

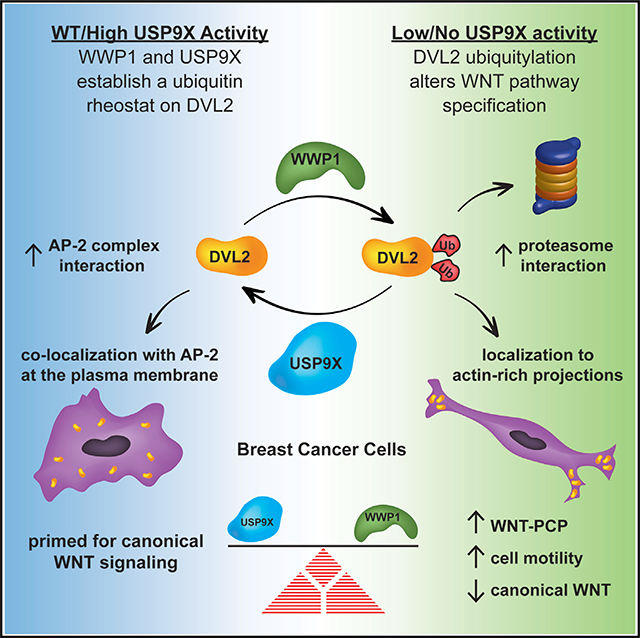
SUMMARY
The WNT signaling network is comprised of multiple receptors that relay various input signals via distinct transduction pathways to execute multiple complex and context-specific output processes. Integrity of the WNT signaling network relies on proper specification between canonical and noncanonical pathways, which presents a regulatory challenge given that several signal transducing elements are shared between pathways. Here, we report that USP9X, a deubiquitylase, and WWP1, an E3 ubiquitin ligase, regulate a ubiquitin rheostat on DVL2, a WNT signaling protein. Our findings indicate that USP9X-mediated deubiquitylation of DVL2 is required for canonical WNT activation, while increased DVL2 ubiquitylation is associated with localization to actin-rich projections and activation of the planar cell polarity (PCP) pathway. We propose that a WWP1-USP9X axis regulates a ubiquitin rheostat on DVL2 that specifies its participation in either canonical WNT or WNT-PCP pathways. These findings have important implications for therapeutic targeting of USP9X in human cancer.
In Brief
DVL2 is a signal transducing protein that participates in canonical and noncanonical WNT signaling relays. Here, Nielsen et al. report that the deubiquitylase USP9X and the E3 ubiquitin ligase WWP1 operate on DVL2 to establish a ubiquitin rheostat that contributes to WNT pathway specification in human breast cancer cells.
Graphical Abstract
INTRODUCTION
The canonical WNT β-catenin signaling pathway is involved in regulating many cellular processes such as cell fate determination during embryonic development, cell proliferation, and adult tissue homeostasis. Thus, it is not surprising that aberrant activation of the canonical WNT pathway is known to occur in many types of cancer (MacDonald et al., 2009; Saito-Diaz et al., 2013). There are also several noncanonical WNT signaling pathways including the WNT-planar cell polarity (WNT-PCP) pathway, which controls cell migration and tissue polarity. Dysregulation of the WNT-PCP pathway has been linked to cancer invasion and metastasis (Katoh, 2005; Luga et al., 2012; Wang, 2009). While the canonical WNT β-catenin pathway and the noncanonical WNT-PCP pathway use divergent effector mechanisms to regulate distinct cellular functions, these pathways share membrane receptor components and the cytoplasmic WNT transducer protein dishevelled (DVL). Despite its key role in both pathways, the mechanisms dictating DVL participation in canonical or noncanonical WNT signaling are yet to be elucidated.
Initiation of the canonical WNT β-catenin pathway occurs when extracellular WNT ligand binds to the co-receptors Frizzled (FZD) and low-density lipoprotein receptor-related protein 5/6 (LRP5/6), leading to recruitment of DVL and AXIN to the WNT ligand receptor complex (MacDonald et al., 2009). This ultimately results in the inhibition of β-catenin ubiquitylation and degradation such that stabilized β-catenin can enter the nucleus to initiate a transcriptional program (MacDonald et al., 2009; Saito-Diaz et al., 2013). On the other hand, core WNT-PCP pathway components Van Gogh-Like 1 (VANGL1), FZD, Prickle (Pk), DVL, and others function to activate RHOA, c-Jun N-terminal kinase (JNK), and nemo-like kinase (NLK) signaling cascades in order to coordinate tissue polarity and cell motility through regulation of actin dynamics (Glinka et al., 2011).
Ubiquitylation is known to be involved in key regulatory steps of both the canonical WNT and noncanonical WNT-PCP pathways. For example, ubiquitin-mediated regulation of cytoplasmic β-catenin stability is well characterized (Marikawa and Elinson, 1998). In addition, other steps of the WNT pathway upstream of β-catenin stabilization undergo regulation by the ubiquitin system. Notably, several members of the NEDD4 family of E3 ubiquitin ligases (SMURF1, ITCH, and NEDD4L) have been found to negatively regulate stability of WNT pathway components. SMURF1 interacts with and ubiquitylates AXIN, inhibiting its interaction with the WNT co-receptor LRP5/6 (Ding et al., 2013; Fei et al., 2013, 2014; Tanksley et al., 2013; Wei et al., 2012). Both ITCH and NEDD4L promote degradation of DVL2 (Cadavid et al., 2000; Ding et al., 2013; Fei et al., 2013, 2014; Wei et al., 2012). Additionally, the deubiquitylase (DUB) USP34 was found to antagonize ubiquitylation of AXIN, promoting its stabilization and function in the canonical WNT pathway (Lui et al., 2011). SMURF1 and SMURF2 negatively regulate the WNT-PCP pathway by targeting WNT-PCP receptor component Prickle1 for degradation (Narimatsu et al., 2009). Furthermore, DVL2 is also known to undergo positive regulation by the ubiquitin system. For example, K63-linked ubiquitylation of the N-terminal DAX domain, which is known to mediate dynamic polymerization of DVL2, has been implicated as a positive regulator of DVL2 signal transduction (Schwarz-Romond et al., 2007; Tauriello et al., 2010). These examples highlight the complex regulation of canonical and noncanonical WNT pathways by ubiquitin conjugation and deconjugation machinery.
The NEDD4 family of E3 ubiquitin ligases is conserved from yeast to humans and has been implicated in the ubiquitin-mediated endocytosis of many plasma membrane (PM) proteins, including surface receptors, ion channels, and nutrient transporters (David et al., 2013; He et al., 2008; Hryciw et al., 2004; Kuratomi et al., 2005; Lee et al., 2011; Lin et al., 2011; MacGurn et al., 2012). There are 9 NEDD4 family E3 ubiquitin ligases encoded in the human genome, each with distinct physiological functions (Rotin and Kumar, 2009). While not all NEDD4 family E3 ubiquitin ligases have been characterized extensively, several have a strong preference for catalyzing K63-linked polyubiquitin chains in vitro (Ingham et al., 2004; Kim and Huibregtse, 2009). Given the broad role of NEDD4 family E3 ubiquitin ligases in the regulation of membrane trafficking and cell signaling, it is not surprising that they are often found to be dysregulated in human diseases, including cancer (Bernassola et al., 2008). NEDD4 family members have a conserved domain structure including an N-terminal C2 domain that binds lipids, a C-terminal HECT ubiquitin ligase domain, and 2–4 internal WW domains. The WW domains have a high affinity for PY (Pro-Pro-x-Tyr) motifs and mediate interactions with substrates, adaptors, and regulatory factors (Ingham et al., 2004). Here, we report that the NEDD4 family member WWP1 interacts with USP9X and DVL2, and we find that these associations are governed by interactions between the WW domains of WWP1 and PY motifs present in both USP9X and DVL2. Importantly, we find that these interactions control a ubiquitylation rheostat on DVL2 that regulates both canonical WNT and noncanonical WNT-PCP activation in human breast cancer cells. Thus, we find that this USP9XDVL2-WWP1 axis is a key regulatory node in the WNT pathway with the potential to influence both canonical and noncanonical WNT signaling in the context of human cancer.
RESULTS
The WW Domains of WWP1 Coordinate Interactions with USP9X and DVL2
In an attempt to identify factors that regulate WWP1 function, we used stable isotope labeling with amino acids in culture (SILAC)-based quantitative proteomics to generate a WWP1 interaction profile in MDA-MB-231 cells (a triple-negative breast cancer cell line). This experiment identified interactions with various PM and PM-associated proteins, factors associated with membrane trafficking, signaling factors, and elements of the ubiquitin-proteasome system (Table S1; Figure S1A). To validate results obtained by quantitative proteomics, we tested for co-purification of a subset of putative WWP1 interacting proteins with FLAG-WWP1 stably expressed in MDA-MB-231 cells (Figure 1A). This approach confirmed FLAG-WWP1 interactions with several putative interactors—including USP9X, DVL2, and SPG20—although some putative interactors such as ARRDC1 and USP24 could not be confirmed by this approach. Considering the possibility that some putative interacting proteins may artificially result from stable overexpression of FLAG-WWP1, we performed native co-immunoprecipitation of WWP1 and identified interactions with USP9X and DVL2 at endogenous expression levels (Figure S1B). Importantly, other studies have reported interactions between USP9X and NEDD4 family E3 ubiquitin ligases—including SMURF1 (Xie et al., 2013) and ITCH (Mouchantaf et al., 2006). Similarly, DVL2 was previously reported to interact with other NEDD4 family E3 ubiquitin ligases—including WWP2 (Mund et al., 2015) and NEDD4L (Ding et al., 2013; Wei et al., 2012). Notably, both USP9X and DVL2 contain multiple PY motifs capable of binding the WW domains of NEDD4 family E3 ubiquitin ligases (Figure 1B).
Figure 1. WWP1 Interacts with USP9X and DVL2 in MDA-MB-231 Human Breast Cancer Cells.

(A) Co-immunoprecipitation analysis testing hits from SILAC-MS analysis in Figure S1A. FLAG-WWP1 was affinity purified from MDA-MB-231 cell lysates. Input and immunoprecipitated (IP) fractions were blotted for the indicated species.
(B) Schematic of known domains in WWP1, DVL2, and USP9X.
(C) Analysis of co-purification of DVL2 and USP9X with WWP1 variants. The indicated WWP1 variants were FLAG affinity purified, and quantitative immunoblots were performed to assess co-purification of interacting factors. The wwp1–4ww mutant indicates that all four WW domains have been mutated to disrupt the ability to bind PY motifs.
(D) Quantification of USP9X co-purification with FLAG-WWP1 variants (normalized to wild-type [WT]) over multiple experiments (n = 3). Red asterisk indicates a significant difference based on Student’s t test analysis.
(E) Analysis of co-purification of DVL2 and USP9X with WWP1 variants. The indicated WWP1 variants were FLAG affinity purified, and quantitative immunoblots were performed to assess co-purification of interacting factors. Each mutant variant of WWP1 contains a single intact WW domain (indicated), while the remaining three are mutated to disrupt PY motif binding.
(F) Quantification of DVL2 co-purification with FLAG-WWP1 variants (normalized to WT) over multiple experiments (n = 3).
(G) Analysis of co-purification of WWP1 and USP9X with DVL2 variants. The indicated mutant DVL2 variants were FLAG affinity purified, and quantitative immunoblots were performed to assess co-purification of interacting factors. Mutant variants of DVL2 included point mutations disrupting the ability of its PY motifs to interact with WW domains.
(H) Analysis of co-purification of WWP1 and DVL2 with USP9X variants. The indicated USP9X variants were FLAG affinity purified, and quantitative immunoblots were performed to assess co-purification of interacting factors. Mutant variants of USP9X included a small C-terminal truncation, which results in deletion of both C-terminal PY motifs.
(I) A schematic model for how USP9× and DVL2 both interact with WWP1 via PY motif interactions. The data also suggest an interaction between USP9X andDVL2 that is independent of WWP1 (dotted red line).
(D and F) Error bars represent SD of the mean.
Based on our findings and previous reports, we decided to further investigate the basis of physical interactions between WWP1, USP9X, and DVL2. Since USP9X and DVL2 both contain multiple PY motifs, we hypothesized that the WW domains of WWP1 might function to scaffold these proteins into a complex. To test this, we generated WWP1 WW domain point mutant variants designed to disrupt PY motif interactions (WxxP → FxxA) (Chen et al., 1997; Ermekova et al., 1997; Gajewska et al., 2001) and analyzed DVL2 and USP9X interaction by co-immunoprecipitation. We found that disruption of individual WW domains within WWP1 did not diminish interaction with USP9X or DVL2 (Figures S1C and S1D). Indeed, we unexpectedly observed that the wwp1-ww2 mutant exhibited significantly greater interaction with DVL2 (Figures S1C and S1D). Although individual WW domains were dispensable for interaction with DVL2 and USP9X, we found that a WWP1 variant with point mutations disrupting each WW domain (wwp1–4ww) exhibited significantly decreased interaction with USP9X and complete loss of interaction with DVL2 (Figures 1C and 1D). Together, these findings indicate that (1) WW domain scaffolding partially contributes to the interaction between WWP1 and USP9X, (2) WW domain interactions are required for WWP1 engagement with DVL2, and (3) the WW domains of WWP1 function redundantly for interaction with both DVL2 and USP9X.
To further probe the WW domain specificity underpinning WWP1 interactions with DVL2 and USP9X, we generated a panel of WWP1 variants containing only a single intact WW domain (designated as wwp1-WW1, wwp1-WW2, wwp1-WW3, and wwp1-WW4). Importantly, we found that any individual WW domain is sufficient to restore interaction with USP9X, suggesting complete redundancy for this interaction (Figure 1E). In contrast, we found that only WW1 and WW3 were sufficient to restore interaction with DVL2 (Figures 1E and 1F), indicating that WW2 and WW4 do not contribute to the interaction between WWP1 and DVL2. Furthermore, the observation that wwp1-WW2 and wwp1-WW4 interact with USP9X but not DVL2 indicates that the USP9X-WWP1 interaction occurs independent of DVL2 engagement. Overall, these findings suggest that the WW domains of WWP1 scaffold interactions between both USP9X and DVL2 in a manner that is redundant for USP9X and partially redundant for DVL2.
PY Motifs in USP9X and DVL2 Promote Interaction with WWP1
Based on the observation that WW domains of WWP1 are important for interacting with USP9X and DVL2, we hypothesized that PY motifs in USP9X and DVL2 are required to engage WWP1. DVL2 contains two PY motifs (FPAY393 and PPPY568) that flank the DEP domain (Figure 1B). To test if either of these PY motifs are required for DVL2 to bind WWP1, we generated DVL2 variants with point mutations at individual PY motifs (FPAY393 → FAAA393 and PPPY568 → PAPA568) and performed co-immunoprecipitation analysis using FLAG-DVL2 as bait. This analysis revealed that PY1 (FPAY393) is dispensable for interaction with WWP1, while PY2 (PPPY568) is required to interact with WWP1 (Figure 1G). Furthermore, these results indicate that disruption of the DVL2-WWP1 interaction does not affect DVL2 engagement with USP9X (Figure 1G). Taken together, our results indicate that the DVL2-WWP1 interaction is governed by binding of the PY2 motif of DVL2 to the WW1 or WW3 domains of WWP1.
We hypothesized that PY motifs in USP9X also contribute to its interaction with WWP1. USP9X contains four PY motifs (QPQY409, HPRY1017, NPQY2437, and APLY2515), with the last two occurring at the C terminus of the protein (Figure 1B). To test if these C-terminal PY motifs are required for interaction with WWP1, we generated a USP9X variant deleted for the C-terminal portion of the protein (USP9X1–2433, or usp9x-Δpy3–4). Importantly, deletion of the two C-terminal PY motifs resulted in complete loss of interaction with WWP1 (Figure 1H). However, point mutations in these C-terminal PY motifs (NPQY2437 → NAQA2437 (py3) and APLY2515 → AALA2515 (py4)) resulted in only partial loss of WWP1 interaction (Figure S1E). Notably, in each experiment loss of USP9X interaction with WWP1 did not affect its interaction with DVL2, indicating that the USP9X-DVL2 interaction occurs independently of WWP1. Thus, WWP1 engages both USP9X (partially) and DVL2 (primarily) via non-exclusive WW-PY interactions, while DVL2 and USP9X exhibit the ability to interact independently of WWP1 (Figure 1I).
WWP1 and USP9X Establish a Ubiquitin Rheostat on DVL2
DVL2 was previously reported to be a substrate of several NEDD4 family E3 ubiquitin ligases, including ITCH, NEDD4L, and NEDL1 (Ding et al., 2013; Miyazaki et al., 2004; Narimatsu et al., 2009; Nethe et al., 2012; Wei et al., 2012), although both positive and negative regulation of DVL2 by NEDD4 family members have been reported. However, WWP1 has not previously been reported to regulate DVL2. Given the physical associations between WWP1 and DVL2 we hypothesized that DVL2 might be subject to ubiquitin conjugation by the E3 activity of WWP1. We found that recombinant WWP1 exhibited ubiquitin conjugation activity toward hemagglutinin (HA)-tagged DVL2 (purified from human STF-293 cell lysates) (Figure 2A; Figures S2A and S2B), and this activity is lost in a lysine-deficient variant of DVL2 (dvl2-K0; Figure S2C). Importantly, we also found that USP9X can reverse the ubiquitylation of DVL2 by WWP1 (Figure 2B), indicating that DVL2 is a substrate for both WWP1 and USP9X. Strikingly, the K48-specific deubiquitylase OTUB1 and the K63-specific deubiquitylase AMSH did not exhibit activity toward WWP1-ubiquitylated DVL2 (Figure 2B), suggesting that WWP1-conjugated ubiquitin on DVL2 is unlikely to contain K63- or K48-linked polymers.
Figure 2. WWP1 and USP9X Operate on DVL2 to Establish a Ubiquitylation Rheostat.

(A) In vitro ubiquitylation reactions were performed using recombinant purified FLAG-WWP1 (60 nM) and HA-DVL2 purified from cultured cells. E3 conjugation reactions were allowed to proceed for 60 min before the conjugation reaction was terminated.
(B) Deconjugation assays were performed using HA-DVL2 ubiquitylated in vitro by WWP1 (as generated in A). Equivalent molar ratios of USP9X, OTUB1, and AMSH (100 nM) were added and each reaction was allowed to proceed for 60 min before the deconjugation reaction was terminated and HA-DVL2 was resolved by SDS-PAGE and immunoblot (bottom panel). For each sample, a ratio of modified:unmodified HA-DVL2 was measured using ImageJ. The modified:unmodified HA-DVL2 ratio was averaged over multiple replicate experiments (n = 3). The red asterisk indicates a statistically significant difference (p < 0.05) compared to DVL2 that has been modified by WWP1 and subsequently treated with OTUB1 or AMSH. Error bars represent SD of the mean.
(C) Representative immunoblot of in vitro ubiquitylation and/or deubiquitylation reactions (replicate experiments shown in Figures S2D and S2E). Experiments were performed using indicated combinations of purified FLAG-WWP1 and 6xHIS-USP9X with HA-DVL2 purified from cultured cells (see Figures S2A and S2B). Reactions were allowed to proceed for 60 min before being terminated and HA-DVL2 was resolved by SDS-PAGE and immunoblot.
(D) Line density plots from the HA-DVL2 immunoblot data in (C) illustrate differences in extent of ubiquitylation in each sample. Replicate experiments are shown in Figures S2D and S2E.
Since WWP1 and USP9X both interact with and operate on DVL2, we hypothesized that the coordination of WWP1 E3 ubiquitin ligase activity and USP9X deubiquitylase activity might serve to establish a ubiquitin rheostat on DVL2. To test this, we incubated DVL2 with different molar ratios of WWP1 and USP9X and analyzed the outcome. Importantly, we found that different molar ratios of WWP1 and USP9X affected the extent of ubiquitylation on DVL2 (Figures 2C, 2D, S2D, and S2E). These findings indicate that coordinated WWP1 and USP9X activities can establish a ubiquitin rheostat on DVL2.
USP9X Promotes Canonical WNT Activation
Given the established function of DVL2 as a key signal transducing protein in the WNT pathway and given that NEDD4 family members have been reported to negatively regulate WNT signaling (Tanksley et al., 2013) potentially via ubiquitylation of DVL2 (Ding et al., 2013), we decided to test if USP9X regulates WNT signaling. We found that knockdown or knockout of usp9x attenuated WNT activation as measured by a TopFLASH reporter assay (Figures 3A and S3A) and by β-catenin stabilization (Figure 3A). The WNT signaling defect observed in usp9x knockout cells was complemented by transient expression of wild-type FLAG-USP9X but not a catalytic dead (C1556V, or CD) variant (Figure 3B), indicating that catalytic activity of USP9X is required for canonical WNT activation. Furthermore, we found that treating cells with WP1130, a small molecule known to inhibit the deubiquitylase activity of USP9X (Kapuria et al., 2010), inhibited canonical WNT activation by WNT3a with a half-maximal inhibitory concentration (IC50) of 2.9 μM (Figure 3C). This result is consistent with our findings that USP9X is required for canonical WNT activation; however, WP1130 is known to target other deubiquitylases (USP24, USP5, USP14, and UCH37) (de Las Pozas et al., 2018; Kapuria et al., 2010; Kushwaha et al., 2015; Peterson et al., 2015). Thus, we cannot exclude the possibility that inhibition of multiple DUB activities drives loss of WNT activation in the presence of WP1130. To further explore the possibility that USP9X activity promotes WNT activation, we transiently transfected expression vectors for FLAG-USP9X (wild-type or CD) and measured WNT activation in the context of WNT3a stimulation. Importantly, we found that transient expression of wild-type FLAG-USP9X, but not a CD variant, resulted in increased activation of the canonical WNT pathway (Figure 3D).
Figure 3. USP9X Promotes Canonical WNT Activation.

(A) Analysis of ligand-stimulated WNT activation in MDA-MB-231 cells (blue bars) and usp9x knockout equivalents (gray bars). TopFLASH luciferase assays were used to measure WNT activation (top panel), and immunoblotting was performed to assess stabilization of nuclear β-catenin. The asterisk (*) indicates WNT activation in usp9x knockout cells is significantly decreased (p < 0.05) compared to sibling MDA-MB-231 cells.
(B) MDA-MB-231 cells (WT and usp9x knockout) were stimulated with WNT3a ligand and R-spondin, and WNT activation was measured by TopFLASH luciferase assays. Prior to activation, cells were transiently transfected with empty vector (EV) or vector expression USP9X (WT or catalytic dead [CD; C1566V]).
(C) Ligand-stimulated WNT activation was measured in the presence of indicated concentrations of WP1130. Based on the data, an IC50 was estimated.
(D) MDA-MB-231 cells were transfected with empty vector or plasmids for CMV-driven expression of WT or CD USP9X.
(E) Ligand-stimulated WNT activation was measured using TopFLASH luciferase assays for MDA-MB-231 cells and usp9x knockout equivalents. Cells were transfected with control siRNA or siRNA targeting knockdown of NEDD4L, WWP1, or WWP2.
(A, B, D, and E) Error bars represent SD of the mean.
We hypothesized that loss of canonical WNT activation in the absence of USP9X may be due to unchecked activity of WWP1 or other NEDD4 family E3 ubiquitin ligases. Importantly, we found that the loss of canonical WNT activation in usp9x knockout cells was suppressed by coordinate knockdown of WWP1 or NEDD4L, but not WWP2 (Figure 3E), suggesting that hyper-activation of multiple NEDD4 family members, including NEDD4L and WWP1, may attenuate WNT signaling in the absence of USP9X. Furthermore, we found that transient overexpression of wild-type WWP1, but not a CD variant (C890S), inhibited TopFLASH activation by WNT3a (Figures S3B and S3C). Unexpectedly, we also found that expression of the wwp1-ww2 variant (harboring point mutations in WW2) inhibited canonical WNT activation to a greater extent than wild-type WWP1 (Figure S3C), while expression of the wwp1-WW2 variant (where all WW domains are disrupted except for WW2) exhibited hyper-activation of canonical WNT in the presence of WNT3a (Figure S3B). Thus, the ability of WWP1 to antagonize canonical WNT activation correlates with its ability to bind DVL2 (Figures 1E, 1F, S1C, and S1D). Taken together, these findings indicate that USP9X promotes canonical WNT activation, while WWP1 antagonizes canonical WNT activation.
USP9X Regulation of WNT Requires DVL2 Deubiquitylation
Given our finding that WWP1 and USP9X interact with and operate on DVL2, we hypothesized that USP9X regulates canonical WNT activation by affecting the ubiquitylation status of DVL2. To address the mechanism of canonical WNT regulation by USP9X, we first tested if USP9X is required for β-catenin stabilization by LiCl—an inhibitor of GSK3. Importantly, LiCl-mediated stabilization did not require USP9X (Figure S4A), indicating USP9X functions upstream of the β-catenin destruction complex. Importantly, we found that knockdown or knockout of USP9X resulted in decreased steady-state abundance of DVL2 protein and mobility shifts by SDS-PAGE consistent with hyper-ubiquitylation (Figures 4A and S4B). To identify post-translational modifications on DVL2 regulated by USP9X, we performed a SILAC-mass spectrometry (MS) on FLAG-DVL2 affinity purified from MDA-MB-231 cells and usp9x knockout equivalents. This analysis resolved many phospho-peptides and ubiquitin remnant (diGly) peptides derived from DVL2 with four internal lysines exhibiting elevated ubiquitylation in the absence of USP9X (Figure 4B; Figures S4C–S4E). Although ubiquitylation at N-terminal lysines of DVL2 has been reported (Madrzak et al., 2015; Tauriello et al., 2010), the ubiquitin modification events described in this study (K343, K428, K477, and K615) have not previously been reported.
Figure 4. USP9X Promotes Canonical WNT Activation via Deubiquitylation of DVL2.

(A) Immunoblotting analysis of FLAG-DVL2 mobility by SDS-PAGE from lysates of MDA-MB-231 cells or usp9x knockout equivalents. A low-exposure α-FLAG blot is shown to illustrate differences in DVL2 abundance, while the higher-exposure α-FLAG is shown to illustrate the higher molecular weight (MW) species evident in usp9X knockout cells.
(B) SILAC-MS profiling of post-translational modification sites (phosphorylation and ubiquitylation) on DVL2. In this experiment, FLAG-DVL2 purified from MDA-MB-231 cells (heavy) was compared to usp9x knockout equivalents (light). A schematic illustrating the domain structure of DVL2 is shown at the top with asterisks to indicate the position of detected ubiquitylation events.
(C) Analysis of ligand-stimulated WNT activation was measured using TopFLASH reporter assays in STF-293 cells transfected with the indicated expression vectors. UL36 is the DUB domain fused to DVL2 in these experiments.
(D) Following siRNA knockdown of USP9X, ligand-stimulated WNT activation was measured using TopFLASH reporter assays in STF-293 cells transfected with the indicated HA-DVL2 expression vectors. DVL2-K0 is a variant where all encoded Lys residues are substituted with Arg residues. The asterisk (*) represents a statistically significant difference when compared to indicated sample (p < 0.05). n.s. represents no significant difference relative to indicated comparison.
(C and D) Error bars represent SD of the mean.
To test if USP9X-mediated regulation of WNT signaling requires DVL2 ubiquitylation, we expressed DVL2 fused to the UL36 DUB domain (from human herpes virus) and found that this (but not a CD UL36 fusion) suppressed the WNT activation defect in the absence of USP9X (Figure 4C). These results indicate that USP9X regulates WNT signaling at (or in very close proximity to) the level of DVL2. To explore the possibility that USP9X-mediated deubiquitylation of DVL2 at specific sites is required for WNT activation, we tested a panel of DVL2 lysine mutants (K343R, K429R, K477R, and K615R) but found expression of these mutants was insufficient to suppress the WNT signaling defect observed in the absence of USP9X (Figure S4F). We next considered the possibility that USP9X may regulate multiple ubiquitylation events on DVL2, as suggested by SILAC-MS analysis (Figure 4B). Importantly, we found that expression of a “lysine-less” DVL2 variant with all encoded lysine residues switched to arginine (DVL2-K0) is sufficient to restore WNT activation in the absence of USP9X (Figure 4D). Taken together, these results indicate that increased ubiquitylation of DVL2 is required for the attenuation of WNT signaling observed in the absence of USP9X. Thus, the USP9X-mediated deubiquitylation of DVL2 is critical for activation of canonical WNT signaling.
DVL2 Ubiquitylation Status Regulates Interactions with Canonical and WNT-PCP Factors
We hypothesized that the altered ubiquitylation status of DVL2 in the absence of USP9X regulates its function as a signal transduction protein. To test this hypothesis, we first characterized the DVL2 interaction network in MDA-MB-231 cells using SILAC-MS (Tables S2 and S3; Figure 5A). These experiments revealed two near-stoichiometric interactions with the AP-2 clathrin adaptor complex and with the noncanonical WNT factors VANGL1 and cofilin, which are known to participate in the PCP pathway (Luga et al., 2012). Additionally, we identified several sub-stoichiometric interactions with NEDD4 family E3 Ub ligases and proteasome subunits (Tables S2 and S3; Figure 5A). Many of these interactions had been previously reported (Angers et al., 2006; Yu et al., 2007). Several interactions were confirmed by co-immunoprecipitation (Figure 5B) and co-localization by immunofluorescence microscopy (Figure S5A). Consistent with previous reports (Yu et al., 2007, 2010), we also found that knockdown of AP-2 complex subunits inhibited canonical WNT activation in a TopFLASH assay (data not shown).
Figure 5. DVL2 Ubiquitylation State Specifies Association with Canonical WNT or WNT-PCP Pathway Factors.

(A) SILAC-MS was used to resolve the DVL2 interaction network in MDA-MB-231 human breast cancer cells. Near-stoichiometric interactions (solid lines) were detected with the AP-2 clathrin adaptor complex and VANGL1-cofilin, which comprise a subcomplex of the WNT-PCP pathway. Sub-stoichiometric interactions (dotted lines) were identified for NEDD4 family E3 ubiquitin ligases and proteasome subunits.
(B) Interactions detected by SILAC-MS were confirmed by co-immunoprecipitation.
(C and D) SILAC-MS analysis was performed to measure how DVL2 interactions are impacted by catalytic active versus CD DUB fusion (C) or by loss of USP9X
(D). Scatterplots depict individual heavy:light (H:L) ratio measurements (LOG2 transformed) for each peptide resolved for the indicated proteins. The mean value and standard deviation for each protein is indicated. For the experiment shown in (C), several peptides with extremely low H:L ratios are not depicted here but are shown in Figure S5B. These outlier peptides correspond to sites of ubiquitin modification (Figure S5C) shown in Figure 4B. Differences between DVL2 (bait) and all interacting proteins (AP2 complex, VANGL1, CFL1, and proteasome subunits) are statistically significant (p < 0.005).
(C and D) Error bars represent SD of the mean.
To quantify how DVL2 interactions are affected by ubiquitylation, we performed SILAC-MS proteomic analysis comparing the interaction profile of catalytic active (light) or CD (heavy) UL36-DVL2 fusions (also used in Figure 4C). This revealed that CD UL36-DVL2 fusion (which can be stably ubiquitylated) interacts more with proteasome subunits and VANGL1/cofilin, while the catalytic active UL36-DVL2 fusion (which is not stably ubiquitylated) interacts more with the AP-2 complex (Figure 5C). Importantly, we noticed in this experiment a small set of peptides derived from the DVL2-UL36 catalytic active fusion protein and absent from the DVL2-UL36 CD fusion protein (Figure S5B). Interestingly, these outlier peptides all correspond to tryptic events that can only occur in the absence of ubiquitylation (at positions K428, K477, and K615) (Figure S5C), indicating that these sites are largely ubiquitin modified in the DVL2-UL36 CD fusion protein. We also used SILAC-MS to quantify how DVL2 interactions are altered in usp9x knockout cells and found similar results: DVL2 interacted more with VANGL1 and cofilin in usp9x knockout cells (where DVL2 ubiquitylation is elevated) but interacted more with AP-2 in the presence of USP9X (where DVL2 is less ubiquitylated) (Figure 5D). These results indicate ubiquitylated DVL2 interacts more with VANGL1/cofilin—components of the WNT-PCP pathway—while deubiquitylated DVL2 interacts more with the AP-2 complex, which is critical for canonical WNT activation.
USP9X Regulates Cellular Distribution of DVL2 and Antagonizes WNT-PCP
Previous studies have reported that VANGL1 and cofilin localize to F-actin rich projections in migrating MDA-MB-231 cells (Hatakeyama et al., 2014) and that DVL2 is required for WNT5a-induced cell migration in these cells (Zhu et al., 2012). Based on our findings that loss of USP9X results in increased ubiquitylation of DVL2 and increased interactions between DVL2 and VANGL1/cofilin, we hypothesized that loss of USP9X may result in re-localization of DVL2 to actin-rich projections in MDA-MB-231 cells. To test this, we used immunofluorescence microscopy to characterize FLAG-DVL2 distribution in MDA-MB-231 cells in the presence or absence of USP9X. In MDA-MB-231 cells treated with control siRNAs, DVL2 exhibited a punctate distribution throughout the cell body, reflective of previous descriptions of DVL2 localization (Figure 6A, top panels) (Schwarz-Romond et al., 2005), whereas MDA-MB-231 siUSP9X knockdown cells displayed a redistribution of DVL2 to actin-rich projections (Figure 6A, middle panels). Quantification of the cellular distribution of DVL2 in the presence and absence of USP9X reveals a significant increase in DVL2 in actin-rich projections upon loss of USP9X (Figure 6B). Importantly, coordinate knockdown of WWP1 prevented this re-distribution of DVL2, indicating that WWP1 activity is required for re-distribution of DVL2 to actin-rich projections in the absence of USP9X (Figure 6A, bottom panel and Figure 6B). Thus, our data indicate that USP9X promotes DVL2 localization to puncta that also contain AP-2 (Figure S5A), while loss of USP9X activity results in WWP1-dependent re-distribution of DVL2 to actin-rich projections in MDA-MB-231 breast cancer cells.
Figure 6. USP9X Regulates DVL2 Localization and Antagonizes WNT-PCP Activation.

(A) Immunofluorescence imaging of FLAG-DVL2 (green) and actin (phalloidin, blue) in MDA-MB-231 human breast cancer cells transfected with control siRNA (top panels), siRNA targeting USP9X (middle panels), or a combination of siRNA targeting USP9X and siRNA targeting WWP1 (bottom panels). Scale bars are 25 μm.
(B) Quantification of DVL2 cellular distribution over a population of MDA-MB-231 cells (n > 100).
(C) WT MDA-MB-231 and usp9x knockout (KO) variant cell lysates were assayed for active Rho by pull-down assay. Addition of GDP or non-hydrolyzable GTP (GTPƔS) to cell lysates served as negative and positive controls, respectively.
(D) Quantification of Rho activation shown in Figure 6C (n = 3) was performed by measuring the amount of active Rho detected in parent and usp9x knockout cells and normalizing to the GTPγS (maximal activation) control. The red asterisk indicates a significant difference when compared to WT cells (p < 0.05).
(E) MDA-MB-231 cells were plated at low density, and cell migration (μm/h) speed was measured. Red asterisk indicates a significant difference between the tested populations (n > 30).
(F) Rose plots showing trajectories of individual MDA-MB-231 cells (left) and usp9x knockout equivalents (right).
(D and E) Error bars represent SD of the mean.
One feature of WNT5a-mediated PCP activation is Rho activation, which is DVL2 dependent and required for induction of cell migration by the WNT-PCP pathway (Zhu et al., 2012). Given our findings, we hypothesized that DVL2 re-distribution to actin-rich projections in MDA-MB-231 cells lacking USP9X might result in Rho activation. Indeed, we found that MDA-MB-231 cells normally exhibit about 35% of maximal Rho activation, while cells lacking USP9X exhibit 50% maximal Rho activation (Figures 6C and 6D). Taken together, these findings indicate that loss of USP9X in MDA-MB-231 cells results in re-distribution of DVL2 to actin-rich projections and Rho activation, which is similar to what occurs during WNT5a-mediated activation of the WNT-PCP pathway in these cells (Luga et al., 2012; Zhu et al., 2012).
USP9X Regulates Cell Motility in a Manner Dependent on DVL2 Ubiquitylation
VANGL1, cofilin, and DVL2 are known to coordinate WNT-PCP activation to regulate actin dynamics and promote cell motility, migration, and metastasis of MDA-MB-231 cells (Katoh, 2005; Luga et al., 2012; Yang and Mlodzik, 2015; Zhu et al., 2012). Based on these previous studies, we hypothesized that USP9X-mediated regulation of DVL2 may antagonize WNT-PCP-mediated cellular motility. Strikingly, we found that MDA-MB-231 breast cancer cells lacking USP9X exhibited a significant increase in motility compared to wild-type equivalents (Figures 6E and 6F). This increase in cell motility was complemented in cells transiently transfected with GFP-USP9X (Figure S6A). Consistent with this observation, we also found that treatment of MDA-MB-231 cells with WP1130—a small-molecule inhibitor of USP9X—also resulted in a significant increase in the speed of cell motility (Figure S6B). Importantly, the increased cell speed observed in usp9x knockout cells was dependent on VANGL1 (Figure S6C), indicating the increased motility phenotype requires an intact WNT-PCP pathway. To test if the increased cell motility observed in usp9x mutant cells was due to increased autocrine signaling, we treated cells with LGK974, a porcupine inhibitor that blocks the secretion of many WNT ligands, and found this had no effect on the speed of usp9x knockout MDA-MB-231 cells (Figure S6D). Taken together, these results indicate that loss of USP9X leads to aberrant activation of PCP-mediated motility independent of autocrine signaling.
We hypothesized that the elevated ubiquitylation of DVL2 might contribute to the increased motility phenotype observed in usp9x knockout MDA-MB-231 cells. Consistent with this hypothesis, we observed that knockdown of WWP1 suppressed the increased motility phenotype of usp9x knockout cells (Figure 7A), although transient overexpression of WWP1 did not alter speed of MDA-MB-231 cells (Figure S7A). Notably, MDA-MB-231 cells transiently expressing GFP-DVL2 did not exhibit any change in cell motility compared to untransfected control cells (Figure S7B); however, expression of either wild-type or lysine-deficient (K0) GFP-DVL2 fully suppressed the increased motility phenotype of usp9x knockout MDA-MB-231 cells (Figures S7C and S7D). This result implicates DVL2 as a mediator of the cell motility phenotype observed for usp9x knockout cells, but it does not inform as to the role of ubiquitylation since both wild-type and lysine-deficient DVL2 fully suppress the usp9x knockout phenotype. To test the role of ubiquitylation, we analyzed cell motility of MDA-MB-231 cells stably expressing DVL2-UL36 (catalytic active and CD) DUB fusion proteins. Strikingly, we found that knockdown of USP9X resulted in increased motility of control cells and cells expressing CD DVL2-UL36 fusions but not of cells expressing catalytic active DVL2-UL36 fusions (Figure 7B). This result reveals that the increased motility phenotype observed in the absence of USP9X requires increased ubiquitylation of DVL2. Finally, we tested if DVL2 ubiquitylation is required for WNT5a-induced cell motility in MDA-MB-231 cells. Interestingly, we found that expression of catalytic active DVL2-UL36 resulted in a significant dampening of WNT5a-induced cell motility compared to control cells expressing CD DVL2-UL36, although the observed inhibition was partial (Figure 7C). These results indicate that USP9X is a critical antagonist of PCP activation and cell motility in MDA-MB-231 cells and that this function is dependent on the ubiquitylation of DVL2. Based on our findings, we propose that the balance of USP9X and WWP1 activities toward DVL2 regulates WNT pathway specification, with USP9X-mediated DVL2 deubiquitylation promoting canonical WNT signaling and WWP1-mediated DVL2 ubiquitylation driving WNT-PCP activation and cell migration (Figure 7D).
Figure 7. USP9X-Mediated Regulation of Cell Motility Requires DVL2 Ubiquitylation.

(A) Migration speed of MDA-MB-231 usp9x KO cells treated with either control siRNA or siRNA targeting WWP1 was measured after plating at low density for the indicated conditions. The red asterisk indicates a significant difference compared to control siRNA-treated cells (p < 0.005).
(B) MDA-MB-231 cell lines stably expressing DVL2-DUB fusions (catalytic active and CD) were treated with control siRNA or siRNA targeting USP9X, and cell speed was measured for 25 cells.
(C) MDA-MB-231 cell lines stably expressing DVL2-DUB fusions (catalytic active and CD) underwent mock or WNT5a treatment, and cell speed was measured for 25 cells. The red asterisk indicates a statistically significant difference (p < 0.005) compared to MDA-MB-231 cells stably expressing the CD DUB-DVL2 fusion treated with WNT5a.
(D) Model for DVL2 regulation by the USP9X-WWP1 axis to establish a ubiquitin rheostat on DVL2 that influences interaction preference with either the clathrin adaptor AP-2 complex, which is important for canonical WNT signaling, or the VANGL1-cofilin complex, which is important for the WNT-PCP pathway. Thus, the USP9X-WWP1 axis regulates DVL2 ubiquitylation to determine WNT pathway specification.
(A–C) Error bars represent SD of the mean.
DISCUSSION
Dishevelled has long been known to play a key role in the transduction of WNT receptor signaling, yet how it is regulated to specify participation in either canonical or noncanonical signaling relays is not well understood. Here, we report that the NEDD4 family E3 ubiquitin ligase WWP1 interacts with DVL2 and the deubiquitylase USP9X and that these interactions govern a ubiquitylation rheostat on DVL2 critical for both canonical WNT and WNT-PCP activation. Specifically, we have shown that (1) USP9X-mediated deubiquitylation of DVL2 promotes canonical WNT activation by enhancing DVL2 interaction with the clathrin adaptor complex AP-2; (2) hyper-ubiquitylation of DVL2 that occurs in the absence of USP9X drives increased DVL2 interaction with WNT-PCP factors VANGL1 and cofilin and re-localization of DVL2 to actin-rich projections; (3) USP9X limits cellular Rho activation and antagonizes noncanonical WNT-PCP activation; (4) USP9X antagonizes cell motility in a WWP1-dependent manner; and (5) USPX-mediated regulation of cell motility requires DVL2 ubiquitylation. Based on the data, we propose that the USP9X-WWP1 axis biases DVL2 engagement with either the canonical WNT pathway or the non-canonical WNT-PCP pathway.
A Regulatory Axis for WNT Pathway Specification
The functional diversity of WNT signaling is a result of the complex network design that interweaves canonical and noncanonical elements to coordinate multiple specific signaling outputs. Thus, it is important to understand the network design features that couple canonical and noncanonical pathways and contribute to pathway specificity. The role of USP9X in WNT pathway specification draws parallels to diversin, an ankyrin repeat protein also implicated in WNT pathway specification. Knockout of diversin in mice resulted in defective WNT-PCP signaling but hyper-activated signaling through the canonical WNT pathway (Allache et al., 2015; Jones et al., 2014)—phenotypes that are opposite to usp9x knockout but suggest a similar mode of pathway coupling. Importantly, diversin has also been reported to be overexpressed in breast cancer cells and contributes to both proliferation and invasion (Yu et al., 2014). Shared use of certain signaling factors—like dishevelled and diversin—between canonical and noncanonical WNT pathways may facilitate mechanisms of pathway specification based on inverse coupling, effectively operating as a “zero sum game.” For example, if a shared component like dishevelled or diversin is limiting for pathway activation, then engagement by one pathway may consequently prevent activation of the coupled pathway. Thus, regulatory mechanisms that impact stability of shared factors or their interaction networks have strong potential to contribute to pathway specification.
DVL2 has been reported to undergo both positive and negative regulation by the ubiquitin system through the action of multiple different E3 ubiquitin ligases and deubiquitylating enzymes. For example, both ITCH and NEDD4L have been shown to negatively regulate DVL2 stability by conjugating K48-linked polyubiquitin chains on DVL2, targeting it to the proteasome for degradation (Ding et al., 2013; Liu et al., 2014; Marikawa and Elinson, 1998; Mukai et al., 2010; Tauriello et al., 2010; Wei et al., 2012). On the other hand, K63-linked polyubiquitin modifications of the DVL2 DIX domain were reported to enhance canonical WNT activation (Tauriello et al., 2010), while other reports have suggested these modifications may antagonize canonical WNT activation (Madrzak et al., 2015). The apparent regulation of DVL2 by multiple members of the NEDD4 family could result from functional redundancy but could also reflect differential regulation based on expression in different contexts or different cell types. Our finding that siRNA targeting of NEDD4L or WWP1 partially rescued WNT activation in the absence of USP9X (Figure 3E) is consistent with a redundancy model, but it does not exclude the possibility that USP9X antagonizes multiple exclusive ubiquitylation events on DVL2 mediated by different E3 ubiquitin ligases. Indeed, our data indicate that functional regulation of DVL2 by USP9X involves reversal of multiple distinct ubiquitylation events (Figure 4B)—possibly mediated by multiple E3 ubiquitin ligases—since no single K → R mutation was capable of suppressing the loss of WNT activation phenotype observed in the absence of USP9X (Figure S4F). Furthermore, the in vitro analysis of USP9X activity presented here suggests that USP9X may be unique in its ability to operate on WWP1-ubiquitylated DVL2 and that it may have activity toward atypical ubiquitin polymers (Figures 2B–2D). These results are consistent with a recent study of USP9X structure and activity that reported a strong preference for K11-linked polyubiquitin chain substrates (Paudel et al., 2019). Although our studies indicate that these ubiquitylation events on DVL2 are important for WNT pathway specification, data presented here do not address the specific polyubiquitin linkage types involved or the contribution of other NEDD4 family ligases. Therefore, additional biochemical and genetic studies will be required to fully elucidate how USP9X operates on distinct ubiquitin modifications on DVL2 that may be mediated by multiple E3 ubiquitin ligases and how the sum of these activities contributes to the ubiquitin rheostat on DVL2 and WNT pathway specification.
Previous studies have reported conflicting roles for USP9X with respect to canonical WNT activation. Specifically, USP9X has previously been reported to positively regulate the stability of β-catenin in glioma cells (Yang et al., 2016). However, USP9X was also reported to negatively regulate the stability of β-catenin in mouse neural progenitor cells (Premarathne et al., 2017). Our studies are consistent with a positive role for USP9X in canonical WNT pathway activation, and our data indicate that USP9X functions upstream of the destruction complex (Figure S4A). Although our findings are consistent with a role for USP9X in the regulation of WNT at the level of DVL2, we cannot exclude the possibility of a context-dependent role for USP9X at other tiers of the WNT pathway. Furthermore, it is possible that expression of different NEDD4 family members, other USP9X-interactors, or substrate adaptors and targeting factors could modify USP9X function in a context-specific manner.
Previous studies reported that WNT5a-mediated activation of the WNT-PCP pathway in human breast cancer cells causes increased cell migration as a result of DVL2/Daam1-dependent activation of RhoA (Zhu et al., 2012). While a role for DVL2 in regulation of WNT-PCP and migration was evident, a role for USP9X in WNT-PCP and migration had not been clearly defined. Data presented here suggest that USP9X and WWP1 interact to provide fine-tuning of DVL2 ubiquitylation state in order to regulate DVL2 localization to actin-rich projections, Rho activation, and motility in breast cancer cells. These studies address how ubiquitylation of DVL2 influences activation of the canonical and noncanonical WNT pathways, but they do not address how activation of WNT and WNT-PCP by WNT3a and WNT5a (respectively) may impact DVL2 ubiquitylation status. Future studies will be required to determine if WNT pathway activation contributes to the ubiquitin rheostat on DVL2, perhaps functioning to tune DVL2 ubiquitylation and provide regulatory feedback.
USP9X: A Complex Factor in Cancer
Given that USP9X has been implicated in the regulation of multiple receptor signaling and trafficking pathways, it is not surprising that in recent years USP9X dysregulation has been linked to many different types of cancer. USP9X has been suggested to exhibit oncogenic behavior in some cancer contexts, including multiple myeloma, melanoma, lung cancer, and breast cancer (Kushwaha et al., 2015; Li et al., 2017; Potu et al., 2017; Schwickart et al., 2010). In contrast, USP9X has also been proposed to have tumor suppressor activity in other cancers, including pancreatic ductal adenocarcinoma (Pérez-Mancera et al., 2012) and colorectal adenocarcinoma (Khan et al., 2018). Recently, USP9X has been shown to be required for the attenuation of EGFR signaling (Savio et al., 2016), which is suggestive of a tumor suppressor function. Taken together, these reports suggest a complex role for USP9X in cancer progression that may be highly context dependent. Our findings suggest that USP9X functions together with WWP1 to regulate a ubiquitin rheostat on DVL2. The ubiquitylation status of DVL2 determines its engagement with either canonical or noncanonical WNT factors. Based on these findings, we hypothesize that in some cellular contexts increased USP9X activity may sensitize cells to activation of the canonical WNT pathway (Figure 3D), which promotes proliferation and tumor growth, while simultaneously antagonizing the noncanonical WNT-PCP pathway that is involved in cell motility and metastasis (Figures 6C–6F). These results indicate that USP9X can function to promote canonical WNT-driven proliferation while simultaneously guarding against migration and metastasis mediated by the WNT-PCP pathway. Based on these findings, we propose that small-molecule inhibitors of USP9X may be effective as antagonists of WNT-based proliferation (Figure 3C), but they may also ultimately promote migration and metastasis (Figure S6B).
Regulation of Cell Signaling Pathways by DUB-E3 Interactions
The ubiquitin proteasome system contributes to the tight spatial and temporal regulation of protein trafficking, localization, and stability. Recently, several examples of interactions between E3 ubiquitin ligases and DUBs have been reported, raising questions about the regulatory significance and design logic of such interactions. In principle, DUB-E3 interactions could facilitate a number of useful design features, including E3 antagonization, E3 deubiquitylation, or polyubiquitin chain editing on substrates. E3 antagonization has the potential to result in a futile cycle, but coordinated regulation of ligase and DUB activities could also establish a ubiquitin rheostat on a shared substrate. Such a rheostat has been reported to occur in the context of endoplasmic reticulum (ER) quality control machinery, where DUB activities can sharpen the distinction between folded and misfolded membrane proteins (Zhang et al., 2013). Another striking example involves the DUB USP7, which was shown to interact with the MAGE-L2-TRIM27 E3 ubiquitin ligase to regulate the ubiquitylation status of WASH, a shared substrate, on the endosome (Wu et al., 2014). Interestingly, USP7 has been reported to interact with multiple E3 ubiquitin ligases (Kim and Sixma, 2017), but the functional significance of many of these interactions remain unclear. E3-DUB interactions have also been reported to protect E3 ubiquitin ligases from degradation, thus promoting E3 activity. For example, the DUB USP19 promotes cellular proliferation by stabilizing KPC1 (also known as RNF123), a ubiquitin ligase for the cyclin-dependent kinase inhibitor p27Kip1 (also known as CDKN1B) (Lu et al., 2009). A striking example of polyubiquitin chain editing occurs with the protein A20, a potent inhibitor of the nuclear factor κB (NF-κB) signaling pathway that contains both a DUB domain and an E3 ubiquitin ligase domain (Wertz et al., 2004). The DUB domain of A20 removes a K63-linked polyubiquitin chain from the Receptor Interacting Protein (RIP), a protein essential for NF-κB signaling. Subsequently, the E3 ubiquitin ligase domain of A20 conjugates a K48-linked polyubiquitin chain to RIP targeting it for degradation by the proteasome, thus inhibiting the NF-κB pathway. Thus, there are numerous examples of E3-DUB interactions that establish ubiquitin rheostats on substrates, protect and stabilize E3 ubiquitin ligases, and coordinate activities to achieve polyubiquitin chain editing in signaling complexes.
We have identified an E3-DUB interaction that functions to provide layered regulation of the WNT pathway. Our results indicate that the activities of WWP1 and USP9X are antagonistic and function to establish a ubiquitin rheostat on DVL2 that is a critical regulator of WNT pathway specification. Functionally, USP9X antagonizes WWP1 activity toward DVL2 (Figures 2C and 2D), although our data cannot exclude the possibility that WWP1-USP9X interactions function in a chain-editing capacity on polyubiquitinated DVL2. Ultimately, rigorous biochemical analysis dissecting mechanisms that regulate the balance of different ubiquitylation and deubiquitylation activities toward DVL2 will be critical to understand how its specification for either canonical or noncanonical WNT signaling pathways is determined.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jason MacGurn (Jason.a.macgurn@vanderbilt.edu). Plasmids generated in this study are available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
HEK293 STF cells (Female) and MDA-MB-231 cells (Female) were purchased from the American Type Culture Collection (ATCC). HEK293 STF cells were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin at 37°C in 5% CO2. MDA-MB-231 cells were cultured in RPMI with 10% FBS and 1% penicillin/streptomycin at 37°C in 5% CO2. MDA-MB-231 cells stably expressing FLAG-WWP1, FLAG-DVL2, FLAG-DUB-DVL2, or FLAG-DUB*-DVL2 were generated using the pQCXIP retroviral vector system. MDA-MB-231 usp9x knockout cells were generated using a CRISPR/Cas-9 genome editing system.
METHOD DETAILS
Assays for measuring WNT activation
Topflash reporter assays in HEK293 STF cells were performed as follows: HEK293 STF cells were seeded on 24-well plates at ~50% confluency. Cells were treated with 100ng/mL purified recombinant WNT3a (R&D Systems) and 100ng/mL purified recombinant R-spondin (R&D systems) 24 hours prior to lysis with 1X Passive Lysis buffer (Promega). Luciferase activity was measured using the Steady-Glo luciferase Assay (Promega). Luciferase signal was normalized to viable cell number using Cell-Titer Glo Assay (Promega). For siRNA knockdown studies, cells were treated for 72 hours prior to lysis, for inhibitor studies cells were treated for 24 hours prior to lysis, and for overexpression studies cells were transfected with plasmid DNA for 48 hours prior to lysis.
Topflash reporter assays in MDA-MB-231 cells were performed as follows: Cells were plated, co-transfected with TOPflash and Renilla expression plasmids at 24 hours and lysed at 48 hours using the Dual-Glo Luciferase Assay system (Promega). Luciferase signal was normalized to co-transfected Renilla expression. Data were normalized to a positive control (set at 100% activation). Assays were performed in triplicate and repeated at least 3 times.
B-catenin stabilization assay: HEK293 STF cells were cultured in Dulbecco’s modified Eagle’s Medium (DMEM) and activated with WNT3A ligand or LiCl for 24 hours. Then cells were lysed and nuclear/cytoplasmic fractionation was performed as previously described (Thorne et al., 2010).
Transfections
Plasmid and siRNA transfections were performed using Lipojet (SignaGen, SL100468) and Lipofectamine LTX with PLUS reagent (Thermo Fisher, 15338100) according to manufacturer’s protocol.
Immunoblots
Whole-cell lysates were generated by lysing cells in Laemmli Sample Buffer (Bio-Rad) with 5% βME and subsequently boiling samples at 95°C for 5 minutes. Proteins were analyzed by SDS-PAGE and immunoblotting using the Li-Cor Odyssey reagents and protocols. Fluorescence signaling was detected using an Odyssey CLx Infrared Scanner (Li-Cor).
Cell lines
HEK293 STF cells and MDA-MB-231 cells were purchased from the American Type Culture Collection (ATCC). HEK293 STF cells were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin at 37°C in 5% CO2. MDA-MB-231 cells were cultured in DMEM with 10% FBS and 1% penicillin/streptomycin at 37°C in 5% CO2. MDA-MB-231 cells stably expressing FLAG-WWP1, FLAG-DVL2, FLAG-DUB-DVL2, or FLAG-DUB*-DVL2 were generated using the pQCXIP retroviral vector system. MDA-MB-231 usp9x knockout cells were generated using a CRISPR/Cas-9 genome editing system.
SILAC-based Quantitative Proteomic Analysis
Quantitative mass spectrometry analysis by SILAC was performed on MDA-MB-231 cells stably expressing the indicated FLAG-tagged substrates. These cells were cultured in the presence of either heavy or light isotopes (lysine and arginine) for at least 6 doublings to ensure incorporation of these isotopes into 100% of the proteome. Affinity purification was performed as previously described (Lee et al., 2017). Eluted proteins were digested with 1 μg Trypsin Gold (Promega) at 37 F overnight. Digested peptides were cleaned-up on a Sep-Pak C18 column. Purified peptides were then dried, reconstituted in 0.1% trifluoroacetic acid, and analyzed by LC-MS/MS using an Orbitrap XL mass spectrometer. Database search and SILAC quantitation were performed using MaxQuant software.
Co-Immunoprecipitation Studies
For coimmunoprecipitation assays, cells were washed with cold PBS and lysed with a nondenaturing lysis buffer (NDLB): 50mM tris (pH 7.4), 300mM NaCl, 5mM EDTA, 1% Triton X-100, cOmplete protease inhibitor cocktail (Roche), Phosphatase inhibitor cocktail (Roche), 1mM PMSF, 1mM Phenanthroline, 10mM Iodoacetamide, and 20 μM MG132. Lysates were diluted to 1mg/mL with NDLB and incubated with Sigma FLAG EZ-view beads for 2 hours with rotation at 4°C. Beads were washed 3 times with cold NDLB and once with cold PBS. Proteins were then eluted from beads using sample buffer and samples were processed for SDS-PAGE followed by immunoblotting.
Fluorescence Microscopy
MDA-MB-231 cells were seeded on coverslips at 50% confluency. At 24 hours after seeding, cells were transfected with FLAG-Dvl2 using LipoJet Transfection Reagent (Signagen). 72 hours after seeding cells were fixed and permeabilized using 4% paraformaldehyde (PFA) and immunofluorescence block buffer (PBS + 0.1% Triton-x + 10% FBS). Cells were then labeled with primary antibodies for 1 hour followed by a 1 hour incubation with Alexa Fluor conjugated secondary antibodies. Coverslips were mounted on slides using ProLong AntiFade mountant (Thermo). Microscopy images were acquired using a DeltaVision Elite system (GE Healthcare) and processed using SoftWoRx software (GE Healthcare, Chicago, IL).
In Vitro Ubiquitin Conjugation and Deubiquitylation Assays
Due to the limited solubility of full-length DVL2 in solution, in all in vitro conjugation and deconjugation assays HA-DVL2 was purified from human HEK293 cells using magnetic anti-HA beads (Pierce #88836) and all conjugation/deconjugation reactions were performed on HA-DVL2 bound to anti-HA beads. Lysis and purification were performed as follows: 24 hours after seeding, HEK293 cells were transfected with HA-Dvl2 and 48 hours post-transfection cells were lysed in NDLB and clarified by centrifugation. Lysates bound anti-HA magnetic beads overnight at 4°C with rotation. Beads were washed 3 times in NDLB and 1 time in cold 1X PBS before being resuspended in conjugation buffer (40 mM Tris pH 7.5, 10 mM MgCl2, and 0.6 mM DTT) for immediate use in conjugation/deconjugation reactions.
For the conjugation reactions, 56 nM GST-Ube1 (Boston Biochem, Cambridge, MA, E-300), 0.77 mM UBE2D3, 2.3 mM Ub, HA-DVL2, and 60 nM FLAG-WWP1 (Sigma, SRP0229) were incubated in conjugation buffer (40 mM Tris pH 7.5, 10 mM MgCl2, and 0.6 mM DTT) with 1 mM ATP, at 37°C. Reactions were initiated by the addition of FLAG-WWP1. Samples were removed at indicated time-points, boiled in 2× Laemmli sample buffer for 10 min, and analyzed by blotting for HA-DVL2 with DVL2 antibody (Cell Signaling Technology, #3216) using goat anti-rabbit secondary (IRDye 800 CW, LI-COR Biosciences, Lincoln, NE). Quantification was performed using ImageStudioLite software (LI-COR).
For deconjugation reactions, polyubiquitinated substrate (HA-DVL2) was generated using the conjugation assay described above within one exception in that 0.1 mM ATP was used. The conjugation reaction was stopped by incubation with 0.75 units Apyrase in a 90 μl reaction at 30°C for 1 hr. For the deconjugation reaction, 100 nM His6-USP9X (Boston Biochem E-552), 100nM OTUB1, or 100nM AMSH was added to conjugated HA-DVL2 as well as 10X DUB buffer (0.5 M Tris pH 7.5, 500 mM NaCl, 50mM DTT) to 1X. Reactions were initiated by the addition of His6-USP9X protein. The reaction was incubated at 37°C. Samples were removed at indicated time-points and boiled in 2× Laemmli sample buffer. HA-DVL2 levels were determined as described in the conjugation assay.
For Rheostat experiments, HA-DVL2 substrate was resuspended in conjugation buffer with 1mM ATP and treated with 56 nM GST-Ube1, 0.77 mM UBE2D3, 2.3 mM Ub, and indicated concentrations of FLAG-WWP1 and His-USP9X for 60 minutes at 37°C. Samples were then boiled in 2× Lammli sample buffer for 10 minutes and analyzed by blotting for HA-DVL2 with DVL2 antibody (CST #3216). Line profile analysis was performed using ImageJ (NIH).
Migration Assays
Cell culture dishes were coated with 5 mg/ml rat tail ColI (Corning #354236) in PBS for 1 h at 37°C then cells were plated at low density and incubated at 37°C for 1 hour in cell culture medium. Cells were kept at 37°C in SFM4MAb medium (HyClone, Logan, UT) supplemented with 2% FBS at pH 7.4 during imaging using phase-contrast and fluorescence microscopy techniques. Images were acquired at 5 min intervals for 6 h, using DeltaVision Elite system software (GE Healthcare) and cell speed was quantified using SoftWoRx software (GE Healthcare, Chicago, IL). Wind rose plots were generated by transposing x,y coordinates of cell tracks to a common origin.
Rho Activation Assays
Rho activation assays were performed using the Rho Activation Assay Kit (Millipore Sigma #17–924). MDA-MB-231 cells (WT and usp9x KO cells) were grown until confluent, washed in ice-cold 1XPBS, lysed in ice-cold 1X Mg2+ Lysis/Wash buffer (MLB) (Millipore Sigma #20–168), scraped and collected in microfuge tubes on ice, and incubated at 4°C for 15 minutes with agitation. Lysates were then cleared of cellular debris by centrifugation at 14,000×g for 5 minutes at 4°C. Clarified lysate was then bound to Rho Assay Reagent (Millipore Sigma #14–383) at 4°C for 45 minutes with gentle agitation. Agarose beads were then washed 3 times with 1X MLB and proteins were eluted in 2× Laemmli reducing sample buffer (BioRad #1610737) by boiling at 95°C for 5 minutes. Samples were fractionated by SDS-PAGE and analyzed by blotting using the Anti-Rho (−A, −B, −C) clone 55 antibody (Millipore Sigma #05–778).
GTPγS/GDP loading for positive and negative controls were prepared as described above with an additional 30 minute incubation at 30°C with agitation of cell lysate in the presence of 10mM EDTA and either 100uM GTPγS (Millipore Sigma #20–176) or 1mM GDP (Millipore Sigma #20–177). This step was performed prior to binding the Rho Assay Reagent.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Detailed statistical analysis can be found in accompanying Statistical Reporting Document (Table S4). For each statistical analysis a Student’s t test was used to test for a statistically significant difference between the means of the two variables of interest. The alpha value for each experiment was set at 0.05 and a p value was calculated using the Student’s t test function in Microsoft Excel to determine statistical significance. Each figure with statistical analysis represents n ≥ 3 for all coIP and luciferase assay experiments, where n represents biological replicates. For quantitation of all microscopy-based data an n ≥ 25 was used where n represents single cells. In all figures including statistics, error bars represent standard deviation of the mean. Details of statistical analysis can also be found in figure legends.
DATA AND CODE AVAILABILITY
Raw data supporting the study is available in Table S4. Mass spectrometry data generated in this study is available from the corresponding author upon request. This study did not generate code.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG M2 antibody produced in mouse | Millipore | Product #: F1804; RRID:AB_262044 |
| USP9X/Y Antibody (E-12) | Santa Cruz | Cat #: sc-365353; RRID:AB_10846088 |
| WWP1 monoclonal antibody (M01), clone 1A7 | Abnova | Catalog #: H00011059-M01; RRID:AB_509107 |
| Dvl2 Antibody #3216 | Cell Signaling Technology | Product # 3216S; RRID:AB_2093338 |
| GAPDH (14C10) Rabbit mAb #2118 | Cell signaling technology | Product # 2118S; RRID:AB_561053 |
| USP24 (S-18) antibody | Cell Signaling Technology | Catalog #: sc-82080; RRID:AB_2212766 |
| Anti-beta Catenin antibody [E247] | Abcam | ab32572; RRID:AB_725966 |
| Tubulin antibody | Vanderbilt Antibody and Protein Resource Core | N/A |
| NEDD4L Antibody #4013 | Cell Signaling Technology | Product # 4013S; RRID:AB_1904063 |
| WWP2 (AIP2) Antibody (A-3) | Santa Cruz | Catalog #: sc-398090; RRID:AB_2288586 |
| HA antibody | Vanderbilt Antibody and Protein Resource Core | N/A |
| Alpha adaptin antibody (AP2A1) AC1-M11 | Thermo Fisher | Cat #: MA3–061; RRID:AB_2056321 |
| Anti-AP2M1 antibody [EP2695Y] | Abcam | ab75995; RRID:AB_1309955 |
| Anti-VANGL1 | Sigma | Product #: HPA025235; RRID:AB_1858718 |
| Cofilin (D3F9) XP Rabbit mAb | Cell Signaling Technology | D3F9; RRID:AB_10622000 |
| Anti-Rho (-A, -B, -C) clone 55 | Millipore Sigma | Catalog # 05–778; RRID:AB_309989 |
| Anti-HA magnetic beads | Pierce | Catalog # 88836 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| WP1130 | UBPBio | Cat #: F2120 |
| MG-132 | APExBIO | Cat #: A2585 |
| Phenanthroline | Sigma-Aldrich | Cat #: P9375 |
| Iodoacetamide | Sigma-Aldrich | Cat #: I1149 |
| LGK-974 | Selleck Chem | Catalog No.S7143 |
| Recombinant Human/mouse Wnt5a | R&D System | Cat #: 645-WN |
| WWP1 Active human recombinant, expressed in baculovirus infected insect cells | Sigma-Aldrich | Prod #: SRP0229 |
| USP9x-His6, isoform 2, human recombinant | Boston Biochem | Cat. # E-552 |
| Recombinant OTUB1 | UBPBio | Catalog #: H3000 |
| AMSH, human recombinant | Boston Biochem | Cat. # E-548B |
| GST-Ubiquitin E1 Enzyme (UBE1), S.cer. recombinant | Boston Biochem | Cat. # E-300 |
| Recombinant Human UbcH5c/UBE2D3 | Boston Biochem | Cat. # E2627 |
| Recombinant Mouse Wnt-3a | R&D Systems | Cat #: 1324-WN |
| Recombinant Mouse R-spondin | R&D Systems | Cat #: 3474-RS |
| Corning® Collagen I, Rat Tail | Corning | Product #: 354236 |
| Heavy Lysine: L-Lysine-13C6,15N2 hydrochloride | Sigma-Aldrich | Product #: 608041 |
| Heavy Arginine: L-Arginine-13C6,15N4 hydrochloride | Sigma-Aldrich | Product #: 608033 |
| Critical Commercial Assays | ||
| Steady Glo Luciferase Assay System | Promega | Catalog #: E2510 |
| Dual Glo Luciferase Assay System | Promega | Catalog #: E2920 |
| Rho Activation Assay Kit | Millipore Sigma | Catalog #: 17–294 |
| Experimental Models: Cell Lines | ||
| Human cells: MDA-MB-231 cells | ATCC | Product #: MDA-MB-231 (ATCC® HTB-26); RRID:CVCL_0062 |
| Human cells: HEK293 cells | ATCC | Product #: 293 [HEK293] (ATCC® CRL-1573; RRID:CVCL_0045 |
| Human cells: HEK293 STF | ATCC | Product #: HEK293 STF (ATCC® CRL-3249); RRID:CVCL_AQ26 |
| Human cells: MDA-MB-231 cells stably expressing FLAG-DVL2, FLAG-DUB-DVL2, or FLAG-DUB*-DVL2 | This paper | N/A |
| Human cells: MDA-MB-231 usp9x KO cells ± FLAG-DVL2 | This paper | N/A |
| Oligonucleotides | ||
| Control siRNA: orKKJ1 s, nonsilencing/pGL2 sense, CGUACGCGGAAUACUUCGAUU | This paper | N/A |
| Usp9x siRNA #1 sequence: AGAAAUCGCUGGUAUAAAUU | This paper | N/A |
| Usp9x siRNA #2 sequence: ACACGAUGCUUUAGAAUUUU | This paper | N/A |
| Nedd4L siRNA #1 sequence: GCUAGACUGUGGAUUGAGUUU | This paper | N/A |
| Nedd4L siRNA #2 sequence: UGAGGAUCAUUUGUCCUACUU | This paper | N/A |
| WWP1 siRNA #1 sequence: GAAGUCAUCUGUAACUAAAUU | This paper | N/A |
| WWP1 siRNA #2 sequence: GCAGAGAAAUACUGUUUAUUU | This paper | N/A |
| WWP2 siRNA #1 sequence: GGGAGAAGAGACAGGACAAUU | This paper | N/A |
| WWP2 siRNA #2 sequence: CAGGAUGGGAGAUGAAAUAUU | This paper | N/A |
| VANGL1 Silencer Select siRNA | Thermo Fisher | Catalog #: 4392420 |
| Recombinant DNA | ||
| Plasmid: FLAG-Wwp1 | This paper | N/A |
| Plasmid: FLAG-Wwp1-ww1 (W377F, P380A) | This paper | N/A |
| Plasmid: FLAG-Wwp1-ww2 (W409F, P412A) | This paper | N/A |
| Plasmid: FLAG-Wwp1-ww3 (W484F, P487A) | This paper | N/A |
| Plasmid: FLAG-Wwp1-ww4 (F524A, P527A) | This paper | N/A |
| Plasmid: FLAG-Wwp1–4ww (W377F, P380A, W409F, P412A, W484F, P487A, F524A, P527A) | This paper | N/A |
| Plasmid: FLAG-Wwp1-WW1 (W409F, P412A, W484F, P487A, F524A, P527A) | This paper | N/A |
| Plasmid: FLAG-Wwp1-WW2 (W377F, P380A, W484F, P487A, F524A, P527A) | This paper | N/A |
| Plasmid: FLAG-Wwp1-WW3 (W377F, P380A, W409F, P412A, F524A, P527A) | This paper | N/A |
| Plasmid: FLAG-Wwpw-WW4 (W377F, P380A, W409F, P412A, W484F, P487A) | This paper | N/A |
| Plasmid: FLAG-DVL2 PY1: FPAY393 → FAAA393 | This paper | N/A |
| Plasmid: FLAG-DVL2 PY2: PPPY568 → PAPA568 | This paper | N/A |
| Plasmid: FLAG-DVL2 2PY: FPAY393 → FAAA393 and PPPY568 → PAPA568 | This paper | N/A |
| Plasmid: USP9X1–2433, or usp9x-Δpy3–4: NPQY2437 → NAQA2437 (py3) and APLY2515 → AALA2515 (py4) | This paper | N/A |
| Plasmid: UL36-DVL2 | Stringer and Piper (2011); Kattenhorn et al. (2005) | N/A |
| Plasmid: UL36 (C65A) catalytic dead-DVL2 | Stringer and Piper (2011); Kattenhorn et al. (2005) | N/A |
| Plasmid: FLAG-DVL2-K0 (all lysines K → R) | This paper | N/A |
| Plasmid: FLAG-USP9X | This paper | N/A |
| Plasmid: FLAG-USP9X (C1556V) catalytic dead | This paper | N/A |
| Plasmid: FLAG-WWP1 (C890S) catalytic dead | This paper | N/A |
| Plasmid: GFP-DVL2 | This paper | N/A |
| Plasmid: GFP-DVL2-K0 (all lysines K → R) | This paper | N/A |
| Plasmid: GFP-WWP1 | This paper | N/A |
| Plasmid: GFP-USP9X | This paper | N/A |
| Software and Algorithms | ||
| SoftWorx | GE | N/A |
| MaxQuant | Max Planck Institute of Biochemistry | N/A |
| ImageJ | NIH | N/A |
| ImageStudioLite software | LICORE | N/A |
Highlights.
USP9X and WWP1 regulate a ubiquitin rheostat on DVL2 in breast cancer cells
USP9X promotes canonical WNT activation via deubiquitylation of DVL2
USP9X is a negative regulator of WNT-PCP activation and cell motility
This ubiquitin rheostat on DVL2 influences WNT pathway specification
ACKNOWLEDGMENTS
We are very grateful to K. Rose for helpful advice regarding technical aspects and analysis of quantitative proteomic data. We are also grateful to E. Lee, K. Saito-Diaz, and V. Ng for reagents and technical advice. We also thank T. Graham, S.M. Ahmed, S. Lee, and N. Hepowit for critical reading of this manuscript and E. MacGurn for assistance with graphic design. This research was supported by NIH grant R00 GM101077 (to J.A.M.) and an NIH SPORE grant in GI Cancer (P50 CA095103).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.083.
REFERENCES
- Allache R, Wang M, De Marco P, Merello E, Capra V, and Kibar Z (2015). Genetic studies of ANKRD6 as a molecular switch between Wnt signaling pathways in human neural tube defects. Birth Defects Res. A Clin. Mol. Teratol 103, 20–26. [DOI] [PubMed] [Google Scholar]
- Angers S, Thorpe CJ, Biechele TL, Goldenberg SJ, Zheng N, Mac-Coss MJ, and Moon RT (2006). The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation. Nat. Cell Biol 8, 348–357. [DOI] [PubMed] [Google Scholar]
- Bernassola F, Karin M, Ciechanover A, and Melino G (2008). The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 14, 10–21. [DOI] [PubMed] [Google Scholar]
- Cadavid AL, Ginzel A, and Fischer JA (2000). The function of the Drosophila fat facets deubiquitinating enzyme in limiting photoreceptor cell number is intimately associated with endocytosis. Development 127, 1727–1736. [DOI] [PubMed] [Google Scholar]
- Chen HI, Einbond A, Kwak SJ, Linn H, Koepf E, Peterson S, Kelly JW, and Sudol M (1997). Characterization of the WW domain of human yes-associated protein and its polyproline-containing ligands. J. Biol. Chem 272, 17070–17077. [DOI] [PubMed] [Google Scholar]
- David D, Nair SA, and Pillai MR (2013). Smurf E3 ubiquitin ligases at the cross roads of oncogenesis and tumor suppression. Biochim. Biophys. Acta 1835, 119–128. [DOI] [PubMed] [Google Scholar]
- de Las Pozas A, Reiner T, De Cesare V, Trost M, and Perez-Stable C (2018). Inhibiting Multiple Deubiquitinases to Reduce Androgen Receptor Expression in Prostate Cancer Cells. Sci. Rep 8, 13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Zhang Y, Xu C, Tao QH, and Chen YG (2013). HECT domain-containing E3 ubiquitin ligase NEDD4L negatively regulates Wnt signaling by targeting dishevelled for proteasomal degradation. J. Biol. Chem 288, 8289–8298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermekova KS, Zambrano N, Linn H, Minopoli G, Gertler F, Russo T, and Sudol M (1997). The WW domain of neural protein FE65 interacts with proline-rich motifs in Mena, the mammalian homolog of Drosophila enabled. J. Biol. Chem 272, 32869–32877. [DOI] [PubMed] [Google Scholar]
- Fei C, Li Z, Li C, Chen Y, Chen Z, He X, Mao L, Wang X, Zeng R, and Li L (2013). Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/β-catenin signaling. Mol. Cell. Biol 33, 4095–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei C, He X, Xie S, Miao H, Zhou Z, and Li L (2014). Smurf1-mediated axin ubiquitination requires Smurf1 C2 domain and is cell cycle-dependent. J. Biol. Chem 289, 14170–14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewska B, Kamińska J, Jesionowska A, Martin NC, Hopper AK, and Zoładek T (2001). WW domains of Rsp5p define different functions: determination of roles in fluid phase and uracil permease endocytosis in Saccharomyces cerevisiae. Genetics 157, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka A, Dolde C, Kirsch N, Huang YL, Kazanskaya O, Ingelfinger D, Boutros M, Cruciat CM, and Niehrs C (2011). LGR4 and LGR5 are R-spondin receptors mediating Wnt/β-catenin and Wnt/PCP signalling. EMBO Rep. 12, 1055–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatakeyama J, Wald JH, Printsev I, Ho HY, and Carraway KL 3rd. (2014). Vangl1 and Vangl2: planar cell polarity components with a developing role in cancer. Endocr. Relat. Cancer 21, R345–R356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Hryciw DH, Carroll ML, Myers SA, Whitbread AK, Kumar S, Poronnik P, and Hooper JD (2008). The ubiquitin-protein ligase Nedd4–2 differentially interacts with and regulates members of the Tweety family of chloride ion channels. J. Biol. Chem 283, 24000–24010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hryciw DH, Ekberg J, Lee A, Lensink IL, Kumar S, Guggino WB, Cook DI, Pollock CA, and Poronnik P (2004). Nedd4–2 functionally interacts with ClC-5: involvement in constitutive albumin endocytosis in proximal tubule cells. J. Biol. Chem 279, 54996–55007. [DOI] [PubMed] [Google Scholar]
- Ingham RJ, Gish G, and Pawson T (2004). The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 23, 1972–1984. [DOI] [PubMed] [Google Scholar]
- Jones C, Qian D, Kim SM, Li S, Ren D, Knapp L, Sprinzak D, Avraham KB, Matsuzaki F, Chi F, and Chen P (2014). Ankrd6 is a mammalian functional homolog of Drosophila planar cell polarity gene diego and regulates coordinated cellular orientation in the mouse inner ear. Dev. Biol 395, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, and Donato NJ (2010). Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 70, 9265–9276. [DOI] [PubMed] [Google Scholar]
- Katoh M (2005). WNT/PCP signaling pathway and human cancer (review). Oncol. Rep 14, 1583–1588. [PubMed] [Google Scholar]
- Kattenhorn LM, Korbel GA, Kessler BM, Spooner E, and Ploegh HL (2005). A Deubiquitinating Enzyme Encoded by HSV-1 Belongs to a Family of Cysteine Proteases that Is Conserved across the Family Herpesviridae. Mol. Cell 19, 547–557. [DOI] [PubMed] [Google Scholar]
- Khan OM, Carvalho J, Spencer-Dene B, Mitter R, Frith D, Snijders AP, Wood SA, and Behrens A (2018). The deubiquitinase USP9X regulates FBW7 stability and suppresses colorectal cancer. J. Clin. Invest 128, 1326–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HC, and Huibregtse JM (2009). Polyubiquitination by HECT E3s and the determinants of chain type specificity. Mol. Cell. Biol 29, 3307–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim RQ, and Sixma TK (2017). Regulation of USP7: A High Incidence of E3 Complexes. J. Mol. Biol 429, 3395–3408. [DOI] [PubMed] [Google Scholar]
- Kuratomi G, Komuro A, Goto K, Shinozaki M, Miyazawa K, Miyazono K, and Imamura T (2005). NEDD4–2 (neural precursor cell expressed, developmentally down-regulated 4–2) negatively regulates TGF-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-beta type I receptor. Biochem. J 386, 461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha D, O’Leary C, Cron KR, Deraska P, Zhu K, D’Andrea AD, and Kozono D (2015). USP9X inhibition promotes radiation-induced apoptosis in non-small cell lung cancer cells expressing mid-to-high MCL1. Cancer Biol. Ther 16, 392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Tumolo JM, Ehlinger AC, Jernigan KK, Qualls-Histed SJ, Hsu PC, McDonald WH, Chazin WJ, and MacGurn JA (2017). Ubiquitin turnover and endocytic trafficking in yeast are regulated by Ser57 phosphorylation of ubiquitin. Elife 6, e29176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Park JS, Kim JH, Jung SM, Lee JY, Kim SJ, and Park SH (2011). Smad6-specific recruitment of Smurf E3 ligases mediates TGF-β1-induced degradation of MyD88 in TLR4 signalling. Nat. Commun 2, 460. [DOI] [PubMed] [Google Scholar]
- Li X, Song N, Liu L, Liu X, Ding X, Song X, Yang S, Shan L, Zhou X, Su D, et al. (2017). USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nat. Commun 8, 14866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A, Hou Q, Jarzylo L, Amato S, Gilbert J, Shang F, and Man HY (2011). Nedd4-mediated AMPA receptor ubiquitination regulates receptor turnover and trafficking. J. Neurochem 119, 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CC, Kanekiyo T, Roth B, and Bu G (2014). Tyrosine-based signal mediates LRP6 receptor endocytosis and desensitization of Wnt/β-catenin pathway signaling. J. Biol. Chem 289, 27562–27570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Adegoke OA, Nepveu A, Nakayama KI, Bedard N, Cheng D, Peng J, and Wing SS (2009). USP19 deubiquitinating enzyme supports cell proliferation by stabilizing KPC1, a ubiquitin ligase for p27Kip1. Mol. Cell. Biol 29, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, Buchanan M, Hosein AN, Basik M, and Wrana JL (2012). Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell 151, 1542–1556. [DOI] [PubMed] [Google Scholar]
- Lui TT, Lacroix C, Ahmed SM, Goldenberg SJ, Leach CA, Daulat AM, and Angers S (2011). The ubiquitin-specific protease USP34 regulates axin stability and Wnt/β-catenin signaling. Mol. Cell. Biol 31, 2053–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, and He X (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGurn JA, Hsu PC, and Emr SD (2012). Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem 81, 231–259. [DOI] [PubMed] [Google Scholar]
- Madrzak J, Fiedler M, Johnson CM, Ewan R, Knebel A, Bienz M, and Chin JW (2015). Ubiquitination of the Dishevelled DIX domain blocks its head-to-tail polymerization. Nat. Commun 6, 6718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marikawa Y, and Elinson RP (1998). beta-TrCP is a negative regulator of Wnt/beta-catenin signaling pathway and dorsal axis formation in Xenopus embryos. Mech. Dev 77, 75–80. [DOI] [PubMed] [Google Scholar]
- Miyazaki K, Fujita T, Ozaki T, Kato C, Kurose Y, Sakamoto M, Kato S, Goto T, Itoyama Y, Aoki M, and Nakagawara A (2004). NEDL1, a novel ubiquitin-protein isopeptide ligase for dishevelled-1, targets mutant superoxide dismutase-1. J. Biol. Chem 279, 11327–11335. [DOI] [PubMed] [Google Scholar]
- Mouchantaf R, Azakir BA, McPherson PS, Millard SM, Wood SA, and Angers A (2006). The ubiquitin ligase itch is auto-ubiquitylated in vivo and in vitro but is protected from degradation by interacting with the deubiquitylating enzyme FAM/USP9X. J. Biol. Chem 281, 38738–38747. [DOI] [PubMed] [Google Scholar]
- Mukai A, Yamamoto-Hino M, Awano W, Watanabe W, Komada M, and Goto S (2010). Balanced ubiquitylation and deubiquitylation of Frizzled regulate cellular responsiveness to Wg/Wnt. EMBO J. 29, 2114–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mund T, Graeb M, Mieszczanek J, Gammons M, Pelham HR, and Bienz M (2015). Disinhibition of the HECT E3 ubiquitin ligase WWP2 by polymerized Dishevelled. Open Biol. 5, 150185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narimatsu M, Bose R, Pye M, Zhang L, Miller B, Ching P, Sakuma R, Luga V, Roncari L, Attisano L, and Wrana JL (2009). Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell 137, 295–307. [DOI] [PubMed] [Google Scholar]
- Nethe M, de Kreuk BJ, Tauriello DV, Anthony EC, Snoek B, Stumpel T, Salinas PC, Maurice MM, Geerts D, Deelder AM, et al. (2012). Rac1 acts in conjunction with Nedd4 and dishevelled-1 to promote maturation of cell-cell contacts. J. Cell Sci 125, 3430–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paudel P, Zhang Q, Leung C, Greenberg HC, Guo Y, Chern YH, Dong A, Li Y, Vedadi M, Zhuang Z, and Tong Y (2019). Crystal structure and activity-based labeling reveal the mechanisms for linkage-specific substrate recognition by deubiquitinase USP9X. Proc. Natl. Acad. Sci. USA 116, 7288–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KA, Grützmann R, Aust D, Rümmele P, et al. ; Australian Pancreatic Cancer Genome Initiative (2012). The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LF, Sun H, Liu Y, Potu H, Kandarpa M, Ermann M, Courtney SM, Young M, Showalter HD, Sun D, et al. (2015). Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood 125, 3588–3597. [DOI] [PubMed] [Google Scholar]
- Potu H, Peterson LF, Kandarpa M, Pal A, Sun H, Durham A, Harms PW, Hollenhorst PC, Eskiocak U, Talpaz M, and Donato NJ (2017). Usp9x regulates Ets-1 ubiquitination and stability to control NRAS expression and tumorigenicity in melanoma. Nat. Commun 8, 14449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premarathne S, Murtaza M, Matigian N, Jolly LA, and Wood SA (2017). Loss of Usp9x disrupts cell adhesion, and components of the Wnt and Notch signaling pathways in neural progenitors. Sci. Rep 7, 8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotin D, and Kumar S (2009). Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol 10, 398–409. [DOI] [PubMed] [Google Scholar]
- Saito-Diaz K, Chen TW, Wang X, Thorne CA, Wallace HA, Page-McCaw A, and Lee E (2013). The way Wnt works: components and mechanism. Growth Factors 31, 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savio MG, Wollscheid N, Cavallaro E, Algisi V, Di Fiore PP, Sigismund S, Maspero E, and Polo S (2016). USP9X Controls EGFR Fate by Deubiquitinating the Endocytic Adaptor Eps15. Curr. Biol 26, 173–183. [DOI] [PubMed] [Google Scholar]
- Schwarz-Romond T, Merrifield C, Nichols BJ, and Bienz M (2005). The Wnt signalling effector Dishevelled forms dynamic protein assemblies rather than stable associations with cytoplasmic vesicles. J. Cell Sci 118, 5269–5277. [DOI] [PubMed] [Google Scholar]
- Schwarz-Romond T, Fiedler M, Shibata N, Butler PJ, Kikuchi A, Higuchi Y, and Bienz M (2007). The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat. Struct. Mol. Biol 14, 484–492. [DOI] [PubMed] [Google Scholar]
- Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O’Rourke K, Bazan F, Eastham-Anderson J, et al. (2010). Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463, 103–107. [DOI] [PubMed] [Google Scholar]
- Stringer DK, and Piper RC (2011). A single ubiquitin is sufficient for cargo protein entry into MVBs in the absence of ESCRT ubiquitination. J. Cell Biol 192, 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanksley JP, Chen X, and Coffey RJ (2013). NEDD4L is downregulated in colorectal cancer and inhibits canonical WNT signaling. PLoS ONE 8, e81514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauriello DV, Haegebarth A, Kuper I, Edelmann MJ, Henraat M, Canninga-van Dijk MR, Kessler BM, Clevers H, and Maurice MM (2010). Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol. Cell 37, 607–619. [DOI] [PubMed] [Google Scholar]
- Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, Jernigan KK, Meyers KC, Hang BI, Waterson AG, et al. (2010). Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol 6, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y (2009). Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol. Cancer Ther 8, 2103–2109. [DOI] [PubMed] [Google Scholar]
- Wei W, Li M, Wang J, Nie F, and Li L (2012). The E3 ubiquitin ligase ITCH negatively regulates canonical Wnt signaling by targeting dishevelled protein. Mol. Cell. Biol 32, 3903–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. (2004). De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430, 694–699. [DOI] [PubMed] [Google Scholar]
- Wu X, Fukushima H, North BJ, Nagaoka Y, Nagashima K, Deng F, Okabe K, Inuzuka H, and Wei W (2014). SCFβ-TRCP regulates osteoclastogenesis via promoting CYLD ubiquitination. Oncotarget 5, 4211–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Avello M, Schirle M, McWhinnie E, Feng Y, Bric-Furlong E, Wilson C, Nathans R, Zhang J, Kirschner MW, et al. (2013). Deubiquitinase FAM/USP9X interacts with the E3 ubiquitin ligase SMURF1 protein and protects it from ligase activity-dependent self-degradation. J. Biol. Chem 288, 2976–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, and Mlodzik M (2015). Wnt-Frizzled/planar cell polarity signaling: cellular orientation by facing the wind (Wnt). Annu. Rev. Cell Dev. Biol 31, 623–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Zhang S, Wang Z, Yang C, Ouyang W, Zhou F, Zhou Y, and Xie C (2016). Deubiquitinase USP9X deubiquitinates β-catenin and promotes high grade glioma cell growth. Oncotarget 7, 79515–79525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Rual JF, Tamai K, Harada Y, Vidal M, He X, and Kirchhausen T (2007). Association of Dishevelled with the clathrin AP-2 adaptor is required for Frizzled endocytosis and planar cell polarity signaling. Dev. Cell 12, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Xing Y, Harrison SC, and Kirchhausen T (2010). Structural analysis of the interaction between Dishevelled2 and clathrin AP-2 adaptor, a critical step in noncanonical Wnt signaling. Structure 18, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Wang M, Dong Q, and Jin F (2014). Diversin is overexpressed in breast cancer and accelerates cell proliferation and invasion. PLoS ONE 9, e98591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZR, Bonifacino JS, and Hegde RS (2013). Deubiquitinases sharpen substrate discrimination during membrane protein degradation from the ER. Cell 154, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Tian Y, Du J, Hu Z, Yang L, Liu J, and Gu L (2012). Dvl2-dependent activation of Daam1 and RhoA regulates Wnt5a-induced breast cancer cell migration. PLoS ONE 7, e37823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data supporting the study is available in Table S4. Mass spectrometry data generated in this study is available from the corresponding author upon request. This study did not generate code.