

Abstract
Recent studies have indicated that circulating noncoding RNAs (ncRNAs) such as miRNAs are stable biomarkers for the diagnosis and prognosis of human diseases. However, due to low concentrations of circulating ncRNAs in blood, data normalization in plasma/serum ncRNA experiments using next-generation sequencing and quantitative real time RT-qPCR is a challenge. We found that the current normalization methods based on synthetic external spiked-in controls or published endogenous miRNA controls are inappropriate as they are not stably expressed and therefore fail to reliably detect differentially expressed ncRNAs. Using the alternative of individual ncRNAs as biomarkers, we considered a ratio-based normalization method calculated taking the ratio of any two ncRNAs in the same sample and used the resulting ratios as biomarkers. We mathematically verified the method to be independent of spiked-in and internal controls, and more robust than existing reference control based normalization methods to identify differentially expressed ncRNAs as potential biomarkers for human diseases. Thus, the ratio-based method can solve the difficult normalization problem for circuiting ncRNA data to identify reliable biomarkers to meet real clinical practice.
Graphical Abstract
MicroRNAs (miRNAs) are endogenous, small noncoding RNAs (ncRNAs), usually 18–25 nucleotides long. They have been found to play crucial roles in post-transcriptional regulation of mRNA.1 MiRNAs play a pivotal role in cell differentiation, proliferation, and apoptosis, and are implicated in many types of disease including cancer,2 diabetes,3 and cardiovascular4 and neural5 diseases. Besides miRNAs, many other small ncRNAs including small-nucleolar RNAs (snoR-NAs), Piwi-interacting RNAs (piRNAs), and tRNAs are important in regulating gene expression at many levels, such as chromatin architecture, transcription, mRNA stability and translation, and are known to be perturbed in cancer and other diseases. For instance, snoRNAs comprise a highly abundant group of small ncRNAs, and a limited number of snoRNAs have been reported to have ncRNA-like functions in gene splicing and silencing.6,7 Recent studies have demonstrated that three snoRNAs displayed altered expression in nonsmall cell lung cancer (NSCLC) patients, and SNORA42 may act as an oncogene in lung tumorigenesis.6
In recent years, a series of studies have shown that miRNAs can also be detected in bodily fluids such as serum,7 plasma,8 saliva,9 milk,10 sputum,11 and urine.12 Circulating miRNAs have been detected packaged in exosomes13 or microvesicles (MVs),14 or bound to specific proteins such as Ago-2.15 Once in the extracellular space, miRNAs could be taken up by other cells (cell-to-cell communication), degenerated by RNases, or excreted.16 Even though the mechanism of secretion and incorporation of miRNAs has not been fully understood, circulating miRNAs may be involved in physiological and pathological events.17
These findings opened a door for circulating ncRNAs as noninvasive biomarkers for diagnostics and prognostics of different kinds of diseases.2,3,15,17 Due to high sensitivity, specificity, and low template requirements, the most common method used for measuring circulating miRNAs is reverse transcription quantitative PCR (RT-qPCR).2 However, due to very low concentrations of circulating RNAs in the body fluids, accurately measuring circulating miRNA expression is a great challenge. Moreover, similar to gene expression analysis, final RT-qPCR results for circulating miRNA are affected by factors of systematic bias such as variations in the amount of starting material, sample collection, RNA isolation, reverse transcription, and PCR amplification. So, normalization reference control molecules are used to normalize circulating miRNA PCR data to minimize biases and fairly evaluate circulating miRNA expression. Current reference control molecules include external and internal endogenous controls. Many researchers choose to use spike-in synthetic RNA sequence (like C. elegans miR-39 and miR-54, or plant miRNAs) as extremal reference controls for normalization of circulating miRNA qPCR analysis.18 A variety of internal controls have been used. For instance, one of the snoRNAs, such as RNU6B was initially utilized to normalize circulating miRNA data,19 but was later found to be deregulated according to particular diseases and tumor prognosis.20 Many studies considered a reference miRNA, like miR-16, that was shown to have variation in plasma samples of cancer patients.21 Due to the lack of consensus normalization methods, data consistency and reproducibility across different studies are often not comparable. Therefore, there is an urgent need to find the best normalization method for the circulating miRNA data.
In this study, we proposed a ratio based normalization method for circulating ncRNA data. We first calculated the ratio of any two ncRNAs in the same sample, and then compared the ratio expression levels between different groups rather than comparing the level of a single ncRNA. Since the two ncRNAs are simultaneously measured in the same sample under the same conditions, the relative expression level in ratio of the two ncRNAs will reflect the true value for comparison by canceling biases.
Using ratio as biomarkers has been applied to some diseases. For instance, the AB42/AB40 ratio has been a promising biomarker for Alzheimer’s disease (AD), and the Apo B/A1 ratio is a much better biochemical indicator for people with obesity. Using the miRNA ratios as a tool for miRNA, RT-qPCR data have been also reported in cancer biomarker papers.22 However, there is no specific method paper to recommend a ratio based normalization method as a good way to normalize circulating ncRNA sequencing and RT-qPCR data. At present, almost 99% of publications involved in circulating ncRNAs (miRNAs) are still using external or internal reference control molecules for normalizing circulating PCR data. Some papers are still desperately searching for better reference controls for normalizing circulating miRNA data.23 It is necessary to have a specialized method article to introduce the advantage of the ratio method.
In the study, we have first mathematically proven that the ratio method is logically correct and independent of any external or internal reference control molecules, and is superior to any currently available external or internal control based normalization methods. This ratio strategy may provide a practical approach in clinical application of circulating ncRNA bio-markers for human diseases. Here, we also presented some initial results for early detection of lung cancer according to the ratio based method.
MATERIALS AND METHODS
Patient Cohorts.
We selected 130 subjects in our Lung Cancer Biorepository at Rush University Medical Center (Chicago, IL) from 2004 to 2010 for the method study. The subjects included 50 patients with early staged (stage I, II) lung adenocarcinoma, 15 squamous cell lung cancer (SCC), 35 benign cases, and 29 normal individuals for this study. Demographic information for these patients and controls is listed in Table S1 of the Supporting Information (SI).
Plasma Samples Collection, RNA Isolation, and Illumina Next Generation Sequencing.
All plasma samples were collected using EDTA-anticoagulative tubes and centrifuged for at 4000 × g for 10 min, followed by a 15 min high-speed centrifugation at 12 000 × g to completely remove cell debris. The supernatant plasma was stored at −80 °C until analysis. All samples were collected at the first diagnosis. In this study, RNA isolation as described previously2 and an Illumina next generation sequencing in plasma samples as described previously24 are provided in the SI.
Reverse Transcription and Real-Time PCR.
NcRNAs were measured using Taqman miRNA assay kits (Applied Biosystems, U.S.A.) according to the manufacturer’s protocol. Briefly, about 30 ng enriched RNA was reverse transcribed with a TaqMan ncRNA Reverse Transcription Kit (Applied Biosystems, U.S.A.) in a 15 μL reaction volume. Abundance levels of ncRNAs were quantified in triplicate by RT-qPCR using human TaqMan MicroRNA Assay Kits (Applied Biosystems, U.S.A.) on Eppendorfiplex 4 system (Eppendorf North America, Hauppauge, NY). To bypass the normalization issue, we use the same ratio strategy instead of normalizing to reduce the experimental variations.
Statistical Analysis.
The ratio was calculated of any two ncRNAs in the same sample for both the sequencing data and RT-qPCR data. For RT-qPCR data, if a CT value is larger than 40, then it was changed to 40. Then abundance levels of ratio of two small ncRNA (ncRNA1/ncRNA2) were evaluated using comparative CT method (2−ΔCT), in which ΔCT = CT ncRNA1 − CT ncRNA2 in the same sample. We used the unpaired T-Test in SPSS 20.0 software to compare mean ncRNA ratios between adeno case, benign patient, and normal control groups after the ncRNA concentrations of plasma, with the significant p-value level set at 0.05. Chi-Square test in SPSS 20.0 software was used to compare the distribution of training and validation stages with regard to gender, race, and tumor stage and t test to age. The significant p-value level was set at 0.05 for all results. Support vector machine recursive feature election (SVM-RFE)25 algorithm was used to select the best ncRNAs (SI).
RESULTS
External Spiked-In C. elegans Cel-miR-54 Is Not a Reliable Control for Normalizing Circulating Small RNA Sequencing.
In order to identify circulating small ncRNA markers for detection of lung cancer, we performed whole genome level small ncRNA sequencing (smRNA-seq) using pooled samples based on human plasma samples to save cost and samples. We first conducted smRNA-seq to identify plasma miRNAs and some other circulating sncRNAs in 7 pooled samples. A total of 30 high-risk healthy individuals (healthy control), 30 individuals with benign nodule lesions, 30 early stage adenocarcinoma lung cancer, and 15 squamous cell lung cancer (SCC) samples were prospectively collected from Rush University Medical Center. These samples were pooled using 15 individual control, benign, and cancer samples of all age, sex, race, and smoking status matched. So, a total of 2 pooled samples were for each control, benign, and adenocarcinoma lung cancer, and only 1 for SCC. The overall experimental design is depicted in Figure S-1. About 500 μL of equally mixed plasma in each pooling sample were used for smRNA-seq This was done using the Illumina next generation sequencing platform at City of Hope (CA). About 20 million reads per sample were generated with about 90% of reads aligned to human genome.
Since C-elegans Cel-miR-5426 is not contained in the human body, it was used as an external spiked-in control for the sequencing. An equal amount of Cel-miR54 was added into the pooled samples before RNA extraction. So we expected to get equal read number of cel-miR-54 in the entire pooled sample. However, the read number for cel-miR-54 was found to be quite different across the 7 pooled samples (Figure 1). The highest number was 220 for one adenocarcinoma lung cancer pooled sample while none for the SCC pooled sample. These variations in data suggested that the external spiked-in control Cel-miR-54 is not a reliable control for normalizing smRNA-seq data, and thus we examined other candidates.
Figure 1.

Read number of spiked-in external control Cel-miR-54. There are a total of 7 pooling plasma samples that were used for small-RNA sequencing. An equal amount of synthesized C. elegans external of Cel-miR-54 was added into the pooling samples before RNA isolating and sequencing. Each pool contained 15 mixed samples. LC represents normal healthy control (2 pooling samples), BE represents Benign (2 pooling samples), AD represents lung adenocarcinoma (2 pooling samples), and SC represents squamous cell lung cancer (1 pooling sample).
External Spiked-In C. elegans Cel-miR-54 Is Not a Reliable Control for Normalizing Circulating Quantitative RT-PCR (RT-qPCR) Small ncRNA Data.
Next we tested if external C. elegans Cel-miR-54 is a good spike-in control for normalizing circulating quantitative RT-PCR (RT-qPCR) small ncRNA data. We selected 129 samples (29 healthy control, 50 adenocarcinoma lung cancer, 35 benign, and 15 SCC) to perform RT-qPCR of Cel-miR-54. Equal amount of Cel-miR-54 was added into the same volume of plasma (200 μL) before RNA was isolated in the individual samples. We found that the CT values of published external control Cel-miR-54 were quite unstable; the CT values ranged from about 16.76 to about 33.13 (Figure 2A). The highest and lowest CT values had difference of 16.37, which is around 84695-fold difference from original data. Surprisingly, we did not get approximately equal CT values across the samples even though the same amount of Cel-miR-54 was added in all samples. These additional experiments further supported the conclusion that external spiked-in Cel-miR-54 is not a consistent control for normalizing circulating RT-qPCR of small ncRNA data either.
Figure 2.
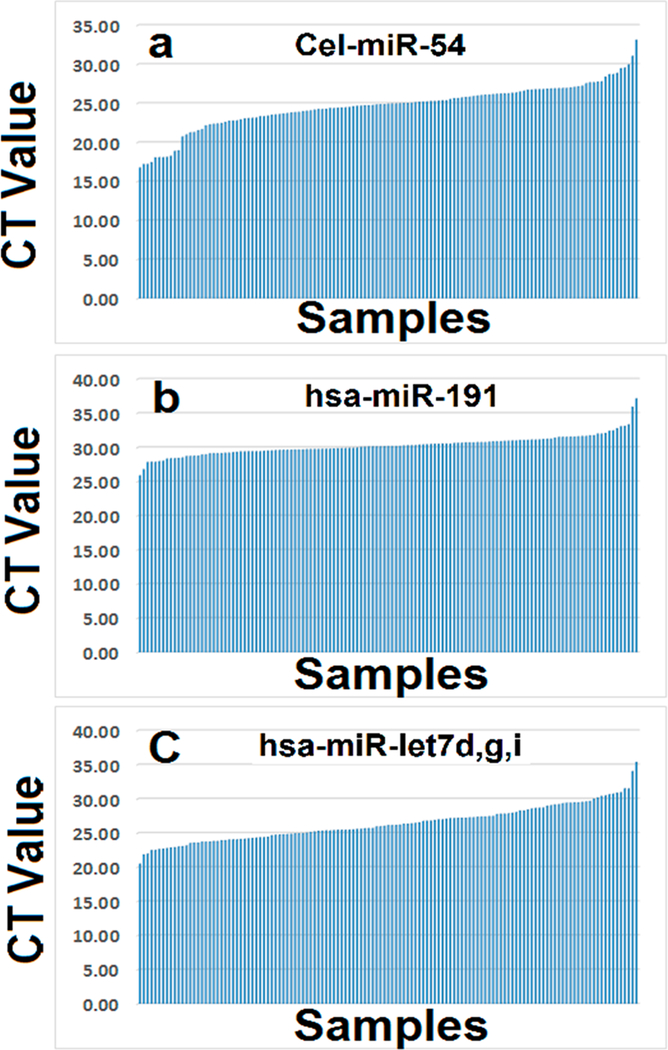
RT-qPCR CT values of external and internal reference controls in cancer and noncancer samples. CT values were sorted based on the number of a total of 129 samples including lung cancer, benign and normal healthy control plasma samples. (a) CT values of external C. elegans Cel-miR-54 across 129 samples. (b) CT values of endogenous reference control hsa-miR-191 across 129 samples. (c) CT values of endogenous reference control of averaged hsa-Let-miR-let 7d,g,i across 129 samples.
Endogenous Controls Are Not Good for Normalizing Circulating Quantitative RT-PCR (RT-qPCR) Small ncRNA Data.
After failing to use external spiked-in control such as Cel-miR-54 to normalize circulating ncRNA RT-qPCR data, we sought whether we could use endogenous controls to normalize circulating ncRNA RT-qPCR data. On the basis of published reports, we chose hsa-miR-191,27 hsa-miRNAs, Let-7d, Let-7g, and Let-7i28 as our endogenous controls. On the basis of the same volume of RNA (about 2 μL) isolated from the equal volume (200 μL) of plasma samples which were the same as we used for external control cel-miR-54 (Figure 2), we conducted RT-qPCR for the endogenous controls in the 129 samples. The CT values of published internal controls including hsa-miR-191(Figure 2B) and averaged hsa-MiRNAs, Let-7d, Let-7g, and Let-7i (Figure 2C) were also ranged quite differently and unstably expressed. Thus, they may not be suitable as reference controls for normalizing circulating ncRNA RT-qPCR data.
Ratio Based Normalization Method for Circulating ncRNA Profiling Data Is Independent of Any Internal or External Normalization Controls.
Failure of external or internal controls (Figure 2) for normalizing circulating ncRNA profiling data led to test a ratio-based normalization method. The method that calculates the ratio of any two ncRNAs in the same sample, and then compares the ratio expression levels between different groups rather than comparing the level of a single ncRNA was found to be reliable. For illustration, if expression values of normal and cancer samples for miRNA1 are 4 and 8 (row 1, Table 1), and those for miRNA2 are 8 and 4 (row 2), then the fold changes between normal and cancer will be 2 (upregulated) and −2 (downregulated), respectively. Likewise, if expression values of normal and cancer internal control 1 (IC 1) be 2 and 4 (row 3) and internal control 2 (IC 2) be 4 and 2 (row 4), then the fold changes between normal and cancer also will be 2 and −2, respectively. In such cases, if miRNA1 is normalized by IC1, the fold change is 1 (no change; row 5) while normalized by IC2, the fold change is 4 (up-regulated in cancer; row 7). Likewise, if miRNA2 is normalized by IC1, the fold change is −4 (down-regulated in cancer; row 6) while normalized by IC2, the fold change is 1 (no change; row 8). However, in the ratio-based normalization method, the ratio of any two miRNAs in the sample will not be changed by any internal controls used (rows 9−11). Thus, the method is not only independent of any unreliable internal or external controls but also cancels out systematic bias factors giving a reliable relative expression level in ratios.
Table 1.
Ratio Based Normalization Method
| row | MiRNAs | normal | cancer | \fold changea |
|---|---|---|---|---|
| 1 | miRNA1 | 4 | 8 | 2 |
| 2 | miRNA2 | 8 | 4 | −2 |
| 3 | Internal Control 1 (IC1) | 2 | 4 | 2 |
| 4 | Internal Control 2 (IC2) | 4 | 2 | −2 |
| 5 | miRNA1/IC1 | 4/2 = 2 | 8/4 = 2 | 1 |
| 6 | miRNA2/IC1 | 8/2 = 4 | 4/4 = 1 | −4 |
| 7 | miRNA1/IC2 | 4/4 = 1 | 8/2 = 4 | 4 |
| 8 | miRNA2/IC2 | 8/4 = 2 | 4/2 = 2 | 1 |
| 9 | (miRNA1/IC1)/(miRNA2/IC1)=miRNA1/miRNA2 | 2/4 = 0.5 | 2/1 = 2 | 4 |
| 10 | (miRNA1/IC2)/(miRNA2/IC2)=miRNA1/miRNA2 | 1/2 = 0.5 | 4/2 = 2 | 4 |
| 11 | miRNA1/miRNA2 | 4/8 = 0.5 | 8/4 = 2 | 4 |
Positive value means upregulation in cancer, and negative value means downregulation in cancer.
Ratio Based Normalization Method Is Mathematically Correct.
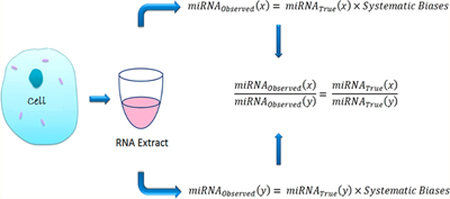
Using miRNA as an example, our ultimate goal is to get the biologically true miRNA expression value (miRNAtrue). The observed miRNA expression value (miRNAobserved) achieved from an experiment is not the true miRNA expression value due to the result of it being impacted by different systematic factors. In the case of RT-qPCR, the systematic factors could include RNA isolation (I), reverse transcription (R), PCR (P), different time (T), and so on. Therefore, in a specific sample, say, sample 1 (S1), observed any miRNA (x) expression value could be set as follows:
| (1) |
Similarly, we assume the systematic factors in the same sample for the miRNA(y) is the same; the observed miRNA expression value for it in the same S1 could also be set as follows:
| (2) |
| (3) |
From eq 3, we see that the ratio of observed two miRNAs in the same sample will equal to the true ratio value of the two true miRNAs. Thus, mathematically, the ratio value of two observed miRNAs in the same sample can reflect the true biological value of the two miRNAs that we want to measure.
CT values of RT-qPCR are logarithmic with base 2. So, we can derive the log ratio values of two miRNAs equal to the difference of two CT values of the two miRNAs (eq 4). The difference of CT values thus makes the calculation much easier and more convenient for clinically practice use based on RT-qPCR data.
| (4) |
Mathematically the Ratio Based Normalization Method Is Better than Internal or External Control Normalization Method.
Even though we have mathematically shown the ratio based normalization method is logically correct, it is only possible under the assumption of systematic factors are same for different miRNAs in the same sample. In theory, it is right because those two miRNAs are in the same samples and should impacted by the same systematic factors. Moreover, the reference such as an internal control (IC) based normalization method does the same. Let us mathematically look at the true expression value of miRNA(x) in sample S1.
We know, observed expression of miRNA(x) and the internal control for S1 can be obtained as follows:
| (5) |
| (6) |
where, FactorS1 = IS1 × RS1 × PS1 × TS1
| (7) |
Similarly for the true expression value of the same miRNA(x) in sample S2, see the following:
| (8) |
| (9) |
where factorS2 = IS2 × RS2 × PS2 × TS2
| (10) |
If,
| (11) |
Then (eq 7) can be considered as follows:
| (12) |
Equation 10 can be considered as follows:
| (13) |
The common external or internal control based normalization method (eqs 12 and 13), that we are currently using is based on two assumptions. First, it assumes that the measured miRNA and the internal control in the same sample are influenced by the same systematic factors (eqs 5, 6, 8, and 9); second, it also assumes that the true internal control values across different samples are the same (eq 11). However, it is hard to know whether the second assumption is true or not. The ratio-based method only assumes different miRNAs in the same sample share the same systematic factors; therefore, we mathematically showed that the ratio based method is better than reference control based normalization methods.
Ratio Based Normalization Method Can Find More Significantly Differentially ncRNA Candidate Markers between Disease Groups.
Originally, we proposed the ratio based normalization method on circulating RT-qPCR data. Yet, the external spiked-in control failed to work for normalizing sequencing data. For example, given miRNA with at least 20 reads for an miRNA, we found 631 mature miRNAs (union) in the sequenced samples. Next, we calculated the ratio of any two miRNAs in a sample and could get 198 765 ratios (Figure 3), which will substantially increase our candidate miRNAs to find different expressed paired ratio markers between disease groups. To provide a list of differentially expressed miRNA ratios, we further did differential expression analysis with comparison between cancer vs control, cancer vs benign, and benign vs control of the pooled samples. On the basis of fold change ≥2 and p-value ≤0.05, we found a large number of significantly altered mature miRNA ratios (miRNA/miRNA) including 30 989 ratios between normal and cancer, 12 701 ratios between normal and benign, and 7044 ratios between benign and cancer. These significantly changed ratio numbers were much higher than the measurements of single miRNAs between the 3 groups based on global median normalization for single miRNA data (Figure 3).
Figure 3.

Differentiated miRNA numbers among 3 groups. X-axis represents the total measurable features (either miRNA of miRNA ratios), the differentiated number of normal healthy control vs lung adenocarcinoma, normal healthy control vs benign, and benign vs lung adenocarcinoma. An unpaired t test was used to identify differentiated miRNA or miRNA ratios. P value ≤0.05 and fold change cut off was 2.0. Ratio was calculated between any two miRNAs in the same sample.
Ratio Based ncRNA Biomarkers Separates Healthy Control from Lung Adenocarcinoma.
To test how these ratio based candidate ncRNAs distinguished lung cancer from noncancer samples, initially we chose about 20 paired significantly ncRNA ratios in the comparison of control vs cancer from sequencing data to conduct RT-qPCR in two stages, adenocarcinoma samples at early stages with age, race, sex, and smoking status matched. The training stage included 50 patients with early stage lung cancer, and 29 high-risk individuals without lung disease (controls) from Rush University Medical Center, and the validation stage included 44 patients with early stage lung cancer, and 51 controls from the Lung Cancer Biospecimen Resource Network at the University of Virginia. We found that a panel of seven small ncRNA pair ratios could differentiate patients with lung cancer from high-risk controls with an area under the curve (AUC) of 100.0%, a sensitivity of 100.0%, and a specificity of 100.0% at the training stage and an AUC of 90.2%, a sensitivity of 91.5%, and a specificity of 80.4% at the validation stage. The results have been published.29
DISCUSSION
Data normalization in plasma/serum ncRNA experiments using RT-qPCR is a challenge. In miRNA, due to the yields of total RNA from small-volume plasma or serum samples (i.e., 100 or 200 μL) are often below the limit of accurate quantification by spectrophotometry, and bias in sample collection, storage and processing affects the accuracy and reliability of the quantitative analysis of circulating miRNA. The inclusion of an external or endogenous reference control molecule is recommended to adjust technical variations in the RNA recovery procedure by the current experiments. Many researchers chose to spiked-in synthetic RNA sequence (like C. elegans miR-39 and miR-54, or plant miRNAs) into the sample for normalization of circulating miRNA qPCR analysis. In our study, we chose C. elegans Cel-miR-54 as an external control, and found it was not a reliable control in both sequencing and RT-qPCR data. This could be due to the synthetic miRNAs added directly to plasma degrades rapidly and are less stable than endogenous miRNAs when added to plasma.30,31 No matter what makes the external spiked-in controls not stable, even a stable spiked-in controls cannot correct sample variability. Some study has found that the exogenous C. elegans miRNAs, including cel-miR-39, cel-miR-54, and cel-miR-238, could not improve assay precision.32 Therefore, spiked-in controls based normalization for circulating ncRNAs is not an ideal method.
Some researchers have made efforts to seek the suitable endogenous control miRNAs (ECM); however, no such suitable ECMs have been established for blood miRNA quantification.33 For example, miR-16 is frequently used as a control,34 but elevated levels of miR-16 in serum correlate with bone metastasis in patients with breast cancer35 and it was reported that endogenous miR-16 was a poor normalizing factor.32 Since Chen X et al.28 reported that let-7d/g/i is a good endogenous control for normalizing circulating miRNA data, we tested using let-7d/g/i in the experiment. We found that they were not stably expressed across our samples. Chen’s samples were only derived from a Chinese population although lung cancer patients were included, which could be a reason why we did not get the similar results. The widely used endogenous control hsa-MiR-19127 did not work as a good control in our experiment either. We could endlessly test more endogenous controls such as U6,36 RNU44,37 RNU48,38 miR-16,39 miR-103,27 and miR-23a40 that have been commonly utilized nowadays. However, Chen’s paper has already found that these controls performed even worse than let-7d/g/i. As we know, ideal endogenous reference control should at least meet the criteria that they are stably expressed across all samples and experimental conditions. It is very hard to prove which candidate endogenous molecule meets the criteria.
Since there are no current consensus normalization external or endogenous factors for normalizing circulating ncRNAs (such as miRNAs), we propose a normalization method by analyzing circulating ncRNA values looking at the reciprocal ratios of ncRNAs in the same sample. We first calculated the ratio of any two ncRNAs in the same sample, then compared the ratio expression levels between different groups rather than compare the level of a single ncRNA. Since the two ncRNAs are simultaneously measured in the same sample under the same conditions such as collection, storage, and isolation, and PCR, or sequencing processing, the relative expression level in ratio of the two ncRNAs will reflect the true value for comparison. We found the ratio based method which is totally independent of any internal of external controls (Table 1). Therefore, on the basis of the method, we do not need to worry about which external or internal control molecules need to be selected and also they are hard to be confirmed as true reference controls.
We have first mathematically proven the ratio based normalization method is logically correct to reflect the true biological value of the two ncRNAs (such as miRNA) that we want to measure. Our mathematical proving demonstrates no need for any additional experiment to validate that the ratio based normalization is correct. The ratio based normalization method is better than any methods based on internal or external control normalization factors. We found that internal or external control normalization based method needs two assumptions. First, it assumes that the measured miRNA and the internal control in the same sample are influenced by the same systematic factors; second, it assumes that the true internal control values across different samples are the same. However, the ratio based method only assumes different miRNAs in the same sample share the same systematic factors, and it is hard to know whether the second assumption is true or not makes the ratio based method is superior than reference control based normalization method. The mathematical demonstration should be more convincing than any additional experimental data to show the ratio method is better than reference control based methods because mathematical validity is the extent to which a concept, conclusion or measurement is well-founded and corresponds accurately to the real world.
We have also compared the ratio based method with the global mean normalization method for miRNA data that were used in the microRNA quality control (miRQC) study (SI). We found the ratio method is better than the global mean normalization method. This is easy to understand because the global mean value will always change when the measured number of miRNA changes for any platforms. Therefore, for a given miRNA in any platforms, the normalized value for the same miRNA in the same sample will always be changed when the measured number of miRNAs changes as mean value differs in each set. However, no matter how many miRNAs are measured, the ratio for any two same miRNAs in the same sample will not be changed, which is certainly true for any platforms.
We are the first to apply the ratio based method to whole genomic level small ncRNA sequencing data. We found that a ratio based normalization method can find more differentially expressed ncRNA candidate markers between disease groups. It is also logically easy to understand, for example, given the ratio miRNA1/miRNA2 in the healthy normal and cancer groups, whether miRNA1 is upregulated or downregulated in cancer vs normal with respect to miRNA2. The fold change miRNA1/miRNA2 between cancer and normal should be larger than that of miRNA1 or miRNA2 alone. So the ratio based method will increase our chance to find clinically useful biomarkers when we may not be able to find significantly changed single markers.
Initially we have found that a panel of circulating 7 paired ncRNA ratios could separate lung adenocarcinoma from normal healthy control with 100% prediction accuracy in the training stage, also were successfully validated in an independent cohort. We not only tested miRNAs but also measured other types of ncRNAs such as snoRNAs and tRNAs.
The ratio based method is superior to any other reference controls such as external or internal control or global mean-based methods for normalizing circulating ncRNA data in terms of biomarker identification. This method is not only applicable to circulating ncRNA data, but also can be used for any types of molecules such as mRNAs, DNAs, proteins, and metabolites at any types of tissues across different organisms. Moreover, this method can be applied in any disease biomarker prediction including diabetes, cardiovascular, neural diseases, and so on.
Declarations Ethics Approval and Consent to Participate.
Human blood samples were obtained from Rush University Medical Center (Chicago, IL). All patient and healthy data were acquired with written informed consent from and in absolute compliance with the institutional review board at Rush University Medical Center (11020405-IRB01)
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institutes of Health (NIH) grants [R21CA164764, 1R01CA223490, 5P30GM114737, P20GM103466, 2U54MD007601, and U54MD007584] and Hawaii Community Foundation to Y.D.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.anal-chem.9b00821.
All data generated or analyzed during this study methods and analysis of MiRQC study samples (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Lee RC; Ambros V Science 2001, 294 (5543), 862–864. [DOI] [PubMed] [Google Scholar]
- (2).Chen H; Liu H; Zou H; Chen R; Dou Y; Sheng S; Dai S; Ai J; Melson J; Kittles RA; et al. J. Cancer 2016, 7 (5), 490–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Marchand L; Jalabert A; Meugnier E; Van den Hende K; Fabien N; Nicolino M; Madec A-M; Thivolet C; Rome S miRNA-375 a Sensor of Glucotoxicity Is Altered in the Serum of Children with Newly Diagnosed Type 1 Diabetes https://www.hindawi.com/journals/jdr/2016/1869082/ (accessed Sep 20, 2018). DOI: 10.1155/2016/1869082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zhou Y; Chen Q; Lew KS; Richards AM; Wang PJ Cardiovasc. Pharmacol. Ther. 2016, 21 (3), 296–309. [DOI] [PubMed] [Google Scholar]
- (5).Topol A; Zhu S; Hartley BJ; English J; Hauberg ME; Tran N; Rittenhouse CA; Simone A; Ruderfer DM; Johnson J; et al. Cell Rep. 2016, 15 (5), 1024–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mei Y-P; Liao J-P; Shen J; Yu L; Liu B-L; Liu L; Li R-Y; Ji L; Dorsey SG; Jiang Z-R; et al. Oncogene 2012, 31 (22), 2794–2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hu Z; Chen X; Zhao Y; Tian T; Jin G; Shu Y; Chen Y; Xu L; Zen K; Zhang C; et al. J. Clin. Oncol. 2010, 28 (10), 1721–1726. [DOI] [PubMed] [Google Scholar]
- (8).Shen J; Todd NW; Zhang H; Yu L; Lingxiao X; Mei Y; Guarnera M; Liao J; Chou A; Lu CL; et al. Lab. Invest. 2011, 91 (4), 579–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Matse JH; Yoshizawa J; Wang X; Elashoff D; Bolscher JGM; Veerman ECI; Bloemena E; Wong DT W. Clin. Cancer Res. 2013, 19 (11), 3032–3038. [DOI] [PubMed] [Google Scholar]
- (10).Simpson MR; Brede G; Johansen J; Johnsen R; Storrø O; Sætrom P; Øien T PLoS One 2015, 10 (12), No. e0143496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Xing L; Su J; Guarnera MA; Zhang H; Cai L; Zhou R; Stass SA; Jiang F Clin. Cancer Res. 2015, 21 (2), 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Mengual L; Lozano JJ; Ingelmo-Torres M; Gazquez C; Ribal MJ; Alcaraz A Int. J. Cancer 2013, 133 (11), 2631–2641. [DOI] [PubMed] [Google Scholar]
- (13).Valadi H; Ekström K; Bossios A; Sjöstrand M; Lee JJ; Lötvall JO Nat. Cell Biol. 2007, 9 (6), 654–659. [DOI] [PubMed] [Google Scholar]
- (14).Camussi G; Deregibus MC; Bruno S; Cantaluppi V; Biancone L Kidney Int. 2010, 78 (9), 838–848. [DOI] [PubMed] [Google Scholar]
- (15).Arroyo JD; Chevillet JR; Kroh EM; Ruf IK; Pritchard CC; Gibson DF; Mitchell PS; Bennett CF; Pogosova-Agadjanyan EL; Stirewalt DL; et al. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (12), 5003–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sozzi G; Pastorino U; Croce CM Cell Cycle 2011, 10 (13), 2045–2046. [DOI] [PubMed] [Google Scholar]
- (17).Kosaka N; Iguchi H; Ochiya T Cancer Sci. 2010, 101 (10), 2087–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Skog J; Wurdinger T; van Rijn S; Meijer D; Gainche L; Sena-Esteves M; Curry WT; Carter RS; Krichevsky AM; Breakefield XO Nat. Cell Biol. 2008, 10 (12), 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ng EKO; Chong WWS; Jin H; Lam EKY; Shin VY; Yu J; Poon TCW; Ng SSM; Sung JJ Y. Gut 2009, 58 (10), 1375–1381. [DOI] [PubMed] [Google Scholar]
- (20).Benz F; Roderburg C; Cardenas DV; Vucur M; Gautheron J; Koch A; Zimmermann H; Janssen J; Nieuwenhuijsen L; Luedde M; et al. Exp. Mol. Med. 2013, 45 (9), No. e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zuo Z; Calin GA; de Paula HM; Medeiros LJ; Fernandez MH; Shimizu M; Garcia-Manero G; Bueso-Ramos CE Blood 2011, 118 (2), 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Boeri M; Verri C; Conte D; Roz L; Modena P; Facchinetti F; Calabrò E; Croce CM; Pastorino U; Sozzi G Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (9), 3713–3718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Niu Y; Wu Y; Huang J; Li Q; Kang K; Qu J; Li F; Gou D Sci. Rep. 2016, 6, 35611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wu X; Somlo G; Yu Y; Palomares MR; Li AX; Zhou W; Chow A; Yen Y; Rossi JJ; Gao H; et al. J. Transl. Med. 2012, 10 (1), 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Pirooznia M; Yang JY; Yang MQ; Deng Y BMC Genomics 2008, 9 (1), S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Nair VS; Pritchard CC; Tewari M; Ioannidis JP A. Am. J. Epidemiol. 2014, 180 (2), 140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Peltier HJ; Latham GJ RNA 2008, 14 (5), 844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chen X; Liang H; Guan D; Wang C; Hu X; Cui L; Chen S; Zhang C; Zhang J; Zen K; et al. PLoS One 2013, 8 (11), e79652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dou Y; Zhu Y; Ai J; Chen H; Liu H; Borgia JA; Li X; Yang F; Jiang B; Wang J; et al. BMC Genomics 2018, 19 (1), 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chen X; Hu Z; Wang W; Ba Y; Ma L; Zhang C; Wang C; Ren Z; Zhao Y; Wu S; et al. Int. J. Cancer 2012, 130 (7), 1620–1628. [DOI] [PubMed] [Google Scholar]
- (31).Schwarzenbach H; Nishida N; Calin GA; Pantel K Nat. Rev. Clin. Oncol. 2014, 11 (3), 145–156. [DOI] [PubMed] [Google Scholar]
- (32).McDonald JS; Milosevic D; Reddi HV; Grebe SK; Algeciras-Schimnich A Clin. Chem. 2011, 57, 833. [DOI] [PubMed] [Google Scholar]
- (33).Brase JC; Johannes M; Schlomm T; Fälth M; Haese A; Steuber T; Beissbarth T; Kuner R; Sültmann H Int. J. Cancer 2011, 128 (3), 608–616. [DOI] [PubMed] [Google Scholar]
- (34).Kroh EM; Parkin RK; Mitchell PS; Tewari M Methods 2010, 50 (4), 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ell B; Mercatali L; Ibrahim T; Campbell N; Schwarzenbach H; Pantel K; Amadori D; Kang Y Cancer Cell 2013, 24 (4), 542–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Corney DC; Flesken-Nikitin A; Godwin AK; Wang W; Nikitin AY Cancer Res. 2007, 67 (18), 8433–8438. [DOI] [PubMed] [Google Scholar]
- (37).Anglicheau D; Sharma VK; Ding R; Hummel A; Snopkowski C; Dadhania D; Seshan SV; Suthanthiran M Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (13), 5330–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Nohata N; Hanazawa T; Kikkawa N; Mutallip M; Sakurai D; Fujimura L; Kawakami K; Chiyomaru T; Yoshino H; Enokida H; et al. J. Hum. Genet. 2011, 56 (8), 595–601. [DOI] [PubMed] [Google Scholar]
- (39).Zhi F; Chen X; Wang S; Xia X; Shi Y; Guan W; Shao N; Qu H; Yang C; Zhang Y; et al. Eur. J. Cancer 2010, 46 (9), 1640–1649. [DOI] [PubMed] [Google Scholar]
- (40).Shen Y; Li Y; Ye F; Wang F; Wan X; Lu W; Xie X Exp. Mol. Med. 2011, 43 (6), 358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.