

Abstract
Accurate modeling of ligand-binding site structures plays a critical role in structure-based virtual screening. However, the structures of ligand-binding site in most predicted protein models are generally of low quality and in need of refinements. In this work, we present a ligand-binding site structure refinement protocol using molecular dynamics simulation with restraints derived from predicted binding site templates. Our benchmark validation shows great performance when tested against 40 diverse set of proteins from the Astex list. The ligand-binding site on modeled protein structures are consistently refined using our method with an average Cα RMSD improvement of 0.90 Å. Comparison of ligand binding modes from ligand docking to initial unrefined and refined structures shows an average of 1.97 Å RMSD improvement in the refined structures. These results demonstrate a promising new method of structure refinement for protein ligand-binding site structures.
Keywords: Ligand-binding site, Structure refinement, Molecular dynamics, Binding modes, Docking
Graphical Abstract
Introduction
The number of protein structures available in the Protein Data Bank (PDB) has increased significantly over the past two decades, reaching over 155,000 structures as of September, 2019.1 Nonetheless, there are still far more protein sequences that lack structures due to experimental limitations.2–4 Computational tools for protein structure predictions play a key role in reducing the gap between available protein sequences and protein structures. Many prediction tools have shown steady progress in providing reliable structures through blind test CASP (critical assessment of structure prediction) contests over the past few years.5, 6 CASP12 reported a 20% increase in average precision of predicted structure and precisions of above 90% with targets that have many homologous sequences.7 On average, structural quality of template-based models tends to fall between 2–10 Å Cα RMSD (root-mean-square deviation) values and GDT-HA (global distance test-high accuracy) scores of 40 to 70 compared to experimental structures.6, 8 Although predicted protein models are useful in many aspects for structural biology studies, higher structural quality with more accurate ligand-binding site structures are needed for structure-based virtual screening (SBVS).9, 10 Previous studies reported that docking accuracy decreased significantly when the ligand-binding site backbone RMSD was greater than 2 Å compared to experimental structures.11–14 In addition, computationally predicted structures are apo proteins that could have significantly different ligand-binding site structures compared to holo proteins, e.g., occluded binding pockets in most predicted protein models could impede ligands from docking correctly.
Protein structure refinement methods are useful to improve the quality of predicted models toward experimental accuracy. A number of protocols that utilize physics-based MD (molecular dynamics) simulations have shown successful results with small but consistent improvements.8, 15–20 Feig and colleagues have consistently better refined CASP targets since CASP8 via extensive sampling with MD simulations.8, 16, 20 Their best results showed average improvement of 0.13 Å Cα RMSD and 3.79 GDT-HA score from 35 targets in CASP11, where they ran 30*40-ns simulation per target.8 Zhang lab reported protein refinement using fragment-guided MD conformational sampling of 181 predicted structures with an average RMSD reduction of 0.031 Å and 0.7-unit increase in GDT-HA.17 More recently, Lee lab has shown that using a series of short MD simulations with positional and distance restraints, they achieved comparable results to other more extensive MD sampling refinement protocols. They showed an average RMSD reduction of 0.013 Å and 1.14-unit increase in GDT-TS score from 54 targets in CASP8, CASP9, and CASP10.19 Although these methodologies have shown consistent results, the small numbers in the overall protein RMSD improvement partly indicates that it is important to focus our refinement efforts on specific regions of the protein that deviate significantly from its native structure.
Ligand-binding site structures are widely accepted as regions of the protein that undergo (significant) conformational changes to facilitate ligand binding.21, 22 Therefore, in predicted protein models, this region could deviate a lot from its native structure. For this reason, structure refinement of the ligand-binding site is an important step to improve the structural quality of the ligand-binding pocket. In this study, we present an MD-based ligand-binding site structure refinement approach that uses restraints derived from predicted binding site templates. We use our local structure alignment tool, G-LoSA (Graph-based Local Structure Alignment) to predict the ligand-binding site on modeled structures and to obtain holo structures binding site templates.23–26 A recent comparative assessment by Brylinski and colleagues showed that G-LoSA outperformed other binding site prediction tools against hard targets from a large and diverse dataset.27–29 In this work, we show a consistent better refinement performance of our method across a set of 40 diverse predicted protein structures from the Astex list.30 Our method selects appropriate binding site templates to derive restraint potentials for MD simulations and shows consistent better refinement of 37 out of 40 targets with an average Cα RMSD improvement of 0.90 Å. In addition, docking of native ligands to both the refined and the predicted structures shows consistent improvements in docking scores and binding modes in the refined structures. These data indicate a promising structure refinement method that can be used to improve SBVS docking accuracy for predicted protein models.
Methods
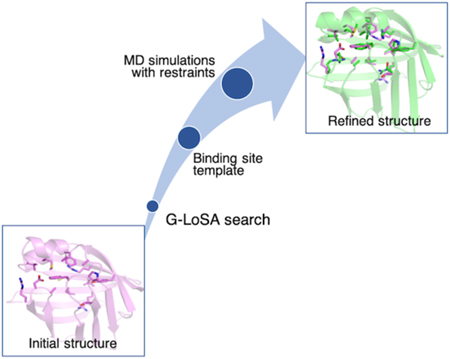
We performed all-atom MD simulations in explicit solvents on 40 protein targets from Astex diverse set.30 Our workflow is shown in Figure 1 and explained in more detail in the following paragraphs. In order to work with targets from the list that can undergo structure prediction in a reasonable amount of time, only proteins with sequences less than 300 amino acid residues were selected. Our ligand-binding site refinement method aims to refine computationally predicted structures, so we used I-TASSER suite to predict the 3D structures of our target protein sequences.31 Homologous sequence option (homoflag) was set to benchmark with an identity cutoff value of 0.3, which excluded homologous templates with greater than 30% sequence identity. The resulting structures are listed in Table S1 and they were separated into four groups based on their overall Cα RMSD relative to the experimental structures: group 1 (1–2 Å), group 2 (2–4 Å), group 3 (4–6 Å), and group 4 (> 6 Å). Their C-scores from I-TASSER results are also listed, which measure the confidence scores for the predicted structures that range from −5 to 2, where a higher score means a better structure.
Figure 1.

Ligand-binding site structure refinement protocol.
G-LoSA is a computational tool for local structure alignment and similarity measurement.23 Using G-LoSA Toolkit (https://compbio.lehigh.edu/GLoSA/toolkit.html) 25 and the latest experimentally solved PDB structures (as of November 2018), we built a ligand/binding site structure library containing at least one protein and one ligand with resolution less than 3.5 Å. We performed G-LoSA search on each of the computationally modeled protein structure to locate their binding sites and obtain ligand-binding site templates. G-LoSA search aligned local structures onto a protein structure in a sequence-independent manner and calculated their similarity using GA-score (G-LoSA Alignment score). GA-score is a scoring function that quantifies the similarity between two local structures. G-LoSA aligned all available ligand-binding site templates in the PDB library onto a whole query protein (without any prior information on their binding sites), and ranked the templates based on their GA-scores. The templates are filtered by selecting the ones with greater than 0.6 GA-scores. We have shown previously that GA-score of 0.6 and above are statistically significant through random local structures analysis.23 Furthermore, we filtered the templates based on their sizes by only including ligand-binding site templates with 11 to 34 amino acid residues. This is because we want to exclude ligand-binding sites that contain invalid ligands from crystallization reagents. The average number of residues for invalid ligands have been reported to be 5.2 residues, whereas valid ligands have 17.7 residues.32 From the selected templates, we identified aligned residues on the query protein as the equivalent residues. Aligned residues are identified using the shortest augmenting path algorithm to solve the linear sum assignment problem.33 We selected one top template based on the number of equivalent residues. We then calculated the distance matrix between Cα atoms of the selected template residues. From this matrix set (M), we derived a harmonic distance restraint potential
| (1) |
where k is the force constant, rij is the distance between ith and jth Cα atoms in the target protein, and r0,ij is the distance between equivalent atoms in the template.
All of our simulation inputs were prepared using CHARMM-GUI Solution Builder.34,35 Entire protein structures were solvated using TIP3P water models extending at least 10 Å from the protein atoms to form a cubic box.36 The systems were neutralized using sodium and chloride ions through 2,000 steps of Monte Carlo simulations. The structures were minimized using steepest descent minimization method for 5,000 steps followed by 1-ns equilibration time. Subsequent production runs were carried out at 300 K and 1 atm with an integration timestep of 2 fs. Periodic boundary conditions were applied in NPT ensemble using langevin thermostat.37 Non-bonded interactions were truncated using a force-based switching function between 10 and 12 Å, and particle-mesh Ewald summation was used to calculate electrostatic interactions. Covalent bonds involving hydrogens were constrained using SHAKE algorithm. The CHARMM36m force field was used for all simulations.38 For each target, we ran 3 × 50-ns production run (with distance restraints) and 3 × 50-ns production run (without distance restraints), each started from the same initial structure but using different initial velocity random seeds. For each target, we ran 300-ns production run and for the total 40 targets, we obtained 12-μs simulation time. Production simulations were carried out using OpenMM.39
The refined structure is the average of the three final conformations from the simulations. In order to remove potentially distorted geometries from structural averaging, a second set of simulation was carried out. The refined structure is resolvated in TIP3P water box and neutralized using sodium and chloride ions. The structure was minimized using steepest descent method for 5,000 steps followed by 25 ps equilibration with restraints of 2 kcal/(mol-Å2) on its backbone atoms and 0.1 kcal/(mol-Å2) on its side chain atoms. Choices of optimal force constants are discussed in Results & Discussion.
Each pair of the initial unrefined and the refined structures were subjected to docking of native ligand from the crystal structure to compare their docking poses and docking scores. AutoDock Vina was used to perform all docking experiments.40 Protein receptors were treated as rigid and ligands were flexible. The search space for each complex was identified based on the coordinate of the native ligand from the crystal structure with 5.0 A padding in each direction. In addition, we performed docking for each native ligand to its protein crystal structure as control.
The ligand-binding site residues were defined as protein residues that are within 4.5 Å of bound ligand in the crystal structure. To compare RMSD of ligand-binding site residues, we performed alignment and RMSD calculation of these residues. Ligand pose comparisons were calculated by aligning ligand-binding site residues and calculating RMSD of the ligand without aligning the ligand.
Results & Discussion
Parameter optimization of MD simulation refinement protocol
In order to optimize the force constant in the distance restraint potential Eq. (1), we randomly selected three representatives from group 1, 2, and 3. We used the ligand-binding site structures from the experimental structures as templates, which was an ideal case, for examining our MD simulation refinement protocol with distance restraints. In this test set, we first performed simulations without Cα positional restraints. Table S2 shows refinement results for ligand-binding site and protein structures of nine representative models with various force constant values applied to their distance restraints. Using our distance restraints at the ligand-binding site, we can see improvement for all three groups in their average ligand-binding site RMSD relative to the experimental structure. Improvements on the ligand-binding site structure increases as we go from a force constant of 0.1 kcal/(mol-Å2) to 1.0 kcal/(mol-Å2). The best results are seen with 1.0 kcal/(mol-Å2) with an average of 0.41 Å Cα RMSD of the ligand-binding sites relative to the experimental structure. However, Cα RMSD of the whole protein gets worse after refinement with an average increase of 0.48 Å. The issue of positionally unrestrained MD simulations has been addressed previously by several groups, where they reported that this approach to refine predicted structures almost always led to worse final structures.16, 41–45 It has also been further explored by Feig and coworkers through their calculation of the energy landscape between predicted models and their native structures. They reported that there were many significant kinetic barriers on microsecond time scales that separated the predicted models from their native structures.46 Often times in unrestrained MD simulations, there are significant off-pathway samplings that prevent productive structure refinement.
In order to optimize our method and prevent the overall protein structure from drifting away from their native structures, we applied weak positional restraints with a force constant of 0.5 kcal/(mol-Å2) on the Cα atoms of the residues that are not part of the ligand-binding sites (Table S3). We find that the weak positional restraint holds the rest of the protein stable while having no significant effect on the improvement of the ligand-binding site using distance restraints. When compared to MD simulations without positional restraints, a similar trend can be seen here, i.e., increased force constants for the distance restraints, going from 0.5 to 1.0 to 1.5 kcal/(mol-Å2), increase the improvement in ligand-binding site structure. At 0.5 kcal/(mol-Å2), average ligand-binding site is improved by 1.20 Å, and as we increase to 1.0 kcal/(mol-Å2), we see better improvement of 1.34 Å and at 1.5 kcal/(mol-Å2), it reaches 1.44 Å. On average, the ligand-binding site RMSD is 0.42 Å away from their experimental structure. This result is comparable to 0.41 Å RMSD in unrestrained MD simulations in Table S2 without negatively affecting the whole protein structure. Instead, the average protein Cα RMSD undergo an average improvement of 0.26 Å (Table S3). Furthermore, we expect that at RMSD value of less than 0.50 Å relative to their native structure, the ligand-binding site structures are properly refined, given that they do not contain ligands.
G-LoSA search to obtain predicted binding templates
For all 40 protein model structures predicted by I-TASSER (Table S1), we ran G-LoSA search and obtained the best template through filtering processes as described in Methods (Figure 1). The structures and their selected templates are listed on Table 1. In order to assess the structural similarity between the model structures and their selected templates, we calculated their TM-scores.47 Our data show that 31 out of 40 model structures have less than 0.5 TM-scores compared to their template structures (Table 1). A value of less than 0.5 indicates that two structures do not have similar global folds.48 The average TM-score for proteins in group 1, 2, 3, and 4 are 0.54, 0.44, 0.31 and 0.18, respectively. It shows that most of our ligand-binding site templates do not originate from proteins with similar global folds to the model structures.
Table 1.
Overview of 40 model structures with their predicted ligand-binding site templates obtained from G-LoSA search
| No. | PDB | LBS residues |
G-LoSA template |
TM- score+ |
GA- score |
Template residues |
Equivalent residues* |
Equivalent residues/LBS residues (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | 1towA | 17 | 4tkhA | 0.93 | 0.75 | 20 | 16 | 94 |
| 2 | 1lpzB | 21 | 2p3uB | 0.94 | 0.79 | 22 | 20 | 95 |
| 3 | 1tz8A | 11 | 4dx90 | 0.14 | 0.76 | 12 | 1 | 9 |
| 4 | 1g9vA | 21 | 4uiqA | 0.21 | 0.70 | 26 | 18 | 86 |
| 5 | 1r55A | 15 | 2w12A | 0.48 | 0.73 | 18 | 15 | 100 |
| Group 1 avg. | 0.54 | 0.75 | 77 | |||||
| 6 | 1hq2A | 25 | 3udvA | 0.88 | 0.74 | 26 | 24 | 96 |
| 7 | 1oweA | 20 | 1qcpA | 0.18 | 0.72 | 21 | 18 | 90 |
| 8 | 1q1gA | 13 | 4tymA | 0.26 | 0.76 | 19 | 4 | 31 |
| 9 | 1ttlA | 15 | 5fhmA | 0.73 | 0.77 | 25 | 15 | 100 |
| 10 | 1ke5A | 15 | 3tjcA | 0.32 | 0.72 | 20 | 13 | 87 |
| 11 | 1oytH | 18 | 1zgvA | 0.19 | 0.67 | 21 | 18 | 100 |
| 12 | 1p62B | 30 | 1p7cA | 0.28 | 0.68 | 31 | 22 | 73 |
| 13 | 1jjeA | 17 | 5fqaA | 0.56 | 0.68 | 14 | 8 | 47 |
| 14 | 2bm2A | 15 | 5pamB | 0.44 | 0.66 | 21 | 14 | 93 |
| 15 | 1ygcH | 21 | 4ngaH | 0.88 | 0.72 | 26 | 20 | 95 |
| 16 | 1s3vA | 17 | 4m2xA | 0.20 | 0.68 | 19 | 16 | 94 |
| 17 | 1k3uA | 21 | 5cgqA | 0.88 | 0.63 | 23 | 20 | 95 |
| 18 | 1u1cA | 14 | 1y1qC | 0.38 | 0.70 | 13 | 11 | 79 |
| 19 | 1unlA | 20 | 5k4jA | 0.27 | 0.71 | 25 | 19 | 95 |
| 20 | 1v0pA | 19 | 5usqA | 0.31 | 0.71 | 20 | 12 | 63 |
| 21 | 1w2gA | 13 | 3tmkA | 0.34 | 0.71 | 32 | 13 | 100 |
| Group 2 avg. | 0.44 | 0.70 | 84 | |||||
| 22 | 1sqnA | 20 | 6chzA | 0.33 | 0.71 | 20 | 19 | 95 |
| 23 | 2br1A | 16 | 3i60A | 0.22 | 0.72 | 26 | 14 | 88 |
| 24 | 1m2zA | 22 | 2p1uA | 0.17 | 0.72 | 24 | 20 | 91 |
| 25 | 1s19A | 24 | 5xplA | 0.21 | 0.70 | 26 | 21 | 88 |
| 26 | 1ia1A | 35 | 4elbC | 0.19 | 0.68 | 22 | 14 | 40 |
| 27 | 1v48A | 18 | 1b8nA | 0.64 | 0.69 | 19 | 15 | 83 |
| 28 | 1u4dA | 14 | 2e2bA | 0.36 | 0.74 | 27 | 12 | 86 |
| 29 | 1n46A | 25 | 1fcyA | 0.20 | 0.75 | 25 | 18 | 72 |
| 30 | 1gkcA | 15 | 4efsA | 0.24 | 0.68 | 22 | 14 | 93 |
| 31 | 1z95A | 24 | 5bnuA | 0.24 | 0.71 | 26 | 18 | 75 |
| 32 | 1kzkA | 15 | 5ah6A | 0.24 | 0.64 | 12 | 12 | 80 |
| Group 3 avg. | 0.31 | 0.70 | 81 | |||||
| 33 | 1sj0A | 21 | 1z95A | 0.21 | 0.72 | 22 | 16 | 76 |
| 34 | 1ig3A | 12 | 1bx6A | 0.17 | 0.63 | 27 | 4 | 33 |
| 35 | 1hnnA | 32 | 2xvmA | 0.15 | 0.69 | 20 | 15 | 47 |
| 36 | 1navA | 23 | 1s19A | 0.21 | 0.70 | 24 | 14 | 61 |
| 37 | 1hvyA | 24 | 4f2vA | 0.21 | 0.64 | 27 | 13 | 54 |
| 38 | 2bsmA | 18 | 4o07A | 0.17 | 0.67 | 23 | 16 | 89 |
| 39 | 1sg0A | 30 | 3lcmA | 0.19 | 0.66 | 21 | 19 | 63 |
| 40 | 1lrhA | 15 | 4qxbA | 0.12 | 0.63 | 12 | 8 | 53 |
| Group 4 avg. | 0.18 | 0.67 | 60 | |||||
| Total avg. | 0.37 | 0.70 | 77 | |||||
Residues on the template structure that are equivalent to the ligand-binding site residues on the model structure
TM-score between model structure and G-LoSA template PDB structure
For all structures, the predicted ligand-binding site templates contain at least one of the equivalent residues on the model structure ligand-binding site. On average, the template residues cover 77% of the ligand-binding site residues on the model structures. Out of 40 templates, 22 have greater than 85% coverage and there are 4 templates with 100% coverage. Moreover, there are only 6 templates that contain less than 50% coverage. Although we have good coverage of binding site residues, the average GA-score is only 0.70 for 40 targets. This is a result of poorly defined ligand-binding site structures from the predicted protein models. It is reasonable to expect that very low resolution apo structures of the predicted models will not align well with binding site templates from holo structures in the PDB library.
Overall, we demonstrate that our method of identifying template binding site structures is consistent and reliable when tested against computationally predicted 40 diverse structures. Accurate prediction of ligand-binding site and reliable identification of good binding site template are integral steps in our workflow of ligand-binding site structure refinement. The ligand-binding site prediction on whole protein structure by G-LoSA has been addressed in our previous study.24 By identifying appropriate binding site templates, we can reliably derive restraint potentials to guide MD simulation to the correct native structure of the ligand-binding site.
Results on 40 refinement targets from Astex diverse set
Using the optimized parameters for distance restraints, we performed all-atom MD simulations for 40 refinement targets from Astex diverse set. For each target, we ran three independent 50-ns simulations with and without distance restraints on the ligand-binding site Cα atoms. A force constant of 1.5 kcal/(mol·Å2) was applied to the distance restraints. All Cα atoms that were not included in the template were positional-restrained with a force constant of 0.5 kcal/(mol·Å2). Final conformations were averaged to obtain the refined structure. In order to remove locally distorted geometries after structural averaging, the structure was resolvated, followed by a short minimization process (see the details in Methods).
Our results show consistent and significant improvement of the ligand-binding site Cα RMSD in 37 out of 40 model structures (Table 2, Figure 2). The average improvement is 0.90 Å (from 2.50 to 1.60 Å). Group 2 (no. 6–21) and group 3 (no. 22–32) show the most improvements in RMSD with averages of 1.15 and 1.20 Å decrease, respectively. Group 1 (no. 1–5) and group 4 (no. 33–40) show modest improvements of 0.41 and 0.30 Å, respectively. Ligand-binding site structures in group 1 show relatively low RMSD improvement because the initial unrefined structures are already of good quality with low RMSD ranging from 0.50 to 1.38 Å. All five structures in group 1 are improved during refinement, and when compared to the control group (MD simulations without distance restraints), they show clear differences in their RMSD relative to the crystal structure (Table 2). Model 1 (fatty-acid binding protein, adipocyte) improves by 0.41 Å in Cα RMSD of its ligand-binding site residues. Most structural adjustments happen on helix 1 binding site residues, including residue F16 (Figure 3A). The best RMSD improvement in group 1 is shown in model 4 (deoxyhemoglobin) that undergoes a decrease of 0.88 A in RMSD after refinement. Conversely, the smallest improvement is shown by model 3 (transthyretin) with a decrease of 0.09 Å in RMSD with refinement. The minimal improvement in this case is partially due to inaccurate template binding site selection that only covers 9% of ligand-binding site residues (Table 1). In addition, we observe that group 1 proteins show an average of 0.12 Å improvement in their overall protein RMSD (Table 2, Figure S1). In contrast, the control group does not show any improvement after the simulations.
Table 2.
Overview of refinement targets from all 40 targets. Group 1 (1–5), group 2 (6–21), group 3 (22–32), and group 4 (33–40). Improved cases with lower RMSD are highlighted in bold.
| No. | PDB | Residues | LBS RMSD (Å) | Protein RMSD (Å) | |||||
|---|---|---|---|---|---|---|---|---|---|
| LBS residues |
Initial | Refined | Control* | Initial | Refined | Control* | |||
| 1 | ItowA | 131 | 17 | 0.87 | 0.46 | 1.39 | 1.20 | 1.07 | 1.41 |
| 2 | 1lpzB | 234 | 21 | 0.73 | 0.35 | 1.25 | 1.69 | 1.64 | 1.72 |
| 3 | 1tz8A | 114 | 11 | 0.50 | 0.41 | 0.48 | 1.69 | 1.68 | 1.70 |
| 4 | 1g9vA | 141 | 21 | 1.38 | 0.50 | 1.55 | 1.72 | 1.35 | 1.78 |
| 5 | 1r55A | 203 | 15 | 0.88 | 0.57 | 1.12 | 1.90 | 1.87 | 1.90 |
| Group | 1 avg. | 0.87 | 0.46 | 1.16 | 1.64 | 1.52 | 1.70 | ||
| 6 | 1hq2A | 158 | 25 | 2.01 | 1.37 | 2.37 | 2.18 | 1.81 | 2.23 |
| 7 | 1oweA | 245 | 20 | 1.44 | 0.55 | 1.91 | 2.44 | 2.34 | 2.44 |
| 8 | 1q1gA | 243 | 13 | 1.16 | 1.29 | 1.47 | 2.51 | 2.49 | 2.48 |
| 9 | 1tt1A | 251 | 15 | 1.07 | 0.58 | 1.17 | 2.53 | 2.17 | 2.35 |
| 10 | 1ke5A | 298 | 15 | 1.15 | 0.85 | 0.82 | 2.54 | 2.43 | 2.40 |
| 11 | 1oytH | 257 | 18 | 3.04 | 0.40 | 3.35 | 2.60 | 2.24 | 2.62 |
| 12 | 1p62B | 241 | 30 | 1.70 | 1.59 | 1.58 | 2.64 | 2.60 | 2.55 |
| 13 | 1jjeA | 220 | 17 | 2.42 | 2.28 | 2.42 | 2.77 | 2.66 | 2.74 |
| 14 2bm2A | 242 | 15 | 3.02 | 1.31 | 3.22 | 3.16 | 2.97 | 3.13 | |
| 15 | lygcH | 254 | 21 | 2.80 | 0.55 | 3.07 | 3.18 | 2.98 | 3.20 |
| 16 | ls3vA | 186 | 17 | 1.70 | 0.64 | 1.39 | 3.22 | 3.19 | 3.20 |
| 17 | lk3uA | 268 | 21 | 5.80 | 0.59 | 5.92 | 3.34 | 1.94 | 3.36 |
| 18 | lulcA | 253 | 14 | 5.08 | 3.54 | 5.12 | 3.35 | 3.16 | 3.39 |
| 19 | 1unlA | 292 | 20 | 1.27 | 0.93 | 1.65 | 3.41 | 3.36 | 3.46 |
| 20 | lv0pA | 286 | 19 | 1.94 | 1.32 | 1.72 | 3.68 | 3.61 | 3.72 |
| 21 | lw2gA | 208 | 13 | 1.54 | 1.00 | 2.26 | 3.73 | 3.82 | 3.81 |
| Group 2 avg. | 2.32 | 1.17 | 2.47 | 2.96 | 2.74 | 2.94 | |||
| 22 | lsqnA | 251 | 20 | 0.95 | 0.43 | 1.00 | 4.02 | 4.01 | 4.01 |
| 23 | 2br1A | 271 | 16 | 1.11 | 0.71 | 1.30 | 4.10 | 4.09 | 4.10 |
| 24 | lm2zA | 255 | 22 | 1.42 | 0.86 | 1.28 | 4.12 | 4.06 | 4.15 |
| 25 | lsl9A | 253 | 24 | 1.96 | 0.80 | 2.14 | 4.14 | 4.01 | 4.26 |
| 26 | 1ia1A | 192 | 35 | 1.81 | 1.64 | 1.93 | 4.41 | 4.34 | 4.44 |
| 27 | lv48A | 283 | 18 | 3.52 | 1.15 | 3.26 | 4.51 | 4.38 | 4.55 |
| 28 | 1u4dA | 273 | 14 | 1.29 | 0.91 | 1.80 | 4.53 | 4.51 | 4.67 |
| 29 | ln46A | 251 | 25 | 1.62 | 1.26 | 1.49 | 4.60 | 4.56 | 4.61 |
| 30 | lgkcA | 163 | 15 | 2.51 | 1.25 | 3.56 | 4.63 | 4.43 | 4.66 |
| 31 | lz95A | 246 | 24 | 1.32 | 1.09 | 1.40 | 5.15 | 4.63 | 4.71 |
| 32 | lkzkA | 99 | 15 | 7.83 | 2.03 | 7.87 | 5.52 | 3.74 | 5.41 |
| Group 3 avg. | 2.30 | 1.10 | 2.46 | 4.52 | 4.25 | 4.51 | |||
| 33 | lsj0A | 245 | 21 | 3.13 | 2.59 | 3.14 | 6.21 | 6.09 | 6.32 |
| 34 | lig3A | 254 | 12 | 1.86 | 1.87 | 1.52 | 6.30 | 5.57 | 5.64 |
| 35 | lhnnA | 261 | 32 | 5.52 | 5.48 | 5.70 | 6.39 | 6.33 | 6.46 |
| 36 | lnavA | 263 | 23 | 1.92 | 1.50 | 1.78 | 6.45 | 6.03 | 6.09 |
| 37 | lhvyA | 288 | 24 | 8.07 | 7.89 | 8.39 | 7.24 | 7.25 | 7.35 |
| 38 | 2bsmA | 208 | 18 | 3.97 | 2.60 | 3.67 | 8.76 | 8.62 | 8.80 |
| 39 | lsg0A | 230 | 22 | 1.27 | 1.08 | 1.20 | 9.41 | 9.48 | 9.45 |
| 40 | llrhA | 160 | 15 | 7.58 | 7.97 | 7.94 | 10.96 | 11.04 | 11.12 |
| Group 4 avg. | 4.17 | 3.89 | 4.17 | 7.72 | 7.55 | 7.65 | |||
| Total avg. | 2.50 | 1.60 | 2.64 | 4.17 | 3.96 | 4.16 | |||
Control = simulations without distance restraints and with positional restraints.
Figure 2.
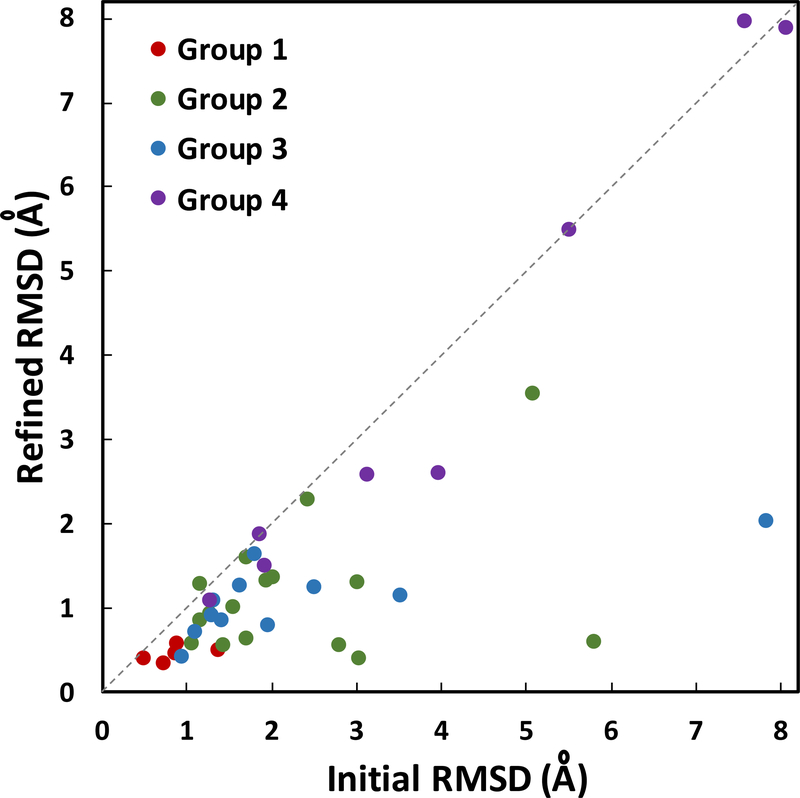
Ligand-binding site Cα RMSD values comparing initial and refined model structures against the experimental structures. The structures are separated into 4 groups based on their initial protein model RMSD to the experimental structures: group 1 (1–2 Å, red), group 2 (2–4 Å, green), group 3 (4–6 Å, blue), and group 4 (>6 Å, purple). The average improvement for ligand-binding site Cα RMSD is 0.90 Å.
Figure 3.
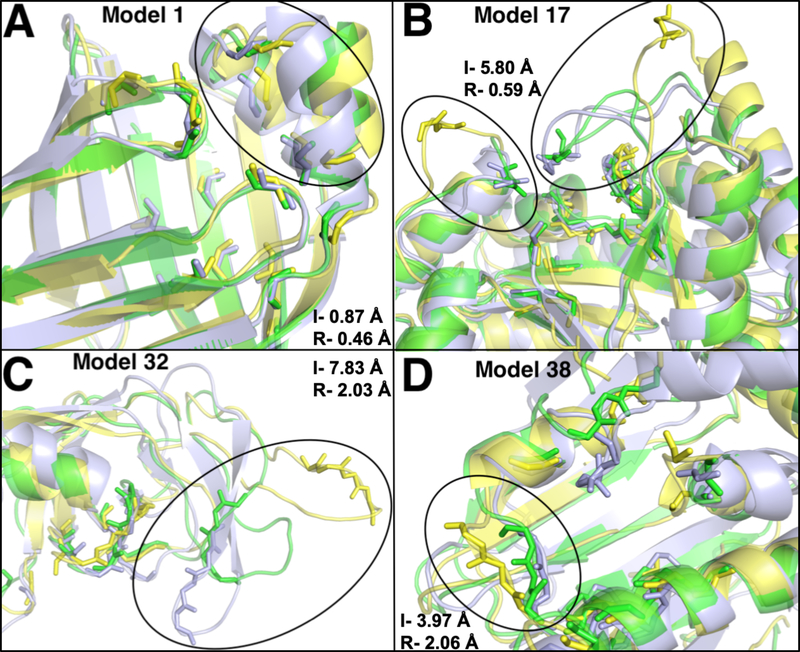
Aligned structures of initial unrefined models (yellow) and refined models (green) on the crystal structures (light blue). The backbones of ligand-binding site residues are shown in sticks. I- is the initial unrefined structure RMSD and R- is the refined structure RMSD relative to the crystal structure. Representatives from each group is shown, (A) group 1: model 1- FABP, adipocyte (PDB 1tow) changed from 0.87 Å to 0.46 Å. (B) Group 2: model 17- tryptophan synthase (PDB lk3u) improved from 5.80 Å to 0.59 Å. (C) Group 3: model 32- HIV protease (PDB 1kzk) changed from 7.83 Å to 2.03 Å. (D) Group 4: model 38-HSP (PDB 2bsm) decreased from 3.97 Å to 2.06 Å.
Group 2 (Table 2, no. 6 to 21) shows significant improvement during ligand-binding site structure refinement with an average decrease of 1.15 Å in RMSD (from 2.32 to 1.17 Å). Out of 16 structures, 15 undergo successful refinements. Five of the structures show greater than 1.50 Å RMSD improvements. None of the initial unrefined ligand-binding site structures in group 2 has RMSD better than 1.00 A relative to their native structures. After refinement, 9 out of 16 structures show RMSDs with less than 1.00 Å away from their crystal structures. The best improvement is shown by model 17 (tryptophan synthase) that undergoes 5.21 Å decrease (from 5.80 Å to 0.59 Å) in RMSD. Major changes at the ligand-binding site originate from nine residues on three different loops that deviate significantly from its native structure (Figure 3B). Assessing its selected binding site template, we observe that model 17 template covers 95% of native binding site residues (Table 1). Other structures with significant improvements are model 11 (thrombin) and model 15 (serine protease factor VII) that undergo 2.67 and 2.25 Å decrease in RMSD, respectively. Both structures have relatively large deviations from the crystal structures at their binding sites with initial RMSDs of 3.04 and 2.80 Å, respectively. Through successful binding site predictions and identification of good templates, with 100% binding site coverage for model 11 and 95% coverage for model 15 (Table 1), these structures are refined to final RMSDs of 0.40 and 0.55 Å, respectively (Figure S2A-B). Our results indicate that these refinements are indeed due to distance restraints derived from predicted binding site templates, because the same simulations without distance restraints in the control group do not show improvements (Table 2). On average, the control group shows 0.15 Å worse ligand-binding site RMSD after MD simulation. The only structure in group 2 that generates unsuccessful final structure is model 8 (purine nucleoside phosphorylase) with 0.13 Å increase in RMSD. In this case, the top template also shows small coverage of ligand-binding site residues, with only 31% (Table 1). Looking at the whole protein structure in group 2, as a result of successful ligand-binding site structure refinement, protein structures are improved with an average decrease of 0.22 Å in RMSDs (Table 2, Figure S1).
Although, the average protein structure RMSD in group 3 (Table 2, no. 22–32) is worse than group 2, their average ligand-binding site RMSD are similar with 2.32 Å in group 2 and 2.30 Å in group 3. All 11 structures in group 3 undergo successful ligand-binding site structure refinement with an average RMSD improvement of 1.20 Å (from 2.30 to 1.10 Å). Model 32 (HIV protease) shows the most dramatic decrease in ligand-binding site RMSD with 5.80 Å (from 7.83 Å to 2.03 Å). This is mainly due to a major error in the positioning of a beta hairpin in the initial model structure. This beta hairpin contains four binding site residues that are located about 20 Å away from the binding pocket. Our refinement method successfully guides the beta hairpin closer to its native state using MD simulations with distance restraints (Figure 3C). Another notable refinement case in group 3 is model 27 (purine nucleoside phosphorylase) that refines to 1.15 Å RMSD from its initial value of 3.52 Å. Successful ligand-binding site structure refinement in group 3 also leads to better overall whole protein structure with 0.27 Å improvement on average (Table 2, Figure S1).
Group 4 contains 8 model structures with major incorrect folds in their overall protein structures, RMSD > 6 Å, that contribute to poorly defined ligand-binding site structures. The average initial ligand-binding site Cα RMSD is 4.17 Å and with structure refinement it decreases to 3.89 Å, where 6 out of 8 structures undergo proper refinements. One of the structures that fails to refine is model 40 (auxin-binding protein 1), which has an incorrect location of its C-terminal helix. It is an important helix because it contains two of the binding site residues that directly interact with the ligand.49 This structure places the helix at about 30 Å away from the binding pocket, which results in an incorrect binding site structure (Figure S2C). Template identification for this structure only picks up 53% of the ligand-binding site residues and does not include the residues on the C-terminal helix. As a result, our method was not able to refine this structure. A similar case is seen in model 37 (thymidylate synthase), where the initial model places the C-terminal loop at about 30 Å away from its experimental holo structure (Figure S2D). Two C-terminal residues, M286 and A287, are binding site residues.50 During MD simulation for refinement, the incorrect C-terminal position stays the same and no significant refinement is gained. Aside from these two cases, there are a few successful refinement cases in group 4, most notably model 38 (heat shock protein hsp90) that gains 1.37 Å RMSD improvement. Our method correctly identifies a good template that covers 89% of the ligand-binding site residues despite the poorly defined protein structure with 8.76 Å RMSD away from its native structure (Table 2). As result, this structure undergoes proper refinement and shows significant adjustments of residues I96, G97, and M98 that are located at the binding loop (Figure 3D). Group 4 shows a few target structures that mislead the proper identification of good templates for MD simulation with distance restraints, including models 37 and 40. Our analysis suggests that hard targets with major incorrect placements of binding site residues around 30 Å will result in unsuccessful refinement with the current approach. It also indicates that similar binding sites might contain slightly different residues depending on which ligand they bind to.
Our overall results show that correct template identification that contains most of the binding site residues lead to successful refinement. Clearly, there is a positive correlation between the percentage of residue coverage in the top template and a successful refinement, where all of the templates with greater than 60 % coverage result in improved refined models (Figure S3). In addition to ligand-binding site structure refinement, the overall protein structure is also improved by 0.21 Å on average (Figure S1, Table 2). This value is comparable to many previously published protein structure refinements using MD simulations, where they reported average Cα RMSD improvement that ranges from 0.013 to 0.13 Å.8, 16, 17, 19, 20 We show that we have a modest but consistent improvement of the overall protein structure as a result of the ligand-binding site refinement using templates derived from predicted binding sites.
Our refinement method models the binding site based on a template structure with bound ligand. In order to test possible effects from a bound ligand, we ran additional simulations to refine the initial structures with bound crystal ligands. The crystal ligands were docked to the initial unrefined structures prior to running the same simulations with restraint potentials. Our results show an average improvement of 0.89 Å, that is similar to 0.90 Å observed in simulations without bound ligands (Table S4). It suggests that our method works well, with and without bound ligands. More discussion on the data is provided in the supporting information.
Docking of native ligands to the refined structures
In order to evaluate the quality of the refined ligand-binding sites, we performed computational docking of native ligands to their initial unrefined and refined structures, as well as to their crystal holo structure. We used AutoDock Vina to run docking calculations with typical rigid ligand-binding sites settings.40 The initial unrefined and refined structures are aligned to the crystal structure prior to docking and the search space for ligand docking is defined based on the crystal structure ligand position. Docking results are compared based on their docking scores and their binding modes by calculating all-heavy-atom ligand RMSD relative to the crystal structure without performing ligand structure alignments (to include RMSD due to ligand translation and rotation). The results indicate that our refined structures improve docking poses of native ligands because their ligand RMSDs are on average 1.97 Å better than those with the initial unrefined models, when compared to their experimental structure (Table 3, Figure 4). Out of 40 structures, 37 show improvement in docking modes. On average, the best results are seen in group 2 with 2.76 Å improvement, follow by group 1, 3, and 4 with 1.82, 1.66 and 0.90 Å, respectively. In addition, the average docking score of native ligands are improved by 4.3 kcal/mol when they are docked to the refined structures as compared to their initial unrefined structures. For the control group, docking of native ligands to their crystal structures are used, where they show an average ligand RMSD of 1.37 Å from 40 structures (Table 3). A general consensus for correct bound structure is within the RMSD cutoff of 2.00 Å.40, 51 In this dataset, 82.5% of the results from AutoDock Vina are within the cutoff, hence showing accurate binding mode prediction. This result is comparable to 78% success rate that was reported by AutoDock Vina using PDBbind dataset.40
Table 3.
Ligand docking score and ligand RMSD relative to the experimental structures. Improved cases with better docking scores and lower RMSD are highlighted in bold.
| Docking scores (kcal/mol) | RMSD (Å) | ||||||
|---|---|---|---|---|---|---|---|
| No. | PDB | Unrefined | Refined | Crystal | Unrefined | Refined | Crystal |
| 1 | 1towA | −7.0 | −8.0 | −8.1 | 3.73 | 2.01 | 1.16 |
| 2 | 1lpzB | −5.1 | −5.5 | −10.2 | 6.60 | 4.97 | 1.69 |
| 3 | 1tz8A | −3.7 | −3.7 | −4.4 | 3.17 | 3.15 | 1.20 |
| 4 | 1g9vA | −0.5 | −8.4 | −12.6 | 6.95 | 1.37 | 1.00 |
| 5 | 1r55A | −4.9 | −4.0 | −6.5 | 3.42 | 3.26 | 1.11 |
| Group 1 avg. | −4.2 | −5.9 | −8.4 | 4.77 | 2.95 | 1.23 | |
| 6 | 1hq2A | −6.4 | −6.8 | −8.5 | 6.17 | 2.40 | 2.90 |
| 7 | 1oweA | 23.7 | −7.8 | −5.5 | 8.41 | 2.30 | 0.30 |
| 8 | 1q1gA | −6.2 | −6.3 | −7.5 | 3.32 | 2.64 | 0.81 |
| 9 | 1tt1A | 2.8 | −1.2 | −7.1 | 4.05 | 3.99 | 1.55 |
| 10 | 1ke5A | −2.3 | −7.0 | −6.9 | 4.51 | 2.47 | 1.38 |
| 11 | 1oytH | −7.1 | −8.4 | −10.0 | 7.41 | 3.28 | 0.41 |
| 12 | 1p62B | −5.8 | −6.5 | −7.6 | 2.50 | 2.44 | 0.22 |
| 13 | 1jjeA | 11.5 | −1.5 | −8.9 | 5.25 | 3.59 | 3.12 |
| 14 | 2bm2A | 3.3 | −7.5 | −8.3 | 8.32 | 2.83 | 2.82 |
| 15 | 1ygcH | 2.9 | −8.9 | −9.7 | 6.54 | 2.21 | 1.19 |
| 16 | 1s3vA | −7.4 | −8.2 | −8.5 | 7.24 | 6.25 | 2.61 |
| 17 | 1k3uA | −6.5 | −6.9 | −9.8 | 3.65 | 1.86 | 1.22 |
| 18 | 1u1cA | −6.8 | −7.0 | −7.7 | 2.76 | 2.47 | 0.47 |
| 19 | 1unlA | −7.0 | −8.4 | −8.3 | 4.48 | 1.82 | 1.26 |
| 20 | 1v0pA | −7.3 | −8.4 | −8.7 | 7.50 | 2.84 | 1.91 |
| 21 | 1w2gA | 1.1 | −0.5 | −8.9 | 4.91 | 0.76 | 0.75 |
| Group 2 avg. | −1.1 | −6.3 | −8.2 | 5.35 | 2.59 | 1.43 | |
| 22 | 1sqnA | −0.5 | −0.8 | −11.9 | 6.51 | 1.44 | 0.33 |
| 23 | 2br1A | −6.9 | −8.7 | −8.3 | 5.83 | 1.58 | 2.21 |
| 24 | 1m2zA | 38.2 | −0.5 | −12.6 | 7.21 | 5.31 | 0.31 |
| 25 | 1s19A | −7.2 | −9.2 | −10.6 | 3.13 | 2.59 | 1.04 |
| 26 | 1ialA | −8.4 | −8.6 | −11.6 | 6.19 | 5.42 | 0.42 |
| 27 | 1v4BA | −5.9 | −7.8 | −8.4 | 6.58 | 3.76 | 0.88 |
| 28 | 1u4dA | −6.5 | −7.0 | −7.5 | 5.81 | 3.57 | 1.06 |
| 29 | 1n46A | 3.9 | 1.9 | −12.8 | 3.82 | 5.00 | 0.84 |
| 30 | 1gkcA | 20.1 | −5.5 | −6.8 | 6.09 | 4.94 | 1.29 |
| 31 | 1z95A | −0.9 | −2.0 | −10.5 | 4.92 | 4.84 | 1.01 |
| 32 | 1kzkA | −5.0 | −5.5 | −7.2 | 7.36 | 6.71 | 4.87 |
| Group 3 avg. | 1.9 | −4.9 | −9.9 | 5.77 | 4.11 | 1.30 | |
| 33 | 1sj0A | 9.9 | 12.2 | −11.7 | 2.10 | 2.72 | 0.96 |
| 34 | 1ig3A | −3.7 | −3.7 | −5.0 | 3.80 | 3.00 | 0.71 |
| 35 | 1hnnA | −6.2 | −6.5 | −8.4 | 4.84 | 3.45 | 0.75 |
| 36 | 1navA | 1.4 | 0.6 | −10.3 | 7.37 | 5.24 | 1.92 |
| 37 | 1hvyA | −6.8 | −7.3 | −8.4 | 6.14 | 5.62 | 3.29 |
| 38 | 2bsmA | 3.7 | −1.4 | −9.1 | 5.95 | 2.84 | 1.15 |
| 39 | 1sg0A | −7.5 | −7.6 | −11.1 | 5.61 | 5.55 | 1.80 |
| 40 | 1lrhA | −5.8 | −5.7 | −7.6 | 3.15 | 4.14 | 0.78 |
| Group 4 avg. | −1.9 | −2.4 | −9.0 | 4.87 | 3.97 | 1.42 | |
| Total avg. | −0.8 | −5.1 | −8.9 | 5.30 | 3.33 | 1.37 | |
Figure 4.

Ligand RMSD values comparing the native ligand binding poses on initial unrefined and refined structures against the experimental binding poses. The ligands were docked using AutoDock Vina. The average improvement for ligand binding poses RMSD is 1.97 Å.
In group 1, all 5 of the refined structures show better ligand binding poses than their initial unrefined structures (Table 3, Figure 4). The average ligand RMSD changes from 4.77 Å to 2.95 Å, with 38% improvement. However, model 3 (transthyretin) and model 5 (human ADAM33 protein) show very small changes of 0.02 and 0.16 Å, respectively. This is because both structures undergo small decrease in RMSD of their ligand-binding site structures with 0.09 and 0.31 Å, respectively. In model 1 (FABP, adipocyte), the refined structure properly orients a binding residue, F16, to interact with the ligand, carbazole butanoic acid, resulting in a better binding mode (Figure 5A). The best improvement in this group is shown by model 4 (deoxyhemoglobin) with 5.58 Å decrease in RMSD. The major incorrect pose in the initial unrefined structure is caused by side chain F98 that blocks the proper orientation of the ligand, which results in a 180° flip of porphyrin’s docking orientation in the binding pocket. Through refinement, F98 moves to adopt its native conformation that allows for correct ligand binding of porphyrin (Figure 5B).
Figure 5.

Ligand binding modes obtained from AutoDock Vina are compared to the crystal structures (light blue). Ligand poses obtained from docking to the initial unrefined structures (yellow) and docking to the refined structure (green). I- is the initial unrefined structure ligand RMSD, and R- refers to refined structure ligand RMSD. Two representatives from each group are shown, (A-B) group 1: model 4 – deoxyhemoglobin (PDB 1g9v) and model 1- FABP, adipocyte (PDB 1tow). (C-D) Group 2: model 15-serine protease factor Vila (PDB 1ygc) and model 17- tryptophan synthase (PDB 1k3u). (E-F) Group 3: model 22- progesterone receptor (PDB 1sqn) and model 27- purine nucleoside phosphorylase (PDB 1v48). (G-H) Group 4: model 33- estrogen receptor (PDB 1sj0) and model 38- HSP (PDB 2bsm).
All 16 structures in group 2 show better ligand binding poses than their initial unrefined structures, with 8 structures showing greater than 2.00 A RMSD improvements (Table 3, Figure 4). The most significant improvements are shown by model 7 (urokinase), model 11 (thrombin), 14 (human beta2 tryptase), 15 (serine protease factor VII), 20 (p. falciparum protein kinase), and 21 (thymidylate kinase) with greater than 4.00 Å decrease in ligand RMSD. In the initial unrefined structure of model 15, a ligand-binding site loop that contains residues W215, G216, Q217, and G219 occludes the binding pocket, effectively blocking the ligand G17905 from reaching and interacting with D189 inside the pocket. After refinement, this loop opens up, allowing for a proper docking of G17905 in the binding pocket and restoring its amidine interaction with D189, as seen in the crystal structure.52 As a result, RMSD of the ligand binding mode improves by 4.33 Å and its docking score by 11.8 kcal/mol (Figure 5C, Table 3). Similar cases are seen in two other structures, model 11 and 14 where their initial unrefined structures contain incorrect binding loop structures that block the binding pockets. After successful refinements, their binding pockets are opened up for better docking of the native ligands. Model 11 and 14 undergo improvements of 4.13 and 5.49 Å RMSD, as well as 1.3 and 10.8 kcal/mol, respectively (Figure S4A, S4B). Model 17 experiences the opposite conformational change during refinement, from a widely opened binding pocket to a closed one, that is led by two large loops around the binding site. Its initial unrefined structure has two of the binding loops away from each other at a distance of about 25 Å, which prevents some binding residues from interacting with the ligand. After refinement, the binding loops move closer to the center of the binding pocket at a distance of about 6 Å from each other, forming a binding pocket that is comparable to the native structure (Figure 3B). Consequently, this leads to an improved ligand binding pose of 1.86 Å relative to the crystal structure (Figure 5D).
There are 10 out of 11 structures in group 3 that undergo ligand RMSD improvements (Table 3). The most dramatic change in ligand binding pose is seen in model 22 (progesterone receptor) that binds norethindrone. Its initial unrefined structure has a small binding pocket, where residues L763 and F778 form a hydrophobic interaction that blocks the proper binding of norethindrone. As a result, norethindrone adopts an incorrect binding mode that is about 90° rotated from its native pose. The refined structure corrects the positions of Cα in the binding pocket by 0.52 Å and breaks up the interaction of L768 and F778, resulting in a bigger pocket for better ligand binding mode (Figure 5E). Ligand RMSD drops from 6.51 Å to 1.44 Å. A similar case, where a hydrophobic residue L15 obstructing the binding pocket is seen in model 23 (serine/threonine protein kinase chk1). Docking to the initial structure results in an incorrect binding of the inhibitor, furanopyrimidine by 5.83 Å. This is corrected in the refined structure that shows 1.58 Å ligand RMSD relative to the crystal structure (Figure S4C). Model 27 (purine nucleoside phosphorylase) gains 2.82 Å in ligand RMSD improvement. The initial model contains three binding loops with incorrect conformations, most notably residues F200 and N243 that cannot interact with the guanine base, resulting in a very different docking mode than in the crystal structure.53 These residues are correctly adjusted in the refined structure, which leads to a better docking pose (Figure 5F). Intriguingly, model 32 (HIV protease), which undergoes the most significant change in ligand-binding site refinement, does not show a dramatic improvement in ligand binding mode during docking. This is because the biological assembly of HIV protease is a homodimer, where the ligand JE-2147 is sandwiched in the dimer interface.54 The absence of the second monomer prevents the proper binding of the ligand (Figure S4D). This also leads to an unsuccessful docking of JE-2147 to the crystal structure, where the ligand RMSD is 4.87 Å relative to the experimental data (Table 3). One case in group 3 that shows worse ligand binding pose is model 29 (thyroid hormone receptor beta-1) with an increase of 1.18 Å RMSD from the initial unrefined structure.
In group 4, 6 out of 8 of the refined structures report better docking results than their initial unrefined structures. The best improvement is shown by model 38 (heat shock protein hsp90) with 3.91 Å decrease in ligand RMSD. Its initial unrefined structure has residue F138 blocking the base of the binding pocket, which prevents correct ligand docking. In the experimental structure, residue F138 is in an open conformation interacting with the resorcinol group on the ligand.55 After structure refinement, this interaction is restored, resulting in a better ligand docking mode (Figure 5H). One of the structures without docking improvement is model 33 (estrogen receptor), where the ligand RMSD increases by 0.62 Å. The refined structure has two incorrect conformations of L346 and L387 that fill the binding pocket and prevent correct binding of benzoxathiin ring on the ligand E4D (Figure 5G).
Many of the docking energetic penalties and incorrect ligand binding modes seen in the initial unrefined models are caused by occluded ligand-binding pockets, e.g., models 4, 11, 14, 15, 22, 23 and 38 that are discussed above. Another problem that comes from incorrect structure of the initial unrefined model is the loss of interactions between the ligand and the binding site residues that are placed too far away. A widely opened binding pocket increases the chance of incorrect docking binding modes, shown in models 1, 17, 21, 27, and 32.
Although the refined structures show significantly better ligand RMSD than the initial unrefined structures, their overall binding modes are still not as good as the crystal structures. On average, ligand RMSD from the crystal structures is 1.96 Å better than the refined models (Table 3). This is in part due to a limitation in our refinement method, where residue side chains are free to adopt energetically favorable conformations during the simulations while interacting with solvent molecules in the absence of a ligand. At the end of the simulations, even if the Cα of ligand-binding site residues are correctly placed, their side chains might not be. Incorrect side chain conformations can block the proper binding modes of native ligands. For example, in model 16, the refined Cα RMSD is 0.64 Å relative to the crystal structure, but ligand RMSD in docking is 6.25 Å away from the crystal structure (Table 2, 3). One difference between the refined and the crystal structure that results in unsuccessful docking is the orientation of the side chain residue, F34.( Nevertheless, this problem can be solved by introducing flexible side chains that are available in most docking methods.40, 56 We expect that a judicious choice of flexible side chains will effectively overcome this problem. It should be noted that the same solution is not effective to be applied to the initial unrefined structures. We have shown here that many of the initial unrefined structures contain incorrect secondary structure placements, including loops that obstruct the binding sites. The addition of flexible side chains cannot solve this problem, because the backbone structures are still incorrect.
Conclusions
Protein structure refinement remains a highly challenging task in the field of computational structural biology. One way to tackle this problem is to focus our target on regions that tend to deviate from the native structure. In here, we are presenting a new protocol for structure refinement that targets the ligand-binding site. Through ligand-binding site structural studies, we have shown previously that similar binding site structures are observed in non-homologous protein structures.23–25 Based on this, we demonstrate successful cases of binding site predictions and careful template selections that lead to effective ligand-binding site refinements through MD simulations with restraints. Our results show consistent improvements in easy, medium, and hard targets. Furthermore, the quality of refined ligand-binding site structures is confirmed through consistent improvement of ligand docking modes compared to the initial unrefined models, in 37 out of 40 cases.
The protocol presented here has a great potential to significantly refine ligand-binding site structures from computationally predicted protein models. As a result, we expect that it will be a valuable tool for docking experiments/virtual screening that do not have experimentally solved structures. A limitation of this protocol is that the local ligand-binding site structure of the refined model is highly correlated to the top template binding site structure. This does not account for the effect of induced fit, where different ligands can induce changes in the ligand-binding site structure. Another limitation is that hard targets, where their initial unrefined structures have more than 6-Å RMSD, can lead to unproductive refinements.
At a time when there are abundant high-resolution experimental protein structures and rapid advances in computational structure prediction tools, our method efficiently harnesses experimental data to improve modeled protein structures. While there is still room to improve our approach, strategically targeting ligand-binding site structure makes this method attractive for structure-based virtual screening. Research in drug discovery seldom use homology models to run virtual screening, because of structural quality concerns. By introducing our structure refinement protocol, we hope to encourage more researchers to use homology models for virtual screening, whenever experimental structures are not available.
Supplementary Material
ACKNOWLEDGEMENT
This work has been supported by NIH GM126140 and XSEDE MCB070009.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge.
Methods; Table S1, List of predicted structures; Table S2–S3, Structures used for force constant optimization; Table S4, Refinement of structures with bound ligands; Figure S1, Protein Cα-RMSD values comparing initial and refined model structures against the experimental structures; Figures S2, Aligned structures of initial and refined models on the crystal structures; Figure S3, Ligand-binding site Cα-RMSD improvements as a function of template coverage of binding site residues; Figures S4, Ligand binding modes obtained from docking are compared to the crystal structures.
The authors declare no competing financial interests.
References
- 1.Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE, The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daga PR; Patel RY; Doerksen RJ, Template-based protein modeling: recent methodological advances. Curr Top Med Chem 2010, 10, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodwin S; McPherson JD; McCombie WR, Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet 2016, 17, 333–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy SE; Myers RM, Advancements in Next-Generation Sequencing. Annu Rev Genomics Hum Genet 2016, 17, 95–115. [DOI] [PubMed] [Google Scholar]
- 5.Dukka BKC, Recent advances in sequence-based protein structure prediction. Brief Bioinform 2017, 18, 1021–1032. [DOI] [PubMed] [Google Scholar]
- 6.Abriata LA; Tamo GE; Monastyrskyy B; Kryshtafovych A; Dal Peraro M., Assessment of hard target modeling in CASP12 reveals an emerging role of alignment-based contact prediction methods. Proteins 2018, 86 Suppl 1, 97–112. [DOI] [PubMed] [Google Scholar]
- 7.Schaarschmidt J; Monastyrskyy B; Kryshtafovych A; Bonvin A, Assessment of contact predictions in CASP12: Co-evolution and deep learning coming of age. Proteins 2018, 86 Suppl 1, 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feig M; Mirjalili V, Protein structure refinement via molecular-dynamics simulations: What works and what does not? Proteins 2016, 84 Suppl 1, 282–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bordogna A; Pandini A; Bonati L, Predicting the accuracy of protein-ligand docking on homology models. J Comput Chem 2011, 32, 81–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wieman H; Tondel K; Anderssen E; Drablos F, Homology-based modelling of targets for rational drug design. Mini Rev Med Chem 2004, 4, 793–804. [PubMed] [Google Scholar]
- 11.Spyrakis F; Cavasotto CN, Open challenges in structure-based virtual screening: Receptor modeling, target flexibility consideration and active site water molecules description. Arch Biochem Biophys 2015, 583, 105–19. [DOI] [PubMed] [Google Scholar]
- 12.Sperandio O; Mouawad L; Pinto E; Villoutreix BO; Perahia D; Miteva MA, How to choose relevant multiple receptor conformations for virtual screening: a test case of Cdk2 and normal mode analysis. Eur Biophys J 2010, 39, 1365–72. [DOI] [PubMed] [Google Scholar]
- 13.Bolstad ES; Anderson AC, In pursuit of virtual lead optimization: the role of the receptor structure and ensembles in accurate docking. Proteins 2008, 73, 566–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erickson JA; Jalaie M; Robertson DH; Lewis RA; Vieth M, Lessons in molecular recognition: The effects of ligand and protein flexibility on molecular docking accuracy. J Med Chem 2004, 47, 45–55. [DOI] [PubMed] [Google Scholar]
- 15.Feig M, Computational protein structure refinement: Almost there, yet still so far to go. Wiley Interdiscip Rev ComputMol Sci 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mirjalili V; Feig M, Protein Structure Refinement through Structure Selection and Averaging from Molecular Dynamics Ensembles. J Chem Theory Comput 2013, 9, 1294–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J; Liang Y; Zhang Y, Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure 2011, 19, 1784–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park H; DiMaio F; Baker D, The Origin of Consistent Protein Structure Refinement from Structural Averaging. Structure 2015, 23, 1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng QY; Joung I; Lee J, A Simple and Efficient Protein Structure Refinement Method. Journal of Chemical Theory and Computation 2017, 13, 5146–5162. [DOI] [PubMed] [Google Scholar]
- 20.Mirjalili V; Noyes K; Feig M, Physics-based protein structure refinement through multiple molecular dynamics trajectories and structure averaging. Proteins 2014, 82 Suppl 2, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gutteridge A; Thornton J, Conformational change in substrate binding, catalysis and product release: an open and shut case? FEBS Lett 2004, 567, 67–73. [DOI] [PubMed] [Google Scholar]
- 22.Gaudreault F; Chartier M; Najmanovich R, Side-chain rotamer changes upon ligand binding: common, crucial, correlate with entropy and rearrange hydrogen bonding. Bioinformatics 2012, 28, i423–i430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee HS; Im W, G-LoSA: An efficient computational tool for local structure-centric biological studies and drug design. Protein Sci 2016, 25, 865–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee HS; Im W, Ligand binding site detection by local structure alignment and its performance complementarity. J Chem Inf Model 2013, 53, 2462–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee HS; Im W, G-LoSA for Prediction of Protein-Ligand Binding Sites and Structures. Methods Mol Biol 2017, 1611, 97–108. [DOI] [PubMed] [Google Scholar]
- 26.Lee HS; Im W, Identification of Ligand Templates using Local Structure Alignment for Structure-Based Drug Design. Journal of Chemical Information and Modeling 2012, 52, 2784–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Govindaraj RG; Brylinski M, Comparative assessment of strategies to identify similar ligand-binding pockets in proteins. BMC Bioinformatics 2018, 19, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao M; Skolnick J, APoc: large-scale identification of similar protein pockets. Bioinformatics 2013, 29, 597–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shulman-Peleg A; Nussinov R; Wolfson HJ, SiteEngines: recognition and comparison of binding sites and protein-protein interfaces. Nucleic Acids Res 2005, 33, W337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hartshorn MJ; Verdonk ML; Chessari G; Brewerton SC; Mooij WT; Mortenson PN; Murray CW, Diverse, high-quality test set for the validation of protein-ligand docking performance. J Med Chem 2007, 50, 726–41. [DOI] [PubMed] [Google Scholar]
- 31.Roy A; Kucukural A; Zhang Y, I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 2010, 5, 725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khazanov NA; Carlson HA, Exploring the composition of protein-ligand binding sites on a large scale. PLoS Comput Biol 2013, 9, e1003321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Derigs U, The shortest augmenting path method for solving assignment problems-motivation and computational experience. Annals of Operations Research 1985, 4, 57–102. [Google Scholar]
- 34.Jo S; Kim T; Iyer VG; Im W, CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 2008, 29, 1859–65. [DOI] [PubMed] [Google Scholar]
- 35.Lee J; Cheng X; Swails JM; Yeom MS; Eastman PK; Lemkul JA; Wei S; Buckner J; Jeong JC; Qi Y; Jo S; Pande VS; Case DA; Brooks CL 3rd; MacKerell AD Jr.; Klauda JB; Im W, CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. JChem Theory Comput 2016, 12, 405–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys 1983, 79, 926–935. [Google Scholar]
- 37.Allen MP, D. J. T., Computer Simulations of Liquids. Clarendon Press: Oxford, 1987. [Google Scholar]
- 38.Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmuller H; MacKerell AD Jr., CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang LP; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS, OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol 2017, 13, e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trott O; Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010, 31, 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen J; Brooks CL 3rd, Can molecular dynamics simulations provide high-resolution refinement of protein structure? Proteins 2007, 67, 922–30. [DOI] [PubMed] [Google Scholar]
- 42.Jagielska A; Wroblewska L; Skolnick J, Protein model refinement using an optimized physics-based all-atom force field. Proc Natl Acad Sci US A 2008, 105, 8268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee MS; Olson MA, Assessment of Detection and Refinement Strategies for de novo Protein Structures Using Force Field and Statistical Potentials. J Chem Theory Comput 2007, 3, 312–24. [DOI] [PubMed] [Google Scholar]
- 44.Fan H; Mark AE, Refinement of homology-based protein structures by molecular dynamics simulation techniques. Protein Sci 2004, 13, 211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raval A; Piana S; Eastwood MP; Dror RO; Shaw DE, Refinement of protein structure homology models via long, all-atom molecular dynamics simulations. Proteins 2012, 80, 2071–9. [DOI] [PubMed] [Google Scholar]
- 46.Heo L; Feig M, Experimental accuracy in protein structure refinement via molecular dynamics simulations. Proc Natl Acad Sci U S A 2018, 115, 13276–13281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y; Skolnick J, Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–10. [DOI] [PubMed] [Google Scholar]
- 48.Xu J; Zhang Y, How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 2010, 26, 889–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woo EJ; Marshall J; Bauly J; Chen JG; Venis M; Napier RM; Pickersgill RW, Crystal structure of auxin-binding protein 1 in complex with auxin. EMBO J 2002, 21, 2877–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phan J; Koli S; Minor W; Dunlap RB; Berger SH; Lebioda L, Human thymidylate synthase is in the closed conformation when complexed with dUMP and raltitrexed, an antifolate drug. Biochemistry 2001, 40, 1897–902. [DOI] [PubMed] [Google Scholar]
- 51.Bursulaya BD; Totrov M; Abagyan R; Brooks CL 3rd, Comparative study of several algorithms for flexible ligand docking. J Comput Aided Mol Des 2003, 17, 755–63. [DOI] [PubMed] [Google Scholar]
- 52.Olivero AG; Eigenbrot C; Goldsmith R; Robarge K; Artis DR; Flygare J; Rawson T; Sutherlin DP; Kadkhodayan S; Beresini M; Elliott LO; DeGuzman GG; Banner DW; Ultsch M; Marzec U; Hanson SR; Refino C; Bunting S; Kirchhofer D, A selective, slow binding inhibitor of factor VIIa binds to a nonstandard active site conformation and attenuates thrombus formation in vivo. J Biol Chem 2005, 280, 9160–9. [DOI] [PubMed] [Google Scholar]
- 53.Luic M; Koellner G; Yokomatsu T; Shibuya S; Bzowska A, Calf spleen purine-nucleoside phosphorylase: crystal structure of the binary complex with a potent multisubstrate analogue inhibitor. Acta Crystallogr D Biol Crystallogr 2004, 60, 1417–24. [DOI] [PubMed] [Google Scholar]
- 54.Reiling KK; Endres NF; Dauber DS; Craik CS; Stroud RM, Anisotropic dynamics of the JE-2147-HIV protease complex: drug resistance and thermodynamic binding mode examined in a 1.09 A structure. Biochemistry 2002, 41, 4582–94. [DOI] [PubMed] [Google Scholar]
- 55.Dymock BW; Barril X; Brough PA; Cansfield JE; Massey A; McDonald E; Hubbard RE; Surgenor A; Roughley SD; Webb P; Workman P; Wright L; Drysdale MJ, Novel, potent small-molecule inhibitors of the molecular chaperone Hsp90 discovered through structure-based design. J Med Chem 2005, 48, 4212–5. [DOI] [PubMed] [Google Scholar]
- 56.Wong CF, Flexible receptor docking for drug discovery. Expert Opin Drug Discov 2015, 10, 1189–200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.