

Abstract
Cytochrome bc1 inhibitors have been broadly studied as human and veterinary medicines, and agricultural fungicides. For the most part, cytochrome bc1 inhibitors compete with ubiquinol at the ubiquinol oxidation (Qo) site or with ubiquinone at the quinone reduction (Qi) site. 4(1H)-Quinolones with 3-position substituents may inhibit either site based on quinolone ring substituents. 4(1H)-Quinolones that inhibit the Qi site are highly effective against toxoplasmosis, malaria and babesiosis and do not inhibit human cytochrome bc1. We tested a series of 4(1H)-Quinolones against wild-type and drug resistant strains of Toxplasma gondii and Plasmodium falciparum. These experiments identified very potent compounds that inhibit T. gondii proliferation at picomolar concentrations. The most potent compounds target the Qo site, and for these compounds, an alkyl side chain confers potency against T. gondii greater than that of bulkier side chains. Our experiments also show that substituents on the quinolone ring influenced selectivity between T. gondii and P. falciparum and between Qo and Qi site-mediated activity. Comparison of the parasite cytochrome b sequences identified amino acids that are associated with drug resistance in P. falciparum that exist naturally in wild-type T. gondii. These underlying differences may influence drug susceptibility. Finally, a Qo site active 4(1H)-quinolone-3-diarylether tested in a murine model of toxoplasmosis was superior to atovaquone, resulting in survival from Type I strain T. gondii infection. These experiments identify highly effective compounds for toxoplasmosis and provide valuable insight into the structure-activity relationship of cytochrome bc1 inhibitors.
Keywords: Toxoplasmosis, Toxoplasma gondii, Plasmodium falciparum, Malaria, Cytochrome bc1, Therapeutics
Graphical Abstract
Toxoplasma gondii is a eukaryotic intracellular parasite estimated to have infected billions of people, placing them at risk for toxoplasmosis. Fetuses and immunocompromised persons are susceptible to severe toxoplasmosis, which can be fatal or lead to permanent ocular or neurologic disability, while healthy people without immunodeficiency are susceptible to ocular disease that may cause vision loss. T. gondii seropositivity has been associated with psychiatric disorders and decreased neurocognitive function in people without overt toxoplasmosis; however, the causal role of T. gondii remains an open question.4–5 T. gondii is a leading cause of deaths attributable to foodborne illness in the United States and is an important veterinary pathogen, causing significant disease and death in wild and domesticated animals.7
Current drugs for toxoplasmosis have high rates of adverse events that have been seen in observational studies as well as clinical trials of pyrimethamine combined with sulfadiazine or clindamycin.8–10 Allergic reactions, which occur at greater rates in people with HIV, are a significant limitation for the lengthy courses that are required for treatment and post treatment prophylaxis.11
Cytochrome bc1 (cyt bc1) inhibition has been a successful strategy for treating infections caused by apicomplexan pathogens. Atovaquone (ATV) is a clinically effective and well-tolerated drug for the treatment and prevention of malaria and babesiosis and an alternate treatment for toxoplasmosis and pneumocystis pneumonia. ATV binds to the Qo site of cyt bc1, stopping Plasmodium growth by disrupting pyrimidine synthesis.1, 12 In other organisms, cyt bc1 inhibition halts growth by blocking cellular respiration.
Endochin-like quinolones (ELQs) are a series of potent inhibitors of the apicomplexan cyt bc1 that are effective as single dose cures and prophylaxis in experimental models of malaria and are currently among the most effective preclinical compounds against acute and chronic toxoplasmosis (Figure 1).13–16 Single atom changes to the quinolone core influence whether quinolones bind to the ubiquinol oxidation (Qo) site or the ubiquinone reduction (Qi) site of cytochrome b (cyt b), as evidenced by differential drug resistance in organisms with cyt b mutations.13–14, 17–18 The discovery of nontoxic Qi site inhibitors has created the potential of combining Qo and Qi site inhibitors to overcome the parasite’s capacity to develop drug resistance. This principle has been demonstrated by the radical cure of babesiosis in immunocompromised mice treated with the combination of a Qi site quinolone and ATV.19
Figure 1.

Structures of quinolone core, ubiquinone, and atovaquone.
In our initial evaluation of endochin derivatives, we discovered that among the lead ELQs with diarylether (DAE) side chains, variations in the quinolone core led to differing activity between parasites. For example, ELQ-300 was less potent against T. gondii and Babesia microti than other quinolones despite having similar in vitro potency with other quinolones against Plasmodium falciparum.19–20 To better understand the structural determinants of ELQ potency as well as parasite and Q site selectivity, we tested a defined set of ELQs against T. gondii and P. falciparum strains with known cyt b drug resistance mutations (Table 1.) This approach is valuable, given that cytochrome b comparisons are limited by the absence of apicomplexan cyt bc1 crystal structures and models are derived from phylogenetically distant organisms that contain variations in key amino acids within the ubiquinone binding sites.17, 21 To evaluate the in vivo efficacy of a Qo site targeting ELQ, we tested compound 8B (ELQ-400) in an acute in vivo model of toxoplasmosis.
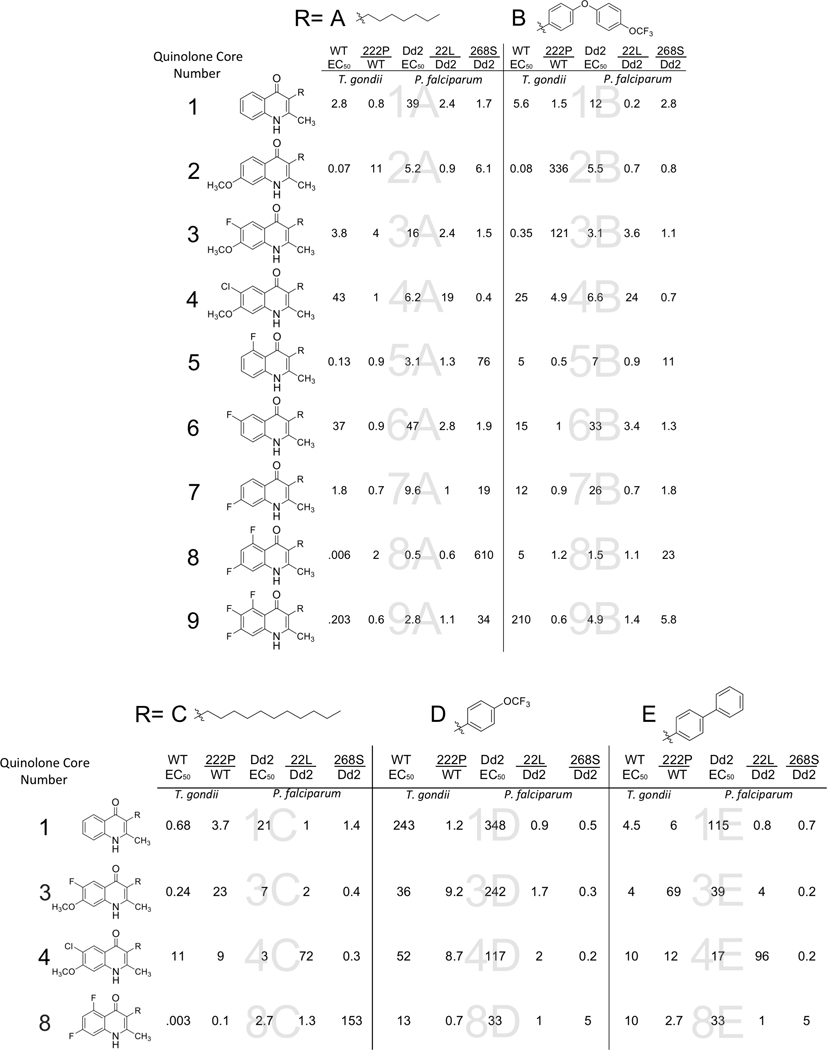
Table 1. Chemical structures of ELQs and EC50s of ELQs against T. gondii and P. falciparum and the ratio of ELQ EC50s against resistant parasites to wild type parasites.
In vitro results of compounds that are the combination of a quinolone core in the left column represented by a number and a side chain at the top represented by a letter. The EC50 of ELQs against wild type T. gondii and P. falciparum Dd2 are listed, followed by the ratio of susceptibility between the resistant and susceptible strains. WT: wild type: T. gondii, 222P: T. gondii strain with threonine to proline substitution at position 222 in cytochrome b, Dd2: P. falciparum strain, 22L: P. falciparum Dd2 strain with isoleucine substituted with leucine at position 22 of cytochrome b, Y268S: P. falciparum strain TM90C2B that has high level resistance to atovaquone caused by tyrosine to serine substitution. All EC50 values are the average of at least 3 experiments.
 |
Results:
In Vitro Efficacy of ELQs against P. falciparum and T. gondii:
We selected a series of 31 ELQs to evaluate 9 different patterns of quinolone substituents combined with varying side chains (Table 1.) Compounds were tested in 72h T. gondii tachyzoite and P. falciparum blood stage proliferation assays to obtain the effective concentration for inhibiting proliferation 50% (EC50.) Compounds were tested against an RH strain of T. gondii expressing β-galactosidase and the Dd2 P. falciparum strain using the SYBR green assay.22 Of the 31 compounds, 9 were highly potent against both T. gondii and P. falciparum with EC50 values below 1 and 10 nM, respectively. Compounds 8A and 8C, both 5,7-difluoro compounds with alkyl side chains, were most effective against T. gondii with EC50 values less than 10 pM. The addition of the larger side chains B, D, and E to these compounds substantially increased the EC50 values (Figure 2A.) A similar shift was seen with the 5-fluoro compounds 5A and 9A losing potency as 5B and 9B. In contrast, the addition of the DAE side chain did not increase the EC50 of the Qi site targeting compounds 2B, 3B, and 4B. However, the phenyl trifluoro methoxy (D) and biphenyl (E) side chain increased the EC50 of the Qi site active compounds, suggesting that the flexibility of the DAE side chain allows for better occupancy of the Qi site. The crystal structure of 8B indicates that the innermost 3-position phenyl group is oriented essentially orthogonal with respect to the quinolone core. All compounds did not inhibit human fibroblast viability at the highest concentration tested as 25 μM with the exception of compound 6B, which inhibited 50% of ATP production at 9 μM. Host cell inhibition by 6B is unlikely to affect T. gondii EC50 values given that the T. gondii EC50 is more than 600 times lower than that of the fibroblast EC50. In general, quinolones have low aqueous solubility, and some ELQs may precipitate over time at the highest concentrations tested in the fibroblast viability assay; however, the ELQs tested remained in solution during serial dilution, allowing for distribution over the 96-well plate. The results of the fibroblast inhibition assay indicate that the T. gondii growth was not inhibited due to fibroblast toxicity.
Figure 2. Comparison of ELQ EC50s against T. gondii and P. falciparum and resistance of T. gondii and P. falciparum with cytochrome b mutations against ELQs.


Figure 2a. The comparison of EC50 values of ELQs in Table 1a and 1b against T. gondii and P. falciparum show that compounds vary in selective potency against parasites. The grey dotted line shows a 1:1 relation for reference. Compound 2C (7-methoxy-2-methyl-3-undecylquinolin-4(1H)-one) T. gondii EC50=0.37nM, Dd2 EC50=1.8nM (not included in Table 1) Figure 2b. Fold difference values from Table 1a and 1b in susceptibility of resistant parasite strains compared to susceptible strains. WT: wild type T. gondii, 222P: T. gondii strain with threonine to proline substitution at position 222 in cytochrome b, Dd2: P. falciparum strain, 22L: P. falciparum Dd2 strain with isoleucine substituted with leucine at position 22 of cytochrome b, Y268S: P. falciparum strain TM90-C2B that has high level resistance to atovaquone caused by tyrosine to serine substitution.
Comparison of ELQ Potency against P. falciparum and T. gondii and cyt b Sequence:
ELQ potencies were different between P. falciparum and T. gondii. The range of EC50s for T. gondii was 0.003 nM to 210 nM, whereas the range for P. falciparum was narrower at 0.5 nM to 348 nM. Within this ELQ series, 39% of the compounds were ≥10 fold more effective against T. gondii, and 1 compound was ≥10 fold less effective against T. gondii. ELQs that were less effective were the 6-chloro, 7-methoxy compounds, 4A, 4B and 4C, and the DAE compounds, 8B and 9B. The decreased activity of the 6-chloro, 7-methoxy compounds 4A, 4B and 4C is primarily due to a substantial loss in efficacy against T. gondii rather than an increase in potency against P. falciparum. The 26L residue in the N-terminal region of T. gondii cyt b (Tg cyt b) Qi site is adjacent to alpha helix A. The homologous position in P. falciparum cyt b (Pf cyt b) is an I22L amino acid substitution that causes resistance to 4A, 4B and 4C in P. falciparum. Compounds 8B and 9B are Qo active compounds with a DAE side chain combined with a 5,7- or 5,6,7-fluorine substituted quinolone core. Each of these drugs maintains activity against P. falciparum but loses activity against T. gondii when the simple carbon side chain is changed to the DAE as mentioned above.
Inherent structural variations in the parasite cyt b are likely to influence drug susceptibility between parasites. Tg cyt b and Pf cyt b have 51% identity and 74% similarity. This degree of homology is also found in the Qi site, which includes part of the A loop and helix as well as parts of the D and E helices and the joining D-E loop. By comparison, cyt b identity and similarity with the human cyt b are approximately 40% and 65%, respectively. Out of the 9 residues that have been shown to influence ubiquinone binding in crystal structures with the bovine cyt b, 3 residues differ between T. gondii and P. falciparum (Figure 3a.)2, 21 The residues involved in pyridone binding to a B. taurus cyt b are conserved between parasites.2
Figure 3. Cytochrome b (cyt b) structure and sequence alignment.

Figure 3a. The Qo site of S. cerevisiae cyt b (pdb 4pd4) with amino acid residues that interact with atovaquone binding are shown in blue and yellow.1 The 279 tyrosine and methionine 295 residues are the S. cerevisiae residues that are analogous to the P. falciparum 268 and 289 residues, respectively. Figure 3b. The Qi site of B. taurus cyt b (pdb 4d6t) with amino acid residues that interact with ubiquinone are shown in blue.2 The 27 isoleucine of B. taurus is analogous to leucine and isoleucine of T. gondii and P. falciparum, respectively. Amino acid residues that confer resistance and are discussed in the text are shown in yellow. Cyt b sequences of relevant organisms were aligned using MUSCLE.3 Amino acids in the sequence alignment that are identical between T. gondii and P. falciparum, and the amino acids of the reference structures that are identical to either T. gondii or P. falciparum are highlighted in light blue. Molecular graphics images were produced using the UCSF Chimera package from the Computer Graphics Laboratory, University of California, San Francisco (supported by NIH P41 RR-01081).6 IMS: intermembrane space, IM: inner membrane. ◆ = blue residues and ★ = yellow residues in cyt b structure.
The Qo pocket is created by helices C, cd1, D, ef and F, as well as the Rieske protein and ef loop that contains the highly conserved PEWY motif. The Tg cyt b and Pf cyt b have greater than 60% identity and 80% similarity in the Qo region. Of the 15 cyt b residues that contact ATV in a Saccharomyces cerevisiae-ATV co-crystallization, 3 residues vary between Tg cyt b and Pf cyt b (Figure 3b.) Tg cyt b Y272, F289 and M290 are F, V and L in PfCYT B, and are in proximity to each other on the ef and F helices as well as the ATV tail group. A V284F substitution in Plasmodium, which corresponds to F289 in Tg cyt b, has previously been shown to cause ATV resistance.23–24 Similar to the I22L variation between Tg cyt b and Pf cyt b, the naturally occurring F284 variation in T. gondii may contribute to decreased susceptibility to ATV, as well as 8B and 9B.
Effect of cyt b Mutations in P. falciparum and T. gondii on ELQ Potency:
Compounds were tested against an RH strain of T. gondii with a wild type cyt b and a strain with a T222P amino acid substitution in the cyt b Qi site that was generated by chemical mutagenesis and selection with compound 3B (ELQ-316).17 Drug-resistant P. falciparum strains included a clone with an I22L substitution that was generated through exposure to compound 4B (ELQ-300) and the ATV resistant strain Tm90-C2B (Table 2A & 2B.)18 The EC50s of these strains were compared to the P. falciparum Dd2 strain, the parental strain of the I22L strain (Figure 2b.) The EC50s of 1–9 A and E against P. falciparum were previously reported, with the exception of 3A, 4A and 7B.18
Other than compound 2A and 8E, no compounds were greater than 2-fold resistant to both the Qo site variants and the Qi site mutations. The presence of a 7-methoxy substituent on 2, 3 and 4 was associated with increased EC50s against the T. gondii T222P strain. The larger side chains, E and B, augmented the degree of resistance in these compounds. Compounds 3 and 4, but not 2 had cross-resistance between TgT222P and PfI22L, indicating that the presence of a substituent at position 6 is affected by changes in the N-terminal region of the Qi site. Increasing the size of the side chain had a less pronounced influence on resistance to PfI22L. Unlike the Qi site mutations, the PfY268S mutation in the Qo site causes greater resistance for the smaller, less rigid, C7 side chain. Interestingly, compounds 1A-E with unsubstituted benzene rings were minimally affected by all 3 resistance mutations.
In Vivo Efficacy of ELQs against T. gondii:
Compound 8B was selected for in vivo evaluation in a model of acute fatal toxoplasmosis based on evidence of Qo activity, predicted bioavailability, and metabolic stability. One day after inoculation with a virulent RH strain of T. gondii that expresses luciferase, compounds were administered at a dose of 5 mg/kg orally to mice for 5 days. Luminescence was assessed 4, 6, 13 and 29 days after T. gondii infection. Mice that demonstrated lethargy or lack of grooming were euthanized prior to the completion of the experiment, and T. gondii DNA was quantified using real time PCR. Mice that received vehicle only were euthanized at days 4 and 6 as they displayed signs of fulminant infection (Figure 4.) Mice that received ATV and 3B were euthanized on day 12 and 13. Mice treated with 3B were euthanized because they were ill appearing but 3 of 4 mice did not have luminescence. All 4 mice treated with 3B had evidence of brain infection by PCR that was similar to controls prior to euthanasia, but no residual spleen infection. By comparison, all mice in the ATV group developed both systemic and brain infection following treatment as was evident by luminescence and PCR. Previously, mice have been treated intraperitoneally with 25 mg/kg/d of compound 3B (ELQ-316) for 16 days and in prodrug form at 10 mg/kg/d for 7 days and observed for more than 5 weeks without developing observable signs of toxicity making it highly unlikely that toxicity contributed to the decreased survival of the 3B treated mice.14, 19 All 4 mice treated with compound 8B survived until day 32 and T. gondii DNA was not detected in their spleen or brain tissue. These results indicate that compounds 3B and 8B are remarkably effective at 5 mg/kg, yet they may differ in tissue distribution or ability to prevent T. gondii from entering the brain.
Figure 4. In vivo efficacy of oral administration of 5mg/kg atovaquone, ELQ-316 (3B) and ELQ-400 (8B.).
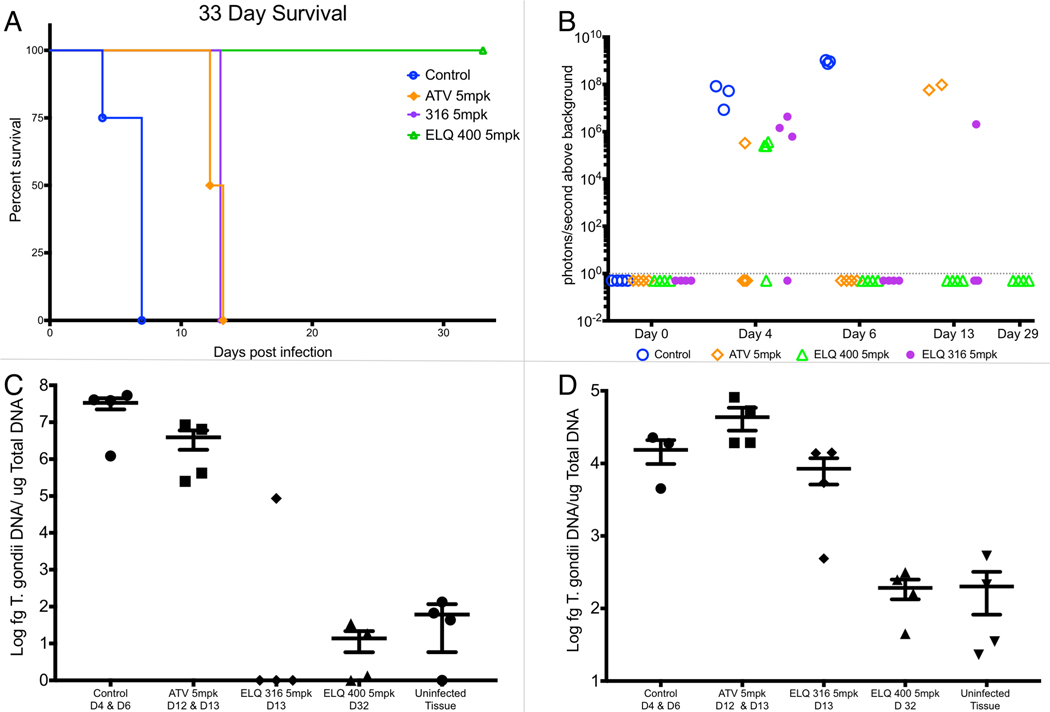
Figure 4A. Survival of treatment groups compared to vehicle-treated controls. All 4 mice treated with ELQ-400 (8B) were alive at the completion of the experiment. The difference in survival between the ELQ-400 (8B) group and control group, ELQ-316 (3B) group and control group and ELQ-316 group and ELQ-400 group were statistically significant (p = .01 by logrank analysis). Figure 4B. Luminescence in mice measured during the experiment. At day 4, luminescence from infection was detectable in each treatment group, but was below the limits of detection at day 6 in treatment groups. At day 13, luminescence was detectable in the 2 surviving mice in the atovaquone group and 1 of 3 mice in the 3B group; however, the remaining 3 mice displayed signs of infection and were euthanized. The dotted line indicates the limits of detection. Figure 4C. Quantitative real-time PCR of T. gondii DNA from splenic tissue. T. gondii DNA was not detected above the limits of detection in the spleen of 3 of 4 mice treated with ELQ-316 and none of the mice treated with ELQ-400. The differences between the ELQ-400 group and control group, and the ELQ-316 group and the control group were statistically significant (p = .02). Figure 4D. Quantitative real-time PCR of T. gondii DNA from brain tissue. All mice treated with ELQ-316 had T. gondii infection in brain tissue, whereas mice treated with ELQ-400 (8B) did not have infection detectable by PCR. The difference between ELQ-400 (8B) and control was statistically significant (p=.02 Student’s t-test). Quantitative real time PCR values from ELQ-400 treated mice were not statistically different from the values from uninfected tissue. D: day, bars and error bars represent the mean and standard error of the mean, respectively.
X-Ray Crystallography of 8B (ELQ-400):
The molecular structure and a fragment of the single-crystal lattice structure of 8B are shown in the Supporting Information. The molecule consists of a central aromatic quinolone ring with a DAE side chain at the 3-position. The central N(1)-C(9) of the quinolone ring is planar within 0.007 Å. The first phenyl ring of the DAE C(11)-C(16) is rotated around the central part with a dihedral angle between the average plane of these 2 rings of 87.75 (5)°. The second phenyl ring C(17)-C(22) of the DAE is rotated by 80.58 (6)° in opposite direction. Thus the dihedral angles between the average planes of the central quinolone fragment and the terminal phenyl ring C(17)-C(22) of the DAE is 9.60 (2)°. The bond distances found for 8B are close to standard values for corresponding bonds. The molecules in the crystal structures form a one-dimensional chain via bifurcated N-H…O, C-O---H and C-H…F hydrogen bonds as shown in the Supporting Information.
Discussion:
The antimalarial drugs, pyrimethamine and ATV, have been effective treatments for toxoplasmosis due to biological similarities between T. gondii and P. falciparum. The in vitro results that we report here demonstrate that despite a high degree of homology between the T. gondii and P. falciparum cyt bc1, ELQs may be more potent in vitro against T. gondii than P. falciparum. The lower EC50s of ELQs against T. gondii compared to P. falciparum may reflect differences in biologic sensitivity to cytochrome bc1 inhibition, structural differences between T. gondii and P. falciparum cyt bc1, additional targets in T. gondii or differences in the characteristics of the assays. The T. gondii inhibition assay used in these experiments measures color change caused by β-galactosidase expressed by T. gondii whereas the P. falciparum inhibition assay measures a fluorescent dye bound to parasite DNA. These assay differences may account for the differences in magnitude of EC50s between parasites; however, EC50s were not uniformly lower in T. gondii, which indicates that assay characteristics do not account for all differences in EC50s. If T. gondii does have greater sensitivity to cyt bc1 inhibition, it may be due T. gondii’s use of cyt bc1 for oxidative phosphorylation, while P. falciparum primarily relies on cyt bc1 for pyrimidine biosynthesis during its erythrocytic cycle.12 It is also possible that T. gondii readily ceases replication and converts to a bradyzoite form as a result of cyt bc1 inhibition, as previously observed with ATV and other respiratory inhibitors, whereas the erythrocytic stages of P. falciparum are not known to convert to a metabolically quiescent form.25 That being said, a greater sensitivity to cyt bc1 inhibition would not fully account for ATV and ELQs that are more potent against P. falciparum. Bidirectional differences in EC50s suggest that parasite specific structural features of cyt b contribute to susceptibility to this series of ELQs.
The I22L and V284F drug resistance mutations in Plasmodium that occur as natural variants in T. gondii are possible structural determinants of drug susceptibility. It is likely that the leucine at position 22, either directly or indirectly by shifting the position of adjacent amino acids, sterically interferes with the chlorine at position 6 compared to the smaller fluorine of compounds in the 3 series or the hydrogen of the 1 and 2 series. Of note, the L22 variant is also present in B. microti, which was less susceptible to compound 4B compared to 3B and 1B.19 The V284F mutation has been associated with ATV resistance in Plasmodium, and like the I22L mutation, F284 is a natural variant in T. gondii. Furthermore, a recent model of ELQ-400 docked in the S. cerevisiae cyt b shows the DAE side chain is in proximity to the analogous position, and an S. cerevisiae cyt b mutant with multiple mutations including a substitution to V284 increased susceptibility to ELQ-400.26 That being said, these variations are subtle and there are likely other significant structural differences in the Qo and Qi sites that contribute to parasite specificity. As a practical matter, the structure-activity relationship observed in these in vitro efficacy experiments provides a basis for further design of T. gondii cyt bc1 inhibitors.
Numerous cyt bc1 structures from vertebrates, yeast and bacteria have been cocrystallized with substrates and inhibitors, providing tremendous insight into cyt bc1 structure and function.1–2, 21 However, these models are limited when evaluating compounds that are specific for the apicomplexan cyt bc1 and do not inhibit the cyt bc1 of the model organisms. Such is the case with the Qi targeting ELQs, 2B, 3B and 4B (previously reported as ELQs 298, 316 and 300,) that do not inhibit human or yeast cyt bc1. These experiments show that a T222P substitution in T. gondii causes resistance to these and similar compounds that possess a methoxy at position 7. In organisms other than apicomplexans, the 222 residue is a lysine that binds to ubiquinone via a water molecule with D223, and mutations in the homologous S. cerevisiae residue decreases inhibition of 1-hydroxy-2-dodecyl-4(1H)quinolone (HDQ.)27 The adjacent aspartate at position 223 is present in all cyt b. This residue in B. taurus has been shown in binding proximity of the quinolone head group carbonyl with pyridones.2 The T222P substitution likely shifts the interaction of the quinolone head group with the conserved aspartate so that the 7-position methoxy hinders binding in the Qi pocket given that unsubstituted compounds 1A-D were not affected to the same extent. Considering that compound 1B, (previously reported as ELQ-271,) has activity against the S. cerevisiae and the human cyt bc1 whereas compounds 2B, 3B and 4B do not, the presence of a substituent at the 7-position is an important feature to limit Qi inhibitor host toxicity in this series of quinolones.13–14
Compounds 8A and 8C stand out as very potent T. gondii inhibitors with EC50s of less than 10 pM. This degree of in vitro efficacy is informative in understanding the structure activity relationship of apicomplexan cyt bc1 inhibition, but these compounds have poor oral bioavailability and metabolic stability.28 The addition of the DAE side chain improved both oral bioavailability and metabolic stability during optimization of ELQs for malaria.13 A previous crystal structure of compound 8A revealed a highly planar structure that is prone to π-π stacking of the central quinolone ring and intermolecular hydrogen bonding.13 By comparison, the crystal structure of compound 8B (ELQ-400) demonstrates that planarity between the central quinolone ring and the DAE ring is disrupted by the 87.75° orientation between the central ring and the first phenyl of the DAE. The different aryl rings of 8B observed in the X-ray crystallography are positioned to minimize the internal steric hindrance between the different substituents (Figure S2.) The X-ray structure of 8B reveals that the DAE side chain results in less crystallinity and thus greater solubility in PEG-400.
The observation that the DAE side chain reduces in vitro efficacy for Qo site inhibitors but has little effect on Qi site inhibitors indicates that there is potential for further optimization of the Qo site inhibitor side chain for T. gondii. Based on additional decreased activity from the addition of phenyl-methoxy or biphenyl side chains, it appears that flexibility and decreased bulk favor binding or access to the Qo pocket. The remarkable potency of the Qo targeting compounds also demonstrates that cyt bc1 inhibition is a highly effective mechanism of stopping T. gondii proliferation. This observation raises interesting questions of whether inhibition of the Qo site more effectively inhibits the cyt bc1, do 8A and 8C reach higher concentrations in the mitochondrial membrane, or do they target additional ubiquinone binding enzymes in the T. gondii electron transport chain? Compound 8B (ELQ-400) has recently been shown to inhibit both the Qo and Qi site of the S. cerevisiae cyt b raising the possibility that 8A and 8C may also act similarly.26 Other ubiquinone binding enzymes that are known T. gondii drug targets include NADH dehydrogenase and dihydroorotate dehydrogenase.29–30 Inhibition of these enzymes by HDQ, a compound that also has been shown to inhibit cyt bc1, raises the possibility that ELQs may interact with multiple enzymes.27 HDQ and its derivatives primarily differ from ELQs by the hydroxylation of the nitrogen and the side chain placement at the 2 position (Figure 1). The marked resistance conferred by Y268S in P. falciparum to compounds 8A and 8C (Figure 2b) suggests that cyt bc1 is the primary target but does not rule out the possibility of other targets for which inhibition may be additive or synergistic with cyt bc1. In addition to these known drug targets, T. gondii expresses succinate dehydrogenase, malate:quinone oxidoreductase and glycerol phosphate dehydrogenase.30 While the possibility of additional ELQ targets is interesting, it should be noted that ELQ resistant parasites obtained in P. falciparum, B. microti and T. gondii by either ELQ selection or mutagenesis plus ELQ selection have all had cyt b mutations.17–19, 31 Accordingly, rational drug design based on cyt b will likely yield the best results.
Despite the decrease in in vitro efficacy for Qo inhibitors, the DAE side chain has enhanced the oral bioavailability and metabolic stability of ELQs to the extent that they are effective in vivo.13–14, 19, 31 Compound 1B and 3B have previously been shown to be considerably more effective than ATV against babesiosis and acute and latent toxoplasmosis. We chose to evaluate compound 8B against toxoplasmosis based on its previous efficacy in a rodent model of malaria.31 At 5 mg/kg, compound 8B decreased acute infection with a virulent type I strain of Toxoplasma to the extent that all mice survived and had no evidence of infection. The strain used in these experiments has previously been shown to be fatal between 8 and 15 days at a low inoculum of 10 parasites.32 The lack of detection of T. gondii in tissue indicates that infection reached a low enough level that it was either immunologically controlled below the limits of detection or that 8B eliminated infection.
Comparatively, the decrease in infection followed by relapse in mice treated with ATV and 3B demonstrates an incomplete clearance of parasites from tissues outside of the brain. Importantly, compound 3B appeared to eliminate infection outside of the brain in 3 out of 4 mice as evidenced by a lack of luminescence and T. gondii in the spleen. Previous in vivo experiments with 3B are consistent with these results in that 3B was superior to ATV in acute infection when administered orally.14 At that time, efficacy against brain tissue cysts was determined in an experiment in which compounds were administered intraperitoneally. Compound 3B and 8B are likely to have similar oral bioavailability based on low aqueous solubility and high crystallinity; however, these experiments indicate that the blood brain barrier penetration and half-life of 3B and 8B may be significantly different. It is also possible that compound 8B inhibits infection more rapidly and prevents T. gondii from reaching the brain; however, luminescence was similar between 3B and 8B at 4 days (Figure 4b) and 8B is less potent than 3B in vitro.
The in vivo efficacy of compound 8B against Plasmodium and Neospora caninum has previously been published as ELQ-400 and MMV671636, respectively.31 For Plasmodium, 8B was curative with a single dose of 1 mg/kg in a mouse model of malaria, and for N. caninum a 10mg/kg dose was effective in reducing infection.33 Pharmacokinetic analysis of MMV671636 administered orally at 5 mk/kg in rats showed modest drug exposure with a maximum concentration of 71.2 ng/ml and an area under the curve (AUClast) of 1090 ng·h/ml.34 Like ATV, 8B has activity against the isolated human cyt bc1; however, in general cell assays for toxicity and in vivo studies for efficacy, toxicity was not observed.31, 33,34 Further in vitro and in vivo toxicity studies will be necessary to determine the safety of 8B. Similar to 4B, 8B is amenable to formulation as carbonate ester or alkoxy carbonate ester prodrugs, which offers a means to overcome the limited oral bioavailability of 8B.35–36
ELQs 3B and 8B are promising compounds that may provide safer more effective treatment for toxoplasmosis or malaria. The cyt bc1 is a sensitive drug target with significant structural differences among apicomplexan parasites that should be considered in designing an antiprotozoal drug with broad activity and limited host toxicity. Analysis of side chain variations coupled with the Q-site targeting substituents of the benzenoid ring reveals key structural side chain characteristics specific to each site. This understanding provides a means for further preclinical development of ELQs as well as the design of more effective cyt bc1 inhibitors.
Methods:
Chemicals and Cells:
ELQs were synthesized as previously described and below, identified by proton nuclear magnetic resonance (1H-NMR), and determined to be >95% pure by reversed-phase high-performance liquid chromatography (HPLC). Atovaquone was obtained from Sigma-Aldrich. Human foreskin fibroblast cells were obtained from the American Type Culture Collection (ATCC). The Type I T. gondii RH strain expressing luciferase and green fluorescence protein (gfp) were kindly provided by Vern Carruthers. The Type I T. gondii RH strain expressing gfp and β-galactosidase was kindly provided by Marilyn Parsons.
Generation of ELQ-316 Resistant T. gondii Expressing Green-Fluorescent Protein and β-Galactosidase:
A clonal strain of ELQ-316 resistant T. gondii that contains a Thr222Pro substitution was created as previously described.17 To adapt this strain for high throughput drug testing, the strain was transfected via electroporation with a DNA cassette containing green fluorescent protein (mGFP5) controlled by the T. gondii GRA2 promoter and a β-galactosidase gene controlled by an α-tubulin promoter.37 Fluorescent plaques were carefully selected from T-25 flasks using an inverted fluorescent microscope at 40x and a 30-gauge needle and syringe and inoculated into a T25 flask containing HFF (human foreskin fibroblast) monolayer. T. gondii expressing gfp and β-galactosidase were then cloned by limiting dilution to one T. gondii tachyzoite per well in a 96-well plate.
In Vitro Potency Assay of ELQs against T. gondii:
Compounds were dissolved in DMSO and diluted across 96-well plates containing human fibroblasts with a final maximum concentration of 0.5% DMSO. Wild type and Thr222Pro RH strain T. gondii expressing β-galactosidase were added to the 96 well plates at 4000 tachyzoites per well and incubated at 37°C and 5% CO2 for 72 hours. T. gondii were quantified by spectrophotometrically measuring the β-galactosidase concentration of each well after cell lysis and exposure to chlorophenol red β-galactopyranose (Sigma-Aldrich, St. Louis, MO) substrate for 4 hours. Each compound was measured at least three times in quadruplicate.
In Vitro Potency Assay of ELQs against P. falciparum:
Experiments were carried out as previously described in triplicate with 2-fold dilutions of each compound across 96 well plates in a total volume of 100 μL and a final RBC concentration of 2% (v/v) and an initial percentage of parasitized RBCs of 0.2%.22 Plates were incubated in chambers for 72 h at 37°C in an atmosphere of 5% CO2, 5% O2, and 90% N2. After this period, a SYBR Green I dye−detergent mixture was added, and after one hour at room temperature in the dark, fluorescence was quantified in a fluorescence plate reader.
Human Cell Toxicity Assay:
ELQ’s, ATV and the positive control mefloquine were tested in triplicate against HFF cells with CellTiter-Glo Kit 2.0 (Promega). 96-well plates containing confluent monolayers of HFF cells in DMEM with 10% FBS media were treated with compounds dissolved in DMSO starting at 20μM and diluted 2-fold across the plate. Plates were incubated for 72 h at 37°C and 5% CO2, and cell viability was quantified by adding 50μl of Promega CellTiter-Glo to each well. After incubating for 30 min at room temperature, luminescence was measured using GloMax®-Multi Detection System. Each compound was measured in 3 independent assays.
In Vivo Efficacy of ELQ-400 against Acute Toxoplasmosis:
CF-1 mice that were 4–6 weeks old were inoculated intraperitoneally with 10,000 virulent Type I T. gondii tachyzoites that express firefly luciferase and gfp.32 After 24 hours, compounds were dissolved and administered in PEG400 via oral gavage daily for five days. Vehicle only control groups and treatment groups consisted of 4 mice per group. Mice were monitored for signs of infection, and underwent bioluminescence imaging on day 4, 6, 13 and 29. Mice were injected i.p. with a dose of 0.1ml of D-luciferin (150mg substrate/kg of body weight) dissolved in PBS. Three minutes after luciferin injection, mice were anesthetized using inhaled isoflurane and positioned ventral side up on a heated platform. Bioluminescent images were obtained using an IVIS Spectrum CT and processed using Living Image software (Perkin Elmer.) Mice that developed signs of severe infection, such as >10% weight loss, lethargy, or lack of self-grooming, or at 33 days, were humanely euthanized and the brain and spleen were collected for quantitative real-time PCR using methods that we have previously published.38 In brief, brain and spleen were collected from infected and non-infected mice and homogenized in PBS using a handheld homogenizer. DNA was isolated with a DNA purification kit (Qiagen, Germantown, MD). 300 ng of total DNA from the brain homogenate and 300 ng of total DNA from the spleen homogenate were analyzed per mouse. A standard curve was generated from DNA purified from T. gondii tachyzoites in 10-fold dilutions from 160 ng to 1.6 fg of DNA. Quantitative real-time PCR was performed in duplicate using a 7300 real-time PCR System (Applied Biosystems, Grand Island, NY) with iTaq SYBR GREEN PCR Supermix (Biorad) and primers for the T. gondii 529-bp repeat element, (sense 5’-AGG AGA GAT ATC AGG ACT GTA G-3’ and anti-sense 5’-GCG TCG TCT CGT CTA GAT CG-3’.)39 Results were quantified as T. gondii DNA per total DNA. Analysis of survival curves and differences of the tissue burden of T. gondii infection were performed using a logrank test and unpaired t-test, respectively. GraphPad Prism 7.0 software was used for statistical analysis. The Institutional Animal Care and Use Committee of the Portland Veterans Administration Medical Center approved this protocol #3276–16.
X-ray Crystallography of 8B (ELQ-400):
Diffraction intensities for 8B were collected at 173 K on a Bruker Apex2 CCD diffractometer using CuKa radiation, l= 1.54178 Å. Space group was determined and assigned based on intensity statistics. Absorption correction was applied by SADABS (Siemens Area Detector Absorption Correction Program.) The structure was solved by direct methods and Fourier techniques and refined on F 2 using full matrix least-squares procedures. All non-H atoms were refined with anisotropic thermal parameters. H atoms were found on the residual density map and refined with isotropic thermal parameters. All calculations were performed by the Bruker SHELXL-2013 package.40
X-ray Crystallography of 8B (ELQ-400) Crystals:
C23 H14 F5 N1 O3, M = 447.35, 0.12 × 0.11 × 0.05 mm, T = 173(2) K, Triclinic, space group P-1, a = 6.7167(4) Å, b = 6.9590(4) Å, c = 20.2321(10) Å, α = 96.770(2)°, β = 91.663(2)°, γ = 91.295(2)°, V = 938.37(9) Å3, Z = 2, D c = 1.583 Mg/m3, μ(Cu) = 1.205 mm−1, F(000) = 456, 2θ max = 134.73°, 12289 reflections, 3328 independent reflections [R int = 0.0447], R1 = 0.0434, wR2 = 0.1225 and GOF = 1.039 for 3328 reflections (345 parameters) with I> 2s(I), R1 = 0.0470, wR2 = 0.1266 and GOF = 1.039 for all reflections, max/min residual electron density +0.429/- 0.499 eÅ −3. The data collection and X-ray analysis are given in the supplemental materials.
Chemical Synthesis:
The synthesis of compounds used in this study have been previously reported except for 1D, 1E, 2C, 3A, 3C, 3D, 3E, 4C, 5A, 5B, 7B, 8C, 8D, 8E and 9A.9,10,14,16,24 The new compounds were synthesized according to published procedures.9,24 All compounds were > 95% pure and their structures were determined by NMR. 1H-NMR spectra were obtained using a Bruker AMX-400 NMR spectrometer operating at 400.14 MHz in DMSO D6. The NMR raw data were analyzed using the iNMR Spectrum Analyst software. 1H chemical shifts were reported in parts million units (ppm), (δ) relative the residual proton at 2.54 ppm in the deuterated DMSO D6. J coupling constants values are in hertz (Hz). Decoupled 19F operating at 376 MHz was also obtained for compounds containing fluorine elements. The 1H-NMR spectra are reported in this manuscript.
NMR of 1D: δ 11.72 (d, J = 0.2 Hz, 1H), 8.08 (d, J = 7.6 Hz, 1H), 7.65 (t, J = 7.3 Hz, 1H), 7.55 (d, J = 8.3 Hz, 1H), 7.41–7.36 (m, 4H), 7.31 (t, J = 7.4 Hz, 1H), 2.25 (s, 3H).
NMR of 1E: δ 11.68 (s, 1H), 8.10 (d, J = 8 Hz, 1H), 7.73–7.63 (m, 5H), 7.56–7.47 (m, 3H), 7.40–7.28 (m, 4H), 2.29 (s, 3H).
NMR of 2C: δ 11.2 (s,1H), 6.8 (m,2H), 7.9 (m, 1H), 3.82 (s, 3H), 2.33 (s, 3H), 2.4 (m, 2H), 1.2–1.4 (m, 18H), 0.85 (t, J = 6.8 Hz, 3H).
NMR of 3A: δ 11.34 (s, 1H), 7.65 (d, J = 11.9 Hz, 1H), 7.02 (d, J = 7.3 Hz, 1H), 3.92 (s, 3H), 2.44 (t, J = 7.2 Hz, 2H), 2.35 (s, 3H), 1.36–1.25 (m, 9H), 0.85 (t, J = 6.3 Hz, 3H).
NMR of 3C: Not sufficiently soluble in DMSO to obtain NMR spectrum resolution. The compound showed 1 spot on TLC.
NMR of 3D: δ 11.78 (s, 1H), 7.72 (d, J = 11.7 Hz, 1H), 7.38 (s, 4H), 7.13 (d, J = 7.3 Hz, 1H), 3.97 (s, 3H), 2.23 (s, 3H).
NMR of 3E: δ 12.52 (s, 1H), 7.87 (d, J = 11.7 Hz, 1H), 7.73 (d, J = 7.7 Hz, 4H), 7.50 (t, J = 7.3 Hz, 2H), 7.39 (t, J = 7.0 Hz, 3H), 7.26 (d, J = 7.1 Hz, 1H), 4.00 (s, 3H), 2.34 (s, 3H).
NMR of 4C: δ 11.4 (s, 1H), 7.96 (s, 1H), 6.99 (s, 1H), 2.35 (s, 3H), 2.42 (m, 2H), 1.2–1.4 (m, 18H), 0.85 (t, J = 7.9 Hz, 3H, ).
NMR of 5A: δ 11.38 (s, 1H), 7.53–7.47 (m, 1H), 7.26–7.23 (m, 1H), 6.91–6.85 (m, 1H), 2.41 (t, J = 6.6 Hz, 2H), 2.34 (s, 3H), 1.39–1.26 (m, 10H), 0.86 (t, J = 5.7 Hz, 3H).
NMR of 5B: δ 11.78 (s, 1H), 7.59 (td, J = 8.1, 5.2 Hz, 1H), 7.42 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 8.4 Hz, 1H), 7.28 (d, J = 8.5 Hz, 2H), 7.17 (d, J = 6.3 Hz, 2H), 7.08 (d, J = 8.5 Hz, 2H), 6.97 (dd, J = 11.9, 8.0 Hz, 1H), 2.22 (s, 3H).
NMR of 7B: δ 11.74 (s, 1H), 8.14 (dd, J = 8.8, 6.5 Hz, 1H), 7.42 (d, J = 8.8 Hz, 2H), 7.30–7.24 (m, 3H), 7.18–7.13 (m, 3H), 7.08 (d, J = 8.5 Hz, 2H), 2.26 (s, 3H).
NMR of 8C: δ 7.01 (ddd, J=12.0, 9.0, 3.0 Hz, 1H), (ddd, J=1.6, 9.6, 3 Hz, 1H), 2.74 (s, 3H), 2.93 (m, 2H), 1.2–1.6 (m, 18H), 0.88 (t, J = 7 Hz, 3H).
NMR of 8D: δ 11.81 (s, 1H), 7.39–7.34 (m, 4H), 7.10–7.02 (m, 2H), 2.18 (s, 3H).
NMR of 8E: δ 11.76 (s, 1H), 7.71 (dd, J = 12 , 8 Hz, 4H), 7.49 (t, J = 8 Hz, 2H), 7.38 (t, J = 8 Hz, 1H), 7.32 (d, J = 8 Hz, 2H), 7.10–7.01 (m, 2H), 2.22 (s, 3H).
NMR of 9A: δ 11.8 (s, 1H), 7.21 (ddd, J =11.0, 6.5, 1.9 Hz, 1H), 2.32 (s, 3H), 2.4 (m, 2H), 1.2–1.4 (m, 10H), 0.85 (t, J = 6.9 Hz, 3H).
Supplementary Material
Figure 1. X-Ray crystallography of 8B (ELQ-400)
Figure 2. X-ray crystallography of 8B (ELQ-400) crystals
Acknowledgements:
We would like to acknowledge Dr. Gustavo Arrizabalaga for providing the plasmid containing β-galactosidase and green fluorescent protein. This work was supported by Career Development Award BX002440 to J. S. D. from the U.S. Department of Veterans Affairs Biomedical Laboratory Research and Development. We also acknowledge support for M. K. R. from NIH R01 AI100569, Peer Reviewed Medical Research Program Project PR130649, and VA Merit Review Funds from the U.S. Department of Veterans Affairs BX003312.
Abbreviations:
- cyt bc1
cytochrome bc1
- ATV
atovaquone
- ELQ
endochin-like quinolones
- cyt b
cytochrome b
- DAE
diarylether
- EC50
50% effective concentration
- WT
wild type
- Tg cyt b
T. gondii cyt b
- Pf cyt b
Pf cytochrome b
- TgT222P
T. gondii T222P
- PfI22L
P. falciparum I22L
- PfY268S
P. falciparum Y268S
- HDQ
1-hydroxy-2-dodecyl-4(1H)quinolone
- AUC
area under the curve
- 1H-NMR
proton nuclear magnetic resonance
- HPLC
high-performance liquid chromatography
- gfp
green fluorescent protein
- ATCC
American Type Culture Collection
- HFF
human foreskin fibroblast
- RBC
red blood cell
- FBS
fetal bovine serum
- DMEM
Dulbecco’s modified Eagle’s medium
- i.p.
intraperitoneally
- PBS
phosphate buffered saline
Footnotes
Conflict of Interest Statement:
The authors declare no competing financial interest.
References:
- 1.Birth D; Kao WC; Hunte C, Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat Commun 2014, 5, 4029 DOI: 10.1038/ncomms5029. [DOI] [PubMed] [Google Scholar]
- 2.Capper MJ; O’Neill PM; Fisher N; Strange RW; Moss D; Ward SA; Berry NG; Lawrenson AS; Hasnain SS; Biagini GA; Antonyuk SV, Antimalarial 4(1H)-pyridones bind to the Qi site of cytochrome bc1. Proc Natl Acad Sci U S A 2015, 112 (3), 755–60. DOI: 10.1073/pnas.1416611112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Edgar RC, MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004, 32 (5), 1792–7. DOI: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yolken R; Torrey EF; Dickerson F, Evidence of increased exposure to Toxoplasma gondii in individuals with recent onset psychosis but not with established schizophrenia. PLoS Negl Trop Dis 2017, 11 (11), e0006040. DOI: 10.1371/journal.pntd.0006040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bharti AR; McCutchan A; Deutsch R; Smith DM; Ellis RJ; Cherner M; Woods SP; Heaton RK; Grant I; Letendre SL, Latent Toxoplasma Infection and Higher Toxoplasma gondii Immunoglobulin G Levels Are Associated With Worse Neurocognitive Functioning in HIV-Infected Adults. Clin Infect Dis 2016, 63 (12), 1655–1660. DOI: 10.1093/cid/ciw655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004, 25 (13), 1605–12. DOI: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 7.Jones JL; Parise ME; Fiore AE, Neglected parasitic infections in the United States: toxoplasmosis. Am J Trop Med Hyg 2014, 90 (5), 794–9. DOI: 10.4269/ajtmh.13-0722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katlama C; De Wit S; O’Doherty E; Van Glabeke M; Clumeck N, Pyrimethamine-clindamycin vs. pyrimethamine-sulfadiazine as acute and long-term therapy for toxoplasmic encephalitis in patients with AIDS. Clin Infect Dis 1996, 22 (2), 268–75. [DOI] [PubMed] [Google Scholar]
- 9.Porter SB; Sande MA, Toxoplasmosis of the central nervous system in the acquired immunodeficiency syndrome. N Engl J Med 1992, 327 (23), 1643–8. DOI: 10.1056/NEJM199212033272306. [DOI] [PubMed] [Google Scholar]
- 10.Dannemann B; McCutchan JA; Israelski D; Antoniskis D; Leport C; Luft B; Nussbaum J; Clumeck N; Morlat P; Chiu J; et al. , Treatment of toxoplasmic encephalitis in patients with AIDS. A randomized trial comparing pyrimethamine plus clindamycin to pyrimethamine plus sulfadiazine. The California Collaborative Treatment Group. Ann Intern Med 1992, 116 (1), 33–43. [DOI] [PubMed] [Google Scholar]
- 11.Lin D; Tucker MJ; Rieder MJ, Increased adverse drug reactions to antimicrobials and anticonvulsants in patients with HIV infection. Ann Pharmacother 2006, 40 (9), 1594–601. DOI: 10.1345/aph.1G525. [DOI] [PubMed] [Google Scholar]
- 12.Painter HJ; Morrisey JM; Mather MW; Vaidya AB, Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446 (7131), 88–91. DOI: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 13.Nilsen A; Miley GP; Forquer IP; Mather MW; Katneni K; Li Y; Pou S; Pershing AM; Stickles AM; Ryan E; Kelly JX; Doggett JS; White KL; Hinrichs DJ; Winter RW; Charman SA; Zakharov LN; Bathurst I; Burrows JN; Vaidya AB; Riscoe MK, Discovery, synthesis, and optimization of antimalarial 4(1H)-quinolone-3-diarylethers. J Med Chem 2014, 57 (9), 3818–34. DOI: 10.1021/jm500147k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doggett JS; Nilsen A; Forquer I; Wegmann KW; Jones-Brando L; Yolken RH; Bordon C; Charman SA; Katneni K; Schultz T; Burrows JN; Hinrichs DJ; Meunier B; Carruthers VB; Riscoe MK, Endochin-like quinolones are highly efficacious against acute and latent experimental toxoplasmosis. Proc Natl Acad Sci U S A 2012, 109 (39), 15936–41. DOI: 10.1073/pnas.1208069109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Montazeri M; Sharif M; Sarvi S; Mehrzadi S; Ahmadpour E; Daryani A , A Systematic Review of In vitro and In vivo Activities of Anti-Toxoplasma Drugs and Compounds (2006–2016). Front Microbiol 2017, 8, 25 DOI: 10.3389/fmicb.2017.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alday PH; Doggett JS, Drugs in development for toxoplasmosis: advances, challenges, and current status. Drug Des Devel Ther 2017, 11, 273–293. DOI: 10.2147/DDDT.S60973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alday PH; Bruzual I; Nilsen A; Pou S; Winter R; Ben Mamoun C.; Riscoe MK; Doggett JS, Genetic Evidence for Cytochrome b Qi Site Inhibition by 4(1H)-Quinolone-3-Diarylethers and Antimycin in Toxoplasma gondii. Antimicrob Agents Chemother 2017, 61 (2). DOI: 10.1128/AAC.01866-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stickles AM; de Almeida MJ; Morrisey JM; Sheridan KA; Forquer IP; Nilsen A; Winter RW; Burrows JN; Fidock DA; Vaidya AB; Riscoe MK, Subtle changes in endochin-like quinolone structure alter the site of inhibition within the cytochrome bc1 complex of Plasmodium falciparum. Antimicrob Agents Chemother 2015, 59 (4), 1977–82. DOI: 10.1128/AAC.04149-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawres LA; Garg A; Kumar V; Bruzual I; Forquer IP; Renard I; Virji AZ; Boulard P; Rodriguez EX; Allen AJ; Pou S; Wegmann KW; Winter RW; Nilsen A; Mao J; Preston DA; Belperron AA; Bockenstedt LK; Hinrichs DJ; Riscoe MK; Doggett JS; Ben Mamoun C., Radical cure of experimental babesiosis in immunodeficient mice using a combination of an endochin-like quinolone and atovaquone. J Exp Med 2016. DOI: 10.1084/jem.20151519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nilsen A; LaCrue AN; White KL; Forquer IP; Cross RM; Marfurt J; Mather MW; Delves MJ; Shackleford DM; Saenz FE; Morrisey JM; Steuten J; Mutka T; Li Y; Wirjanata G; Ryan E; Duffy S; Kelly JX; Sebayang BF; Zeeman AM; Noviyanti R; Sinden RE; Kocken CH; Price RN; Avery VM; Angulo-Barturen I; Jimenez-Diaz MB; Ferrer S; Herreros E; Sanz LM; Gamo FJ; Bathurst I; Burrows JN; Siegl P; Guy RK; Winter RW; Vaidya AB; Charman SA; Kyle DE; Manetsch R; Riscoe MK, Quinolone-3-diarylethers: a new class of antimalarial drug. Sci Transl Med 2013, 5 (177), 177ra37. DOI: 10.1126/scitranslmed.3005029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia D; Esser L; Tang WK; Zhou F; Zhou Y; Yu L; Yu CA, Structural analysis of cytochrome bc1 complexes: implications to the mechanism of function. Biochim Biophys Acta 2013, 1827 (11–12), 1278–94. DOI: 10.1016/j.bbabio.2012.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smilkstein M; Sriwilaijaroen N; Kelly JX; Wilairat P; Riscoe M, Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother 2004, 48 (5), 1803–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siregar JE; Kurisu G; Kobayashi T; Matsuzaki M; Sakamoto K; Mi-ichi F; Watanabe Y; Hirai M; Matsuoka H; Syafruddin D; Marzuki S; Kita K, Direct evidence for the atovaquone action on the Plasmodium cytochrome bc1 complex. Parasitol Int 2015, 64 (3), 295–300. DOI: 10.1016/j.parint.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 24.Syafruddin D; Siregar JE; Marzuki S, Mutations in the cytochrome b gene of Plasmodium berghei conferring resistance to atovaquone. Mol Biochem Parasitol 1999, 104 (2), 185–94. [DOI] [PubMed] [Google Scholar]
- 25.Tomavo S; Boothroyd JC, Interconnection between organellar functions, development and drug resistance in the protozoan parasite, Toxoplasma gondii. Int J Parasitol 1995, 25 (11), 1293–9. [DOI] [PubMed] [Google Scholar]
- 26.Song Z; Iorga BI; Mounkoro P; Fisher N; Meunier B, The antimalarial compound ELQ-400 is an unusual inhibitor of the bc1 complex, targeting both Qo and Qi sites. FEBS Lett 2018. DOI: 10.1002/1873-3468.13035. [DOI] [PubMed] [Google Scholar]
- 27.Vallieres C; Fisher N; Antoine T; Al-Helal M; Stocks P; Berry NG; Lawrenson AS; Ward SA; O’Neill PM; Biagini GA; Meunier B, HDQ, a potent inhibitor of Plasmodium falciparum proliferation, binds to the quinone reduction site of the cytochrome bc1 complex. Antimicrob Agents Chemother 2012, 56 (7), 3739–47. DOI: 10.1128/AAC.00486-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Winter R; Kelly JX; Smilkstein MJ; Hinrichs D; Koop DR; Riscoe MK, Optimization of endochin-like quinolones for antimalarial activity. Exp Parasitol 2011, 127 (2), 545–51. DOI: 10.1016/j.exppara.2010.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin SS; Kerscher S; Saleh A; Brandt U; Gross U; Bohne W, The Toxoplasma gondii type-II NADH dehydrogenase TgNDH2-I is inhibited by 1-hydroxy-2-alkyl-4(1H)quinolones. Biochim Biophys Acta 2008, 1777 (11), 1455–62. DOI: 10.1016/j.bbabio.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Hegewald J; Gross U; Bohne W, Identification of dihydroorotate dehydrogenase as a relevant drug target for 1-hydroxyquinolones in Toxoplasma gondii. Mol Biochem Parasitol 2013, 190 (1), 6–15. DOI: 10.1016/j.molbiopara.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 31.Stickles AM; Ting LM; Morrisey JM; Li Y; Mather MW; Meermeier E; Pershing AM; Forquer IP; Miley GP; Pou S; Winter RW; Hinrichs DJ; Kelly JX; Kim K; Vaidya AB; Riscoe MK; Nilsen A, Inhibition of cytochrome bc1 as a strategy for single-dose, multi-stage antimalarial therapy. Am J Trop Med Hyg 2015, 92 (6), 1195–201. DOI: 10.4269/ajtmh.14-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kafsack BF; Pena JD; Coppens I; Ravindran S; Boothroyd JC; Carruthers VB, Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science 2009, 323 (5913), 530–3. DOI: 10.1126/science.1165740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muller J; Aguado A; Laleu B; Balmer V; Ritler D; Hemphill A, In vitro screening of the open source Pathogen Box identifies novel compounds with profound activities against Neospora caninum. Int J Parasitol 2017, 47 (12), 801–809. DOI: 10.1016/j.ijpara.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Medicines For Malaria Venture https://www.pathogenbox.org (accessed March 1st).
- 35.Miley GP; Pou S; Winter R; Nilsen A; Li Y; Kelly JX; Stickles AM; Mather MW; Forquer IP; Pershing AM; White K; Shackleford D; Saunders J; Chen G; Ting LM; Kim K; Zakharov LN; Donini C; Burrows JN; Vaidya AB; Charman SA; Riscoe MK, ELQ-300 prodrugs for enhanced delivery and single-dose cure of malaria. Antimicrob Agents Chemother 2015, 59 (9), 5555–60. DOI: 10.1128/AAC.01183-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frueh L; Li Y; Mather MW; Li Q; Pou S; Nilsen A; Winter RW; Forquer IP; Pershing AM; Xie LH; Smilkstein MJ; Caridha D; Koop DR; Campbell RF; Sciotti RJ; Kreishman-Deitrick M; Kelly JX; Vesely B; Vaidya AB; Riscoe MK, Alkoxycarbonate Ester Prodrugs of Preclinical Drug Candidate ELQ-300 for Prophylaxis and Treatment of Malaria. ACS Infect Dis 2017, 3 (10), 728–735. DOI: 10.1021/acsinfecdis.7b00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Siemering KR; Golbik R; Sever R; Haseloff J, Mutations that suppress the thermosensitivity of green fluorescent protein. Curr Biol 1996, 6 (12), 1653–63. [DOI] [PubMed] [Google Scholar]
- 38.Vidadala RS; Rivas KL; Ojo KK; Hulverson MA; Zambriski JA; Bruzual I; Schultz TL; Huang W; Zhang Z; Scheele S; DeRocher AE; Choi R; Barrett LK; Siddaramaiah LK; Hol WG; Fan E; Merritt EA; Parsons M; Freiberg G; Marsh K; Kempf D; Carruthers VB; Isoherranen N; Doggett JS; Van Voorhis WC; Maly DJ, Development of an Orally Available and Central Nervous System (CNS)-Penetrant Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) Inhibitor with Minimal Human Ether-a-go-go-Related Gene (hERG) Activity for the Treatment of Toxoplasmosis. J Med Chem 2016. DOI: 10.1021/acs.jmedchem.6b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reischl U; Bretagne S; Kruger D; Ernault P; Costa JM, Comparison of two DNA targets for the diagnosis of Toxoplasmosis by real-time PCR using fluorescence resonance energy transfer hybridization probes. BMC Infect Dis 2003, 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheldrick GM, A short history of SHELX. Acta Crystallogr A 2008, 64 (Pt 1), 112–22. DOI: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1. X-Ray crystallography of 8B (ELQ-400)
Figure 2. X-ray crystallography of 8B (ELQ-400) crystals