

Abstract
Minimalist enzymes designed to catalyze model reactions provide useful starting points for creating catalysts for practically important chemical transformations. We have shown that Kemp eliminases of the AlleyCat family facilitate conversion of leflunomide (an immunosupressor pro-drug) to its active form teriflunomide with outstanding rate enhancement (nearly four orders of magnitude) and catalytic proficiency (more than seven orders of magnitude) without any additional optimization. This remarkable activity is achieved by properly positioning the substrate in close proximity to the catalytic glutamate with very high pKa.
Keywords: allostery, Kemp elimination, protein design, calmodulin, Nuclear Magnetic Resonance
Graphical Abstract
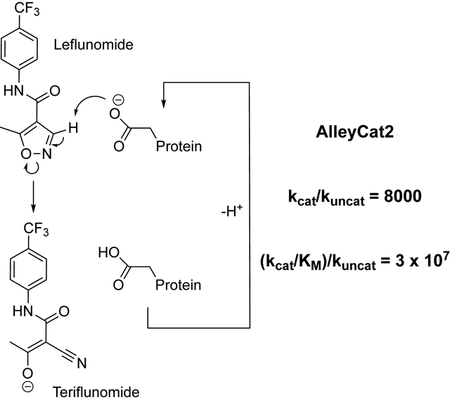
Kemp eliminases of the AlleyCat family, designed using minimalist approach, facilitate activation of an immunosupressor leflunomide with outstanding rate enhancement and catalytic proficiency.
Enzymes facilitate chemical transformations with high efficiency and extraordinary stereo- and regioselectivity. Thus, it is hardly surprising that much effort has been dedicated to designing enzyme-like catalysts for reactions that are not observed in nature. Past decades yielded spectacular successes in repurposing naturally occurring enzymes for new reactions,[1] however completely de novo approaches, where a catalyst is designed from first principles, have been much less productive.[2] As disappointing as our advances may be, the challenge of creating a highly sophisticated catalyst to rival those found in nature is a formidable one. The protein at a minimum has to bind substrate, adapt proper geometry for the transition state and then release the product. All of these steps have to be appropriately matched in energy, and the dynamics of the protein have to be adapted accordingly. Thus, the failure of even most sophisticated computational methods to produce highly efficient enzymes de novo, or even using an existing catalytic scaffold does not seem to be very surprising.
In a minimalistic approach to enzyme design, few, ideally one, mutations are introduced to obtain initial (often low) activity that is subsequently improved via directed evolution.[3] The simplicity of the approach allows for identification of a large number of potential catalysts for a particular chemical transformation that can be later characterized experimentally. This approach has been already shown to produce catalysts for model reactions with efficiencies rivalling those of catalytic antibodies after directed evolution. [4]
But can the lessons obtained for model reactions be expanded to practically useful catalysis? Here we can take inspiration from nature. Evolution of protein function occurs largely through enzymatic promiscuity (including progressive enrichment of already existing high energy conformational states).[5] An enzyme optimized to catalyze a particular “primary” chemical reaction can also facilitate similar “secondary” chemical transformation(s) and given enough selection pressure can evolve to improve its efficiency for the “secondary” reaction. The question is then can we take the enzymes designed to promote model reactions and use them as starting points for subsequent directed evolution for other chemical transformations? Recent advances in protein design[6] suggest affirmative answer to this question, but so far, designed Kemp eliminases have not been established to perform other reactions or perform ring-opening on non-benzisoxazole substrates. Since proteins designed using minimalist computational approaches have inherently low specificity for the intended substrate they are expected to be more promiscuous as compared to the ones developed using sophisticated computational tools.
Kemp elimination (Scheme 1) has been arguably the most well studied model reaction used to test essentially every protein design methodology providing a large library of Kemp eliminases that can be used as starting points for identification of new reactivities.[2a]
Scheme 1.

Kemp elimination (left) and base-promoted leflunomide conversion to teriflunomide (right).
Mechanistically, Kemp elimination presents a simple case of acid-base catalysis, where a hydrogen atom of a benzisoxazole ring is abstracted by a base. The carbon-bound hydrogen is relatively acidic, especially if strong electron-withdrawing substituents are present in the ring. While so far ring opening in isoxazoles has been studied in the context of mechanistic physical organic chemistry it can be of practical significance. Leflunomide, an isoxazole-containing pro-drug is converted in vivo through a redox-process[7] to an immunosupressor teriflunomide (Scheme 1). Proton abstraction from an nonactivated isoxazole ring is a more difficult reaction as compared to nitro-benzisoxazoles – the corresponding kuncat value is more than an order of magnitude lower (Figure S1).
In this paper, we focused on the question if previously designed Kemp eliminases evolved for generic proton abstraction can catalyze ring opening in leflunomide, for which, to our knowledge no catalysts that employ acid-base mechanism have been reported.
We chose two sets of previously designed Kemp eliminases to test for leflunomide ring opening. The first set contains AlleyCat, an allosterically regulated, minimalist Kemp eliminase obtained by introducing a single mutation into calmodulin, a non-enzymatic protein, as well as its seven derivatives (AlleyCat1-AlleyCat7) obtained via directed evolution. The second set included KE07, a Kemp eliminase designed from a thermostable imidazole-3-glycerolphosphate synthase using the theozyme approach,[8] as well as its evolved versions KE07 R7 1/3H and KE07 R7 10/11G.[9] The activities of the starting designs for Kemp elimination (AlleyCat and KE07) are similar to each other, the same is true for the activities of the evolved versions (AlleyCat7 and KE07 R7 1/3H). Finally, we have included a recently reported mutant of ketosteroid isomerase (KSI D38N) that showed high efficiency in Kemp elimination [10].
We have established a UV-Vis assay for leflunomide ring-opening by monitoring teriflunomide formation at 300 nm. Just like in the case of Kemp elimination, base promoted leflunomide conversion to teriflunomide proceeds cleanly without any side reactions (Figure S2). Ability of the designed enzymes to promote leflunomide ring-opening and subsequent turnover to teriflunomide was hence spectroscopically assayed in 96-well plates (Figure S3 – Figure S11). KE07 as well as its evolved versions KE07 R7 1/3 H, KE07 R7 10/11G and KSI D38N showed essentially no activity in leflunomide ring opening. The inability of the KE07 family enzymes to promote leflunomide ring opening is quite striking as imidazole-3-glycerolphosphate synthase has been shown to have promiscuous activity (ester hydrolysis)[11] and serve as a scaffold for successful computational design of retroaldolases in addition to Kemp eliminases.[12] At the same time, each of the evolved AlleyCats had some degree of activity towards teriflunomide formation. Since the AlleyCat proteins were evolved to catalyze Kemp elimination and not leflunomide ring opening, it is not surprising that there is no clear trend in the catalytic efficiency in leflunomide ring opening vs. the directed evolution round (Table 1).
Table 1.
Catalytic efficiency of various designed proteins in conversion of leflunomide to teriflunomide. Conditions (unless stated otherwise): 25 mM HEPES pH 7.4, 10 mM CaCl2, 100 mM NaCl.
| Protein | kcat × 10−3 /min−1 | KM /μM | kcat/KM /M−1 min−1 |
|---|---|---|---|
| AlleyCat[b] | --- | --- | 2.14 ± 0.12 |
| AlleyCat1 | 5.45 ± 0.61 | 263.8 ± 55 | 20.64 ± 4.86 |
| AlleyCat2 | 15.63 ± 0.62 | 236.8 ± 18 | 66.02 ± 5.73 |
| AlleyCat2[a] | 46.46 ± 3.9 | 553.8 ± 65 | 83.89 ± 12.10 |
| AlleyCat2 E92Q | --- | --- | No activity |
| AlleyCat3 | 12.22 ± 0.93 | 731.1 ± 74 | 16.71 ± 2.12 |
| AlleyCat4 | 12.56 ± 0.45 | 308.3 ± 19 | 40.74 ± 2.89 |
| AlleyCat5 | 6.88 ± 1.24 | 462.0 ± 125 | 14.88 ± 4.84 |
| AlleyCat6[b] | --- | --- | 6.75 ± 0.31 |
| AlleyCat7[b] | --- | --- | 7.35 ± 0.52 |
| KE07 | --- | --- | No activity |
| KE07 R7 1/3 H | --- | --- | No activity |
| KE07 R7 10/11G[b] | --- | --- | 0.32 ± 0.04 |
| KSI D38N | --- | --- | No activity |
in 25 mM TAPS pH 9.0, 10 mM CaCl2, 100 mM NaCl.
individual kcat and KM values could not be determined.
Importantly, the mechanism of the ring opening is still acid-base as confirmed by the lack of the activity shown by the E92Q mutant of AlleyCat2. In agreement with the design, AlleyCat2 and AlleyCat4 are allosterically regulated and only catalyze ring opening in the presence of calcium (Figure 1).
Figure 1.

Leflunomide ring-opening by AlleyCat2 (sky blue, black) and AlleyCat2 E92Q (lavender). Experimental conditions: 25 μM enzyme, 100 mM NaCl, 25 mM HEPES pH 7.4, 0.3 mM substrate. The sample with calcium contained 10 mM CaCl2 (sky blue dots for AlleyCat2 and lavender dots for AlleyCat2 E92Q), the sample without calcium contained 50 μM EDTA (black dots for AlleyCat2).
To determine the origins of AlleyCat2’s high activity, we performed in depth characterization of the protein and its interactions with the substrate. AlleyCat2’s circular dichroism (CD) is indicative of the typical alpha-helical calmodulin fold (Figure S12) and the mutations introduced in the course of directed evolution do not substantially disrupt the overall fold of the CaM scaffold (Figure S13).
In our earlier work[4h], we observed the proteins from AlleyCat family had high effective pKa of the active site glutamate. The apparent pKa of the catalytic base is 7.06 ± 0.02, determined from a pH activity profile of AlleyCat2 in leflunomide ring-opening (Figure S14), is close to the previously reported pKa value in Kemp elimination (6.73 ± 0.03).[4h] Nonetheless this apparent value is a product of many coupled equilibria associated with active base deprotonation, substrate deprotonation, etc. NMR spectroscopy provides a tool for direct measurement of the active residue’s pKa in the absence of any complicating factors. Sadly, except for one notable case[13] no direct measurement of active residue pKa has been performed in designed proteins. Therefore, we decided to determine the pKa of the active Glu92 residue directly. The chemical shift of the side chain carboxylate carbon is the most sensitive parameter that changes upon deprotonation. The positions of the side chain CO resonances were determined using a 2D CBCG(CO) spectrum at pH 7.0 (Figure 2A).[14] The Cα, Cβ and Cγ assignments for all the residues were obtained using traditional backbone and side chain-based HNcoCACB and HNCACB experiments. Unfortunately, the Cγ carbon of Glu92 does not appear in the spectrum of AlleyCat2, thus for the pKa studies we decided to use AlleyCat that has a very similar effective pKa (6.9 ± 0.1) determined from a pH profile of Kemp elimination and for which an NMR structure is available. While original AlleyCat does not efficiently promote leflunomide ring opening it can still provide correlation between the dependence of the enzymatic rate on pH and the pKa of the active base.
Figure 2.

pKa of the catalytic side chain measured by NMR. A) 2D CBCG(CO) spectrum of AlleyCat (pink) and AlleyCat2 (green) at pH 6.99, showing Cβ-CO and Cγ-CO side chain connectivities for Asp and Glu, respectively. All Glu and Asp residues of AlleyCat were assigned; B) Dependence of chemical shift of Cδ resonances of Glu82 and Glu114 residues in AlleyCat in the absence (black filled circle for Glu82 and red filled triangle for Glu114) and in the presence (black empty circle for Glu82 and red empty triangle for Glu114) of 5-nitrobenzotriazole; C) Dependence of chemical shift of Cδ resonances of Glu92 residue in AlleyCat in the absence (filled circle) and in the presence (empty circle) of 5-nitrobenzotriazole. Spectra were collected at 298 K on a Bruker 800 MHz NMR instrument in the buffer containing 10 mM citrate, 10 mM MES, 10 mM HEPES, 10 mM Tricine, 10 mM CaCl2 and 50 mM NaCl.
The Cγ carbon of Glu92 signal in the 2D CBCG(CO) spectrum of AlleyCat is clearly resolved (Figure 2A) and allows for direct determination of Glu92’s pKa upon changing the pH of the buffer. The pH dependence of the chemical shifts of the Cγ residues in various glutamates and aspartates are given in Figure 2B/C. Interestingly, the pKa value for Glu92 of 5.10 is not significantly higher than those of the side chains not facing the hydrophobic pocket (Figure S15, Table S1). Puzzled by this observation, we hypothesized that in the substrate-free state the side chain of Glu92 swings out of the hydrophobic core and is solvent accessible, consistent with some of the models in the previously determined NMR structure of AlleyCat.[4a] To mimic the pKa of the residue in the substrate-bound state we performed NMR pH titration in the presence of 5-nitrobenzotriazole, a competitive inhibitor of Kemp elimination. While competitive inhibitors may not fully represent the true transition state, they do offer the simplest way of characterizing it. The observed titration curve cannot be explained by a single protonation event and is consistent with a two-state model outlined in Scheme 2, with two conformations of Glu92 and two corresponding pKa values (5.14 ± 0.05 and 7.71 ± 0.04). The first pKa value is identical to the one observed in the absence of the substrate and the second value is very high ensuring strong basicity of the active glutamate when the substrate is bound. The pKa of the other Glu/Asp residues were not perturbed by inhibitor binding (Figure 2B and Figure S15).
Scheme 2.

A two-state model describing deprotonation behaviour of catalytic Glu92 in AlleyCat.
While we have independently validated the high pKa of the active residue in AlleyCat and established that the pH profile of the activity does not necessarily directly represent the pKa of the active residue, the question remains as to what is determining the high activity of AlleyCat2 relative to the other mutants. We hypothesized that substrate binding pocket reshaping over multiple rounds of directed evolution can be responsible for the observed differences in reactivity. In order to test this hypothesis, we performed computational docking of leflunomide into the NMR structure of AlleyCat[4a] and a computational model of AlleyCat2, built as described in the Methods section. Algorithms for docking studies include sampling of multiple side chain rotamers, while allowing backbone Ca atoms move up to 0.3 Å.
We have generated 4,000 docking poses and ranked them based on their overall Rosetta score and Rosetta score for the docking interface energy. We picked the 20 lowest-scoring models and determined the most probable representation of the ligand in the substrate binding pocket by clustering the lowest-scoring decoys using a previously established approach.[15] The docking poses of leflunomide in the two cases are strikingly different (Figure 3). The average distance between the catalytic base of Glu92 and the acidic carbon of leflunomide is 8.3 Å in the lowest scoring cluster of leflunomide poses docked into AlleyCat. Similar results are obtained for KE07, the substrate associates with the protein but not in a productive conformation (Figure S18). When the same procedure has been performed on AlleyCat2 the O(Glu92)-H(C5)(leflunomide) distance decreases to 2.5 Å, therefore the substrate is well positioned for proton abstraction. The same trend is visible in the interface energies obtained from the docking calculations (Figure S19, S20, Tables S3–S10). Binding of leflunomide to AlleyCat2 is ~2.4 REU more favourable than that to AlleyCat. The role of the M144R mutation is also supported by the mutational data. This mutation uniquely introduced in AlleyCat1 leads to a ten-fold increase in catalytic efficiency. Additionally, we have performed NMR chemical shift perturbation studies on both AlleyCat and AlleyCat2 upon addition of leflunomide (Figure S17). Addition of the substrate to AlleyCat2 leads to large changes in chemical shifts for the vast majority of residues (including R144), whereas backbone resonances for AlleyCat are essentially unaffected by the presence of leflunomide.
Figure 3.

Docking of leflunomide into AlleyCat and AlleyCat2. (A) Ligand docking into AlleyCat shows that isoxazole group of leflunomide is positioned away from Glu92, which serves as the base for Kemp elimination. The abstracted proton (H5) for this reaction is 8.3 Å away from the catalytic residue. (B) Model of the more active AlleyCat2 shows the proximity (2.5 Å) of H5 to Glu92, which allows Kemp elimination to proceed. Hydrogen bonding interactions (black) may play a role in stabilizing and positioning the ligand.
The results of the docking studies are consistent with the notion that the increased activity of AlleyCat2 is due to the conformational selection of a more beneficial mode of substrate binding. The protein reshaping as a result of directed evolution has a large effect not only on the original substrate (5-nitrobenzisoxazole), but also on other potential substrates providing opportunities for development.
In conclusion, we have demonstrated that libraries of computationally designed minimalist catalysts provide fertile ground for finding new catalysts for various chemical reactions. The activity of the AlleyCat family of catalysts in leflunomide ring opening is remarkable (reaching the rate enhancement kcat/kuncat of ~ 8,000 and catalytic proficiency[16] (kcat/KM)/kuncat of more than 3 × 107 M−1) considering that these catalysts were not evolved with any consideration for alternative substrates. To our knowledge, this is the first catalyst of leflunomide ring opening that proceeds using an acid-base mechanism. We observe that proteins designed using minimalist approach appear to be more promiscuous than those created by other approaches likely due to the fact that precise substrate fit is not a major consideration in the minimalist approach, but a much larger study would be necessary to confirm this observation. From a practical perspective, our results show that screening relatively small libraries of designed enzymes for various catalytic reactivities can be quite successful. We have also demonstrated that the pKa values of the active residues can be dramatically different from those determined from simple fits of pH-rate profiles. Finally, using computational studies we have determined that the activity of AlleyCat2 in leflunomide ring opening stems from a combination of high pKa of the active base and proper positioning of the substrate in the hydrophobic cleft of the enzyme.
Supplementary Material
Acknowledgements
We thank Prof. Dr. Patrick J. Bednarsky (University of Greifswald) for bringing chemical similarity of benzisoxazoles and leflunomide to our attention. The plasmid encoding the tKSI (Comamonas testosteroni) D38N gene was a generous gift from the Herschlag lab (Stanford University). This work was supported by the NIH (grants GM119634, RR016461, OD012254 and GM103499), the NSF (grants 1332349, 1750462 and 1229559), the College of Charleston [Summer Undergraduate Research with Faculty grants], the Howard Hughes Medical Institute [grant 52007537 through the Pre-college & Undergraduate Science Education Program], the Ralph E. Powe Junior Enhancement Award to C.A.C, the Nappi Family Award and the Alexander von Humboldt Foundation.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Olga V. Makhlynets, Email: ovmakhly@syr.edu.
Ivan V. Korendovych, Email: ikorendo@syr.edu.
References
- [1].a) Bornscheuer UT, Huisman G, Kazlauskas RJ, Lutz S, Moore J, Robins K, Nature 2012, 485, 185–194; [DOI] [PubMed] [Google Scholar]; b) Gumulya Y, Sanchis J, Reetz MT, ChemBioChem 2012, 13, 1060–1066; [DOI] [PubMed] [Google Scholar]; c) Reetz MT, Angew. Chem. Int. Ed 2011, 50, 138–174; [DOI] [PubMed] [Google Scholar]; d) Fasan R, Chen MM, Crook NC, Arnold FH, Angew. Chem., Int. Ed 2007, 46, 8414–8418. [DOI] [PubMed] [Google Scholar]
- [2].a) Korendovych IV, DeGrado WF, Curr. Opin. Struct. Biol 2014, 27, 113–121; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Makhlynets OV, Korendovych IV, Nat. Chem 2016, 8, 823–824. [DOI] [PubMed] [Google Scholar]
- [3].DeGrado WF, Wasserman ZR, Lear JD, Science 1989, 243, 622–628. [DOI] [PubMed] [Google Scholar]
- [4].a) Korendovych IV, Kulp DW, Wu Y, Cheng H, Roder H, DeGrado WF, Proc. Nat. Ac. Sci. U.S.A 2011, 108, 6823–6827; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mack KL, Moroz OV, Moroz YS, Olsen AB, McLaughlin JM, Korendovych IV, J Biol Inorg Chem 2013, 18, 411–418; [DOI] [PubMed] [Google Scholar]; c) Makhlynets OV, Korendovych IV, Biomolecules 2014, 4, 402–418; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Makhlynets OV, Raymond EA, Korendovych IV, Biochemistry 2015, 54, 1444–1456; [DOI] [PubMed] [Google Scholar]; e) Raymond EA, Mack KL, Yoon JH, Moroz OV, Moroz YS, Korendovych IV, Protein Sci. 2015, 24, 561–570; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Merski M, Shoichet BK, Proc. Nat. Acad. Sci. U.S.A 2012, 109, 16179–16183; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Wei Y, Liu T, Sazinsky SL, Moffet DA, Pelczer I, Hecht MH, Protein Sci 2003, 12, 92–102; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Moroz OV, Moroz YS, Wu Y, Olsen AB, Cheng H, Mack KL, McLaughlin JM, Raymond EA, Zhezherya K, Roder H, Korendovych IV, Angew. Chem. Int. Ed 2013, 52, 6246–6249; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Farid TA, Kodali G, Solomon LA, Lichtenstein BR, Sheehan MM, Fry BA, Bialas C, Ennist NM, Siedlecki JA, Zhao ZY, Stetz MA, Valentine KG, Anderson JLR, Wand AJ, Discher BM, Moser CC, Dutton PL, Nature Chemical Biology 2013, 9, 826–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Campbell E, Kaltenbach M, Correy GJ, Carr PD, Porebski BT, Livingstone EK, Afriat-Jurnou L, Buckle AM, Weik M, Hollfelder F, Tokuriki N, Jackson CJ, Nature Chemical Biology 2016, 12, 944; [DOI] [PubMed] [Google Scholar]; b) Maria-Solano MA, Serrano-Hervás E, Romero-Rivera A, Iglesias-Fernández J, Osuna S, Chemical Communications 2018, 54, 6622–6634; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Romero-Rivera A, Garcia-Borràs M, Osuna S, ACS Catalysis 2017, 7, 8524–8532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Xavier G, Tobias B, Donald H, Angewandte Chemie International Edition 2015, 54, 5609–5612;25777153 [Google Scholar]; b) Garrabou X, Verez R, Hilvert D, Journal of the American Chemical Society 2017, 139, 103–106; [DOI] [PubMed] [Google Scholar]; c) Garrabou X, Wicky BIM, Hilvert D, Journal of the American Chemical Society 2016, 138, 6972–6974; [DOI] [PubMed] [Google Scholar]; d) Garrabou X, Macdonald Duncan S, Hilvert D, Chemistry – A European Journal 2017, 23, 6001–6003. [DOI] [PubMed] [Google Scholar]
- [7].Yu J, Folmer JJ, Hoesch V, Doherty J, Campbell JB, Burdette D, Drug Metabolism and Disposition 2011, 39, 302–311. [DOI] [PubMed] [Google Scholar]
- [8].Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D, Nature 2008, 453, 190–195. [DOI] [PubMed] [Google Scholar]
- [9].Khersonsky O, Rothlisberger D, Dym O, Albeck S, Jackson CJ, Baker D, Tawfik DS, J. Mol. Biol 2010, 396, 1025–1042. [DOI] [PubMed] [Google Scholar]
- [10].Lamba V, Sanchez E, Fanning LR, Howe K, Alvarez MA, Herschlag D, Forconi M, Biochemistry 2017, 56, 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Taglieber A, Höbenreich H, Carballeira J, Mondière RJG, Reetz MT, Angew. Chem. Int. Ed 2007, 46, 8597–8600. [DOI] [PubMed] [Google Scholar]
- [12].Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, Hilvert D, Houk KN, Stoddard BL, Baker D, Science 2008, 319, 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Katharina H, Zsófia L, C. A. C, O. V. M, Proteins 2017, 85, 1656–1665.28544090 [Google Scholar]
- [14].Castaneda CA, Fitch CA, Majumdar A, Khangulov V, Schlessman JL, Garcia-Moreno BE, Proteins 2009, 77, 570–588. [DOI] [PubMed] [Google Scholar]
- [15].a) DeLuca S, Khar K, Meiler J, PLoS One 2015, 10, 1–19; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li SC, Ng YK, BMC Bioinformatics 2010, 11, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Radzicka A, Wolfenden R, Science 1995, 267, 90–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.