

Abstract
Patient: Female, 33
Final Diagnosis: VIPoma
Symptoms: Diarrhea
Medication: —
Clinical Procedure: Laparoscopy
Specialty: Gastroenterology and Hepatology
Objective:
Rare disease
Background:
VIPomas are rare neuroendocrine tumors typically located in the pancreas. The majority of cases autonomously secret vasoactive intestinal polypeptide (VIP), which can result in profuse, refractory, watery diarrhea. The fluid and electrolyte imbalance can progress to dehydration and profound hypokalemia, resulting in the watery diarrhea, hypokalemia, achlorhydria (WDHA) syndrome. One previous case of a pancreatic VIPoma progressing to hypokalemic rhabdomyolysis has been described.
Case Report:
A 33-year-old woman presented with 3 months of progressive, refractory diarrhea and weakness. Her serum VIP level was elevated and imaging discovered a mass in the region of the pancreatic tail. Laparoscopic partial pancreatic resection was performed and a 3.7-cm diameter, solitary stage T2 N0 M0, well-differentiated carcinoma was removed.
Conclusions:
A high index of suspicion is important when diagnosing chronic diarrhea. Minimally invasive surgery is an option in the surgical treatment of pancreatic VIPoma.
MeSH Keywords: Neuroendocrine Tumors, Rhabdomyolysis, Vipoma
Background
Pancreatic neuroendocrine tumors (pNETs) are non-beta cell lesions that account for 1–2% of all pancreatic tumors [1]. Up to half of all pNETs are functional tumors that produce hormones autonomously. A VIPoma is a rare variety of pNET that produces vasoactive intestinal polypeptide (VIP). The mean age of patients with VIPoma is 49 years, but it can occur in children. In adults, 80–90% of VIPomas are located in the pancreas and are typically solitary. Up to 75% are in the pancreatic tail and 50–75% are malignant, with 37–68% having hepatic metastases at diagnosis [2,3]. VIPoma has a reported incidence of less than 1 case per million per year [4]. VIP is a brain and gut protein hormone that is a potent biologic mediator responsible for stimulation of vasodilation and secretion of fluid and electrolytes into the bowel lumen [5]. The unregulated overproduction of VIP results in large-volume diarrhea, often severe enough to cause hypokalemia, dehydration, and hypochlorhydria. This watery diarrhea, hypokalemia, achlorhydria (WDHA) syndrome caused by VIP-producing tumors is a well-known effect of VIPoma, but progression of the metabolic disturbance to the point of hypokalemic rhabdomyolysis has only been reported on 3 other occasions, with 2 of those cases involving the adrenal gland [6,7]. The only reported case originating in the pancreas also had hepatic metastasis [8]. We present a case of a solitary, well-differentiated pancreatic VIPoma causing rhabdomyolysis treated with fluid and electrolyte replacement, and laparoscopic pancreatic resection.
Case Report
A 33-year-old woman with a past medical history of gastroesophageal reflux disease presented with multiple watery, non-bloody bowel movements per day over 3 months. Her symptoms were associated with intermittent mid-abdominal pain, nausea, vomiting, generalized weakness, and a 50-pound weight loss. Prior to presenting, she underwent an extensive workup at an outside hospital, which included endoscopy, colonoscopy, multiple stool studies, and bloodwork. The evaluation was negative for infection, malignancy, celiac disease, inflammatory bowel disease, carcinoid tumor, and gastrinoma. The endoscopic biopsy revealed gastritis and the patient was discharged home with an oral proton pump inhibitor. Her symptoms progressed to nearly 8 bowel movements a day associated with severe polyarthralgia and weakness, with difficulty walking. On arrival at our Emergency Department, she was afebrile and vital signs were stable. A physical exam revealed mild epigastric tenderness to palpation. The remainder of the exam was unremarkable other than severe generalized weakness and myalgia. Her potassium was 1.7 mEq/L (3.5–5.1 mEq/L), bicarbonate 20 mEq/L (21–31 mEq/L), phosphorous 1.6 mg/dL (2.4–4.7 mg/dL), calcium 9.5 mg/dL (8.6–10.3 mg/dL), C reactive protein <.05 mg/L (≤9.9 mg/L), and CK (creatine kinase) was 1110 U/L (30–223 U/L). The chloride was 102 mmol/L (98–107 mmol/L), blood glucose was 179 mg/dL (65–100 mg/dL), AST 48 U/L (13–39 U/L), ALT 67 U/L (7–52 U/L), and alkaline phosphatase 111 U/L (34–104). Her urinalysis was negative other than 30 mg/dL of protein. Her aldosterone was 6.9 ng/dL (≤21 ng/dL) and renin activity was 18 ng/mL/hr (2.9–24). Her troponin I was 0.05 ng/mL (0.00–0.05 ng/mL) and her electrocardiogram (EKG) was negative, with the exception of a right bundle branch block, which was still present weeks later. Her serum gastrin was 67 pg/mL (<100 pg/mL). Other routine chemistries, complete blood counts, and thyroid panel were unremarkable. Stool studies were negative and cultures did not grow any pathologic organisms. She was admitted for aggressive potassium replacement and workup. After stabilization of her potassium, a CT (computed tomography) scan of the abdomen and pelvis without contrast revealed a well-circumscribed hyperdense rounded structure in the left upper quadrant, measuring 4.0 cm, and possibly involving the tail of the pancreas (Figure 1). Further imaging, including a CT enterography (Figure 2) and MRI (magnetic resonance imaging) of the abdomen, confirmed a mass inseparable from the tail of the pancreas in the region of the splenic hilum. Additional records later received from the outside hospital revealed a VIP (vasoactive intestinal peptide) level of 378 pg/mL (normal range <75 pg/mL, radioimmunoassay technique, Mayo Clinic in Rochester, MN). The serum VIP was repeated at our facility and was 430 pg/mL. On hospital day 4, her CK peaked at 2472 U/L. To rule out an accessory spleen, a technetium 99 scan was performed and no uptake was identified correlating to the mass along the inferior aspect of the spleen; therefore, the mass was more likely to be neoplastic. The next day, a nuclear medicine somatostatin receptor scintigraphy (OctreoScan) revealed a mass disturbance in the region of the splenic hilum that was compatible with a neuroendocrine tumor. The patient underwent an endoscopic ultrasound, which revealed a 30×35 mm well-circumscribed hypoechoic mass at the tail of the pancreas, with microvascularity suggesting a NET. An ultrasound-guided biopsy confirmed a low-grade NET. One day later, a laparoscopic distal pancreatectomy with intraoperative ultrasound was performed. The distal pancreas confirmed the tumor was a well-differentiated pancreatic neuroendocrine carcinoma (Figure 3) with no invasion of the muscular proprialis and metastasis identified, and a low proliferation index (Figure 4). The 1 node that was present was negative for tumor. The total size was 3.7 cm, the biologic behavior was low-grade, and the stage was T2 N0 M0. Immunohistochemistry was performed and showed immuno-reactivity to cytokeratin AE1/AE3, cytokeratin CAM 5.2, and CD 56, (Figures 5–7) and was non-reactive to trypsin and chymotrypsin. Four days after surgery, the patient’s diarrhea had resolved, her VIP level was less than 50, and her electrolytes normalized rapidly. The final histopathology was a low-grade NET that was a well-differentiated carcinoma. Three months after surgery she was seen in follow-up and had no diarrhea and was off treatment.
Figure 1.

Well-circumscribed hyperdense rounded structure in the left upper quadrant (arrows) measuring 4.0 cm. A lesion involving the tail of the pancreas could not be excluded.
Figure 2.

CT enterography demonstrates a normal appearance to bowel structures, but a pancreatic tail mass (arrows),
Figure 3.

Synaptophysin immunostaining is positive, demonstrating the neuroendocrine nature of the tumor cells.
Figure 4.

The proliferation marker Ki-67 highlights 1–2% of the neoplastic cells (arrow), consistent with a low proliferation index.
Figure 5.

CD-56 IHC stain is positive, with membranous staining in the neoplastic population.
Figure 6.

Cytokeratin AE1/AE3 IHC stain shows membranous positivity in the tumor cells.
Figure 7.
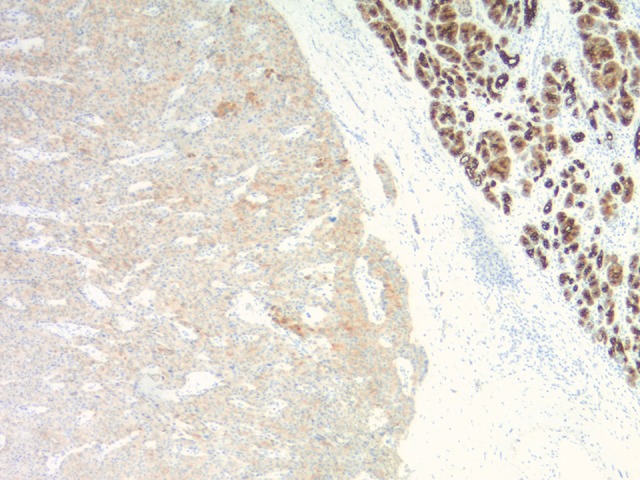
The Keratin CAM 5.2 stain shows patchy positivity within the tumor cells.
Discussion
This case is important for 3 reasons. First, a pancreatic VIPoma is an extremely rare neuroendocrine tumor. It secretes a biologically active peptide that stimulates intestinal water and electrolyte secretion into the intestinal lumen. The unregulated release of VIP results in unexplained high-volume secretory diarrhea despite fasting. Depending on the duration of symptoms, patients may have serious fluid and electrolyte disturbances, resulting in the watery diarrhea, hypokalemia, and achlorhydria or hypochlorhydria (WDHA) syndrome. If the disorder is undiagnosed and untreated it will progress, and this case illustrates the consequences of a delayed diagnosis. By the time the patient arrived for her second workup, her disease had progressed to the point of constitutional collapse.
The second important element to this case is that the patient’s WDHA progressed to rhabdomyolysis. We performed a MEDLINE search of the English language literature via PubMed using the MeSH terms and keywords “VIPoma”, “watery diarrhea, hypokalemia, achlorhydria syndrome” and “hypokalemic rhabdomyolysis” and only 1 case was found [8]. In both the previous report and our case, persistent WDHA eventually resulted in muscle necrosis and the release of intracellular muscle elements into the circulation. One key difference between the 2 cases, however, was in the presenting symptoms. Our patient presented with abdominal pain, which is common in rhabdomyolysis [9] but is not typical in symptomatic VIPoma with WHDA. Our patient’s pain is difficult to explain since she did not have metastatic disease, but she did have over 3 months of refractory, explosive diarrhea. Nonetheless, abdominal pain clearly can be part of the clinical presentation of a solitary pancreatic VIPoma with WDHA in a patient who is developing a more serious constitutional destructive rhabdomyolytic process. In the case of VIPomas, the likely etiology of the muscle necrosis is the hypokalemia. Significant potassium depletion can impair the function of striated muscle, which will often cause the observed weakness and immobility of that muscle. When the hypokalemia is severe or prolonged, frank rhabdomyolysis can develop [10]. The clinical syndrome can vary from pigmenturia to severe multi-organ complications, which includes renal failure, cardiac arrhythmias, and disseminated intravascular coagulation. Early identification is critical to a successful resolution [11]. Early identification, however, can be difficult, depending on the extent of diarrhea. In fact, 16% of patients with pancreatic VIPoma have no WDHA at all [7]. Consequently, diagnosing the tumor requires a high index of suspicion of the condition as well as a serum VIP concentration in excess of 75 pg/ml [8]. Most tumors are >3 cm when discovered, and thus typically can be identified and localized on abdominal CT and MRI [12]. Somatostatin receptor scintigraphy (OctreoScan) using radio-labeled octreotide (Indium-111 [111-In]) is a whole-body scan that can detect somatostatin receptors and is helpful in staging the tumor [8]. Our patient’s scan demonstrated her tumor was confined to the primary location. Endoscopic ultrasound can detect tumors <3 mm and is also helpful in the preoperative planning stage [13]. Finally, positron emission tomography (PET) tracers can be used for functional imaging in patients with somatostatin receptor-positive NETs.
The third important element to this case concerns the treatment. Surgical resection of a pancreatic VIPoma is the definitive treatment. Partial pancreatectomy performed by laparotomy was the procedure of choice for tumors located in the tail or body of the pancreas until the mid-2000’s [14]. However, over the last decade, a minimally invasive approach has become standard [15]. Laparoscopic surgical resection of a pancreatic VIPoma is not unique, but this is the first case that had not only a malignant VIPoma and WDHA but also rhabdomyolysis that was managed with minimally invasive technique. Other select treatments for symptomatic VIPoma include a somatostatin analog. which can acutely lower VIP and has even been used for long-term therapy [16]. The tumor is usually well-differentiated and slow-growing; therefore, prolonged palliation is possible. In aggressive, recurrent, or refractory tumors, molecularly targeted therapy may play a role. Our patient did not receive a somatostatin analog because she had a complete remission after surgery. Moreover, no chemotherapy was given since published opinion suggests there is controversy whether these tumors respond to systemic chemotherapy [17]. Her postoperative management consisted of serial follow-up serum VIP levels and abdominal imaging. Her VIPoma was an early-stage, well-differentiated carcinoma; therefore, a partial pancreatic resection was performed using minimally invasive techniques.
Conclusions
The progression of WDHA to rhabdomyolysis caused by the unregulated release of VIP is a life-threatening condition. An awareness of the disease and its diagnosis can allow medical mitigation before there is constitutional damage from severe fluid and electrolyte imbalance and possibly even metastasis. This case illustrates that a delay in diagnosis can result in severe consequences. We also suggest that a minimally invasive approach with laparoscopy is desirable, even if the disease process has progressed past symptomatic dehydration to life-threatening rhabdomyolysis.
Footnotes
Conflict of interest
None.
References:
- 1.Klöppel G, Perren A, Heitz PU. The gastroenteropancreatic neuroendocrine cell system and its tumors: The WHO classification. Ann NY Acad Sci. 2004;1014:13–27. doi: 10.1196/annals.1294.002. [DOI] [PubMed] [Google Scholar]
- 2.Ayub A, Zafar M, Abdulkareem A, et al. Primary hepatic vipoma. Am J Gastroenterol. 1993;88(6):958–61. [PubMed] [Google Scholar]
- 3.Smith SL, Branton SA, Avino AJ, et al. Vasoactive intestinal polypeptide secreting islet cell tumors: A 15-year experience and review of the literature. Surgery. 1998;124(6):1050–55. doi: 10.1067/msy.1998.92005. [DOI] [PubMed] [Google Scholar]
- 4.Modlin IM, Lewis JJ, Ahlmann H, et al. Management of unresectable malignant endocrine tumors of the pancreas. Surg Gynecol Obstet. 1993;176:507–18. [PubMed] [Google Scholar]
- 5.Yoshioka M, Sakazume M, Fukagawa M, et al. A case of the watery diarrhea-hypokalemia-achlorhydria syndrome: Successful preoperative treatment of watery diarrhea with a somatostatin analogue. Jpn J Clin Oncol. 1989;19(3):294–98. [PubMed] [Google Scholar]
- 6.Onozawa M, Fukuhara T, Minoguchi M, et al. Hypokalemic rhabdomyolysis due to WDHA syndrome caused by VIP-producing composite pheochromocytoma: A case in neurofibromatosis type 1. Jpn J Clin Oncol. 2005;35(9):559–63. doi: 10.1093/jjco/hyi139. [DOI] [PubMed] [Google Scholar]
- 7.Ende K, Henkel B, Brodhun M, et al. A 45-year-old female with hypokalemic rhabdomyolysis due to VIP-producing composite pheochromocytoma. Z Gastroenterol. 2012;50(6):589–94. doi: 10.1055/s-0031-1299111. [DOI] [PubMed] [Google Scholar]
- 8.Kibria R, Ahmed S, Ali SA, Barde CJ. Hypokalemic rhabdomyolysis due to watery diarrhea, hypokalemia, achlorhydria (WDHA) syndrome caused by vipoma. South Med J. 2009;102(7):761–64. doi: 10.1097/SMJ.0b013e3181a5cead. [DOI] [PubMed] [Google Scholar]
- 9.Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: Pathophysiology and diagnosis. Eur J Intern Med. 2007;18(2):90–100. doi: 10.1016/j.ejim.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 10.Gennari FJ. Hypokalemia. N Eng J Med. 1998;339:451–58. doi: 10.1056/NEJM199808133390707. [DOI] [PubMed] [Google Scholar]
- 11.Rupert SA. Pathogenesis and treatment of rhabdomyolysis. J Am Acad Nurse Pract. 2002;14(2):82–87. doi: 10.1111/j.1745-7599.2002.tb00095.x. [DOI] [PubMed] [Google Scholar]
- 12.Kirkwood KS, Debas HT. Neuroendocrine tumors: Common presentations of uncommon diseases. Compr Ther. 1995;21(12):719–25. [PubMed] [Google Scholar]
- 13.Rosch T, Lightdale CJ, Botet JF, et al. Localization of pancreatic endocrine tumors by endoscopic ultrasonography. N Engl J Med. 1992;326:1721–26. doi: 10.1056/NEJM199206253262601. [DOI] [PubMed] [Google Scholar]
- 14.Chu QD, Hill HC, Douglass HO, Jr, et al. Predictive factors associated with long-term survival in patients with neuroendocrine tumors of the pancreas. Ann Surg Oncol. 2002;9(9):855–62. doi: 10.1007/BF02557521. [DOI] [PubMed] [Google Scholar]
- 15.Cesaretti M, Bifulco L, Costi R, et al. Pancreatic resection in the era of laparoscopy: State of Art. A systematic review. Int J Surg. 2017;44:309–16. doi: 10.1016/j.ijsu.2017.07.028. [DOI] [PubMed] [Google Scholar]
- 16.Debas HT, Gittes G. Somatostatin analog therapy in functioning neuroendocrine gut tumors. Digestion. 1993;54:68–71. doi: 10.1159/000201080. [DOI] [PubMed] [Google Scholar]
- 17.Joyce DL, Hong K, Fishman EK, et al. Multi-visceral resection of pancreatic VIPoma in a patient with sinistral portal hypertension. World J Surg Oncol. 2008;6:80. doi: 10.1186/1477-7819-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]