

SUMMARY
Regulation of chromatin structure is essential for controlling access of DNA to factors that require association with specific DNA sequences. Here we describe the development and validation of engineered chromatin remodeling proteins (E-ChRPs) for inducing programmable changes in nucleosome positioning by design. We demonstrate that E-ChRPs function both in vitro and in vivo to specifically reposition target nucleosomes and entire nucleosomal arrays. We show that induced, systematic positioning of nucleosomes over yeast Ume6 binding sites leads to Ume6 exclusion, hyperacetylation, and transcriptional induction at target genes. We also show that programmed global loss of nucleosome-free regions at Reb1 targets is generally inhibitory with mildly repressive transcriptional effects. E-ChRPs are compatible with multiple targeting modalities, including the SpyCatcher and dCas9 moieties, resulting in high versatility and enabling diverse future applications. Thus, engineered chromatin remodeling proteins represent a simple and robust means to probe and disrupt DNA-dependent processes in different chromatin contexts.
In Brief
Donovan et al. develop a versatile approach to alter local or genome-wide nucleosome positions in vivo through engineered chromatin remodeling proteins (E-ChRPs). These alterations in chromatin structure affect downstream processes including histone modification and transcription. E-ChRPs represent a powerful method of investigating the causes and consequences of chromatin states.
Graphical Abstract
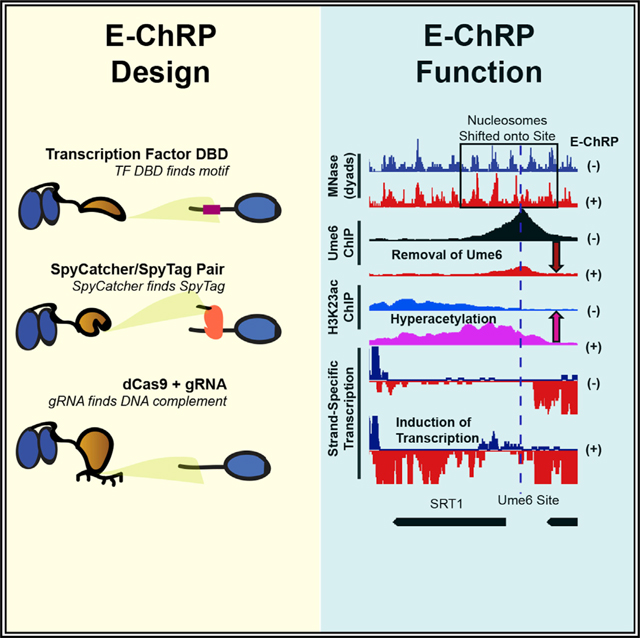
INTRODUCTION
The nucleosome is the fundamental repeating unit of chromatin, composed of DNA wrapped around an octamer of histone proteins. Although nucleosomes are dynamic structures that are constantly assembled, disassembled, and repositioned in the genome, their positions at gene-regulatory elements such as transcription start sites (TSSs) show characteristic organization (Lai and Pugh, 2017). Thus, nucleosome positions are thought to have regulatory implications for DNA-dependent processes such as transcription, replication, and DNA repair (Hauer and Gasser, 2017; MacAlpine and Almouzni, 2013; Venkatesh and Workman, 2015). Because positions of nucleosomes in the genome play a major role in determining DNA sequence accessibility, the ability to precisely manipulate nucleosome positions would have profound implications for investigating and controlling DNA-dependent processes in vivo.
ATP-dependent chromatin remodeling factors couple the hydrolysis of ATP to the movement of nucleosomes along a fragment of DNA (Cairns et al., 1996; Fazzio and Tsukiyama, 2003; Längst et al., 1999; Smith and Peterson, 2005; Stockdale et al., 2006; Tsukiyama et al., 1994). By altering the positions of nucleosomes, this family of enzymes controls the accessibility of underlying DNA in vivo, thereby regulating DNA-dependent processes. The CHD and ISWI families of chromatin remodelers contain a conserved catalytic ATPase that drives chromatin remodeling by binding and hydrolyzing ATP (Zhou et al., 2016) and a C-terminal region that interacts with extranucleosomal DNA to modify the direction of nucleosome repositioning (Gangaraju and Bartholomew, 2007; Hota et al., 2013; McKnight et al., 2011; Ryan et al., 2011).
Previous work established that chromatin remodeling by S. cerevisiae Chd1 can be targeted to specific nucleosomes by replacing the native, nonspecific Chd1 DNA binding domain (DBD) with sequence-specific DBDs (McKnight et al., 2011; Nodelman and Bowman, 2013). We previously showed that hybrid Chd1 fusions with exogenous, sequence-specific DBDs predictably move nucleosomes onto their recruitment sequences in vitro (McKnight et al., 2011). We recently demonstrated that fusion of Chd1 to the Zn2Cys6 DBD from Ume6, a meiotic repressor from yeast, allows directed nucleosome positioning at target genes across the S. cerevisiae genome (McKnight et al., 2016).
Here we have simplified and greatly expanded the customizable design and validated the function of sequence-targeted chromatin remodeling proteins using diverse targeting strategies. These engineered chromatin remodeling proteins (E-ChRPs) work with a wide variety of targeting domains and can occlude target DNA sequences by precisely repositioning nucleosomes onto recruitment motifs. We show that E-ChRPs possessing transcription factor (TF) DBDs can incorporate TF binding sites into nucleosomes to block binding of and prevent signaling by endogenous TFs genome wide. E-ChRPs can also be directly recruited to DNA-associated TFs through SpyTag/SpyCatcher pairs (Zakeri et al., 2012), allowing identification and occlusion of TF-bound genomic loci. Finally, we show that positioning of nucleosomes can be achieved by a dCas9-targeted E-ChRP using both canonical and noncanonical gRNAs.
RESULTS
The core E-ChRP design was inspired by previous work (McKnight et al., 2011, 2016) where individual sequence-specific DBDs replaced the C-terminal nonspecific DBD of a functional S. cerevisiae Chd1 chromatin remodeler fragment (Figure 1A). Yeast Chd1 is an ideal enzyme for engineered chromatin remodeling because it is monomeric, displays robust nucleosome positioning activity on nucleosome substrates derived from multiple organisms, and is less influenced by histone modifications than other chromatin remodelers (Ferreira et al., 2007; Hauk et al., 2010). Following the Chd1 catalytic module, we incorporated restriction sites flanking the targeting domain in vectors allowing recombinant expression in E. coli, constitutive expression from ADH1 or GPD promoters in S. cerevisiae (Mumberg et al., 1995), or galactose-inducible expression after integration at the HO locus in S. cerevisiae (Voth et al., 2001). This scaffold allows easy swapping of the C-terminal targeting domain, resulting in a simple method to design chromatin remodelers that can be localized to desired nucleosomes. To demonstrate the versatility of the approach, we incorporated and assessed engineered chromatin remodeling through multiple TF DBDs, through SpyCatcher/SpyTag pairs, and through dCas9 targeting (Figure 1B). We first assessed the ability of different E-ChRPs to reposition target-containing mononucleosomes in a purified biochemical assay (Eberharter et al., 2004). To validate in vivo function, we introduced E-ChRPs into S. cerevisiae and measured global nucleosome positions using MNase sequencing (MNase-seq). Functional E-ChRPs can position targeted nucleosomes onto recruitment motifs as measured using mononucleosome sliding toward recruitment sequences in vitro or target motif occlusion by nucleosomes in vivo (Figures 1C–1F).
Figure 1. Strategies for Optimizing Targeted Nucleosome Positioning by E-ChRPs.

(A) Architecture of the E-ChRP core where the yeast Chd1 catalytic domain is linked to a targeting domain with a flexible linker.
(B) Summary of targeting methods used in this work, including sequence-specific DBD targeting to a recognition motif (top), SpyCatcher domain covalently attaching to a SpyTag-containing chromatin-bound protein (middle), and dCas9-bound gRNA interacting with a complementary sequence (bottom).
(C) Predicted outcome from targeted E-ChRPs, indicating that select nucleosomes are positioned by the E-ChRP onto the recruitment site.
(D) Nucleosome repositioning in vitro by Chd1-Ume6 with and without 11 repeats of glycine-glycine-serine between the Chd1 catalytic domain and Ume6 DBD. Nucleosomes with the Ume6 recognition motif (URS1) located 20 or 40 bp from the nucleosome edge were incubated with Chd1-Ume6(DBD) or Chd1-GGSx11-Ume6(DBD), and nucleosomes were resolved using native PAGE (top). Nucleosome positions before and after remodeling were resolved using 6% native PAGE. Summary of repositioning of each substrate by Chd1 fusions (bottom).
(E) Genome Browser image showing nucleosome dyad positions at a representative Ume6 binding site (URS1) for a parental strain lacking endogenous Ume6 (black), after introduction of Chd1-Ume6 without (yellow) or with (red) a flexible linker. Dashed line indicates the location of the Ume6 binding motif. Arrow indicates nucleosome that is more efficiently positioned with a Chd1-GGSx11-Ume6(DBD) fusion.
(F) Average nucleosome positioning (dyad signal) at genomic Ume6 binding sites showing Chd1-GGSx11-Ume6(DBD) more readily positions nucleosomes distal to the recognition element than Chd1-Ume6(DBD) lacking a flexible linker
Development and Optimization of a Targeted Remodeler Core
We previously demonstrated that fusion of a foreign DBD to the Chd1 catalytic core leads to occlusion of a recruitment motif by targeted and directional repositioning of nucleosomes (McKnight et al., 2011, 2016). Although functional both in vitro and in vivo, remodeler fusions in which the DBD was directly fused to the Chd1 core resulted in a limited “reach,” and nucleosomes residing further than 20 bp from the DNA recognition element were not efficiently moved. In addition, the creation of new remodeler fusion proteins was previously cumbersome and lacked versatility. To address these limitations, we first created the E-ChRP scaffold (Figure 1A), which consists of the catalytic core of the yeast Chd1 protein followed by a flexible linker including 11 repeats of the glycine-glycine-serine sequence, which was previously shown to extend the Chd1 reach in vitro (Nodelman and Bowman, 2013). We next created an array of plasmids for recombinant bacterial expression or yeast constitutive or inducible expression, allowing one-step cloning of a desired fusion domain (Figure 1A).
We examined whether the addition of a flexible linker between the Chd1 remodeler core and DBD increased the reach of these E-ChRPs. We tested the ability of an E-ChRP with a DBD from the S. cerevisiae meiotic repressor Ume6 to move mononucleosomes containing a recognition motif, URS1 (Park et al., 1992), 20 or 40 bp from the nucleosome edge (Figure 1D). Without a flexible linker, the Chd1-Ume6 E-ChRP was strongly stimulated only when the motif was 20 bp away (compare lane 1 with lane 3 and lane 2 with lane 4). In contrast, the addition of 11 repeats of glycine-glycine-serine (GGSx11) allowed the remodeler to efficiently mobilize both nucleosome substrates (compare lane 1 with lane 5 and lane 2 with lane 6). Additionally, the final location of the positioned nucleosomes was dependent on the location of the recognition motif (Figure 1D, compare lanes 5 and 6). Consistent with this increased reach in vitro, the Ume6 E-ChRP containing a GGSx11 linker positioned a larger fraction of distal nucleosomes onto target sequences across the S. cerevisiae genome as measured using paired-end MNase-seq (Figures 1E and 1F). Because the flexible linker led to more robust E-ChRP activity and our design was compatible in vitro and in vivo, we used this scaffold in all subsequent experiments.
Remodeler Fusions Are Highly Versatile In Vitro and In Vivo
We next tested mononucleosome targeting of multiple E-ChRPs with various DBDs. We fused the DBD from E. coli AraC, S. pombe Res1, D. melanogaster engrailed, or R. norvegicus glucocorticoid receptor to the E-ChRP scaffold. To determine if these E-ChRPs were functional on target nucleosomes in vitro, we generated end-positioned mononucleosomes assembled on the 601-positioning sequence (Lowary and Widom, 1998) with 125 bp of flanking DNA. The extranucleosomal DNA either contained or lacked a consensus binding motif corresponding to each different fusion tested. E-ChRPs were able to mobilize nucleosomes possessing well-defined recruitment motifs for each distinct E-ChRP DBD (Ades and Sauer, 1994; Alroy and Freedman, 1992; Ayté et al., 1995; Niland et al., 1996) as measured using a native PAGE nucleosome sliding assay (Figure 2A). These E-ChRPs were inactive on nucleosomes lacking their respective motifs (Figure 2A, lanes 22–27), demonstrating specificity for target substrates in vitro. Fusion of the native, sequence-nonspecific DBD from Chd1 to our E-ChRP scaffold showed no apparent discrimination against DNA sequences and was capable of fully mobilizing the nonspecific mononucleosome control (Figure 2A, lanes 28–30).
Figure 2. E-ChRPs with Distinct TF DBDs Specifically Position Target Nucleosomes In Vitro and In Vivo.

(A) Nucleosome sliding assay demonstrating functionality of Increasing concentrations of E-ChRPs containing AraC DBD (lanes 1–5 and 22–24), Res1 DBD (lanes 6–10 and 25–27), engrailed DBD (lanes 11–15), glucocorticoid receptor DBD (lanes 16–20), or Chd1 endogenous DBD (lanes 28–30) in vitro. Nucleosomes in lanes 1–20 possess recognition motifs in extranucleosomal DNA for the respective E-ChRP (Ades and Sauer, 1994; Alroy and Freedman, 1992; Anderson et al., 1995; Ayté et al., 1995; Khan et al., 2018; Niland et al., 1996), while lanes 21–30 have no recognition motif. Lower electrophoretic mobility indicates repositioning of nucleosomes away from their end positions.
(B) Yeast genomic nucleosome dyad positions are shown at representative Ume6 targets (URS1, top) or engrailed targets (TAAT, bottom) in the presence or absence of Ume6 E-ChRP or engrailed E-ChRP. Motif-proximal nucleosomes are highlighted next to indicated motifs, with blue arrows showing direction of nucleosome movement. Cartoon representations of nucleosome positions are provided for each locus.
(C) Nucleosome dyad signal at a representative locus in yeast demonstrating positioning of nucleosomes toward recruitment motif (dashed line) by galactose-inducible Ume6 E-ChRP or a constitutively expressed E-ChRP under the ADH1 promoter.
See also Figure S1.
To determine whether E-ChRPs can be differentially targeted to specific subsets of nucleosomes in vivo, we introduced E-ChRPs into S. cerevisiae on a constitutive, ADH1-driven expression plasmid. When the E-ChRP possessed a Ume6 DBD, nucleosomes were repositioned toward Ume6 binding motifs across the genome (Figure 2B). Although no nucleosome changes were detected at other genomic loci, we cannot rule out the possibility that these E-ChRPs possessed low-level nonspecific nucleosome positioning activity throughout the genome, as MNase-seq detects population-average nucleosome positions. Similarly, an E-ChRP containing the engrailed DBD moved nucleosomes onto engrailed motifs in the yeast genome without altering nucleosome positions at Ume6 binding motifs (Figure 2B). We also introduced Ume6 and engrailed E-ChRPs into yeast under the high-expression GPD (TDH3) promoter on a 2 μm plasmid (Mumberg et al., 1995). Expression of the Ume6 E-ChRP from this construct resulted in positioned nucleosomes at target sites without detected off-target activity, similar to an ADH1-driven E-ChRP (Figure S1A). Again, we cannot rule out the possibility of low-level, global off-target activity that does not give rise to reproducibly shifted nucleosomes at specific sites. However, introduction of this higher expression plasmid containing an engrailed E-ChRP only produced viable transformants in which the E-ChRP construct was deleted, truncated, or mutated. This obligate inactivation of the engrailed E-ChRP at high expression levels may result from promiscuous action of the engrailed E-ChRP at tens of thousands of potential target sequences, which would presumably disrupt global nucleosome positioning in a pleiotropic manner. Importantly, even when driven from the GPD (TDH3) promoter, neither the Ume6 E-ChRP nor the engrailed E-ChRP was active at target sites when the Chd1 remodeler core contained a catalytically inactive Walker B (D513N) substitution (Hauk et al., 2010; Walker et al., 1982) (Figures S1A and S1B).
To gain temporal control of E-ChRPs in vivo, we introduced the Ume6 E-ChRP under a galactose-inducible promoter integrated at the HO locus in yeast (Voth et al., 2001). Prior to addition of galactose, endogenous Ume6 associates with its consensus sequence across the genome and cooperates with the ISW2 complex to position motif-proximal nucleosomes, leaving 30 bp between the nucleosome edge and URS1 motif (Goldmark et al., 2000; McKnight et al., 2016). After galactose-driven transcriptional induction of the Ume6 E-ChRP, a majority of nucleosomes nearest the URS1 site are efficiently repositioned to occlude the URS1 motif within 2 h (Figure 2C). This galactose-inducible approach allows a larger fraction of nucleosomes to be repositioned in a population compared with the same E-ChRP under the control of a constitutively active ADH1 promoter, potentially commensurate with differing expression levels under these distinct promoters (Figure 2C). Taken together, these results suggest that E-ChRPs can be specifically targeted with temporal regulation using multiple distinct DBDs that recognize sequence motifs with high or low complexity both in vitro and in vivo.
E-ChRPs Can Inducibly Remove Transcription Factors
After E-ChRP activity, the population average of positioned nucleosomes displays a maximum nucleosome dyad signal 49 nt from the recruitment motif center (distribution from 73 to 15 nt), which corresponds to the motif being buried within the 145 bp nucleosomal footprint by approximately 25 bp. This matches what we have mapped previously in vitro (McKnight et al., 2011), where we see occlusion of motifs by 20–30 bp in the presence of E-ChRPs. We reasoned that because the post-induction nucleosome position results in the Ume6 recruitment motif becoming buried within nucleosomal DNA, remodeling by the Ume6 E-ChRP should interfere with binding of endogenous Ume6 (Figure 3A). To test this possibility, we tagged endogenous Ume6 with a FLAG epitope and measured Ume6-FLAG binding by chromatin immunoprecipitation sequencing (ChIP-seq) before and after induction of the Ume6 E-ChRP. Prior to induction, reproducible Ume6-FLAG binding was observed at URS1 sites across the genome (Figure S2A). After induction of the Ume6 E-ChRP, which shifted nucleosomes over URS1 sites, Ume6 binding (as measured by Ume6-FLAG ChIP signal) was strongly reduced or eliminated at many genomic locations (Figures 3B and S2A). When we sorted Ume6 binding sites on the basis of whether proximal nucleosome positions were shifted after Ume6 E-ChRP induction, we noticed that loss of Ume6-FLAG signal was strikingly reduced where nucleosomes were shifted but minimally reduced where nucleosomes were not shifted (Figures 3B and S2B). To verify that this reduction in Ume6-FLAG signal was not due to direct binding competition between endogenous Ume6-FLAG and the E-ChRP Ume6 DBD, we measured Ume6-FLAG ChIP signal in the presence of a catalytically inactive Ume6 E-ChRP. This construct, which cannot move nucleosomes but retains the Ume6 DBD, did not similarly reduce Ume6-FLAG signal (Figures 3B and S2A). Together with the observation that Ume6 was removed only where nucleosome shifts were observed (Figures 3B and S2B) but not where nucleosomes remained, we therefore believe the detected loss of Ume6 is due to site occlusion by E-ChRP activity rather than direct competition for DNA binding by the E-ChRP DBD. Thus, E-ChRPs can inducibly move nucleosomes over target sequences to restrict access of the underlying DNA to endogenous DNA binding factors.
Figure 3. E-ChRPs Can Inducibly Remove Endogenous Ume6 from Chromatin.

(A) Cartoon depiction of Ume6 E-ChRP activity blocking association of endogenous Ume6 at target sites.
(B) Representative examples of E-ChRP targets where endogenous Ume6 is removed (top) or not removed (bottom) after galactose induction of Ume6 E-ChRP. The catalytic null E-ChRP retains a Ume6 DBD but has a Walker B (D513N) substitution in the Chd1 catalytic core.
(C and D) Representative Genome Browser images showing E-ChRP-dependent nucleosome dyad movement (within black rectangles) onto Ume6 sites (dashed line) with associated reduction of Ume6-FLAG ChIP, increase in acetylation, and transcriptional induction for the SRT1 locus (C) and MEI5 locus (D).
See also Figure S2.
We next assessed the functional consequences of endogenous Ume6 removal by E-ChRP induction. Because Ume6 recruits the Rpd3 histone deacetylase to strongly repress target genes (Goldmark et al., 2000; Kadosh and Struhl, 1997), we tested whether Ume6 displacement could lead to increased histone acetylation (as measured using H3K23ac ChIP) and transcriptional induction (as measured using strand-specific RNA sequencing [RNA-seq]). Strikingly, at sites where E-ChRP induction led to loss of Ume6-FLAG signal, we detected strong increases in histone acetylation and associated increases in transcription (Figures 3C, 3D, S2B, and S2C). Importantly, the increased histone acetylation was observed only at targets where nucleosome positions were altered and Ume6-FLAG was removed, indicating that these effects were likely not due to simple competition between the E-ChRP and endogenous Ume6. These highly correlated results, where temporally regulated nucleosome positioning onto Ume6 target sites led to Ume6 removal and loss of Ume6 mediated repression, demonstrate that E-ChRPs can be used for disrupting the binding and activity of transcriptional regulators at target sites.
SpyCatcher E-ChRPs Allow Simple Targeting to Chromatin-Bound Loci
One limitation of the above-described E-ChRPs is their need to compete with endogenous factors for binding sites. To circumvent this problem, we created an E-ChRP in which the SpyCatcher protein is fused in place of a DBD in the Chd1 E-ChRP scaffold. SpyCatcher specifically recognizes a short (~1 kDa) SpyTag epitope, forming an isopeptide linkage that allows covalent protein fusions to be created in vitro and in vivo (Zakeri et al., 2012). This fusion provides two major improvements to the E-ChRP system. First, by simply appending SpyTag to different chromatin-binding factors of interest, nucleosome positioning can be achieved by a single SpyCatcher E-ChRP without the need to design new DBD fusions (Figure 4). Second, by tagging a TF at its endogenous locus, nucleosomes can only become targetable for the SpyCatcher E-ChRP when the TF is bound to chromatin (Figure 4A). This bypasses the requirement of a vacant DNA binding site to target a DBD-containing E-ChRP, allowing access to sequences in the genome that could otherwise be blocked by a stably bound TF. In sum, this strategy produces a single SpyCatcher E-ChRP that can be targeted to any chromatin-bound protein of interest in the genome by simple attachment of a short SpyTag.
Figure 4. Development and Validation of E-ChRPs Containing SpyCatcher/SpyTag Pairs.

(A) Cartoon representation for introducing a Chd1-SpyCatcher E-ChRP into cells containing SpyTagged, chromatin-bound proteins. The SpyCatcher domain forms a covalent isopeptide bond with SpyTag, allowing localization of E-ChRP activity to endogenously bound chromatin proteins.
(B) Nucleosome sliding assay demonstrating that a single SpyCatcher E-ChRP cannot position nucleosomes without a SpyTag-containing DBD (lanes 2–4) but can use a SpyTagged AraC DBD (lanes 5–7) or engrailed DBD (lanes 8–10) to reposition nucleosomes containing respective DBD recognition motifs. The AraC and engrailed recognition elements are located 7 and 11 nt from the nucleosome edge, respectively.
(C) Representative motif in yeast where ADH1-driven SpyCatcher E-ChRP can reposition nucleosomes (dyads) at a Ume6 binding site in the presence of Ume6-SpyTag (left) and genomic analysis of nucleosome dyad positioning by SpyCatcher E-ChRP at 202 intergenic instances of the Ume6 recognition sequence in cells containing SpyTagged Ume6 (right).
(D) Genomic analysis of nucleosome dyad positions in Reb1-SpyTagged cells (left) or Ume6-SpyTagged cells (right) before and after 2 h induction of galactose-inducible SpyCatcher E-ChRP. Heatmaps show change in nucleosome dyad signal after induction of SpyCatcher E-ChRP, and individual traces show average positions of nucleosomes in each cluster before and after SpyCatcher E-ChRP induction for each SpyTag-DBD strain.
See also Figure S3.
To validate the function of the SpyCatcher E-ChRP design, we purified recombinantly expressed Chd1-SpyCatcher and two SpyTag-containing DBDs. Mononucleosomes harboring recognition sequences for each DBD were incubated with the Spy-Catcher E-ChRP with and without addition of SpyTag-engrailed(DBD) or SpyTag-AraC(DBD). The SpyCatcher E-ChRP has no activity on nucleosome substrates in the absence of a Spy-Tag-DBD pair (Figure 4B, lanes 2–4), because SpyCatcher has no intrinsic DNA binding affinity. However, addition of either Spy-Tag-AraC(DBD) (Figure 4B, lanes 5–7) or SpyTag-engrailed(DBD) (Figure 4B, lanes 8–10) resulted in robust repositioning of mononucleosomes in vitro, demonstrating the versatility of this system. We next introduced the SpyCatcher E-ChRP under a constitutive ADH1 promoter into S. cerevisiae cells in which a C-terminal SpyTag was added to full-length Ume6 at the endogenous locus. As expected, we observed repositioned nucleosomes at URS1 sites across the genome, indicating chromatin remodeling at Ume6-bound loci (Figure 4C).
To achieve temporal control of this modular system in vivo, we appended SpyTag to the C terminus of either Ume6 or Reb1, a yeast general regulatory factor, in a strain harboring a galactose-inducible SpyCatcher E-ChRP at the HO locus. After induction of SpyCatcher E-ChRP expression, nucleosomes were shifted toward Ume6 binding sites in cells containing Ume6-Spy-Tag or toward Reb1 binding sites in cells containing Reb1-Spy-Tag (Figures 4D, S3A, and S3B). Interestingly, the fraction of shifted nucleosomes was generally low at Ume6 binding sites in Ume6-SpyTag cells but comparatively higher at Reb1 binding sites in Reb1-SpyTag strains (Figure 4D). This difference could be explained by higher occupancy or stability of Reb1 than Ume6 binding at target sites, which would allow a greater fraction of Reb1-tethered SpyCatcher to mobilize motif-proximal nucleosomes. Consistent with this possibility, the cellular abundance of Ume6 is significantly lower than that of Reb1 (Kulak et al., 2014). For Reb1-SpyTag strains, the positioning of a single motif-proximal nucleosome by the SpyCatcher E-ChRP initiated the shift of an entire array of nucleosomes toward the target motif (Figure S3C), consistent with previous observations that the positioning of a “barrier nucleosome” influences and constrains positions of an entire array of nucleosomes (Mavrich et al., 2008; McKnight et al., 2016).
Interestingly, the positioning of nucleosomes appeared to occur on only the 5′ side of the Reb1 recognition sequence, suggesting the orientation of Reb1 binding affects the ability of Chd1 to reach nucleosomes near binding sites (Figures 4D and S3C). This restriction could be explained by a constrained C terminus of Reb1 when bound to chromatin, which is consistent with similarly constrained Reb1-MNase cleavage patterns seen in previous chromatin endogenous cleavage sequencing (ChEC-seq) experiments (Zentner et al., 2015). Importantly, the nucleosome shifts observed are not due to the inability of Reb1 to associate with target sites, because the changes in nucleosome positions are distinct from bidirectional nucleosome repositioning observed when Reb1 is depleted from the nucleus using the anchor-away method (Kubik et al., 2015) (Figure S4A). In aggregate, induction of the SpyCatcher E-ChRP in a Reb1-SpyTag strain leads to nucleosome depleted region (NDR) occlusion at roughly 650 TSSs (Figure S4B). Unexpectedly, the fraction of nucleosomes shifted at individual Reb1 binding sites varied greatly in our dataset, with some sites exhibiting repositioning of nearly 100% of motif-proximal nucleosomes in the population and others having much smaller fractions moved (Figures 5A–5C). These differences are not explained by initial nucleosome occupancy or location differences (Figure 5B) but are possibly related to relative Reb1 occupancy at different genomic locations.
Figure 5. E-ChRP Targeting to Chromatin-Bound Reb1 Provides Differential Occupancy Information at Reb1 Motifs.

(A) Nucleosome dyad signal at 943 intergenic Reb1 binding motifs in Reb1-SpyTag strains before (left) and after (right) 2 h induction of SpyCatcher E-ChRP. Rows are ordered by change in nucleosome positioning after galactose induction. Purple shading highlights the region to which nucleosomes are moved by SpyCatcher E-ChRP in the Reb1-SpyTag strain.
(B) The purple mobile fraction from (A) was split into deciles (~50 motifs per decile) showing average positioning by SpyCatcher E-ChRP for each decile. Dashed lines indicate the pre-induction, unremodeled position (red) or post-induction, remodeled position (black). Ume6-SpyTag control traces are provided for the top and bottom deciles demonstrating that SpyCatcher E-ChRP cannot function at Reb1 sites in the presence of Ume6-SpyTag instead of Reb1-SpyTag.
(C) Genome Browser images for representative loci showing nucleosome dyad positioning by SpyCatcher E-ChRP in a Reb1-SpyTag strain for the top, middle, and bottom deciles. Purple shading indicates the motif-proximal, repositioned nucleosomes. Dashed lines indicate the location of Reb1 motif.
See also Figures S4 and S5.
To further characterize the ability of SpyCatcher E-ChRP to identify fractional Reb1 occupancy at Reb1 binding sites, we compared our dataset with crosslinking ChIP-seq, CUT&RUN (Skene and Henikoff, 2017), ORGANIC (Kasinathan et al., 2014), and ChEC-seq (Zentner et al., 2015) datasets. There was striking correlation among our data, ORGANIC, and ChEC-seq, with some motifs exclusively showing Reb1 occupancy when measured using these three methods (Figure S5). Minimally, the observation that all nucleosomes are shifted at some Reb1 binding sites in a population of cells argues that some Reb1 sites are nearly 100% occupied, as E-ChRP-derived nucleosome movement cannot be observed without Reb1 binding (Figure 5C). Although these Reb1 occupancy estimates are conflated with presence, accessibility and relative occupancy of motif-proximal nucleosomes, our data suggest that Spy-Catcher E-ChRPs can serve as a relative measure of protein localization in cells that is orthogonal to ChIP, allowing a lower limit estimate of SpyTagged protein occupancy at individual binding motifs in the genome.
Loss of Nucleosome Depleted Regions at Reb1 Sites Is Mildly Repressive
Because we observed gross movement of nucleosomes toward Reb1 binding sites across the genome, we wondered whether there would be severe transcriptional consequences associated with loss of Reb1-created nucleosome-depleted regions (NDRs). We measured RNA abundance 13 h after SpyCatcher E-ChRP induction (+galactose) compared with a raffinose-treated control. Despite the occlusion of > 600 NDRs, we noted only very mild growth and transcriptional defects associated with nucleosome positioning toward Reb1 sites. Although several genes were repressed when their respective NDRs were occluded by E-ChRP activity at Reb1 sites, the extent of transcriptional repression was generally very low (Figures 6A and 6C). Surprisingly, transcription of many genes was not altered even when their NDR sizes were grossly reduced by E-ChRP activity (Figure 6B). Globally, the loss of NDR is generally repressive at Reb1 target genes, though the extent of repression is typically less than 3-fold and cannot be predicted on the basis of NDR loss alone. Inclusion of an external RNA spike-in control (Figure 6C) supports the conclusion that no gross transcriptional changes were detected, though we cannot rule out RNA buffering processes that could correct gross transcriptional changes (Sun et al., 2013). These surprisingly small transcriptional defects associated with large reduction in NDR size at Reb1 target genes suggests that the presence of an NDR is not intimately linked to transcription output.
Figure 6. Loss of NDR by Reb1-Tethered SpyCatcher E-ChRP Is Generally Repressive.

(A) Representative Genome Browser images showing loss of NDR and associated transcriptional reduction by SpyCatcher E-ChRP at Reb1-bound loci.
(B) Representative Genome Browser images showing significant reduction of NDR size by SpyCatcher E-ChRP without detectable changes in transcription at Reb1-bound loci.
(C) Volcano plots showing transcription changes (x axis) and statistical significance (y axis) associated with SpyCatcher E-ChRP induction in Reb1-SpyTag strains for all genes (left) or genes containing Reb1 binding motifs in their promoter regions (right). Loci from (A) and (B) are labeled. Vertical dashed lines indicate 4-fold transcription change. Horizontal dashed line represents a corrected p value threshold of 0.01. p values were calculated in DEseq2 using the default Wald test. Most motif-containing genes with significant changes in transcription move in the repressive (left) direction. Scatterplot showing no change in global transcription as measured by External RNA Controls Consortium (ERCC) spike-in RNA control is provided (center).
Custom Nucleosome Positioning with a dCas9 E-ChRP
Although the E-ChRPs described above show robust nucleosome positioning activity when targeted through various DBDs or through SpyCatcher/SpyTag pairs, their ability to alter nucleosome positions depends on the interaction between pre-existing DBDs with defined DNA motifs. To overcome this limitation and allow targeted positioning of single nucleosomes by design, we created a dCas9 (Gilbert et al., 2013; Qi et al., 2013) E-ChRP (Figure 7). This construct allows versatile targeting to specific nucleosomes by designing proximal guide RNAs (gRNAs). We recombinantly expressed the dCas9 E-ChRP in E. coli and purified the ~300 kDa fusion protein. To test its ability to move gRNA-targeted nucleosomes, we reconstituted end-positioned mononucleosomes and designed gRNAs with or without complementarity to the extranucleosomal DNA. Successful gRNA-stimulated chromatin remodeling would result in movement of the nucleosome toward the target sequence, producing a slower migrating, centrally positioned nucleosome (Figure 7A). Although nucleosomes were efficiently moved toward the center of DNA fragments with control Chd1 protein, introduction of Chd1-dCas9 and complementary gRNA resulted in supershifted complexes with unresolved nucleosome positions. Even in the presence of 1,000-fold competitor DNA for 3 days, the Chd1-dCas9 fusion protein would not release from gRNA-targeted nucleosomes (Figure 7B). This inability of dCas9 to release from target sequences is consistent with the ability of dCas9 to specifically bind and interfere with transcription in cells because of stable R-loop formation (Jinek et al., 2012; Laughery et al., 2019; Qi et al., 2013). Importantly, the inability of the dCas9 E-ChRP to release from substrate prevents its utility in vitro and could possibly limit use for precise, gRNA-targeted nucleosome positioning in cells.
Figure 7. Remodeling Can Be Targeted Using a dCas9 E-ChRP with Canonical and Noncanonical gRNA Substrates.

(A) Cartoon depiction of dCas9-targeted chromatin remodeling and predicted repositioning of target nucleosome.
(B) Nucleosome sliding assay showing irreversible association of the dCas9 E-ChRP with target nucleosomes and free DNA. The “slid ctrl” includes ATP and a control Chd1 protein capable of positioning nucleosomes toward the center of the DNA fragment. Excess unlabeled competitor DNA was added 3 days prior to loading. Nucleosome concentration was 25 nM, and E-ChRP concentration was 75 nM.
(C) Cartoon depiction of canonical and nonstandard gRNA protospacers.
(D) Nucleosome sliding assay demonstrating robust positioning of nucleosomes by a dCas9 E-ChRP targeted with nonstandard gRNAs in single-turnover (top) or multi-turnover (bottom) conditions. The slid ctrl (lanes 2 and 12) includes ATP and a control Chd1 protein. The “no gRNA” lanes (3 and 13) contain a dCas9 E-ChRP, ATP, and no gRNA. No-target gRNA samples (lanes 4 and 14) contain a dCas9 E-ChRP and a gRNA without any sequence complementarity to the substrate nucleosome. Note that the order of lanes between the upper and lower panels is similar except for loading of hp-gRNA (lanes 5, 6, 17, and 18) and mm-gRNA (lanes 7, 8, 15, 16).
(E) Nucleosome sliding assay demonstrating lack of stable association of dCas9 E-ChRPs with target nucleosomes or DNA in the presence of nonstandard gRNAs. For the upper gel, competitor DNA was added prior to loading. The bottom gel contains the identical reactions in the same order as the upper gel, but no competitor DNA was added before loading.
(F) Genome Browser image showing both canonical and mismatched gRNAs allow repositioning of nucleosome dyads (note appearance of dyad signal in blue rectangle regions) by a dCas9 E-ChRP at the ALP1 locus when the gRNA sequence is exposed but not when gRNA sequence is occluded in the absence of dCas9 E-ChRP.
See also Figure S6.
To promote release of the dCas9 E-ChRP from nucleosome substrates, we used gRNAs with noncanonical structures (Figure 7C), including a truncated gRNA (tru-gRNA) (Fu et al., 2014) containing only 14 nt of complementarity to target sequences, a gRNA with a PAM-distal hairpin (Josephs et al., 2015) that has predicted self-annealing capacity and reduced affinity for target sequences, and a 20 nt gRNA with a PAM-distal 3 nt mismatch (mm-gRNA) that would result in an R-loop with a frayed end. Both the tru-gRNA and the mm-gRNA allowed efficient targeted repositioning of nucleosomes toward the gRNA binding site either through direct Chd1-dCas9 fusion or introduction of Chd1-SpyCatcher and dCas9-SpyTag pairs (Figure 7D, lanes 1–10; Figure S6). These noncanonical gRNAs promoted multi-turnover catalysis by the dCas9 E-ChRP, demonstrating that the weakened dCas9/gRNA complexes were stable enough to promote specific enzymatic activity but weak enough to readily and repeatedly disengage from its substrate (Figure 7D, lanes 11–20). Strikingly, dCas9-Chd1 targeted through weakened gRNAs did not require any competitor DNA to disengage from nucleosome substrates (Figure 7E). We believe this ability to readily dissociate from DNA targets while providing enough dwell time and specificity for targeted nucleosome positioning could provide a facile method to alter nucleosome positions by design. To test whether the dCas9 E-ChRP is functional in cells, we targeted the ALP1 locus with either perfect match or 3 nt mismatched gRNAs targeting a region that was either nucleosome occluded or within the nucleosome linker region. Both the perfect-match and mismatched gRNA allowed repositioning of a nucleosome array in the targeted region (Figure 7F), though only a fraction of nucleosomes in the population were moved in both cases. Nucleosome repositioning was observed only with a gRNA targeted within the nucleosome linker region but not when targeted to nucleosome-occluded DNA. These results suggest that dCas9-targeted nucleosome positioning can be achieved in cells but may require additional optimization; gRNA success is likely dependent on initial nucleosome positioning, and unlike what was observed in vitro, mm-gRNAs behave similarly to perfect-match gRNAs in a cellular context.
DISCUSSION
Although the E-ChRPs described in this work are highly versatile, allowing multiple targeting schemes, there are some limitations in the ability of E-ChRPs to position target nucleosomes. First, to create a functional Chd1-TF(DBD) fusion, the boundary of the DBD for the specific TF must be known, and it must fold in the context of the fusion protein. Although all fusions we have tested have been functional to date, we focused on stable and well-studied DBDs. Second, there is still a limitation on how far E-ChRPs can “reach.” On the basis of our in vivo mapping results, if a nucleosome edge is initially beyond ~75 bp from the E-ChRP recruitment site, nucleosome repositioning activity is less favorable. Moreover, E-ChRPs do not appear to have any de novo nucleosome deposition activity, so exaggerated nucleosome-free regions of the genome would not permit nucleosome positioning. Although we are very interested in the ability of SpyCatcher E-ChRPs to position different fractions of nucleosomes at different sites in the genome and we speculate that this is due to relative SpyTagged TF occupancy, our method is blind to TF binding sites where there are no motif-proximal nucleosomes. Furthermore, it would be challenging to use E-ChRPs in isolation to define the TF binding landscape because of the relatively noisy signal generated from fractional repositioned nucleosomes. This is especially true for sites at which small fractions of nucleosomes are repositioned. Finally, although the determination that dCas9 E-ChRPs work readily with mismatched gRNAs may allow better assessment of targeted chromatin modification effects (because dCas9 does not remain stably associated with the target sequence), we note that mismatched gRNAs naturally possess a lower capacity for target specificity. Despite these limitations, we believe E-ChRPs are versatile and powerful tools for studying direct consequences of chromatin changes on DNA-dependent processes.
We used a Ume6 E-ChRP to move nucleosomes onto Ume6 recruitment sequences across the yeast genome and found that Ume6 site occlusion resulted in loss of endogenous Ume6 binding and led to histone hyperacetylation and transcriptional induction of Ume6-repressed genes. This result demonstrates that E-ChRPs are capable of occluding DNA accessibility at targets to inactivate factors that cannot bind DNA in the context of a nucleosome. We were also able to assess the contribution of NDR formation to transcriptional regulation at Reb1 targets by tethering Chd1 directly to Reb1-bound loci. Not surprisingly, we found that loss of NDR is generally repressive, though the extent of repression is quite modest and not uniform at Reb1 target promoters. Interestingly, Reb1 is an essential gene in S. cerevisiae (Giaever et al., 2002) that is required for NDR formation, “roadblock” termination of transcription, H2AZ incorporation, TBP binding, and transcriptional regulation (Colin et al., 2014; Hartley and Madhani, 2009; Kubik et al., 2015; Kubik et al., 2018). We detected no gross changes in transcription termination at Reb1 sites at which NDRs were reduced by E-ChRP activity, likely because of redundant regulation by the TRAMP-exosome pathway (Colin et al., 2014). We speculate that the small changes in transcription noted when NDRs were lost at Reb1 sites may be due to the atypical cyclical nature required for NDR occlusion. In our system, Reb1 must bind and initiate NDR formation prior to E-ChRP occlusion of one side of the NDR. This means that after each cell cycle, Reb1 is likely able to bind and transiently create a full NDR at target sites. The SpyCatcher-tethered Chd1 then fills in one side of the NDR by positioning a nucleosome toward the Reb1 binding motif. Such transient NDR creation may allow initial regulatory events to occur, potentially including H2AZ incorporation, initial polymerase recruitment/PIC formation, and other events. In addition, NDR loss through Reb1-tethered Chd1 does not shrink NDR size as fully as Reb1 loss (Kubik et al., 2015) (Figure S4A). We believe this temporally controlled, cyclical, and robust approach to reduce NDR size will allow a versatile strategy to dissect function of GRFs and NDRs in transcriptional regulation in subsequent experiments.
In conclusion, we have created and validated the use of E-ChRPs as an easy and versatile method for altering the positions of specific nucleosomes both in vitro and in vivo. We have demonstrated that E-ChRPs have widespread compatibility with various DBDs and have created a single SpyCatcher E-ChRP that can be inducibly attached to chromatin-associated factors to move adjacent nucleosomes. We have shown that induced positioning of nucleosomes by E-ChRPs can establish new nucleosomal arrays, occlude TF binding motifs across the genome to block regulatory function, and report on relative TF occupancy at target motifs. Finally, we have developed a functional dCas9-targeted E-ChRP that works in vitro and in cells using canonical or noncanonical gRNAs with PAM-distal mismatches to robustly position and release from targeted nucleosomes in vitro.
We envision that future research can use E-ChRPs to probe questions directly relating the position of nucleosomes to downstream biological processes and can lead to insight into how cells can tolerate or correct ectopic nucleosome positioning events. We suspect that weakened mismatch-gRNAs may provide an alternative strategy to more transiently target specific changes in histone modification or nucleosome positioning changes in cells while limiting indirect consequences of stably or irreversibly bound dCas9 in specific systems (Laughery et al., 2019). We note, however, that dCas9-targeted Chd1 behaves differently in cells (in which nucleosome shifts are observed with both canonical and mismatched gRNAs) compared with our biochemical assay, where Chd1-dCas9 is functional only with weakened gRNAs. Finally, the ability to position nucleosomes onto target sequences may potentially lead to the development of E-ChRPs that block oncogenic or other disease-related TFs from accessing binding sites genome wide.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffrey McKnight (jmcknig2@uoregon.edu). All plasmids and strains generated in this study are available from the Lead Contact without restriction.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast (S. cerevisiae W303 RAD5+) strains used in this study are described in Table S1. Unless otherwise indicated, cells were grown at 30°C and 160 rpm in YPD (yeast extract-peptone-2% glucose) medium. Strains were streaked from glycerol stocks onto 2% agar YPD plates and grown at 30°C for 2–3 days. An isolated colony was then grown overnight in 25 mL of YPD. This pre-culture was used to inoculate 25 mL of YPD at an OD600 of 0.2 which was grown to an OD600 of 0.6–0.8 for chromatin analysis. Yeast containing non-integrating plasmids (p416- or p426-) were grown in SD-Ura overnight, diluted to OD600 = 0.2 in YPD and grown to OD600 = 0.6–0.8 for chromatin analysis. For galactose induction of E-ChRPs, cells were grown in YP media with 2% raffinose as the sole carbon source. In mid-log phase, raffinose (−induction) or galactose (+ induction) was added to a final concentration of 2% and cells were grown for 2 additional hours at 30°C with shaking. Cells were then fixed and harvested for chromatin analysis.
METHOD DETAILS
Yeast Strains and Plasmids
All yeast strains and plasmids used in this study are listed in Tables S1 and S2, respectively. Gene deletions were made by replacing the gene of interest with antibiotic resistance markers amplified from pAG vectors listed in Table S2. E-ChRPs were introduced to yeast either through plasmid transformation of a p416 or p426 vector containing the E-ChRP, or by inserting the E-ChRP at the HO locus through homologous recombination. To make Spytagged yeast strains, a C-terminal 3x-FLAG tag followed by the Spytag sequence (AHIVMVDAYKPTK) was cloned into a pFA6a vector. Tags were then inserted at the endogenous locus of interest by homologous recombination of PCR products from the respective tagging vectors listed in Table S2, using selectable drug markers. To recombinantly express Spytagged DNA binding domains for biochemical analysis, domains of interest were amplified by PCR from source (yeast or fly genomic DNA) followed by restriction cloning into a pDEST-MCS-Spytag vector.
The initial Ume6 E-ChRP scaffold was prepared by Gibson assembly (Gibson et al., 2009) in p416-ADH (Mumberg et al., 1995) to include an N-terminal NLS (KKKRK), residues 118–1000 of S. cerevisiae Chd1, nine repeats of glycine-glycine-serine, residues 1001–1014 of S. cerevisiae Chd1, two additional repeats of glycine-glycine-serine, an Afel restriction site, residues 764–836 from S. cerevisiae Ume6 and a HindIII restriction site. An analogous backbone was also created with residues 118–1000 of S. cerevisiae Chd1 linked directly to the two glycine-glycine-serine repeats followed by Afel, Ume6 DBD, and HindIII site. Cloning vectors were created in pDEST17, p416-ADH, p426-GPD and HO-pGAL-poly-KanMX4-H) (Voth et al., 2001) (a gift from David Stillman, Addgene 51664) to swap the C-terminal Ume6 domain with other targeting domains using Afel/HindIII restriction cloning, sticky-end PCR, or Gibson cloning (Gibson et al., 2009; Walker et al., 2008). Fusions used in this study include S. cerevisiae Ume6 (residues 764–836, cloned from yeast genomic DNA), D. melanogaster Engrailed (residues 454–543, cloned from fly genomic DNA), S. pombe Res1 (residues 1–147, cloned from a gBlock), R. norvegicus Glucocorticoid Receptor (residues 428–513, cloned from a gBlock), E. coli AraC (residues 175–281, provided by Gregory Bowman) and the Spycatcher domain (Zekeri et al.; 2012) (a gift from Mark Howarth, Addgene 35044), and dCas9 (subcloned from Addgene 49013, a gift from Timothy Lu (Farzadfard et al., 2013)).
Protein Purification
Chd1 constructs were expressed from pDEST17 vectors (Invitrogen) as previously described (Hauk et al., 2010). Briefly, proteins were expressed in BL21(DE3) cells, with the RIL plasmid (Stratagene) to aid expression and a plasmid expressing the Trigger Factor chaperone (a kind gift from Li Ma and Guy Montelione) to improve solubility. After induction with 300uM isopropyl-D-thiogalactopyr-anoside (IPTG) and growth at 18°C for 16 h, cells were lysed by sonication in 500 mM NaCl, 10% glycerol, and 25 mM Tris, pH 7.8. Lysate was clarified by centrifugation at 25,000×g and soluble protein was purified using Co2+ affinity chromatography (TALON column, GE Healthcare) followed by anion-exchange chromatography (Q-FF, GE Healthcare).
Nucleosome Sliding Assay
Recombinant yeast histones were purified as previously described (Luger et al., 1999) and dialyzed by gradient salt dialysis onto the Widom 601 positioning sequence (Lowary and Widom, 1998). Nucleosome sliding was performed at 25°C in sliding buffer (50 mM KCl, 15 mM HEPES, pH 7.8, 10 mM MgCl2, 0.1 mM EDTA, 5% sucrose, 0.2 mg/ml bovine serum albumin [BSA], with or without 5 mM ATP) by incubating 0, 10, 20, 40, or 60nM purified E-ChRP with 30nM reconstituted mononucleosomes for 100 minutes. Spycatcher reactions included 25, 50, or 100nM Chd1-Spycatcher and 0, 50 or 100nM of the Spytagged DNA binding domain with 30nM of labeled mononucleosomes. Unless specifically indicated, mononucleosomes contained E-ChRP recognition sequences in the extranucleosomal DNA. Reactions were quenched by diluting 1:2 with solution containing 3 uM competitor DNA (with E-ChRP recognition sequences) and 5% sucrose. Native PAGE (6%) was used to separate the positioning of the mononucleosomes, with Cy5.5-labeled nucleosomal DNA detected by a Li-Cor Odyssey FC imager. Chd1-dCas9 experiments were performed with indicated concentrations of nucleosomes and remodeler in the presence or absence of 5mM ATP for 100 minutes. Guide RNAs were synthesized from a T7 promoter using the SureGuide gRNA Synthesis Kit (Agilent). gRNA was added in a 2-fold excess, concentrated Chd1-SpyCatcher was pre-incubated with SpyTag-dCas9 for 1 hour prior to dilution and reaction initiation.
Micrococcal Nuclease Digestions and Library Construction
Micrococcal nuclease digestions were performed with a minimum of two biological replicates as previously described (Rodriguez et al., 2014). Briefly, 200ml cells were grown to mid-log phase and fixed with 1% formaldehyde. Chromatin was digested with 10, 20, and 40 units of MNase for 10 minutes and quenched with excess EDTA and SDS. Proper nuclease digestion of DNA was analyzed by agarose gel and samples with approximately 80% mononucleosomes were selected for library construction. After crosslink reversal, RNase treatment, Calf Intestine Phosphatase (CIP, NEB) treatment and Proteinase K digestion, mononucleosome-sized fragments were gel-purified and used to construct libraries with the NuGEN Ovation Ultralow kit per the manufacturer’s instructions. Libraries were sequenced at the University of Oregon’s Genomics and Cell Characterization Core Facility on an Illumina NextSeq 500 on the 37 cycle, paired-end, High Output setting, yielding approximately 20 million paired reads per sample.
Chromatin Immunoprecipitation and Library Construction
Chromatin immunoprecipitation was performed with biological replicates as previously described (Rodriguez et al., 2014). Briefly, cells were grown to mid-log phase, fixed with 1% formaldehyde, and lysed by bead-beating in the presence of protease inhibitors. Chromatin was fragmented by shearing in a Bioruptor sonicator (Diagenode) for a total of 30 minutes (high output, 3 × 10’ cycles of 30 s. on, 30 s. off). Sonication conditions were optimized to produce an average fragment size of ~300 basepairs. FLAG-tagged protein was immunoprecipitated using FLAG antibody (Sigma) and Protein G magnetic beads (Invitrogen). After crosslink reversal and Proteinase K digestion, DNA was purified using QIAGEN MinElute columns and quantified by Qubit High-Sensitivity fluorometric assay. Libraries were prepared using the NuGEN Ovation Ultralow kit by the manufacturer’s instructions and sequenced at the University of Oregon’s Genomics and Cell Characterization Core Facility on an Illumina HiSeq4000 with 50 or 100 cycles of single-end setting, yielding approximately 15 million reads per sample.
RNA Extraction and Library Construction
For RNA-Seq (minimum two biological replicates), RNA was purified by hot acid phenol extraction followed by rRNA depletion (Illumina RiboZero Gold Yeast). Strand-specific libraries were created using the NuGEN Universal Plus mRNA Kit according to the manufacturer’s instructions and sequenced on an Illumina NextSeq 500 on the 37 cycle, paired-end, High Output setting. ERCC Spike-in RNA controls were incorporated prior to rRNA depletion to account for potential global differences in RNA levels. Paired end reads were quality filtered for adaptor contamination and low quality ends using trimmomatic (Bolger et al., 2014). After quality filtering an average of 10.5 million reads per paired end sample remained. Surviving reads were mapped to the S. cerevisiae reference genome (Cunningham et al., 2015) using STAR (V.2.5.3) (Dobin et al., 2013). Gene counts were quantified from uniquely aligning reads using HTSeq (V.0.9.1) (Anders et al., 2015). Differential gene expression was performed using DESeq2(V.1.22.2) (Love et al., 2014), and expression graphs were generated using ggplot2 (Wickham, 2016).
QUANTIFICATION AND STATISTICAL ANALYSIS
MNase sequencing data were analyzed as described previously (McKnight et al., 2015, 2016; McKnight and Tsukiyama, 2015). Briefly, paired-end reads were aligned to the S. cerevisiae reference genome (Cunningham et al., 2015) with Bowtie 2 (Langmead and Salzberg, 2012), and filtered computationally for unique fragments between 100 and 200 bp. Dyad positions were calculated as the midpoint of paired reads, then dyad coverage was normalized across the S. cerevisiae genome for an average read/bp of 1.0. Note that dyad coverage is what is displayed in all figures. Nucleosome alignments to transcription factor binding sites were performed by taking average dyad signal at each position relative to all intergenic instances of a motif center. Motifs were obtained from the JASPAR database (Khan et al., 2018) and intergenic instances were found using the Saccharomyces Genome Database Pattern Matching tool (https://www.yeastgenome.org/nph-patmatch). Specifically, the Reb1 motif was defined as TTACCC(G/T) and Ume6 motif was WNGGCGGCWW. For ChIP-Seq data, single-end reads were aligned to the S. cerevisiae reference genome with Bowtie 2 and total read coverage was normalized such that the average read at a genomic location was 1.0. ChIP peaks were called using a 400 bp sliding window with a threshold average enrichment within the window of 4.0. Reb1 ORGANIC data (Kasinathan et al., 2014) were from the “80mM IP” sample (SRX263794); Reb1 CUT&RUN data (Skene and Henikoff, 2017) were from merged “cut-and-run 8s” and “cut-and-run 16s” samples (SRX2009989 and SRX2009990); Reb1 ChEC-seq data (Zentner et al., 2015) were from “Reb1 ChEC-seq 30s” (SRX974362); Reb1 anchor away and respective vehicle control MNase-seq data (Kubik et al., 2015) were from “Reb1veh1” (SRX1274605), “Reb1veh02” (SRX1274608), “Reb1rapa1” (SRX1274606) and “Reb1rapa02” (SRX1274607). Data were visualized using Integrated Genome Browser (Freese et al., 2016). RNA-seq p values (plotted as y axis of volcano plots; Figure 6C; Figure S2C) were calculated in DEseq2 using the default Wald test. Adjusted p values were calculated using a Benjamini and Hochberg test for multiple comparisons. Volcano plots where produced using ggplot2.
DATA AND CODE AVAILABILITY
The datasets generated during this study are available at GEO under accession code GSE123239.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal ANTI-FLAG M2 | Sigma-Aldrich | Cat# F1084; RRID:AB_262044 |
| Rabbit polyclonal anti-acetyl-Histone H3 (Lys23) | Millipore | Cat# 07–355 RRID:AB_310546 |
| IRDye 680RD Goat anti-rabbit (IgG H+L) | LI-COR Biosciences | Cat# LIC-926–32210; RRID:AB_621842 |
| IRDye 800CW Goat anti-Mouse (IgG H+L) | LI-COR Biosciences | Cat# LIC-926–32210; RRID:AB_621842 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 37% Formaldehyde | Sigma Aldrich | 252549 |
| Protease inhibitor cocktail EDTA-FREE | Expedion | 44214 |
| Acid washed glass beads (450–600um) | Sigma-Aldrich | G8772 |
| Dynabeads Protein G | Thermo Fisher Scientific | 10004D |
| RNase A | Fisher BioReagents | BP25391 |
| Proteinase K | Invitrogen | AM2544 |
| Glycogen, RNA grade | Thermo Scientific | FERR0551 |
| Glycogen, molecular biology grade | Thermo Scientific | FERR0551 |
| NEBuffer 2 | New England Biolabs | B7002S |
| Nuclease, Micrococcal | Worthington Biochemical | LS004798 |
| Zymolyase (R) 100T | AMS Bio | 120493–1 |
| Exonuclease III | NEB | M0206S |
| Calf Intestinal Phosphatase (CIP) | NEB | M02090L |
| Salmon Sperm DNA | Sigma Aldrich | D1626 |
| Agencourt AMPure XP beads | Beckman Coulter | A63881 |
| D-Raffinose pentahydrate | Gold Bio | R-030–500 |
| D-Galactose (low glucose) | USBiological | G1030 |
| IPTG | Gold Bio | I2481C |
| Oligo (dT)25 cellulose beads | NEB | S1408S |
| Critical Commercial Assays | ||
| MinElute PCR Purification Kit | QIAGEN | 28006 |
| RNeasy Mini Kit | QIAGEN | 74104 |
| Rnase-free Dnase Set | QIAGEN | 79254 |
| Ovation Ultralow System V2 | NuGEN | 0344NB-32 |
| Universal Plus mRNA-Seq | NuGEN | 0508–32 |
| Ribo-Zero Gold rRNA Removal Kit (yeast) | Illumina | MRZY1324 |
| Quant-IT PicoGreen dsDNA Assay Kit | Invitrogen | P7589 |
| SureGuide gRNA Synthesis Kit | Agilent | 5190–7719 |
| Deposited Data | ||
| MNase-Seq, RNA-Seq, and ChIP-Seq data generated in this manuscript | This study | GEO: GSE123239 |
| Reb1 ORGANIC | Kasinathan et al., 2014 | SRA: SRX263794 |
| Reb1 CUT&RUN | Skene and Henikoff, 2017 | SRA: SRX2009989 and SRX2009990 (merged) |
| Reb1 ChEC-Seq | Zentner et al., 2015 | SRA: SRX974362 |
| Reb1 anchor away | Kubik et al., 2015 | SRA: SRX1274605, SRX1274608, SRX1274606, SRX1274607 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: Strain background W303–1A | Laboratory of Rodney Rothstein | ATCC: 208352 |
| Yeast strains, see Table S1 | N/A | N/A |
| Recombinant DNA | ||
| pDEST17 | Thermo Fisher Scientific | 11803012 |
| p416-ADH1 | Mumberg, et al., 1995 | N/A |
| p426-GPD | Mumberg, et al., 1995 | N/A |
| HO-pGAL-poly-KanMX4-HO | Voth, et al., 2001 | Addgene 51664 |
| pDEST14-SpyCatcher | Zakeri, et al., 2012 | Addgene 35044 |
| Other plasmids, see Table S2 | N/A | |
| Software and Algorithms | ||
| trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/index.php?page=trimmomatic |
| STAR (V.2.5.3) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| HTSeq (V.0.9.1) | Anders et al., 2015 | https://htseq.readthedocs.io/en/release_0.9.1/install.html |
| DESeq2 (V.1.22.2) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ggplot2 | Wickham, 2016 | https://ggplot2.tidyverse.org/ |
| Bowtie 2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| JASPAR database | Khan et al., 2018 | http://jaspar.genereg.net/ |
| SGD Pattern Match Tool | https://www.yeastgenome.org/nph-patmatch | |
| Integrated Genome Browser | Freese et al., 2016 | https://www.bioviz.org/ |
Highlights.
Developed engineered chromatin remodeling proteins (E-ChRPs) as molecular tools
E-ChRPs induce targeted, sequence-specific nucleosome positioning in S. cerevisiae
Remodeled nucleosomes can disrupt native transcription factor binding and function
Modular targeting strategies enable investigation of chromatin-dependent processes
ACKNOWLEDGMENTS
We thank the Bowman lab (Johns Hopkins University)for histone plasmids and Chd1 constructs, the Tsukiyama lab (Fred Hutchinson Cancer Research Center) and Zentner lab (Indiana University) for epitope tagging vectors, and the Doe lab (University of Oregon) for a fly DNA extraction protocol. This work was supported by NIH training grants T32 GM007759 (to D.A.D. and O.G.B.B.) and T32 GM007413 (to D.A.D. and V.T.), and by NIGMS grant R01 GM129242 (J.N.M.) and the Donald and Delia Baxter Foundation (J.N.M.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.10.046.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ades SE, and Sauer RT (1994). Differential DNA-binding specificity of the engrailed homeodomain: the role of residue 50. Biochemistry 33, 9187–9194. [DOI] [PubMed] [Google Scholar]
- Alroy I,and Freedman LP (1992). DNA binding analysis of glucocorticoid receptor specificity mutants. Nucleic Acids Res. 20, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SF, Steber CM, Esposito RE, and Coleman JE (1995). UME6, a negative regulator of meiosis in Saccharomyces cerevisiae, contains a C-terminal Zn2Cys6 binuclear cluster that binds the URS1 DNA sequence in a zinc-dependent manner. Protein Sci. 4, 1832–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayté J, Leis JF, Herrera A, Tang E, Yang H, and DeCaprio JA (1995). The Schizosaccharomyces pombe MBF complex requires heterodimerization for entry into S phase. Mol. Cell. Biol 15, 2589–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns BR, Lorch Y, Li Y, Zhang M, Lacomis L, Erdjument-Bromage H, Tempst P, Du J, Laurent B, and Kornberg RD (1996). RSC, an essential, abundant chromatin-remodeling complex. Cell 87, 1249–1260. [DOI] [PubMed] [Google Scholar]
- Colin J, Candelli T, Porrua O, Boulay J, Zhu C, Lacroute F, Steinmetz LM, and Libri D (2014). Roadblock termination by reb1p restricts cryptic and readthrough transcription. Mol. Cell 56, 667–680. [DOI] [PubMed] [Google Scholar]
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. (2015). Ensembl 2015. Nucleic Acids Res. 43, D662–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberharter A, Längst G, and Becker PB (2004). A nucleosome sliding assay for chromatin remodeling factors. Methods Enzymol. 377, 344–353. [DOI] [PubMed] [Google Scholar]
- Farzadfard F, Perli SD, and Lu TK (2013).Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS Synth. Biol 2, 604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzio TG, and Tsukiyama T (2003). Chromatin remodeling in vivo: evidence for a nucleosome sliding mechanism. Mol. Cell 12, 1333–1340. [DOI] [PubMed] [Google Scholar]
- Ferreira H, Flaus A, and Owen-Hughes T (2007). Histone modifications influence the action of Snf2 family remodelling enzymes by different mechanisms. J. Mol. Biol 374, 563–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freese NH, Norris DC, and Loraine AE (2016). Integrated genome browser: visual analytics platform for genomics. Bioinformatics 32, 2089–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, and Joung JK (2014). Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol 32, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangaraju VK, and Bartholomew B (2007). Dependency of ISW1a chromatin remodeling on extranucleosomal DNA. Mol. Cell. Biol 27, 3217–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, et al. (2002). Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391. [DOI] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA 3rd, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmark JP, Fazzio TG, Estep PW, Church GM, and Tsukiyama T (2000). The Isw2 chromatin remodeling complex represses early meiotic genes upon recruitment by Ume6p. Cell 103, 423–433. [DOI] [PubMed] [Google Scholar]
- Hartley PD, and Madhani HD (2009). Mechanisms that specify promoter nucleosome location and identity. Cell 137, 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer MH, and Gasser SM (2017). Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 31, 2204–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauk G, McKnight JN, Nodelman IM, and Bowman GD (2010). The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol. Cell 39, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hota SK, Bhardwaj SK, Deindl S, Lin YC, Zhuang X, and Bartholomew B (2013). Nucleosome mobilization by ISW2 requires the concerted action of the ATPase and SLIDE domains. Nat. Struct. Mol. Biol 20, 222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs EA, Kocak DD, Fitzgibbon CJ, McMenemy J, Gersbach CA, and Marszalek PE (2015). Structure and specificity of the RNA-guided endo-nuclease Cas9 during DNA interrogation, target binding and cleavage. Nucleic Acids Res. 43, 8924–8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadosh D, and Struhl K (1997). Repression by Ume6 involves recruitment of a complex containing Sin3 corepressor and Rpd3 histone deacetylase to target promoters. Cell 89, 365–371. [DOI] [PubMed] [Google Scholar]
- Kasinathan S, Orsi GA, Zentner GE, Ahmad K, and Henikoff S (2014). High-resolution mapping of transcription factor binding sites on native chromatin. Nat. Methods 11, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni SR, Tan G, et al. (2018). JAS-PAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46 (D1), D1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubik S, Bruzzone MJ, Jacquet P, Falcone JL, Rougemont J, and Shore D (2015). Nucleosome stability distinguishes two different promoter types at all protein-coding genes in yeast. Mol. Cell 60, 422–434. [DOI] [PubMed] [Google Scholar]
- Kubik S, O’Duibhir E, de Jonge WJ, Mattarocci S, Albert B, Falcone JL, Bruzzone MJ, Holstege FCP, and Shore D (2018). Sequence-directed action of RSC remodeler and general regulatory factors modulates +1 nucleosome position to facilitate transcription. Mol. Cell 71, 89–102.e5. [DOI] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, and Mann M (2014). Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11, 319–324. [DOI] [PubMed] [Google Scholar]
- Lai WKM, and Pugh BF (2017). Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol 18, 548–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Längst G, Bonte EJ, Corona DF, and Becker PB (1999). Nucleosome movement by CHRAC and ISWI without disruption or trans-displacement of the histone octamer. Cell 97, 843–852. [DOI] [PubMed] [Google Scholar]
- Laughery MF, Mayes HC, Pedroza IK, and Wyrick JJ (2019). R-loop formation by dCas9 is mutagenic in Saccharomyces cerevisiae. Nucleic Acids Res. 47, 2389–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, and Widom J (1998). New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol 278, 19–42. [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, and Richmond TJ (1999). Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol 119, 1–16. [DOI] [PubMed] [Google Scholar]
- MacAlpine DM, and Almouzni G (2013). Chromatin and DNA replication. Cold Spring Harb. Perspect. Biol 5, a010207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrich TN, loshikhes IP, Venters BJ, Jiang C, Tomsho LP, Qi J, Schuster SC, Albert I, and Pugh BF (2008). A barrier nucleosome model for statistical positioning of nucleosomes throughout the yeast genome. Genome Res. 18, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight JN, and Tsukiyama T (2015). The conserved HDAC Rpd3 drives transcriptional quiescence in S. cerevisiae. Genom. Data 6, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight JN, Jenkins KR, Nodelman IM, Escobar T, and Bowman GD (2011). Extranucleosomal DNA binding directs nucleosome sliding by Chd1. Mol. Cell. Biol 31, 4746–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight JN, Boerma JW, Breeden LL, and Tsukiyama T (2015). Global promoter targeting of a conserved lysine deacetylase for transcriptional shutoff during quiescence entry. Mol. Cell 59, 732–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight JN, Tsukiyama T, and Bowman GD (2016). Sequence-targeted nucleosome sliding in vivo by a hybrid Chd1 chromatin remodeler. Genome Res. 26, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Müller R, and Funk M (1995). Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122. [DOI] [PubMed] [Google Scholar]
- Niland P, Huhne R, and Müller-Hill B (1996). How AraC interacts specifically with its target DNAs. J. Mol. Biol. 264, 667–674. [DOI] [PubMed] [Google Scholar]
- Nodelman IM, and Bowman GD (2013). Nucleosome sliding by Chd1 does not require rigid coupling between DNA-binding and ATPase domains. EMBO Rep. 14, 1098–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HD, Luche RM, and Cooper TG (1992). The yeast UME6 gene product is required for transcriptional repression mediated by the CAR1 URS1 repressor binding site. Nucleic Acids Res. 20, 1909–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, and Lim WA (2013). Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, McKnight JN, and Tsukiyama T (2014). Genome-wide analysis of nucleosome positions, occupancy, and accessibility in yeast: nucleosome mapping, high-resolution histone ChIP, and NCAM. Curr. Protoc. Mol. Biol. 108, 21.28.1–21.28.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan DP, Sundaramoorthy R, Martin D, Singh V, and Owen-Hughes T (2011). The DNA-binding domain of the Chd1 chromatin-remodelling enzyme contains SANT and SLIDE domains. EMBO J. 30, 2596–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, and Henikoff S (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife 6, e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, and Peterson CL (2005). A conserved Swi2/Snf2 ATPase motif couples ATP hydrolysis to chromatin remodeling. Mol. Cell. Biol. 25, 5880–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockdale C, Flaus A, Ferreira H, and Owen-Hughes T (2006). Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J. Biol. Chem. 281, 16279–16288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Pirkl N, Maier KC, Schenk A, Failmezger H, Tresch A, and Cramer P (2013). Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol. Cell 52, 52–62. [DOI] [PubMed] [Google Scholar]
- Tsukiyama T, Becker PB, and Wu C (1994). ATP-dependent nucleosome disruption at a heat-shock promoter mediated by binding of GAGA transcription factor. Nature 367, 525–532. [DOI] [PubMed] [Google Scholar]
- Venkatesh S, and Workman JL (2015). Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol 16, 178–189. [DOI] [PubMed] [Google Scholar]
- Voth WP, Richards JD, Shaw JM, and Stillman DJ (2001). Yeast vectors for integration at the HO locus. Nucleic Acids Res. 29, E59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JE, Saraste M, Runswick MJ, and Gay NJ (1982). Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker A, Taylor J, Rowe D, and Summers D (2008). A method for generating sticky-end PCR products which facilitates unidirectional cloning and the one-step assembly of complex DNA constructs. Plasmid 59, 155–162. [DOI] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag; ). [Google Scholar]
- Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, and Howarth M (2012). Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. U S A 109, E690–E697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Kasinathan S, Xin B, Rohs R, and Henikoff S (2015). ChEC-seq kinetics discriminates transcription factor binding sites by DNA sequence and shape in vivo. Nat. Commun. 6, 8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CY, Johnson SL, Gamarra NI, and Narlikar GJ (2016). Mechanisms of ATP-dependent chromatin remodeling motors. Annu. Rev. Biophys. 45, 153–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at GEO under accession code GSE123239.