

Abstract
Lithium perchlorate and acetic anhydride were the key additives for the multi-component reaction between 3-aminocyclohex-2-enones, formaldehyde, and malonates yielding adducts that were annulated under acidic conditions to afford bicyclic 2,5-dioxooctahydroquinoline-3-carboxylates. When methyl cyanoacetate was subjected to the same reaction conditions in the presence of a catalytic amount of triphenylphosphine, the bicyclic 2,5-dioxooctahydroquinoline-3-carbonitriles were obtained in a one-flask reaction.
Keywords: Multicomponent reaction; catalysis; lithium perchlorate; acetic anhydride; triphenylphosphine; 2,5-dioxooctahydroquinolines
Graphical abstract

1. Introduction
1,4-Dihydropyridines, 1,4-dihydropyridine-2-pyridones, pyridines, and piperidines are closely related structural motifs found in many natural products and bioactive molecules. 1,4-Dihydropyridines are considered to be privileged structures, because they bind to multiple ion channels and receptors.1 For example, the 4-aryl-1,4-dihydropyridine moiety is a component of the cardiovascular drug nifedipine (1, Figure 1), which is a calcium channel antagonist.2, 3 The annulated 1,4-dihydropyridine analogue 2 (Figure 1) initiates cardiac differentiation in mouse embryonic stem cells by inhibiting TGFβ signaling – a mechanism unrelated to calcium channel blockade.4 These analogues represent a new class of agents for potential use in cardiac regenerative medicine.5 The structurally related 1,4-dihydropyridine-2-pyridone scaffold is also of medicinal interest. The 2-pyridone moiety is a component of clinically used phosphodiesterase-3 (PDE3) inhibitors such as milrinone (3, Figure 1) and related derivatives,6, 7 including the annulated PDE3 inhibitor 4 (Figure 1).8 2-Pyridones are also found as structural elements in a variety of other bioactive compounds including natural products.7, 9 Octahydro-2-pyridones such as LY191704 and the related tetrahydro analogues were found to be specific inhibitors of human type I steroid 5-α-reductase.10-12 Despite the fact that these structural types have displayed interesting bioactivities, 3,4-dihydropyridinones and their annulated analogues, 2,5-dioxooctahydroquinoline-3-carboxylates 5 (target scaffold 5, Figure 1) have not been examined extensively yet in terms of synthesis, biological activity, and structure-activity relationships.
Figure 1.
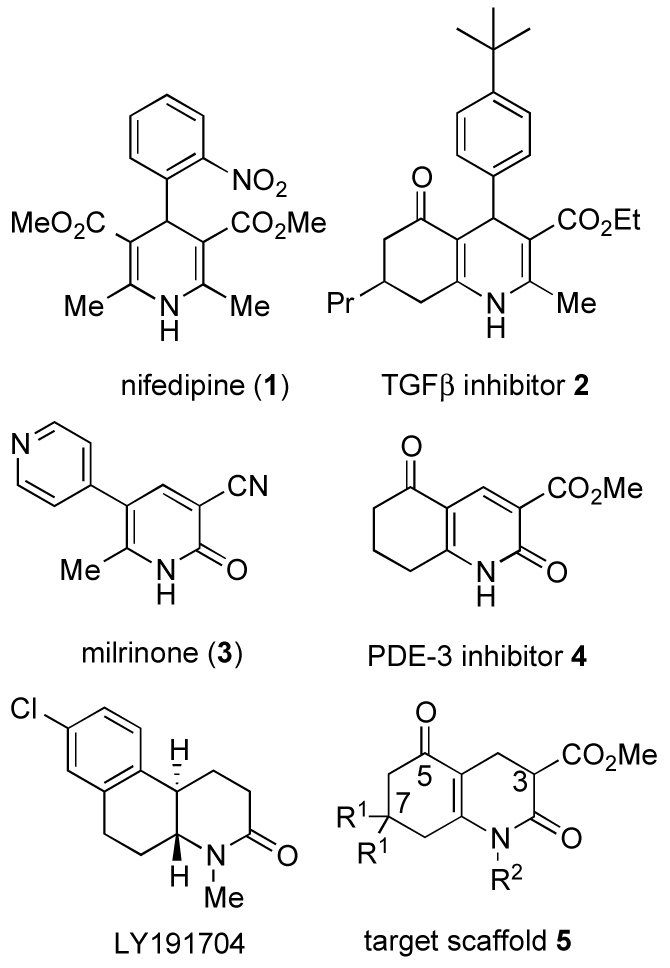
Representative structures of cardiovascular drugs, 1,4-dihydropyridine nifedipine (1), a calcium channel blocker, TGFb inhibitor 2, milrinone (3), annulated 2-pyridone phosphodiesterase-3 inhibitor 4, human type I steroid 5-α-reductase inhibitor LY191704, and target scaffold 5.
It is well known that 1,3-diketones, including cyclic ketones,13 react via enamine formation (Scheme 1, eq 1) to produce substituted 1,4-dihydropyridines (Hantzsch reaction). Similar to the Hantzsch-type reaction that is used to synthesize annulated 1,4-dihydropyridines (Scheme 1, eq 1), formal [3+3] cycloaddition reactions were employed to construct dihydro-2-pyridones including annulated core structures.9, 14-17 In these multi-component reactions 1,3-cyclohexanediones, malonates or Meldrum’s acid, aldehydes, and amines or ammonia are reacted in one flask, as is depicted in Scheme 1, eq 2. In the example shown in eq 2, ZnO serves as an efficient catalyst to effect the reaction with aromatic aldehydes in excellent yields, but apparently does not work with formaldehyde or aliphatic aldehydes.18 When Meldrum’s acid is used in the multicomponent reaction, formaldehyde and aliphatic aldehydes can be employed, but the reaction products lack the 3-carboxylic acid moiety (Scheme 1, eq 3), which is lost by decarboxylation.17 In related chemistry (Scheme 1, eq 4), hydrazinyl enamines of 1,3-cyclohexanediones, and the arylidenemalonates are preformed and then reacted in the presence of butyl lithium to form the Michael addition products that are subsequently cyclized to yield 4-aryl-2,5-dioxooctahydroquinoline-3-carboxylates.19
Scheme 1.

Multicomponent literature methods for the synthesis of 5-oxohexahydroquinoline-3-carboxylates (eq 1), 2,5-Dioxooctahydroquinoline-3-carboxylates (eq 2 and 3) and a stepwise reaction to form 2,5-dioxooctahydroquinoline-3-carboxylates (eq 4).
2. Results and discussion
In our efforts to prepare structurally diverse and novel nitrogen-containing heterocyclic compounds with privileged structures for biological screening, we targeted the synthesis of bicyclic 1,4-dihydropyridine-2-pyridones 5 (Figure 1) that do not contain a substituent at C4 but an ester moiety at C3 (2,5-dioxooctahydroquinoline-3-carboxylates). While aromatic aldehydes have been employed in the multicomponent reaction shown in eq 2 in Scheme 1, the reaction apparently did not take place when formaldehyde was used.18 The availability of a method that provides the target compounds 5 without a substituent at C4 is of importance to expand the scope of this chemistry. Once an aryl or aliphatic group is present at C4 it will require multiple steps for removal if unsubstituted analogues are desired for biological investigations.
We started our investigations by carrying out a one-flask reaction similar to the one shown in Scheme 1, eq 3, by following a reported procedure17 using 1,3-cyclohexanedione, formaldehyde, dimethyl malonate and benzylamine, but failed to obtain the desired bicyclic reaction product. We then decided to explore the possibility of synthesizing the target molecule in a step-wise manner in order to simplify the reaction system. 3-(Benzylamino)cyclohex-2-enone (6a), easily prepared by condensation of 1,3-cyclohexanedione and benzylamine,20 paraformaldehyde, and dimethyl malonate, were envisaged to form an adduct, followed by a subsequent cyclization to yield the target molecule. Yet, when a mixture of the enaminone, formaldehyde, and dimethyl malonate were heated in a reaction vessel, only little of adduct 7a formed and instead bis-addition of the enaminone to formaldehyde took place and 8a was isolated as the major product in 74% (Scheme 2, eq 1). Clearly, this side reaction needed to be suppressed in order to allow the desired multi-component reaction to become the major reaction pathway and yield the desired adduct 7a. Since LiClO4 has been introduced as a useful reagent to facilitate Mannich-type reactions,21-25 we tested this Lewis acid as an additive in the reaction (Scheme 2, eq 2). The reaction was heated to 90 °C in acetonitrile in the presence of one equivalent of LiClO4, and the desired adduct 7a was isolated as the major product in 77%, along with the bis-addition side product 8 in 13% as the minor product.
Scheme 2.
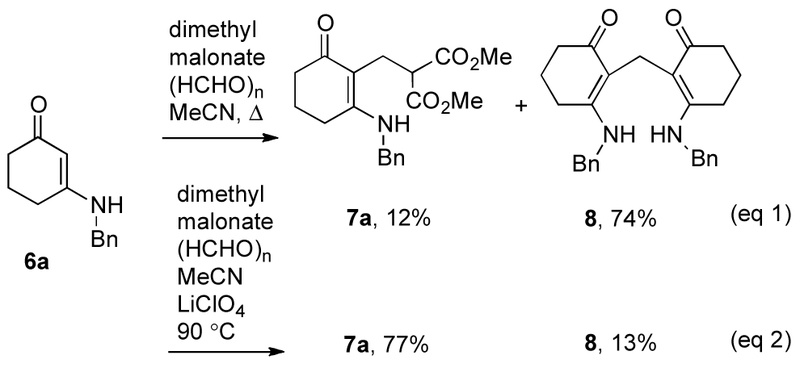
Multi-component reactions of 3-(benzylamino)cyclohex-2-enone (6a) with and without LiClO4 as additive.
Optimization of the reaction conditions (Table 1) showed that an equimolar amount of LiClO4 was crucial to obtain good yields (Table 1, entries 1-3) and that excess LiClO4 was unnecessary (entries 4 and 5). In an attempt to annulate the adduct in the same flask, acetic anhydride was added as an additive. Whereas no annulated product was detected, the yield for the adduct formation of 7a improved to 93% (Table 1, entry 6). When acetic anhydride was added, the reaction could be completed at a lower temperature (60 °C instead of 90 °C) in a comparable amount of time (0.5 - 3.0 h). Several other mild Lewis acids (Table 1, entries 7-9) were also tested, but the results were not satisfactory.
Table 1.
Optimization of the LiClO4-assisted formation of 7a
 | ||||
|---|---|---|---|---|
| entry | temp (°C) |
LiClO4 (equiv) |
Ac2O (equiv) |
yield (%) |
| 1 | 90 | 0 | 0 | 12 |
| 2 | 90 | 0.9 | 0 | 63 |
| 3 | 90 | 1.0 | 0 | 77 |
| 4 | 90 | 1.9 | 0 | 75 |
| 5 | 90 | 10.0 | 0 | 62 |
| 6 | 60 | 1.0 | 1.0 | 93a |
| 7 | 60 | Li+TFA− 1.0 | 1.0 | 57 |
| 8 | 60 | Ti(OiPr)4 1.0 |
1.0 | 0b |
| 9 | 60 | MgBr2•OEt2 | 1.0 | 0b |
used as the standard condition;
bis-addition product observed,
none of the desired product was observed.
In order to explore the scope of the reaction we subjected a variety of N-substituted 3-aminocyclohex-2-enones and malonates to the reaction under the optimized conditions (Table 2). Most of the reactions yielded the corresponding adducts in moderate to excellent yields (66%-97%). Attempts to replace formaldehyde with benzaldehyde were not successful. No product formation was detected when a sterically hindered malonate tert-butyl ester (Table 2, entry 16) or dimethyl methylmalonate (Table 2, entry 17) was employed in the reaction. Methyl acetoacetate, methyl cyanoacetate, and triethyl phosphonoacetate were tested as surrogates of malonates, but no product was detected under the standard conditions. We propose that methyl acetoacetate forms an intermediate similar to 9a through Knoevenagel reaction,26 however such an intermediate would be a more reactive Michael acceptor than 9a, due to the higher electrophilicity provided by the ketone carbonyl versus the ester carbonyl. Thus, this intermediate would get consumed rapidly via self-polymerization,26 without reacting with 6a. We have provided the 1H NMR of 9a (see supplementary material), showing its rapid self-polymerization. The Michael acceptor intermediate derived from methyl acetoacetate can be expected to polymerize even faster.26, 27
Table 2.
Scope of the LiClO4-assisted multi-component reaction
 |
|||||||
|---|---|---|---|---|---|---|---|
| entry | compound | n | R1 | R2 | R3 | R4 | yield (%) |
| 1 | 7b | 1 | H | Ph | Me | Me | 90 |
| 2 | 7c | 1 | H | Ph | Bn | Bn | 97 |
| 3 | 7d | 1 | H | Bn | Me | Bn | 73 |
| 3 | 7e | 1 | H | Bn | Bn | Bn | 90 |
| 5 | 7f | 1 | H | nBu | Me | Me | 66 |
| 6 | 7g | 1 | H | nBu | Bn | Bn | 80 |
| 7 | 7h | 1 | H | morpholine | Me | Me | 72 |
| 8 | 7i | 1 | H | morpholine | Bn | Bn | 86 |
| 9 | 7j | 1 | Me | Ph | Me | Me | 83 |
| 10 | 7k | 1 | Me | Ph | Bn | Bn | 87 |
| 11 | 7l | 1 | Me | Bn | Me | Me | 95 |
| 12 | 7m | 1 | Me | Bn | Bn | Bn | 88 |
| 13 | 7n | 1 | Me | nBu | Me | Me | 88 |
| 14 | 7o | 0 | H | Ph | Me | Me | 96 |
| 15 | 7p | 0 | H | Bn | Me | Me | 94 |
| 16 | 7q | 1 | H | Bn | tBu | Et | np |
| 17 | 7r | 1 | H | Bn | dimethyl methylmalonate | npa | |
| 18 | 7s | 1 | H | Bn | methyl acetoacetate | np | |
| 19 | 7t | 1 | H | Bn | triethyl phosphonoacetate | np | |
| 20 | 7u | 1 | H | Bn | benzaldehyde, dimethyl malonate | np | |
np = no product.
Similar to what has been proposed in the literature17 and is shown in Scheme 1, eq 4,19 we assume that product 7a formed via a conjugate addition of the enaminone to an intermediate generated from an in situ Knoevenagel reaction between dimethyl malonate and formaldehyde, followed by an imine-enamine tautomerism (Scheme 3). To investigate the proposed pathway, dimethyl methylenemalonate (9a) was prepared following a literature procedure,28 and the conjugate addition by the enaminone successfully yielded 7a in 90% without the assistance of LiClO4 and acetic anhydride. Thus, LiClO4 and acetic anhydride facilitate the formation of intermediate 9a and thereby, favoring the Michael addition of the enamine 6a to intermediate 9a over the formation of the bis-addition product 8, although the detailed mechanism is not clear.
Scheme 3.
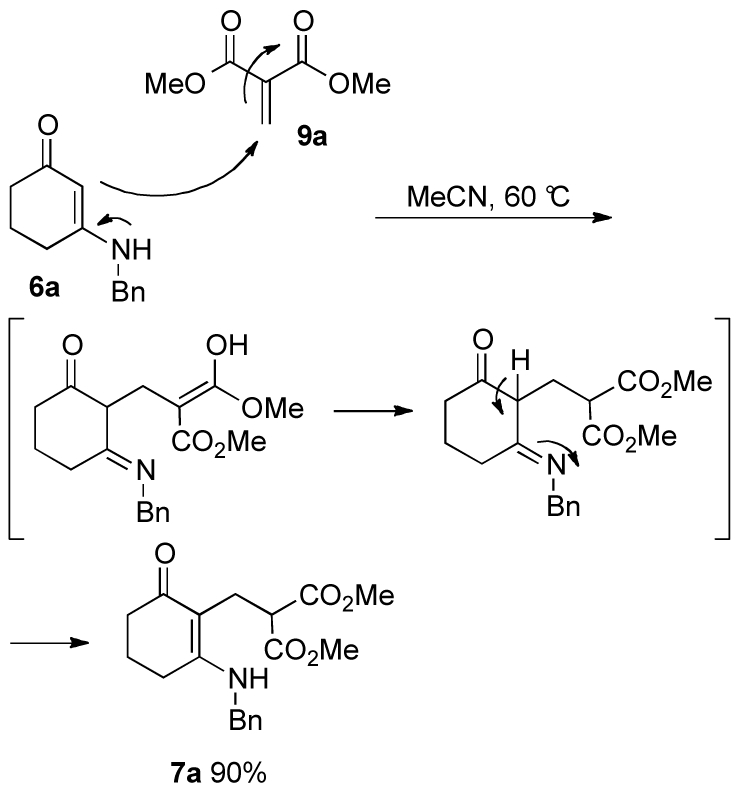
Reaction of enaminone 6a with Knoevenagel intermediate 9a.
No product formation was detected when benzaldehyde was used in the LiClO4-assisted reaction (Table 2, entry 20). Based on the proposed mechanism, the difficulty in the corresponding conjugate addition to the sterically more demanding intermediate and/or a difference in electronic properties could be the cause of the lack of reactivity. To confirm that hypothesis, dimethyl benzylidenemalonate (9b)29 was prepared and subjected to the reaction with the enaminone. Indeed, the enaminone did not add to dimethyl 2-benzylidenemalonate (9b) in the presence of LiClO4 or without it.
The observed rate enhancement after the addition of acetic anhydride most likely facilitates the LiClO4-catalyzed Knoevenagel reaction by in situ formation of the more reactive geminal formaldehyde diacetate,30 similar to what has been described for the iodine-catalyzed Knoevenagel reaction in the presence of acetic anhydride (Scheme 4).31
Scheme 4.
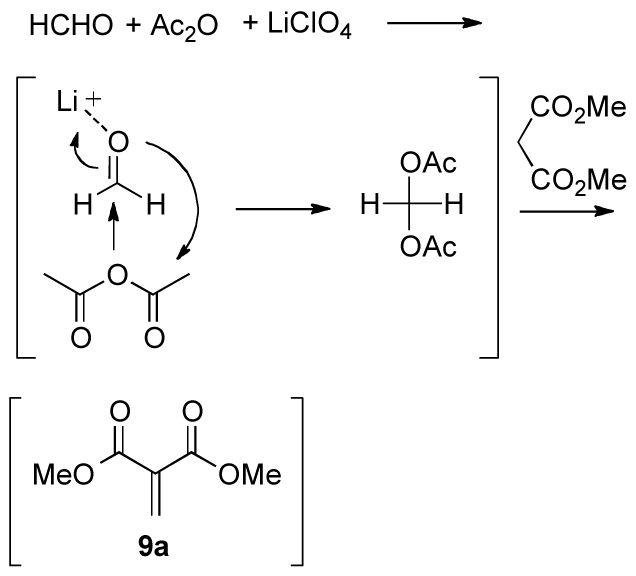
Proposed mechanism for the LiClO4-catalyzed Knoevenagel reaction in the presence of acetic anhydride.
Intermediates 7 were next treated with trifluoroacetic acid at 90 °C in dichloroethane or toluene in a sealed reaction vessel to yield the desired 2,5-dioxooctahydroquinoline-3-carboxylate esters 5 in mostly moderate yields as shown in Table 3.32-34 The cyclization of dimethylmalonate adducts was carried out in dichloroethane, while toluene was a better choice of solvent for the dibenzylmalonate adducts, with an approximately 10% improvement in yields on average. Adducts containing a 1-morpholino group (7h and 7i, Table 2) decomposed under the acidic conditions, furnishing none of the annulated products.
Table 3.
Synthesis of dioxooctahydroquinoline esters 5
 |
||||||
|---|---|---|---|---|---|---|
| entry | compound | n | R1 | R2 | a R3 | yield (%) |
| 1 | 5a | 1 | H | Bn | Me | 79 |
| 2 | 5b | 1 | H | Bn | Bn | 70 |
| 3 | 5c | 1 | H | Ph | Me | 84 |
| 4 | 5d | 1 | H | Ph | Bn | 49 |
| 5 | 5e | 1 | H | nBu | Me | 57 |
| 6 | 5f | 1 | H | nBu | Bn | 60 |
| 7 | 5g | 1 | Me | Bn | Me | 67 |
| 8 | 5h | 1 | Me | Bn | Bn | 64 |
| 9 | 5i | 1 | Me | Ph | Me | 64 |
| 10 | 5j | 1 | Me | Ph | Bn | 72 |
| 11 | 5k | 1 | Me | nBu | Me | 62 |
| 12 | 5l | 0 | H | Bn | Me | 62 |
| 13 | 5m | 0 | H | Ph | Me | 49 |
reaction performed in dichloroethane
when R3 = Me, in toluene when R3 = Bn
We next subjected methyl cyanoacetate (instead of a malonate ester) to the same reaction conditions but no reaction product was obtained. However, when a catalytic amount of triphenylphosphine was added to the reaction mixture, the annulated products 10a-e (Table 4) were formed directly.
Table 4.
Synthesis of dioxooctahydroquinoline-3-carbonitriles 10
 | ||||
|---|---|---|---|---|
| entry | compound | R1 | R2 | yield (%) |
| 1 | 10a | H | Bn | 85 |
| 2 | 10b | H | Ph | 65 |
| 3 | 10c | H | Bu | 73 |
| 4 | 10d | Me | Pr | 75 |
| 5 | 10e | Me | MeO(CH2)2- | 67 |
The triphenylphosphine catalyst presumably plays multiple roles in this reaction. It is well known that triphenylphosphine catalyzes the Knoevenagel condensation between aldehydes and cyanoacetates to form α-cyanoacrylonitriles, but not with malonates.35 To probe whether triphenylphosphine catalyzed this reaction in our case, we carried out the reaction in the presence of triphenylphosphine but without LiClO4. We did not obtain any of the desired product and only bis-addition product 8 was found. We therefore concluded that LiClO4 is responsible for the formation of the presumed methyl 2-cyanoacrylate intermediate, rather than triphenylphosphine.
Triphenylphosphine is a strong nucleophile that adds to electron-deficient double and triple bonds.36-39 Scheme 5 shows a possible mechanism for the formation of compounds 10, which is similar to the mechanism reported by Steward et al. for the triphenylphosphine catalyzed conjugate addition of alcohols and water to enones.40 Triphenylphosphine adds to methyl 2-cyanoacrylate and forms phosphonium enolate 11, which abstracts a proton from 6a forming ion pair 12, which then adds to an equivalent of methyl 2-cyanoacrylate. The new ion pair 13 abstracts a proton from another molecule of 6a to form intermediate 14 that cyclizes to yield product 10a. Deprotonated 6a and regenerated 11 reform ion pair 12. (Steward et al. were able to rule out a direct proton abstraction by triphenylphosphine.40)
Scheme 5.

Proposed mechanism for the triphenylphosphine catalyzed synthesis of 10a.
The cyclization reaction is most likely catalyzed by triphenylphosphine as well since it has been reported that triphenylphosphine can be a catalyst for transesterifications,41 although an influence of steric and/or electronic effects of the nitrile group on the cyclization cannot be ruled out. We propose that triphenylphosphine adds to the ester 14 and forms acylphosphonium intermediate 15 that would cyclize to form compound 10a and regenerate PPh3.
Formaldehyde was the only viable aldehyde in this reaction. Malonates were also subjected to such reaction conditions in order to obtain 5 in one step, but no cyclized product was detected after 12 h. Instead the bis-addition product 8 was the major product, and only a small amount of the conjugated addition adduct 7a was detected.
The syntheses of structurally related monocyclic 4-aryl-2-oxo-tetrahydropyridine-3-carbonitriles from 4H-pyrans has recently been reported.42, 43 The 4H-pyrans were prepared from aromatic aldehydes, malononitrile, and ethyl acetoacetate in the presence of ZnO as the catalyst.43 Another recent publication describes a three-component reaction that employed aromatic aldehydes (and one aliphatic aldehyde) with either malononitrile or 2-cyanoacetamide in the presence of PEG-SO3H or vitamin B1, respectively, to provide bicyclic 4-substituted-2,5-dioxooctahydroquinolone-3-carbonitriles.44 Thus, our technique complements existing methods for the synthesis of these types of scaffolds in that we are employing formaldehyde rather than aromatic or aliphatic aldehydes in the multi-component reaction and thereby providing a method for the synthesis of 4-unsubstituted analogues.
The compounds were submitted to the NIH Molecular Libraries Program and screened in several assays that are reported in PubChem.45
3. Conclusion
In summary, a LiClO4-assisted multi-component reaction of 3-aminocyclohex-2-enones 6 with malonates or cyanoacetates, and formaldehyde in the presence of acetic anhydride is described, providing access to 4-unsubstituted 2,5-dioxooctahydroquinoline-3-carboxylates and 3-carbonitriles.
4. Experimental section
4.1. General
Proton nuclear magnetic resonance spectra were recorded at 400 MHz and carbon nuclear magnetic resonance spectra were recorded at 100 MHz. All chemical shifts were recorded as parts per million (ppm), and all samples were dissolved in CDCl3 using the residual solvent peak as internal standard. Mass spectra were obtained from a ZAB HS mass spectrometer equipped with an 11/250 data system. Fast-atom bombardment mass spectrometry (FAB-MS) experiments were performed with a Xenon gun operated at 8 Kev energy and 0.8 mA emission at the Institute for Therapeutic Discovery and Development at University of Minnesota. Fast-atom bombardment high resolution mass spectra (FAB-HRMS) were recorded at 1:10,000 resolution using linear voltage scans under data system control and collected in a multi-channel analyzer mode (MCA). A Recording Infrared Spectrophotometer or a FT-IR was used to record infrared spectra. Melting points are uncorrected. Solvents and reagents that are commercially available were used without further purification. Acetonitrile, dichloroethane, and toluene were distilled freshly from calcium hydride under argon. Silica gel (230 - 400 mesh) was used for flash column chromatography. All compounds were concentrated using a standard rotary evaporator and high-vacuum techniques.
General Method for LiClO4-assisted Reaction of Enaminones with Malonates and Formaldehyde to Form Adducts 7 (Table 2).
A suspension of the enaminone, LiClO4 (1.0 equiv), the malonate diester (1.5 equiv), and acetic anhydride (1.0 equiv) in acetonitrile (10 mL, ~ 35 mL/mmol) was stirred and pre-heated in a sealed screw-cap vial until the reaction mixture turned into a clear solution after approximately 5 min. To the reaction medium was then added paraformaldehyde (2.0 equiv). The reaction was then sealed, stirred at 60 °C, and monitored by TLC. In general, the reaction reached completion within 0.5 to 3 h, and was quenched subsequently with H2O (20 mL, ~70 mL/mmol). The aqueous phase was extracted with EtOAc (3 × 10 mL, ~35 mL/mmol), and then, the organic layers were combined, and washed with brine, and dried over MgSO4. The organic solvent was then removed under reduced pressure and the resulting residue was purified by flash column chromatography on silica gel, using 50% acetone/hexanes as the eluent.
Dimethyl 2-((2-(Benzylamino)-6-oxocyclohex-1-enyl)methyl)malonate (7a):
The N-benzyl-enaminone (50 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dimethyl malonate (0.042 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous colorless oil (80 mg, 0.23 mmol, 93%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.36 - 7.34 (m, 2H), 7.30 - 7.26 (m, 3H), 6.35 (m, 1H), 4.43 (d, J = 6.1 Hz, 2H), 3.80 (t, J = 7.2 Hz, 1H), 3.69 (s, 6H), 2.83 (d, J = 7.2 Hz, 2H), 2.39 (t, J = 6.2 Hz, 2H), 2.26 (t, J = 6.2 Hz, 2H), 1.84 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 194.9, 171.1, 162.8, 138.4, 129.0, 127.8, 127.0, 105.8, 52.7, 50.6, 47.2, 36.3, 25.8, 23.6, 21.3; ESI-HRMS: calc’d m/e for [M+Na+] C19H23NNaO5: 368.1474, found 368.1484; IR (neat, NaCl, cm−1): 2951, 1733, 1575, 1436, 1409, 1357, 1299, 1246, 1202, 1158, 1118, 1031.
Dimethyl 2-((6-Oxo-2-(phenylamino)cyclohex-1-enyl)methyl)malonate (7b):
The N-phenyl-enaminone (47 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dimethyl malonate (0.042 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions and yielded a viscous colorless oil (74 mg, 0.22 mmol, 90%). 1H NMR (CDCl3, 400 MHz, ppm): δ 8.03 (s, 1H), 7.33 – 7.35 (m, 2H), 7.16 (m, 1H), 7.06 – 7.09 (m, 2H), 3.82 (t, J = 7.2 Hz, 1H), 3.74 (s, 6H), 2.91 (d, J = 7.2 Hz, 2H), 2.50 (t, J = 6.0 Hz, 2H), 2.37 (t, J = 6.0 Hz, 2H), 1.86 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.9, 171.3, 160.6, 139.3, 129.3, 125.0, 124.1, 109.0, 52.9, 50.9, 36.9, 27.4, 23.8, 22.2; ESI-HRMS: calc’d m/e for [M+H+] C18H22NO5: 332.1498, found 332.1505; IR (neat, NaCl, cm−1): 2593, 1733, 1567, 1499, 1435, 1398, 1306, 1245, 1204, 1080, 1038.
Dibenzyl 2-((6-Oxo-2-(phenylamino)cyclohex-1-enyl)methyl)malonate (7c):
The N-phenyl-enaminone (47 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dibenzyl malonate (0.070 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous light yellow oil (120 mg, 0.24 mmol, 97%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.89 (s, 1H), 7.35-7.29 (m, 12H), 7.15 (m, 1H), 7.01 (m, 2H), 5.17 (q-like, J = 12.4 Hz, 4H), 3.95 (t, J = 7.4 Hz, 1H), 2.95 (d, J = 7.4 Hz, 2H), 2.39 (t, J= 6.0 Hz, 2H), 2.32 (t, J = 6.0 Hz, 2H), 1.76 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 196.0, 170.6, 160.8, 139.2, 135.7, 129.3, 128.7, 128.4, 128.2, 125.1, 124.2, 108.6, 67.4, 51.1, 36.8, 27.3, 23.7, 22.0; ESI-HRMS: calc’d m/e for [M+H+] C30H30NO5: 484.2124, found 484.2118; IR (neat, NaCl, cm−1): 2948, 1736, 1578, 1498, 1454, 1431, 1396, 1304, 1233, 1199, 1153, 1080, 1030, 1003.
1-Benzyl 3-Methyl 2-((2-(Benzylamino)-6-oxocyclohex-1-en-1-yl)methyl)malonate (7d):
The N-benzyl-enaminone (50 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), benzyl methyl malonate (0.070 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous light yellow oil (77 mg, 0.18 mmol, 73%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.37 - 7.24 (m, 10H), 6.24 (t, J = 6.2 Hz, 1H), 5.17 (q-like, J = 12.4 Hz, 2H), 4.36 (d, J = 6.1 Hz, 2H), 3.89 (t, J = 7.6 Hz, 1H), 3.67 (s, 3H), 2.84 (m, 2H), 2.33 (m, 2H), 2.24 (m, 2H), 1.81 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 194.9, 170.9, 170.7, 162.7, 138.4, 129.0, 128.6, 128.4, 128.2, 127.0, 105.8, 67.2, 54.2, 52.7, 50.71, 47.2, 36.4, 25.8, 23.6, 21.3; ESI-HRMS: calc’d m/e for [M+H+] C25H28NO5: 422.1967, found 422.1973; IR (neat, NaCl, cm−1): 2949, 1732, 1575, 1497, 1455, 1436, 1409, 1378, 1356, 1297, 1242, 1202, 1154, 1119, 1079, 1029.
Dibenzyl 2-((2-(Benzylamino)-6-oxocyclohex-1-enyl)methyl)malonate (7e):
The N-benzyl-enaminone (50 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dibenzyl malonate (0.070 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous light yellow oil (112 mg, 0.23 mmol, 90%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.36 - 7.22 (m, 15H), 6.20 (t, J = 6.1 Hz, 1H), 5.12 (q-like, J = 12.4 Hz, 4H), 4.30 (d, J = 6.1 Hz, 2H), 3.95 (t, J = 7.3 Hz, 1H), 2.87 (d, J = 7.3 Hz, 2H), 2.28 (t, J = 6.2 Hz, 2H), 2.22 (t, J = 6.3 Hz, 2H), 1.77 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): 194.9, 170.4, 162.7, 138.3, 135.7, 129.0, 128.6, 128.3, 128.1, 127.7, 127.0, 105.6, 67.2, 50.7, 47.1, 36.3, 25.7, 23.5, 21.1; ESI-HRMS: calc’d m/e for [M+H+] C31H32NO5: 498.2280, found 498.2275; IR (neat, NaCl, cm−1): 2945, 1731, 1577, 1497, 1456, 1434, 1372, 1356, 1295, 1200, 1149, 1028.
Dimethyl 2-((2-(Butylamino)-6-oxocyclohex-1-enyl)methyl)malonate (7f):
The N-butyl-enaminone (42 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dimethyl malonate (0.042 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous colorless oil (52 mg, 0.17 mmol, 66%). 1H NMR (CDCl3, 400 MHz, ppm): δ 5.88 (s, 1H), 3.75 (t, J = 6.2 Hz, 1H), 3.69 (s, 6H), 3.18 (m, 2H), 2.76 (d, J = 7.1 Hz, 2H), 2.41 (t, J = 6.2 Hz, 2H), 2.26 (t, J = 6.3 Hz, 2H), 1.87 (m, 2H), 1.57 (m, 2H), 1.40 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 194.4, 171.2, 163.0, 104.9, 52.7, 50.6, 43.2, 36.3, 32.4, 25.8, 23.4, 21.3, 20.1, 13.9; ESI-HRMS: calc’d m/e for [M+H+] C16H26NO5: 312.1811, found 312.1801; IR (neat, NaCl, cm−1): 2954, 1733, 1570, 1436, 1417, 1368, 1301, 1243, 1203, 1155, 1100.
Dibenzyl 2-((2-(Butylamino)-6-oxocyclohex-1-enyl)methyl)malonate (7g):
The N-butyl-enaminone (42 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dibenzyl malonate (0.070 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous colorless oil (92 mg, 0.20 mmol, 80%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.32 - 7.26 (m, 10H), 5.69 (t, J = 4.8 Hz, 1H), 5.13 (q-like, J = 12.4 Hz, 4H), 3.92 (t, J = 7.4 Hz, 1H), 3.04 (m, 2H), 2.81 (d, J = 7.3 Hz, 2H), 2.28 (t, J = 6.2 Hz, 2H), 2.22 (t, J = 6.1 Hz, 2H), 1.81 (m, 2H), 1.50 (m, 2H), 1.37 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 194.4, 170.6, 162.8, 135.8, 128.6, 128.3, 128.1, 104.7, 67.1, 50.7, 43.1, 36.3, 32.3, 25.8, 23.4, 21.2, 20.1, 13.79; ESI-HRMS: calc’d m/e for [M+H+] C28H34NO5: 464.2437, found 464.2444; IR (neat, NaCl, cm−1): 2955, 1731, 1575, 1438, 1413, 1377, 1198, 1152.
Dimethyl 2-((2-(Morpholinoamino)-6-oxocyclohex-1-enyl)methyl)malonate (7h):
The N-morpholino-enaminone (49 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dimethyl malonate (0.042 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous light yellow oil (58 mg, 0.17 mmol, 72%). 1H NMR (CDCl3, 400 MHz, ppm): δ 6.78 (s, 1H), 3.77 (t, J = 7.2 Hz, 1H), 3.76 (br, 4H), 3.71 (s, 6H), 2.76 (br, 4H), 2.74 (d, J = 7.2 Hz, 2H), 2.64 (t, J = 6.2 Hz, 2H), 2.27 (t, J = 6.2 Hz, 2H), 1.86 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.6, 171.1, 162.6, 103.6, 66.7, 56.9, 52.7, 50.6, 36.7, 25.2, 23.2, 21.4; ESI-HRMS: calc’d m/e for [M+H+] C16H25N2O6: 341.1713, found 341.1699; IR (neat, NaCl, cm−1): 2953, 2857, 1733, 1583, 1438, 1403, 1303, 1267, 1243, 1201, 1157, 1111, 1074, 1042, 1028.
Dibenzyl 2-((2-(Morpholinoamino)-6-oxocyclohex-1-enyl)methyl)malonate (7i):
The N-morpholino-enaminone (49 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dibenzyl malonate (0.070 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous light yellow oil (106 mg, 0.22 mmol, 86%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.33 - 7.25 (m, 10H), 6.64 (s, 1H), 5.14 (q-like, J = 12.5 Hz, 4H), 3.94 (t, J = 7.4 Hz, 1H), 3.90 - 3.40 (br, 4H), 2.78 (d, J = 7.3 Hz, 2H), 2.58 (s, 4H), 2.54 (t, J = 6.2 Hz, 2H), 2.25 (t, J = 6.2 Hz, 2H), 1.82 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.5, 170.4, 162.6, 135.8, 128.6, 128.3, 127.9, 103.5, 67.2, 66.6, 56.7, 50.8, 36.7, 25.2, 23.1, 21.3; ESI-HRMS: calc’d m/e for [M+H+] C28H33N2O6: 493.2339, found 493.2342; IR (neat, NaCl, cm−1): 2917, 2850, 1732, 1584, 1497, 1455, 1402, 1385, 1266, 1233, 1200, 1152, 1112, 1076.
Dimethyl 2-((4,4-Dimethyl-6-oxo-2-(phenylamino)cyclohex-1-enyl)methyl)-malonate (7j):
The N-phenyl-enaminone (54 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dimethyl malonate (0.042 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous colorless oil (75 mg, 0.21 mmol, 83%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.94 (s, 1H), 7.35 (m, 2H), 7.16 (m, 1H), 7.06 (m, 2H), 3.86 (t, J = 7.4 Hz, 1H), 3.75 (s, 6H), 2.93 (d, J = 7.4 Hz, 2H), 2.36 (s, 2H), 2.23 (s, 2H), 0.97 (s, 6H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.5, 171.3, 158.4, 139.3, 129.4, 124.9, 123.9, 108.0, 52.9, 50.7, 50.5, 40.8, 32.8, 28.1, 23.8; ESI-HRMS: calc’d m/e for [M+H+] C20H26NO5: 360.1811, found 360.1801; IR (neat, NaCl, cm−1): 2954, 1735, 1579, 1498, 1436, 1395, 1321, 1270, 1243, 1150.
Dibenzyl 2-((4,4-Dimethyl-6-oxo-2-(phenylamino)cyclohex-1-enyl)methyl)-malonate (7k):
The N-phenyl-enaminone (54 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dibenzyl malonate (0.070 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous light yellow oil (111 mg, 0.22 mmol, 87%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.90 (s, 1H), 7.34 (m, 2H), 7.29 (m, 10H), 7.16 (m, 1H), 7.00 (m, 2H), 5.16 (dt, J = 3.2, 14.0 Hz, 4H), 3.99 (t, J = 7.5 Hz, 1H), 2.95 (d, J = 7.5 Hz, 2H), 2.27 (s, 2H), 2.18 (s, 2H), 0.91 (s, 6H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.5, 170.5, 158.4, 139.3, 135.5, 129.3, 128.7, 128.4, 128.2, 124.9, 124.0, 107.9, 67.4, 51.0, 50.5, 40.7, 32.7, 28.1, 23.7; ESI-HRMS: calc’d m/e for [M+H+] C32H34NO5: 512.2437, found 512.2431; IR (neat, NaCl, cm−1): 2956, 1733, 1579, 1498, 1455, 1395, 1384, 1321, 1271, 1227, 1147, 1071.
Dimethyl 2-((2-(Benzylamino)-4,4-dimethyl-6-oxocyclohex-1-enyl)methyl)-malonate (7l):
The N-benzyl-enaminone (57 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dimethyl malonate (0.042 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous colorless oil (81 mg, 0.24 mmol, 95%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.37 (m, 2H), 7.29 (m, 3H), 6.20 (m, 1H), 4.43 (d, J = 6.2 Hz, 2H), 3.82 (t, J = 7.4 Hz, 1H), 3.69 (s, 6H), 2.84 (d, J = 7.3 Hz, 2H), 2.24 (s, 2H), 2.16 (s, 2H), 0.96 (s, 6H); 13C NMR (CDCl3, 100 MHz, ppm): δ 194.4, 171.1, 161.0, 138.5, 129.0, 127.8, 126.9, 104.5, 52.7, 50.5, 50.0, 47.1, 39.3, 31.8, 28.5, 23.4; ESI-HRMS: ESI-HRMS: calc’d m/e for [M+H+] C21H28NO5: 374.1967, found 374.1964; IR (neat, NaCl, cm−1): 2953, 1732, 1576, 1436, 1407, 1357, 1325, 1271, 1245, 1150, 1118, 1036.
Dibenzyl 2-((2-(Benzylamino)-4,4-dimethyl-6-oxocyclohex-1-enyl)methyl)malonate (7m):
The N-benzyl-enaminone (57 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dibenzyl malonate (0.070 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous light yellow oil (115 mg, 0.22 mmol, 88%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.37 - 7.23 (m, 15H), 6.10 (t, J = 6.5 Hz, 1H), 5.11 (q-like, J = 12.4 Hz, 4H), 4.32 (d, J = 6.3 Hz, 2H), 3.95 (t, J = 7.4 Hz, 1H), 2.87 (d, J = 7.4 Hz, 2H), 2.13 (s, 2H), 2.10 (s, 2H), 0.92 (s, 6H); 13C NMR (CDCl3, 100 MHz, ppm): δ 194.4, 170.4, 160.9, 138.5, 135.6, 129.0, 128.6, 128.3, 128.2, 127.7, 126.9, 104.3, 67.2, 50.8, 49.9, 47.0, 39.2, 31.8, 28.5, 23.3; ESI-HRMS: calc’d m/e for [M+H+] C33H36NO5: 526.2593, found 526.2586; IR (neat, NaCl, cm−1): 2955, 1731, 1577, 1497, 1455, 1407, 1356, 1324, 1271, 1233, 1147, 1028.
Dimethyl 2-((2-(Butylamino)-4,4-dimethyl-6-oxocyclohex-1-enyl)methyl)malonate (7n):
The N-butyl-enaminone (49 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dimethyl malonate (0.042 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous colorless oil (75 mg, 0.22 mmol, 88%). 1H NMR (CDCl3, 400 MHz, ppm): δ 5.73 (s, 1H), 3.77 (t, J = 7.3 Hz, 1H), 3.69 (s, 6H), 3.18 (m, 2H), 2.77 (d, J = 7.3 Hz, 2H), 2.25 (s, 2H), 2.16 (s, 2H), 1.57 (m, 2H), 1.40 (m, 2H), 1.02 (s, 6H), 0.96 (t, 7.3 Hz, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 193.8, 171.1, 161.1, 103.5, 52.6, 50.5, 49.9, 43.2, 39.5, 32.7, 31.8, 28.6, 23.3, 20.1, 13.9; ESI-HRMS: calc’d m/e for [M+H+] C18H30NO5: 340.2124, found 340.2126; IR (neat, NaCl, cm−1): 2953, 1733, 1576, 1436, 1407, 1357, 1325, 1271, 1245, 1150, 1118, 1036.
Dimethyl 2-((5-Oxo-2-(phenylamino)cyclopent-1-enyl)methyl)malonate (7o):
The N-phenyl-enaminone (43 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dimethyl malonate (0.042 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous colorless oil (76 mg, 0.24 mmol, 96%). 1H NMR (CDCl3, 400 MHz, ppm): δ 8.20 (s, 1H), 7.34 (t, J = 7.7 Hz, 2H), 7.14 (m, 3H), 3.75 (s, 6H), 3.72 (t, J = 7.2 Hz, 1H), 2.78 (d, J = 7.0 Hz, 2H), 2.70 (s, 2H), 2.37 (s, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 202.9, 171.5, 170.8, 139.3, 129.5, 124.9, 122.3, 111.6, 52.9, 50.3, 33.2, 26.3, 21.7; ESI-HRMS: calc’d m/e for [M+H+] C17H20NO5: 318.1341, found 318.1344; IR (neat, NaCl, cm−1): 2953, 1734, 1667, 1581, 1499, 1435, 1410, 1340, 1293, 1240, 1158, 1079, 1063.
Dimethyl 2-((2-(Benzylamino)-5-oxocyclopent-1-enyl)methyl)malonate (7p):
The N-benzyl-enaminone (47 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (16 mg, 0.50 mmol), dimethyl malonate (0.042 mL, 0.37 mmol), and acetic anhydride (0.024 mL, 0.25 mmol) were subjected to the standard reaction conditions, and yielded a viscous colorless oil (78 mg, 0.24 mmol, 94%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.36 (m, 2H), 7.29 (m, 3H), 6.43 (s, 1H), 4.47 (d, J = 6.4 Hz, 2H), 3.71 (t, J = 6.9 Hz, 1H), 3.69 (s, 6H), 2.69 (d, J = 7.2 Hz, 2H), 2.54 (s, 2H), 2.33 (s, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 202.3, 174.1, 170.6, 138.0, 129.0, 127.9, 127.0, 108.6, 52.8, 50.3, 47.3, 32.9, 24.9, 21.8; ESI-HRMS: calc’d m/e for [M+H+] C18H22NO5: 332.1498, found 332.1492; IR (neat, NaCl, cm−1): 2952, 1733, 1669, 1594, 1578, 1496, 1436, 1353, 1279, 1238, 1156, 1118, 1077, 1056, 1030.
2,2′-Methylenebis(3-(benzylamino)cyclohex-2-enone) (8):
The N-benzyl enaminone (50 mg, 0.25 mmol) was dissolved in acetonitrile (10 mL), and paraformaldehyde (16 mg, 0.50 mmol), and dimethyl malonate (0.042 mL, 0.37 mmol) were added sequentially. The reaction was stirred at 60 °C for 2 h, the solvent was then removed under reduced pressure, and the crude material was purified by flash column flash column chromatography on silica gel, using 50% acetone:hexanes (v/v) as the eluent. After purification, a light yellow oil was obtained (38 mg, 74%). 1H NMR (CDCl3, 400 MHz, ppm): δ 9.51 (s, 2H), 7.33 - 7.20 (m, 10H), 4.48 (d, J = 6.4 Hz, 4H), 3.77 (s, 2H), 2.36 (br, 4H), 2.27 (br, 4H), 1.82 (m, 4H); 13C NMR (CDCl3, 100 MHz, ppm): δ 194.9, 166.9, 139.1, 128.8, 127.3, 126.7, 108.9, 63.8, 46.8, 35.7, 25.6, 21.5; ESI-HRMS: calc’d m/e for [M+H+] C27H31N2O2: 415.2386, found 415.2390; IR (neat, NaCl, cm−1): 3262, 3171, 3030, 2946, 1734, 1610, 1560, 1478, 1433, 1355, 1317, 1201, 1127, 1106, 1051, 1011.
Dimethyl 2-Methylenemalonate (9a):
Dimethyl malonate (11.5 mL, 100.0 mmol) was dissolved in acetic acid (45 mL), Cu(OAc)2 (2.0 g, 11.2 mmol) was added, and the reaction mixture was heated at 100 °C (external bath temperature) for 4 h. After cooling to ambient temperature, the salts were filtered off, and the acetic acid was removed using bulb-to-bulb distillation (bp 50-60 °C, 100 mmHg). Further bulb-to-bulb distillation (bp 120-130 °C, 20 mm Hg) gave a colorless oil (3.7 g, 25.7 mmol, 26%). The product tended to polymerize rapidly and was therefore, taken to the reaction with the enaminone immediately after isolation. 1H NMR (CDCl3, 400 MHz, ppm): δ 6.56 (s, 2H), 3.83 (s, 6H); 13C NMR (CDCl3, 100 MHz, ppm): δ 167.1, 135.2, 134.5, 52.7; ESI-HRMS: calc’d m/e for [M+H+] C6H9O4: 145.0501, found 145.0500.
Dimethyl 2-Benzylidenemalonate (9b):
To a solution of benzaldehyde (6.6 mL, 60 mmol) in DMSO (20 mL), proline (690.0 mg, 6.0 mmol) was added. The mixture was stirred for 5 min before the addition of dimethyl malonate (13.7 mL, 120 mmol). After stirring at ambient temperature for 24 h, the reaction was diluted with EtOAc (60 mL) and washed with water (60 mL × 2). The combined organic layer was dried over Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel using 50% acetone:hexanes (v/v) as the eluent to furnish a viscous oil (12.8 g, 98%): 1H NMR (CDCl3, 400 MHz, ppm): δ 7.78 (s, 1H), 7.42 - 7.40 (m, 5H), 3.86 (s, 3H), 3.85 (s, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 167.3, 164.6, 143.1, 132.9, 130.8, 129.5, 129.0, 125.6, 52.87, 52.86; ESI-HRMS: calc’d m/e for [M+H+] C12H13O4: 221.0814, found 221.0805.
Four reactions were set up in parallel, utilizing the enaminone (3-(benzylamino)cyclohex-2-enone) to react with dimethyl 2-methylenemalonate 9a and dimethyl 2-benzylidenemalonate 9b synthesized above, respectively:
a) A solution of enaminone (50.2 mg, 0.25 mmol), LiClO4 (30 mg, 0.25 mmol), and dimethyl 2-methylenemalonate (54 mg, 0.38 mmol) was heated in a sealed screw-cap vial at 60 °C;
b) A solution of enaminone (50.2 mg, 0.25 mmol) and dimethyl 2-methylenemalonate (54 mg, 0.38 mmol) was heated in a sealed screw-cap vial at 60 °C;
c) A solution of enaminone (50.2 mg, 0.25 mmol), LiClO4 (30 mg, 0.25 mmol), and dimethyl 2- benzylidenemalonate (83 mg, 0.38 mmol) was heated in a sealed screw-cap vial at 60 °C;
d) A solution of enaminone (50.2 mg, 0.25 mmol) and dimethyl 2-benzylidenemalonate (83 mg, 0.38 mmol) was heated in a sealed screw-cap vial at 60 °C.
The reactions were monitored by TLC for product formation. After the starting material was completely consumed, the reaction medium was then allowed to cool to ambient temperature and was subsequently washed with H2O (20 mL). The aqueous phase was extracted with EtOAc (3 × 10 mL). The organic layers were combined, washed with brine, and dried with MgSO4. The organic solvent was then removed under reduced pressure, and the residue was purified by flash column chromatography on silica gel. Reactions a and b gave the desired adduct in a yield of 90%, while only starting material was recovered for reactions c) and d).
General Method for the Synthesis of 2,5-Dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylates 5 (Table 3).
The adduct 7 was dissolved in the corresponding solvent (30 mL/mmol) in a screw-cap vial. Dichloroethane was selected as the solvent for the adducts derived from dimethyl malonate, and toluene was used for those derived from dibenzyl malonate. To the solution was slowly added trifluoroacetic acid (30 equiv). The vial was sealed and stirred at 90 °C for 0.5 to 3 hr. The reaction medium was cooled to ambient temperature and washed with saturated NaHCO3 solution (20 mL). The aqueous phase was extracted with CH2Cl2 or EtOAc (3 × 10 mL). The organic layers were combined, washed with brine, and dried over MgSO4. The organic solvent was then removed under reduced pressure, and the residue was purified by flash column chromatography on silica gel, using 20% EtOAc:hexanes (v/v) as the eluent.
Methyl 1-Benzyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5a):
The adduct (80 mg, 0.23 mmol) was dissolved in dichloroethane (7 mL); then TFA (0.54 mL, 7.0 mmol) was added, and the reaction was treated under the standard conditions to yield a light yellow oil (58 mg, 0.18 mmol, 79%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.27 (m, 2H), 7.20 (m, 1H), 7.11 (m, 2H), 4.96 (q-like, J = 16.4 Hz, 2H), 3.70 (s, 3H), 3.57 (t, J = 8.3 Hz, 1H), 3.00 (dd, J = 8.4, 16.9 Hz, 1H), 2.78 (dd, J = 8.5, 16.9 Hz, 1H), 2.42 (t, J = 6.3 Hz, 2H), 2.28 (t, J = 6.2 Hz, 2H), 1.88 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.8, 169.6, 167.2, 154.7, 136.7, 129.0, 127.7, 126.2, 115.0, 52.8, 47.4, 45.7, 36.0, 26.5, 21.8, 20.8; ESI-HRMS: calc’d m/e for [M+H+] C18H20NO4: 314.1392, found 314.1385; IR (neat, NaCl, cm−1): 2953, 1743, 1692, 1654, 1622, 1497, 1454, 1437, 1392, 1365, 1326, 1299, 1280, 1259, 1195, 1167, 1128.
Benzyl 1-Benzyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5b):
The adduct (108 mg, 0.22 mmol) was dissolved in toluene (7 mL), TFA (0.50 mL, 6.5 mmol) was added, and the reaction mixture was then treated under the standard conditions to yield a light yellow oil (60 mg, 0.15 mmol, 70%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.37 -7.24 (m, 8H), 7.16 (m, 2H), 5.20 (q-like, J = 12.2 Hz, 2H), 5.00 (q-like, J = 16.5 Hz, 2H), 3.68 (t, J = 7.0 Hz, 1H), 3.15 (dd, J = 7.5, 16.8 Hz, 1H), 2.80 (dd, J = 6.6, 16.8 Hz, 1H), 2.41 (m, 2H), 2.26 (m, 2H), 1.84 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.6, 168.9, 167.1, 154.7, 136.6, 135.4, 129.0, 128.6, 128.4, 128.3, 127.5, 126.1, 115.0, 67.3, 47.5, 45.6, 35.9, 26.3, 21.6, 20.8; ESI-HRMS: calc’d m/e for [M+H+] C24H24NO4: 390.1705, found 390.1700; IR (neat, NaCl, cm−1): 2953, 1738, 1691, 1652, 1622, 1497, 1455, 1428, 1390, 1364, 1297, 1258, 1194, 1164, 1126.
Methyl 2,5-Dioxo-1-phenyl-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5c):
The adduct (57 mg, 0.17 mmol) was dissolved in dichloroethane (5 mL), TFA (0.40 mL, 5.2 mmol) was added, and the reaction mixture was treated under the standard conditions to yield a white solid (43mg, 0.14 mmol, 84%, mp: 120 - 121 °C). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.48 - 7.40 (m, 3H), 7.17 (s, 2H), 3.76 (s, 3H), 3.68 (t, J = 7.6 Hz, 1H), 3.18 (dd, J = 8.4, 17.0 Hz, 1H), 2.90 (dd, J = 8.6, 17.0 Hz, 1H), 2.39 (t, J = 6.0 Hz, 2H), 2.09 (t, J = 6.0 Hz, 2H), 1.93 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 196.0, 169.6, 167.1, 154.8, 137.0, 129.7, 129.0, 128.8, 113.9, 52.8, 47.5, 36.3, 28.0, 21.9, 21.1; ESI-HRMS: calc’d m/e for [M+H+] C17H18NO4: 300.1236, found 300.1228; IR (neat, NaCl, cm−1): 2953, 1742, 1702, 1652, 1625, 1594, 1493, 1380, 1271, 1244, 1154.
Benzyl 2,5-Dioxo-1-phenyl-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5d):
The adduct (117 mg, 0.24 mmol) was dissolved in toluene (7 mL), TFA (0.55 mL, 7.2 mmol) was added, and the reaction mixture was treated under the standard conditions to yield a light yellow solid (44 mg, 0.12 mmol, 49%, mp: 117 - 118 °C). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.43 (m, 3H), 7.35 (m, 5H), 7.09 (m, 2H), 5.20 (q-like, J = 12.1 Hz, 2H), 3.73 (t, J = 6.5 Hz, 1H), 3.30 (dd, J = 6.5, 17.0 Hz, 1H), 2.85 (dd, J = 6.6, 16.9 Hz, 1H), 2.33 (m, 2H), 1.99 (m, 2H), 1.81 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.8, 168.8, 167.0, 154.8, 136.9, 135.3, 129.6, 128.9, 128.6, 128.5, 128.4, 113.8, 67.4, 47.6, 36.1, 27.9, 21.7, 21.2; ESI-HRMS: calc’d m/e for [M+H+] C23H22NO4: 376.1549, found 376.1546; IR (neat, NaCl, cm−1): 2951, 1740, 1704, 1652, 1624, 1595, 1491, 1455, 1379, 1350, 1311, 1271, 1244, 1197, 1155.
Methyl 1-Butyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5e):
The adduct (42 mg, 0.14 mmol) was dissolved in dichloroethane (4 mL), TFA (0.31 mL, 3.8 mmol) was added, and then, the reaction mixture was treated under the standard conditions to yield a viscous yellow oil (24 mg, 0.077 mmol, 57%). 1H NMR (CDCl3, 400 MHz, ppm): δ 3.74 (s, 3H), 3.69 (m, 2H), 3.48 (dd, J = 6.8, 9.3 Hz, 1H), 2.94 (dd, J = 9.3, 17.0 Hz, 1H), 2. 79 (dd, J = 6.8, 16.6 Hz, 1H), 2.60 (m, 2H), 2.40 (t, J = 6.2 Hz, 2H), 2.07 (m, 2H), 1.54 (m, 2H), 1.35 (m, 2H), 0.94 (t, J = 7.3 Hz, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.7, 169.6, 166.8, 154.3, 114.7, 52.6, 47.3, 42.6, 36.0, 31.2, 26.2, 21.8, 20.6, 20.1, 13.8; ESI-HRMS: calc’d m/e for [M+H+] C15H22NO4: 280.1549, found 280.1544; IR (neat, NaCl, cm−1): 2957, 1744, 1690, 1652, 1618, 1435, 1393, 1279, 1245, 1201, 1121.
Benzyl 1-Butyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5f):
The adduct (96 mg, 0.21 mmol) was dissolved in toluene (6 mL), TFA (0.48 mL, 6.2 mmol) was added, and then, the reaction mixture was treated under the standard conditions to yield a viscous colorless oil (44 mg, 0.13 mmol, 60%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.36 - 7.29 (m, 5H), 5.16 (q-like, J = 12.2 Hz, 2H), 3.68 (m, 2H), 3.52 (dd, J = 6.6, 8.0 Hz, 1H), 3.04 (dd, J = 8.0, 16.7 Hz, 1H), 2.73 (dd, J = 6.4, 17.2 Hz, 1H), 2.51 (m, 2H), 2.34 (m, 2H), 1.96 (m, 2H), 1.50 (m, 2H), 1.34 (m, 2H), 0.93 (t, J = 7.2 Hz, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.6, 168.9, 166.7, 154.5, 135.4, 128.6, 128.4, 128.3, 114.7, 67.2, 47.5, 42.6, 35.9, 31.2, 26.1, 21.7, 20.8, 20.1, 13.7; ESI-HRMS: calc’d m/e for [M+H+] C21H26NO4: 356.1862, found 356.1861; IR (neat, NaCl, cm−1): 2958, 1741, 1691, 1652, 1618, 1498, 1456, 1394, 1278, 1245, 1197, 1172, 1121.
Methyl 1-Benzyl-7,7-dimethyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5g):
The adduct (52 mg, 0.14 mmol) was dissolved in dichloroethane (5 mL), TFA (0.32 mL, 4.2 mmol) was added, and then, the reaction mixture was treated under the standard conditions to yield a viscous colorless oil (32 mg, 0.093 mmol, 67%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.34 (m, 2H), 7.26 (m, 1H), 7.17 (m, 2H), 5.09 (q-like, J = 16.4 Hz, 2H), 3.75 (s, 3H), 3.64 (t, J = 6.9 Hz, 1H), 3.14 (dd, J = 7.2, 16.8 Hz, 1H), 2.80 (dd, J = 6.6, 16.8 Hz, 1H), 2.34 (s, 2H), 2.22 (s, 2H), 0.95 (s, 3H), 0.92 (s, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.7, 169.6, 167.4, 152.9, 136.8, 129.1, 127.7, 126.1, 114.1, 52.8, 49.8, 47.5, 45.6, 40.1, 33.2, 28.9, 27.5, 20.6; ESI-HRMS: calc’d m/e for [M+H+] C20H24NO4: 342.1705, found 342.1698; IR (neat, NaCl, cm−1): 2956, 1741, 1693, 1653, 1625, 1497, 1454, 1439, 1386, 1303, 1280, 1252, 1204, 1168.
Benzyl 1-Benzyl-7,7-dimethyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5h):
The adduct (117 mg, 0.22 mmol) was dissolved in toluene (7 mL), TFA (0.46 mL, 6.7 mmol) was added, and then, the reaction mixture was treated under the standard conditions to yield a viscous colorless oil (60 mg, 0.14 mmol, 64%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.35 - 7.25 (m, 8H), 7.16 (m, 2H), 5.18 (q-like, J = 12.2 Hz, 2H), 5.00 (q-like, J = 16.6 Hz, 2H), 3.69 (t, J = 6.8 Hz, 1H), 3.19 (dd, J = 7.0, 16.8 Hz, 1H), 2.79 (dd, J = 4.7, 16.8 Hz, 1H), 2.27 (q-like, J = 10.2 Hz, 2H), 2.15 (q-like, J = 16.3 Hz, 2H), 0.90 (s, 3H), 0.84 (s, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.6, 169.0, 167.3, 153.0, 136.7, 135.4, 129.0, 128.7, 128.5, 128.4, 127.6, 126.0, 114.0, 67.4, 49.7, 47.6, 45.5, 40.0, 33.0, 28.6, 27.7, 20.7; ESI-HRMS: calc’d m/e for [M+H+] C26H28NO4: 418.2018, found 418.2021; IR (neat, NaCl, cm−1): 2959, 1739, 1694, 1653, 1625, 1497, 1455, 1386, 1369, 1303, 1252, 1213, 1169, 1124.
Methyl 7,7-Dimethyl-2,5-dioxo-1-phenyl-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5i):
The adduct (54 mg, 0.21 mmol) was dissolved in toluene (6 mL), TFA (0.48 mL, 6.2 mmol) was added, and then, the reaction mixture was treated under the standard conditions to yield a white solid (44 mg, 0.12 mmol, 64%, mp: 123 - 124 °C). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.50 - 7.42 (m, 3H), 7.21 - 7.10 (m, 2H), 3.75 (s, 3H), 3.69 (t, J = 5.8 Hz, 1H), 3.25 (dd, J = 6.6, 16.9 Hz, 1H), 2.86 (dd, J = 6.6, 16.9 Hz, 1H), 2.26 (s, 2H), 1.95 (q-like, J = 17.6 Hz, 2H), 0.97 (s, 3H), 0.95 (s, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.8, 169.5, 167.2, 153.0, 136.9, 129.7, 128.9, 128.5, 113.0, 52.7, 49.9, 47.5, 41.5, 33.2, 28.8, 27.4, 20.8; ESI-HRMS: calc’d m/e for [M+H+] C19H22NO4: 328.1549, found 328.1540; IR (neat, NaCl, cm−1): 2956, 1740, 1704, 1654, 1628, 1596, 1492, 1437, 1380, 1317, 1292, 1263, 1239, 1144.
Benzyl 7,7-Dimethyl-2,5-dioxo-1-phenyl-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5j):
The adduct (111 mg, 0.22 mmol) was dissolved in toluene (7 mL), TFA (0.50 mL, 6.5 mmol) was added, and then, the reaction mixture was treated under the standard conditions to yield a white solid (63 mg, 0.16 mmol, 72%, mp: 126 - 127 °C). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.43 (m, 3H), 7.34 (m, 5H), 7.07 (m, 2H), 5.18 (q-like, J = 12.2 Hz, 2H), 3.73 (t, J = 6.3 Hz, 1H), 3.33 (dd, J = 5.8, 16.9 Hz, 1H), 2.84 (dd, J = 6.3, 16.9 Hz, 1H), 2.19 (q-like, J = 16.4 Hz, 2H), 1.88 (q-like, J = 17.0 Hz, 2H), 0.94 (s, 3H), 0.86 (s, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.7, 168.8, 167.2, 152.9, 136.9, 135.2, 129.62, 129.61, 128.9, 128.7, 128.52, 128.47, 112.8, 67.5, 49.8, 47.6, 41.4, 33.0, 28.6, 27.6, 20.9; ESI-HRMS: calc’d m/e for [M+H+] C25H26NO4: 404,1862, found 404.1864; IR (neat, NaCl, cm−1): 2958, 1740, 1705, 1654, 1628, 1596, 1492, 1455, 1379, 1351, 1317, 1292, 1240, 1262, 1173, 1143.
Methyl 1-Butyl-7,7-dimethyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carboxylate (5k):
The adduct (75 mg, 0.22 mmol) was dissolved in dichloroethane (7 mL), TFA (0.51 mL, 6.6 mmol) was added, and then, the reaction mixture was treated under the standard conditions to yield a viscous light yellow oil (42 mg, 0.14 mmol, 62%). 1H NMR (CDCl3, 400 MHz, ppm): δ 3.71 (s, 3H), 3.68 (m, 2H), 3.48 (dd, J = 6.6, 8.0 Hz, 1H), 3.00 (dd, J = 8.1, 16.7 Hz, 1H), 2.72 (dd, J = 6.6, 16.8 Hz, 1H), 2.42 (s, 2H), 2.27 (s, 2H), 1.53 (m, 2H), 1.35 (m, 2H), 1.10 (s, 3H), 1.07 (s, 3H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.6, 169.7, 167.1, 152.6, 113.8, 52.7, 49.8, 47.4, 42.5, 40.1, 33.2, 31.4, 29.0, 27.9, 20.5, 20.2, 13.9; ESI-HRMS: calc’d m/e for [M+H+] C17H26NO4: 308.1862, found 308.1868; IR (neat, NaCl, cm−1): 2958, 2872, 1745, 1692, 1653, 1623, 1439, 1391, 1310, 1279, 1248, 1206, 1173, 1118, 1032, 1009.
Methyl 1-Benzyl-2,5-dioxo-2,3,4,5,6,7-hexahydro-1H-cyclopenta[b]pyridine-3-carboxylate (5l):
The adduct (51 mg, 0.15 mmol) was dissolved in dichloroethane (5 mL), TFA (0.36 mL, 4.6 mmol) was added, and then, the reaction mixture was treated under the standard conditions to yield a white solid (29 mg, 0.095 mmol, 62%, mp: 134 - 135 °C). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.37 - 7.29 (m, 3H), 7.24 (m, 2H), 4.96 (q-like, J = 15.8 Hz, 2H), 3.77 (s, 3H), 3.72 (t, J = 7.6 Hz, 1H), 2.96 (dd, J = 7.3, 17.3 Hz, 1H), 2.75 (dd, J = 7.9, 17.0 Hz, 1H), 2.65 (m, 2H), 2.46 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 201.5, 169.6, 168.4, 167.4, 136.3, 129.1, 128.0, 126.9, 116.9, 53.0, 47.5, 46.5, 34.3, 25.4, 19.8; ESI-HRMS: calc’d m/e for [M+H+] C17H18NO4: 300.1236, found 300.1230; IR (neat, NaCl, cm−1): 2953, 2928, 1742, 1687, 1640, 1496, 1437, 1412, 1363, 1322, 1281, 1253, 1213, 1167, 1137, 1123, 1027, 1083.
Methyl 2,5-Dioxo-1-phenyl-2,3,4,5,6,7-hexahydro-1H-cyclopenta[b]pyridine-3-carboxylate (5m):
The adduct (76 mg, 0.24 mmol) was dissolved in dichloroethane (7 mL), TFA (0.55 mL, 7.2 mmol) was added, and then, the reaction mixture was treated under the standard conditions to yield a white solid (34 mg, 0.12 mmol, 49%, mp: 100 - 101 °C). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.50 - 7.44 (m, 3H), 7.24 (m, 2H), 3.79 (t, J = 6.4 Hz, 1H), 3.78 (s, 3H), 3.06 (dd, J = 6.3, 17.1 Hz, 1H), 2.83 (dd, J = 7.8, 17.1 Hz, 1H), 2.45 (m, 2H), 2.33 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 201.9, 169.7, 168.6, 167.3, 136.1, 129.8, 129.4, 128.3, 116.1, 53.1, 47.6, 34.3, 26.0, 20.1; ESI-HRMS: calc’d m/e for [M+H+] C16H16NO4: 286.1079, found 286.1074; IR (neat, NaCl, cm−1): 2954, 2929, 1743, 1690, 1641, 1595, 1564, 1492, 1439, 1395, 1352, 1317, 1293, 1257, 1140, 1024, 1000.
General Procedure for the Preparation of 2,5-Dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carbonitriles 10 (Table 4).
The enaminone 6 was dissolved in acetonitrile (100 mL/mmol) and stirred at ambient temperature. To the solution, LiClO4 (1 equiv) was added, followed by the addition of PPh3 (0.5 equiv), methyl cyanoacetate (1.5 equiv), and paraformaldehyde (1.0 equiv) in sequence. The reaction was heated to 60 °C in a sealed vessel for 14 h (monitored by TLC). The reaction was quenched with saturated NaHCO3 solution, upon consumption of starting material. The mixture was then extracted with CH2Cl2 (10 mL × 2), and the organic layers were combined, dried over MgSO4, and concentrated in vacuo. The crude product was purified by flash column chromatography on silica gel, using 25% EtOAc:hexanes (v/v) as the eluent.
1-Benzyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carbonitrile (10a):
The N-benzyl enaminone (50 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (8 mg, 0.25 mmol), methyl cyanoacetate (0.033 mL, 0.37 mmol), and triphenylphosphine (33 mg, 0.13 mmol) in acetonitrile (25 mL) were subjected to the standard conditions and yielded a colorless oil (60 mg, 0.21 mmol, 85%). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.37 - 7.27 (m, 3H), 7.13 (m, 2H), 5.12 (d, J = 16.3 Hz, 1H), 4.91 (d, 16.3 Hz, 1H), 3.75 (dd, J = 6.3, 11.3 Hz, 1H), 3.17 (ddd, J = 1.4, 4.8, 15.3 Hz, 1H), 2.87 (m, 1H), 2.53 (m, 2H), 2.38 (t, J = 6.2 Hz, 2H), 1.99 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.1, 163.0, 154.6, 135.8, 129.2, 127.9, 126.1, 115.6, 114.6, 46.0, 35.7, 34.4, 26.3, 21.8, 21.5; ESI-HRMS: calc’d m/e for [M+H+] C17H17N2O2: 281.1290, found 281.1281; IR (neat, NaCl, cm−1): 2959, 2890, 2256, 1756, 1700, 1655, 1627, 1497.
2,5-Dioxo-1-phenyl-1,2,3,4,5,6,7,8-octahydroquinoline-3-carbonitrile (10b):
The N-phenyl enaminone (47 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (8 mg, 0.25 mmol), methyl cyanoacetate (0.033 mL, 0.37 mmol), and triphenylphosphine (33 mg, 0.13 mmol) in acetonitrile (25 mL) were subjected to the standard conditions and yielded a yellow solid (43 mg, 0.16 mmol, 65%, mp: 158 - 160 °C). 1H NMR (CDCl3, 400 MHz, ppm): δ 7.57 - 7.46 (m, 3H), 7.15 (m, 2H), 3.83 (dd, J = 6.4, 11.1 Hz, 1H), 3.24 (dd, J = 6.3, 15.2 Hz, 1H), 2.97 (m, 1H), 2.42 (dt, J = 1.9, 6.4 Hz, 2H), 2.13 (m, 2H), 1.96 (m, 2H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.3, 162.7, 154.8, 136.1, 129.9, 129.4, 128.5, 115.5, 113.6, 36.0, 34.7, 27.9, 22.1, 21.6; ESI-HRMS: calc’d m/e for [M+H+] C16H15N2O2: 267.1134, found 267.1134; IR (neat, NaCl, cm−1): 2940, 2260, 1765, 1710, 1655, 1627, 1500.
1-Butyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carbonitrile (10c):
The N-butyl enaminone (42 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (8 mg, 0.25 mmol), methyl cyanoacetate (0.033 mL, 0.37 mmol), and triphenylphosphine (33 mg, 0.13 mmol) in acetonitrile (25 mL) were subjected to the standard conditions and yielded a colorless oil (44 mg, 0.18 mmol, 73%). 1H NMR (CDCl3, 400 MHz, ppm): δ 3.77 (m, 1H), 3.63 (m, 2H), 3.11 (dd, J = 6.3, 16.5 Hz, 1H), 2.74 (m, 1H), 2.61 (m, 2H), 2.43 (t, J = 7.1 Hz, 2H), 2.11 (m, 2H), 1.53 (m, 2H), 1.34 (m, 2H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.0, 162.5, 154.4, 115.7, 114.3, 43.1, 35.8, 34.4, 31.2, 26.2, 21.8, 21.7, 20.0, 13.7; ESI-HRMS: calc’d m/e for [M+H+] C14H19N2O2: 247.1447, found 247.1449; IR (neat, NaCl, cm−1): 2964, 2897, 2256, 1756, 1700, 1655, 1627.
7,7-Dimethyl-2,5-dioxo-1-propyl-1,2,3,4,5,6,7,8-octahydroquinoline-3-carbonitrile (10d):
The N-propyl enaminone (45 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (8 mg, 0.25 mmol), methyl cyanoacetate (0.033 mL, 0.37 mmol), and triphenylphosphine (33 mg, 0.13 mmol) in acetonitrile (25 mL) were subjected to the standard conditions and yielded a white solid (49 mg, 0.19 mmol, 75%, mp: 136 - 137 °C). 1H NMR (CDCl3, 400 MHz, ppm): δ 3.71 (m, 1H), 3.62 (m, 2H), 3.03 (dd, J = 6.3, 16.6 Hz, 1H), 2.81 (m, 1H), 2.43 (m, 2H), 2.29 (d, J = 16.9 Hz, 2H), 1.53 (m, 2H), 1.08 (s, 3H), 1.06 (s, 3H), 0.88 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 194.0, 161.7, 151.5, 114.8, 112.2, 48.5, 43.6, 38.9, 33.4, 32.1, 27.5, 27.3, 21.5, 20.5, 10.0; ESI-HRMS: calc’d m/e for [M+H+] C15H21N2O2: 261.1603, found 261.1612; IR (neat, NaCl, cm−1): 2962, 2903, 2252, 1754, 1702, 1652, 1628.
1-(2-Methoxyethyl)-7,7-dimethyl-2,5-dioxo-1,2,3,4,5,6,7,8-octahydroquinoline-3-carbonitrile (10e):
The N-methoxyethyl enaminone (49 mg, 0.25 mmol), LiClO4 (27 mg, 0.25 mmol), paraformaldehyde (8 mg, 0.25 mmol), methyl cyanoacetate (0.033 mL, 0.37 mmol), and triphenylphosphine (33 mg, 0.13 mmol) in acetonitrile (25 mL) were subjected to the standard conditions and yielded a white solid (47 mg, 0.17 mmol, 67%, mp: 77 - 79 °C). 1H NMR (CDCl3, 400 MHz, ppm): δ 3.91 (m, 2H), 3.63 (dd, J = 6.2, 11.1 Hz, 1H), 3.52 (m, 2H), 3.28 (s, 3H), 3.08 (dd, J = 6.2, 16.3 Hz, 1H), 2.80 (m, 1H), 2.61 (d, J = 17.2 Hz, 1H), 2.49 (d, J = 17.2 Hz, 1H), 2.28 (d, J = 3.0 Hz, 2H), 1.10 (s, 3H), 1.08 (s, 3H); 13C NMR (CDCl3, 100 MHz, ppm): δ 195.4, 163.1, 153.6, 115.7, 112.6, 70.3, 59.1, 49.5, 43.3, 40.2, 34.4, 33.0, 28.4, 28.3, 21.5; ESI-HRMS: calc’d m/e for [M+H+] C15H21N2O3: 277.1552, found 277.1558; IR (neat, NaCl, cm−1): 2957, 2889, 2252, 1770, 1715, 1655, 1627, 1249, 1207.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
1H NMR and 13C NMR spectra for all new compounds are available.
Notes
The authors declare no competing financial interest.
References
- 1.Triggle DJ Cell. Mol. Neurobiol 2003, 23, 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rose U Arch. Pharm 1990, 323, 281–286. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi D; Oyunzul L; Onoue S; Ito Y; Uchida S; Simsek R; Gunduz MG; Safak C; Yamada S Biol. Pharm. Bull 2008, 31, 473–479. [DOI] [PubMed] [Google Scholar]
- 4.Willems E; Cabral-Teixeira J; Schade D; Cai W; Reeves P; Bushway PJ; Lanier M; Walsh C; Kirchhausen T; Izpisua BJC; Cashman J; Mercola M Cell Stem Cell 2012, 11, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schade D; Lanier M; Willems E; Okolotowicz K; Bushway PJ; Wahlquist C; Gilley C; Mercola M; Cashman JR J. Med. Chem 2012, 55, 9946–9957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Movsesian MA; Kukreja RC, Phosphodiesterase Inhibition in Heart Failure In Phosphodiesterases as Drug Targets, Francis SH; Conti M; Houslay MD, Eds. Springer: Berlin, 2011; pp 237–250. [DOI] [PubMed] [Google Scholar]
- 7.Gorobets NY; Yousefi BH; Belaj F; Kappe CO Tetrahedron 2004, 60, 8633–8644. [Google Scholar]
- 8.Fossa P; Menozzi G; Dorigo P; Floreani M; Mosti L Bioorg. Med. Chem 2003, 11, 4749–4759. [DOI] [PubMed] [Google Scholar]
- 9.Noguez MO; Marcelino V; Rodriguez H; Martin O; Martinez JO; Arroy GA; Perez FJ; Suarez M; Miranda R Int. J. Mol. Sci 2011, 12, 2641–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hirsch KS; Jones CD Preparation of Hexahydrobenzo [f]quinolinones as 5-α-Reductase Inhibitors. 1992–307636 531026, 19920820., 1993. [Google Scholar]
- 11.Jones CD; Audia JE; Lawhorn DE; McQuaid LA; Neubauer BL; Pike AJ; Pennington PA; Stamm NB; Toomey RE; Hirsch KS J. Med. Chem 1993, 36, (3), 421–423. [DOI] [PubMed] [Google Scholar]
- 12.Astleford BA; Audia JE; Deeter J; Heath PC; Janisse SK; Kress TJ; Wepsiec JP; Weigel LO J. Org. Chem 1996, 61, (13), 4450–4454. [DOI] [PubMed] [Google Scholar]
- 13.Ko S; Sastry MNV; Lin C; Yao C Tetrahedron Lett. 2005, 46, 5771–5774. [Google Scholar]
- 14.Sun J; Xia E-Y; Wu Q; Yan C-G ACS Comb. Sci 2011, 13, 421–426. [DOI] [PubMed] [Google Scholar]
- 15.Fan XS; Li YZ; Zhang XY; Qu GR; Wang JJ; Hu XY Heteroatom. Chem 2006, 17, 382–388. [Google Scholar]
- 16.Tu S; Zhu X; Zhang J; Xu J; Zhang Y; Wang Q; Jia R; Jiang B; Zhang J; Yao C Bioorg. Med. Chem. Lett 2006, 16, 2925–2928. [DOI] [PubMed] [Google Scholar]
- 17.Suárez M; Ochoa E; Verdecia Y; Pita B; Morán L; Martín N; Quinteiro M; Seoane C; Soto J; Novoa H; Blaton N; Peters OM Tetrahedron 1999, 55, 875–884. [Google Scholar]
- 18.Azzam SHS; Siddekha A; Pasha MA Tetrahedron Lett. 2012, 53, (46), 6306–6309. [Google Scholar]
- 19.Enders D; Demir AS; Puff H; Franken S Tetrahedron Lett. 1987, 28, (33), 3795–3798. [Google Scholar]
- 20.Wang G; Miao C Green Chem. 2006, 8, 1080–1085. [Google Scholar]
- 21.Saidi MR; Azizi N; Zali-Boinee H Tetrahedron 2001, 57, 6829–6832. [Google Scholar]
- 22.Azizi N; Saidi MR Tetrahedron 2003, 59, 5329–5332. [Google Scholar]
- 23.Saidi MR; Azizi N; Naimi-Jamal MR Tetrahedron Lett. 2001, 42, 8111–8113. [Google Scholar]
- 24.Saidi MR; Azizi N Tetrahedron: Asymmetry 2002, 13, 2523–2527. [Google Scholar]
- 25.Aryanasab F; Saidi MR Synthetic Commun. 2008, 38, 4036–4044. [Google Scholar]
- 26.Hoye TR; Caruso AJ; Magee AS J. Org. Chem 1982, 47, (21), 4152–6. [Google Scholar]
- 27.Nenajdenko VG; Statsuk AV; Balenkova ES Tetrahedron 2000, 56, (35), 6549–6556. [Google Scholar]
- 28.Cannon JG J. Am. Pharmaceut. Assoc 1956, 45, 430. [DOI] [PubMed] [Google Scholar]
- 29.Smith AB; Liu Z Org. Lett 2008, 10, 4363–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deka N; Kalita DJ; Borah R; Sarma JC J. Org. Chem 1997, 62, (5), 1563–1564. [Google Scholar]
- 31.Li Z-X; Liu X-P; Qiu Z; Xu D; Yu X-J J. Chem. Res 2011, 35, (1), 35–36. [Google Scholar]
- 32.Hourcade S; Ferdenzi A; Retailleau P; Mons S; Marazano C Eur. J. Org. Chem 2005, 2005, 1302–1310. [Google Scholar]
- 33.Gray D; Gallagher T Angew. Chem. Int. Ed 2006, 45, 2419–2423. [DOI] [PubMed] [Google Scholar]
- 34.Barnett CJ; Wilson TM; Evans DA; Somers TC Tetrahedron Lett. 1997, 38, 735–738. [Google Scholar]
- 35.Yadav JS; Reddy BVSR; Basak AK; Visali BN, A.V.; Nagagaiah K Eur. J. Org. Chem 2004, 546–551. [Google Scholar]
- 36.Lu X; Zhang C; Xu Z Acc. Chem. Res 2001, 34, 535–544. [DOI] [PubMed] [Google Scholar]
- 37.Nair V; Menon RS; Sreekanth AR; Abhilash N; Biju AT Acc. Chem. Res 2006, 39, 520–530. [DOI] [PubMed] [Google Scholar]
- 38.Methot JL; Roush WR Adv. Synth. Catal 2004, 346, 1035–1050. [Google Scholar]
- 39.Liu H; Zhang Q; Wang L; Tong X Chem. Commun 2010, 46, 312–314. [DOI] [PubMed] [Google Scholar]
- 40.Stewart IC; Bergman RG; Toste FD J. Am. Chem. Soc 2003, 125 (29) 8696–8697 [DOI] [PubMed] [Google Scholar]
- 41.Yadav JS; Reddy BVS; Krishna AD; Reddy CS; Narsaiah AV J. Mol. Catal. A: Chem 2007, 261, (1), 93–97. [Google Scholar]
- 42.Dyachenko VD; Chernega AN Russ. J. Gen. Chem 2005, 75, 952–960. [Google Scholar]
- 43.Bhattacharyya P; Pradhan K; Paul S; Das AR Tetrahedron Lett. 2012, 53, (35), 4687–4691. [Google Scholar]
- 44.Pradhan K; Bhattacharyya P; Paul S; Das AR Tetrahedron Lett. 2012, 53, (44), 5840–5844. [Google Scholar]
- 45.http://pubchem.ncbi.nlm.nih.gov.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.