/A |
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| 7-AAD | Calbiochem | Cat# 129935 |
| rmIL-12 | R&D systems | Cat# 419ML-010 |
| rhIL-2 | NCI repository | N/A |
| IMDM | Life Technologies | Cat# 12440-053 |
| FBS | Life Technologies | Cat# 26140-079 |
| Penicillin/Streptomycin | Life Technologies | Cat# 15140122 |
| 2-mercaptoethanol | Sigma | Cat# M3148 |
| iQSybr Green Mix | Biorad | Cat# 1708882 |
| Rnase, Dnase-free, High Concentration | Sigma-Aldrich | Cat# 11579681001 |
| Proteinase K | Clontech | Cat#9034 |
| Buffer NTB | Clontech | Cat# 740595.150 |
| GoTaq DNA Polymerase | Promega | Cat# M3005 |
| Critical Commercial Assays | ||
| MagCellect Mouse CD4+ T Cell Isolation Kit | R&D systems | Cat# MAGM202 |
| MagCellect Mouse CD8+ T Cell Isolation Kit | R&D systems | Cat# MAGM203 |
| Gentra Puregene Cell Kit | Qiagen | Cat# 158767 |
| Nucleospin RNA kit | Clontech | Cat# 740955.50 |
| PrimeScript first strand synthesis | Clontech | Cat# 6110A |
| TOPO TA Expression Kit | Invitrogen | Cat# K4800-01 |
| Nucleospin Gel and PCR Clean-Up Kit | Clontech | Cat# 740609.50 |
| Quant-iT dsDNA Assay kit, high sensitivity | Invitrogen | Cat# Q33120 |
| Deposited Data | ||
| RNA-seq | NCBI Gene Expression Omnibus | GSE129575 |
| ChIP-seq | NCBI Gene Expression Omnibus | GSE129576 |
| SuperSeries | NCBI Gene Expression Omnibus | GSE129577 |
| Experimental Models: Cell Lines | ||
| N/A | ||
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratory; Bred at UAB facility with IACUC approval | Cat# 000664 |
| B6.SJL-PtprcaPepb/BoyJ | Jackson Laboratory; Bred at UAB facility with IACUC approval | Cat# 002014 |
| SJL/J | Jackson Laborarory | Cat# 000686 |
| C57BL/6NCrl | Charles River | Cat# 027 |
| B6.SJL-PtprcaPepcb/BoyCrCrl | Charles River | Cat# 564 |
| B6.SJL-Ptprca/BoyAiTac | Taconic Biosciences | Cat# 4007 |
| Germ-free B6.SJL-PtprcaPepcb/Boy | UAB Gnotobiotic Core | NA |
| F1: C57BL/6J x B6.SJL-PtprcaPepcb/BoyJ | Jackson Laboratory; Bred at UAB facility with IACUC approval | Cat# 000664, Cat# 002014 |
| B6.129S6-Tbx21tm1Glm/J | Jackson Laboratory; Bred at UAB facility with IACUC approval | Cat# 004648 |
| B6.129P2-Tcrbtm1Mom Tcrdtm1Mom/J | Jackson Laboratory | Cat# 002122 |
| Oligonucleotides | ||
| Atrnl1 Forward | Eurofins | CTACCGAGAGGATCTTCAGG |
| Atrnl1 Reverse | Eurofins | GACACAAGTTCCTTGACGTG |
| Ctse Forward | Eurofins | GAGCCTGACAGGAATCATTGG |
| Ctse Reverse | Eurofins | CAGACCCAGAATCCCATCAAAC |
| Ctse-(LTR) Forward | Eurofins | GTTTCCACACAGAGCTCAGC |
| Ctse-(LTR) Reverse | Eurofins | CAGACCTCTCGTTGTACTGC |
| Glul Forward | Eurofins | CATTCCAGGGAACTGGAATG |
| Glul Reverse | Eurofins | GAATGTGGTACTGGTGCCTC |
| Mfsd13a Forward | Eurofins | GCTCCTCTTTGGCATGGTTG |
| Mfsd13a Reverse | Eurofins | GATTGCTGAAAAAGGTCGTG |
| Additional oligonucleotide sequences can be found in Table S4 | ||
| Recombinant DNA | ||
| N/A | ||
| Software and Algorithms | ||
| GraphPad Online | GraphPad Software; http://www.graphpad.com/quickcalcs/ | N/A |
| FlowJo | FlowJo, LLC | v. 10 |
| TrimGalore! | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ | v. 0.4.4 |
| SAMtools | Li H et al., 2009 | v. 1.6.0 |
| BEDtools | Quinlan AR et al., 2010 | v. 2.26.0 |
| Bowtie2 | Langmead et al., 2009 | v 2.2.9 |
| Picard | http://broadinstitute.github.io/picard | v. 2.18.26 |
| MACS2 | Zhang et al., 2008 | v. 2.1.1 |
| deepTools | https://github.com/fidelram/deepTools | v. 3.1.3 |
| EnrichedHeatmap | https://github.com/jokergoo/EnrichedHeatmap | v. 1.12.0 |
| GREAT analysis | McLean et al., 2010 | v. 3.0.0 |
| STAR aligner | Dobin A et al., 2013 | v. 2.5.4b |
| Rsubread | Liao Y et al., 2014 | v. 1.32.2 |
| DESeq2 | https://github.com/mikelove/DESeq2 | v. 1.22.2 |
| Circlize | https://github.com/jokergoo/circlize | v. 0.4.5 |
| ComplexHeatmap | https://github.com/jokergoo/ComplexHeatmap | v. 1.20.0 |
| Other | ||
Highlights.
Developed alternative processing strategies for NGS data to define genetic variation
Substantial differences in SJL genetic content found in B6.SJL-CD45.1 mice by vendor
Genetic variation in B6-CD45 congenics impacts gene regulation and H3K27Ac elements
Genetic variation in B6-CD45 congenics impacts Cxcr4 and cathepsin E expression
Acknowledgements
We thank the Weinmann lab for discussions, the Gnotobiotic Core Facility at UAB for providing germ-free mice, the National Institutes of Health for funding (AI061061 to A.S.W.; CA216234 and AI109962 to T.R.), and the NCI preclinical repository for IL-2.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no competing interests.
References
- Basu S, Ray A, and Dittel BN (2013). Differential representation of B cell subsets in mixed bone marrow chimera mice due to expression of allelic variants of CD45 (CD45.1/CD45.2). Journal of immunological methods 396, 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisolm DA, Savic D, Moore AJ, Ballesteros-Tato A, Leon B, Crossman DK, Murre C, Myers RM, and Weinmann AS (2017). CCCTC-Binding Factor Translates Interleukin 2- and alpha-Ketoglutarate-Sensitive Metabolic Changes in T Cells into Context-Dependent Gene Programs. Immunity 47, 251–267 e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisolm DA, and Weinmann AS (2018). Connections Between Metabolism and Epigenetics in Programming Cellular Differentiation. Annu Rev Immunol 36, 221–246. [DOI] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, and Feschotte C (2017). Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet 18, 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwir O, Kansas GS, and Alon R (2000). An activated L-selectin mutant with conserved equilibrium binding properties but enhanced ligand recognition under shear flow. J Biol Chem 275, 18682–18691. [DOI] [PubMed] [Google Scholar]
- Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, and Littman DR (2004). An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol 5, 64–73. [DOI] [PubMed] [Google Scholar]
- Estes JD, Thacker TC, Hampton DL, Kell SA, Keele BF, Palenske EA, Druey KM, and Burton GF (2004). Follicular dendritic cell regulation of CXCR4-mediated germinal center CD4 T cell migration. J Immunol 173, 6169–6178. [DOI] [PubMed] [Google Scholar]
- Gezsi A, Bolgar B, Marx P, Sarkozy P, Szalai C, and Antal P (2015). VariantMetaCaller: automated fusion of variant calling pipelines for quantitative, precision-based filtering. BMC genomics 16, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Ramirez-Valle F, Xu Y, Wu S, Wu Z, Karjalainen KE, and Cyster JG (2013). Deficiency in IL-17-committed Vgamma4(+) gammadelta T cells in a spontaneous Sox13-mutant CD45.1(+) congenic mouse substrain provides protection from dermatitis. Nat Immunol 14, 584–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Kim E, Lee I, and Marcotte EM (2015). Systematic comparison of variant calling pipelines using gold standard personal exome variants. Scientific reports 5, 17875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafri S, Moore SD, Morrell NW, and Ormiston ML (2017). A sex-specific reconstitution bias in the competitive CD45.1/CD45.2 congenic bone marrow transplant model. Scientific reports 7, 3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y, Gerbec ZJ, Won T, Choi B, Podsiad A, B BM, Malarkannan S, and Laouar Y (2018). Cutting Edge: Check Your Mice-A Point Mutation in the Ncr1 Locus Identified in CD45.1 Congenic Mice with Consequences in Mouse Susceptibility to Infection. J Immunol 200, 1982–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, and Haussler D (2002). The human genome browser at UCSC. Genome research 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AG, Mendoza A, Hemmers S, Moltedo B, Niec RE, Schizas M, Hoyos BE, Putintseva EV, Chaudhry A, Dikiy S et al. (2017). Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 546, 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Link VM, Duttke SH, Chun HB, Holtman IR, Westin E, Hoeksema MA, Abe Y, Skola D, Romanoski CE, Tao J, et al. (2018). Analysis of Genetically Diverse Macrophages Reveals Local and Domain-wide Mechanisms that Control Transcription Factor Binding and Function. Cell 173, 1796–1809 e1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert E, Yowe DL, Gonzalo JA, Justice JP, Webster JM, Fedyk ER, Hodge M, Miller C, Gutierrez-Ramos JC, Borrego F v et al. (2003). Role of regulator of G protein signaling 16 in inflammation-induced T lymphocyte migration and activation. J Immunol 171, 1542–1555. [DOI] [PubMed] [Google Scholar]
- Mahajan VS, Demissie E, Mattoo H, Viswanadham V, Varki A, Morris R, and Pillai S (2016). Striking Immune Phenotypes in Gene-Targeted Mice Are Driven by a Copy-Number Variant Originating from a Commercially Available C57BL/6 Strain. Cell reports 15, 1901–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nature biotechnology 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier FE, Sykes DB, and Scadden DT (2016). Single Targeted Exon Mutation Creates a True Congenic Mouse for Competitive Hematopoietic Stem Cell Transplantation: The C57BL/6-CD45.1(STEM) Mouse. Stem cell reports 6, 985–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oestreich KJ, Read KA, Gilbertson SE, Hough KP, McDonald PW, Krishnamoorthy V, and Weinmann AS (2014). Bcl-6 directly represses the gene program of the glycolysis pathway. Nat Immunol 15, 957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan UT, Waldron TT, and Springer TA (2006). Remodeling of the lectin-EGF-like domain interface in P- and L-selectin increases adhesiveness and shear resistance under hydrodynamic force. Nat Immunol 7, 883–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purtha WE, Swiecki M, Colonna M, Diamond MS, and Bhattacharya D (2012). Spontaneous mutation of the Dock2 gene in Irf5−/− mice complicates interpretation of type I interferon production and antibody responses. Proc Natl Acad Sci U S A 109, E898–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo R, Romanish MT, and Mager DL (2012). Transposable elements: an abundant and natural source of regulatory sequences for host genes. Annu Rev Genet 46, 21–42. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nature biotechnology 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR Jr., et al. (2008). Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558. [DOI] [PubMed] [Google Scholar]
- Shen FW, Saga Y, Litman G, Freeman G, Tung JS, Cantor H, and Boyse EA (1985). Cloning of Ly-5 cDNA. Proc Natl Acad Sci U S A 82, 7360–7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, and Glimcher LH (2002). Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science 295, 338–342. [DOI] [PubMed] [Google Scholar]
- Tacchini-Cottier F, Weinkopff T, and Launois P (2012). Does T Helper Differentiation Correlate with Resistance or Susceptibility to Infection with L. major? Some Insights From the Murine Model. Frontiers in immunology 3, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JJ, Pape KA, and Jenkins MK (2012). A germinal center-independent pathway generates unswitched memory B cells early in the primary response. J Exp Med 209, 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpson NJ, Greenwood CMT, Soranzo N, Lawson DJ, and Richards JB (2018). Genetic architecture: the shape of the genetic contribution to human traits and disease. Nat Rev Genet 19, 110–124. [DOI] [PubMed] [Google Scholar]
- Vahedi G, Kanno Y, Furumoto Y, Jiang K, Parker SC, Erdos MR, Davis SR, Roychoudhuri R, Restifo NP, Gadina M, et al. (2015). Super-enhancers delineate disease-associated regulatory nodes in T cells. Nature 520, 558–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Hulpiau P, Martens L, Vandenbroucke RE, Van Wonterghem E, Perry SW, Bruggeman I, Divert T, Choi SM, Vuylsteke M, et al. (2015). Passenger Mutations Confound Interpretation of All Genetically Modified Congenic Mice. Immunity 43, 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterstrat A, Liang Y, Swiderski CF, Shelton BJ, and Van Zant G (2010). Congenic interval of CD45/Ly-5 congenic mice contains multiple genes that may influence hematopoietic stem cell engraftment. Blood 115, 408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SY, Coffre M, Ramanan D, Hines MJ, Gomez LE, Peters LA, Schadt EE, Koralov SB, and Cadwell K (2018). B Cell Defects Observed in Nod2 Knockout Mice Are a Consequence of a Dock2 Mutation Frequently Found in Inbred Strains. J Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ, Galan C, Belkaid Y, Bonneau R, and Littman DR (2018). c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin B, Wong K, Agam A, Goodson M, Keane TM, Gan X, Nellaker C, Goodstadt L, Nicod J, Bhomra A, et al. (2011). Sequence-based characterization of structural variation in the mouse genome. Nature 477, 326–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin B, Wong K, Bhomra A, Goodson M, Keane TM, Adams DJ, and Flint J (2012). The fine-scale architecture of structural variants in 17 mouse genomes. Genome biology 13, R18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Accession Numbers
Next-generation sequencing data are publicly available on the NCBI GEO website with the accession code for the SuperSeries (GSE129577). The individual sequencing types are also available with the following accession codes: RNA-seq (GSE129575) and ChIP-seq (GSE129576). ImmGen Consortium datasets were from the GEO accession code (GSE60337).
All software used for data analysis is publicly available.