

Star-like block copolymer nanoreactor enables the crafting of dual-shelled perovskite nanocrystals with enhanced stabilities.
Abstract
The past few years have witnessed rapid advances in the synthesis of high-quality perovskite nanocrystals (PNCs). However, despite the impressive developments, the stability of PNCs remains a substantial challenge. The ability to reliably improve stability of PNCs while retaining their individual nanometer size represents a critical step that underpins future advances in optoelectronic applications. Here, we report an unconventional strategy for crafting dual-shelled PNCs (i.e., polymer-ligated perovskite/SiO2 core/shell NCs) with exquisite control over dimensions, surface chemistry, and stabilities. In stark contrast to conventional methods, our strategy relies on capitalizing on judiciously designed star-like copolymers as nanoreactors to render the growth of core/shell NCs with controlled yet tunable perovskite core diameter, SiO2 shell thickness, and surface chemistry. Consequently, the resulting polymer-tethered perovskite/SiO2 core/shell NCs display concurrently a stellar set of substantially improved stabilities (i.e., colloidal stability, chemical composition stability, photostability, water stability), while having appealing solution processability, which are unattainable by conventional methods.
INTRODUCTION
Perovskites with a generic chemical formula ABX3 [A = monovalent organic cation, such as CH3NH3+, CH(NH2)2+, or Cs; B2+ = Pb2+ or Sn2+; and X = Cl, Br, or I] have been widely recognized as an emerging material for photovoltaics (1), light emitting dioxides (LEDs) (2), and scintillators (3) due to their outstanding optical, excitonic, and charge transport properties (4). The practical application of perovskite materials is, however, largely hindered by their intrinsic structural instability due to their ionic crystal characteristics, thereby leading to degradation upon the exposure to external stimuli, such as moisture, polar solvents, ultraviolet (UV) light, and oxygen (5). Their inherent instability becomes a more pronounced issue as the dimension of perovskites reduces to the nanometer length scale as a result of the increased surface-to-volume ratio (6). Seminal works on the solution-based synthesis of perovskite nanocrystals (PNCs) have been reported (7–10), yielding high-quality PNCs with uniform size, high photoluminescence (PL) quantum yield, narrow full width at half maximum of emission, and wide tunable bandgap over the entire visible range (11, 12). Nonetheless, the ability to greatly improve the stability of PNCs remains a prerequisite for creating solution-processed, high-performance PNC-based optoelectronic devices such as solar cells (13), LEDs (14), and lasers (15). In this context, several approaches have emerged to enhance the stability of PNCs, including the incorporation of PNCs in silica, silica/alumina monolith, or amorphous alumina (16–20), in situ formation of PNCs within mesoporous silica (21, 22), swelling-deswelling microencapsulation in a polymer matrix (23), stabilization by surface ligands (13, 24, 25), and passivation with protective ligands (26, 27). It is notable that the routes noted above have inherent limitations associated with the formation of complex hybrid materials as opposed to well-defined structures (e.g., core/shell) and decreased colloidal dispersity and solution processability in nonpolar solvents.
Synthesis of PNCs, particularly organolead halide PNCs, necessitates the use of highly polar solvents such as dimethylformamide (DMF), owing to the poor solubility of lead halide in most organic solvents. This likely introduces surface defects or intrinsic halide vacancies to PNCs (23), thereby leading to their increased propensity to decompose when exposed to air and/or polar solvents (28). Furthermore, PNCs synthesized by conventional approaches (7–10) generally involve the use of weak protic solvent (e.g., alcohols) or polar solvent as precipitant during the purification process, which inevitably and negatively affects the structural integrity of PNCs (29) because of dissolution of PNCs induced by the polar solvent. This dissolution, in particular, makes it challenging to obtain high-quality organolead halide PNCs as trace amounts of polar solvent is sufficient to decompose the PNCs (28). Moreover, polar solvents introduced during the purification process can result in the loss of PL and decreased colloidal stability due to highly dynamic binding (i.e., association-dissociation) of short-chain ligands (e.g., octylamine, oleylamine, and/or oleic acid) on the surface of PNCs (30). An effective synthetic strategy for creating organolead halide PNCs that overcomes all of the stability issues described above while having excellent solution processability, controlled dimensions, and tunable surface chemistry on a single nanocrystal (NC) level remains elusive (31).
Here, in an attempt to address the stability challenges, we developed an unconventional, robust strategy by capitalizing on a class of rationally designed star-like triblock copolymers as nanoreactors to create dual-shelled organolead halide PNCs with delicate control over dimensions, surface chemistry, and, more importantly, an array of greatly improved stabilities. These PNCs are coated with inorganic SiO2 shells, followed by intimate and permanent ligating with polymer shells, that is, polymer-capped perovskite/SiO2 core/shell NCs. Two types of multiarm star-like amphiphilic triblock copolymers, namely, poly(4-vinylpyridine)-block-poly(tert-butyl acrylate)-block-poly(ethylene oxide) (denoted P4VP-b-PtBA-b-PEO) and poly(4-vinylpyridine)-block-poly(tert-butyl acrylate)-block-polystyrene (denoted P4VP-b-PtBA-b-PS), are first synthesized by combining two sequential atom transfer radical polymerizations (ATRP) with a click reaction and by performing three consecutive ATRP, respectively. Subsequently, these amphiphilic star-like P4VP-b-PtBA-b-PEO and P4VP-b-PtBA-b-PS are used as nanoreactors to craft PEO- and PS-tethered organolead halide perovskite/SiO2 core/shell NCs, respectively, via strong coordination interaction between precursors with hydrophilic blocks of star-like triblock copolymers. Specifically, methylammonium lead bromide (MAPbBr3) is selected as a representative organolead halide perovskite to demonstrate the effectiveness of our nanoreactor strategy. Compared with well-established conventional hot injection (7) and ligand-assisted reprecipitation (LARP) (8) approaches, the star-like triblock copolymer nanoreactor strategy confers a collection of improved stabilities (i.e., colloidal, chemical composition, photo, and water stabilities) and tunable surface chemistry while retaining good solution processability as a direct consequence of dual coating of organolead halide PNC core with a uniform inorganic SiO2 shell, followed by a polymeric shell on a single NC level.
RESULTS
Our amphiphilic star-like triblock copolymer nanoreactor strategy carries several noteworthy advantages over the traditional routes. First, it eliminates the use of either high–boiling point solvents [e.g., octadecene used in the hot injection method for synthesizing all-inorganic PNCs (7)] or nonvolatile solvents [e.g., DMF used in the LARP method (8) for preparing organic-inorganic hybrid PNCs] for synthesis of PNCs. All solvents (i.e., chloroform, isopropanol alcohol, and methanol) involved in our nanoreactor strategy have low boiling point, are volatile, and have low toxicity, which are beneficial for potential device fabrication at low temperature, dispensing with the need for high-temperature treatment by eliminating the use of high–boiling point or nonvolatile solvents as in conventional approaches. Second, it affords an amine-free route to controlled synthesis of high-quality PNCs, as amines have been found in several recent studies to drive the dissolution of PNCs and their transformation into an insulating phase (32). Third, it offers a practical route to purifying PNCs by solely using a nonpolar solvent of extremely low polarity [e.g., n-hexane used as a precipitant in our strategy as opposed to function as a solvent in conventional approaches (7, 8)], thereby minimizing the potential decomposition of PNCs during the purification process, preserving their structural integrity, and, ultimately, retaining the PL of PNCs. This is in sharp contrast to conventional methods [e.g., hot injection (7) and LARP (8)] with which as-synthesized PNCs are typically stabilized with short organic ligands (e.g., octylamine, oleylamine, and/or oleic acid) having high solubility in a wide range of nonpolar solvents. As a result of high organic solubility, conventional methods require the use of polar solvents (e.g., acetone or butanol) as precipitants for PNC purification, which can lead to the degradation of PNCs, and thus quench their emission. Fourth, it renders tunable surface chemistry of PNCs (i.e., surface capping ligands can be readily regulated from PEO to hydrophobic PS), which has not been demonstrated by conventional methods. Notably, as star-like triblock copolymers are synthesized by either sequential ATRP or a combination of ATRP with click reaction, our strategy can be readily extended to graft virtually any functional polymer (e.g., stimuli responsive) as the outer block of star-like triblock copolymers by these reactions, thereby further imparting the tailoring of surface chemistry of PNCs (e.g., forming an exciting variety of stimuli-responsive polymer-capped PNC/SiO2 NCs with temperature-sensitive, pH-sensitive, and light-responsive characteristics). Fifth, it imparts excellent solution processability and long-term dispersion of perovskite/SiO2 core/shell NCs due to intimate and permanent tethering of polymer ligands (PEO or PS) on the surface of perovskite/SiO2 NCs as a result of original covalent bonding between outer PEO (or PS) block and star-like P4VP-b-PtBA. This contrasts with the issue of ligand dissociation over a long period of time that leads to the aggregation of NCs, as largely encountered in copious past work. Sixth, it enables the synthesis of organolead halide PNCs through an in situ rapid conversion of lead halide NCs. This provides the possibility of crafting a variety of organolead halide PNCs under ambient condition via preferential coordination interaction between the metal moieties of precursors of organolead halide PNCs of interest and the pyridyl groups of P4VP block in star-like triblock copolymers. The advantageous attributes of the nanoreactor strategy noted above endow a set of outstanding stabilities to PNCs. These include colloidal stability owing to the versatile surface chemistry offered by the outer block of the star-like triblock copolymer nanoreactor; chemical composition, photo-, and water stabilities due to the dual protection rendered by inorganic SiO2 shell and hydrophobic organic PS shell; as well as structural stability attributed to the elimination of DMF or aliphatic amines used in the synthesis.
Starting from β-cyclodextrin (β-CD) with 21 hydroxyl groups, the addition of 2-bromo isobutyl bromide yielded nearly 100% conversion of these hydroxyl groups into ω-bromide terminals (i.e., ATRP initiation sites) as confirmed by 1H and 13C nuclear magnetic resonance (NMR) (fig. S1, A and B; bromination efficiency calculation can be found in the Supplementary Materials). The resulting brominated β-CD (denoted 21-Br-β-CD; upper second panel in Fig. 1) was then used as a macroinitiator to prepare multiarm star-like amphiphilic triblock copolymers using copper (I) chloride (CuCl) and copper (II) chloride (CuCl2) as the catalysts. It is worth noting that the commonly used catalyst for metal-mediated ATRP [e.g., copper (I) bromide (CuBr) and copper (II) bromide CuBr2] does not work well for preparing copolymer containing pyridyl groups due to the significant tendency for the coupling reaction between ω-bromide terminals of 21-Br-β-CD and pyridyl groups of P4VP during polymerization. The use of CuBr or CuBr2 as catalysts in ATRP resulted in not only low solubility of copolymer in organic solvent but also intermolecular coupling (fig. S1C), leading to ill-defined polymer gel or polymer network. In addition, instead of using N,N,N′,N′′,N′′-pentamethyldiethylenetriamine or 2,2′-bipyridyl [commonly used ligands for ATRP of tert-butyl acrylate (tBA) and styrene (St)], the preparation of a star-like copolymer via chain extension reaction (i.e., growth of the additional block) or copper(I)-catalyzed alkyne-azide cycloaddition (i.e., CuAAC) click reaction was achieved in the presence of tris[2-(dimethylamino)ethyl]amine (Me6TREN), which served as ligand owing to its bridged cyclam structure and strong coordination ability to a copper catalyst compared with the pyridine groups in P4VP block, thereby leading to high equilibrium constant of the Cu(I)Cl/Me6TREN complex (i.e., high polymerization rate). The substitution of CuBr and CuBr2 with CuCl and CuCl2 substantially suppressed the tendency for intermolecular coupling, leading to a well-defined star-like P4VP homopolymer (upper third panel in Fig. 1 and fig. S1D). The amphihilic star-like diblock copolymer P4VP-b-PtBA was then obtained by the second ATRP of the tBA monomer (upper fourth panel in Fig. 1). The terminal Cl groups were then converted into azide functionalities (N3) by reacting with NaN3, yielding star-like P4VP-b-PtBA-N3. Separately, alkyne-terminated PEO methyl ether (mPEO-≡) was prepared by nucleophilic substitution of the hydroxyl group on mPEO. The click reaction between star-like P4VP-b-PtBA-N3 and mPEO-≡ using Cu(I)Cl/Cu(II)Cl2 and Me6TREN as catalysts and ligands, respectively, yielded the amphiphilic star-like triblock copolymer P4VP-b-PtBA-b-PEO (fig. S2, A and B).
Fig. 1. Stepwise representation of the synthetic route to PS-capped MAPbBr3/SiO2 core/shell NCs and PEO-capped MAPbBr3/SiO2 core/shell NCs by exploiting star-like P4VP-b-PtBA-b-PS and P4VP-b-PtBA-b-PEO as nanoreactors, respectively.
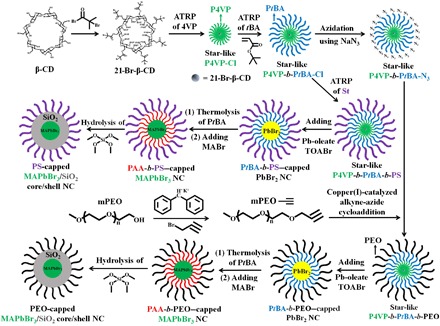
CD, cyclodextrin; BMP, 2-bromo-2-methylpropionate; and TOABr, tetraoctylammonium bromide.
Notably, all star-like polymers (i.e., P4VP homopolymer, P4VP-b-PtBA diblock copolymer, and P4VP-b-PtBA-b-PEO triblock copolymer) exhibited the monomodal gel permeation chromatograph (GPC) traces, suggesting there was no intermolecular coupling or residual homopolymers in the purified products. The successful formation of the unimolecular star-like P4VP-b-PtBA-b-PEO triblock copolymer (lower fourth panel in Fig. 1) in organic solvent (e.g., DMF) was further confirmed by dynamic light scattering (DLS), showing a unimolecular micelle with a narrow size distribution (fig. S2, C and D).
Figure 1 depicts the synthetic route to organolead halide perovskite MAPbBr3/SiO2 core/shell NCs capped with different polymer ligands by capitalizing on the respective judiciously designed amphiphilic multiarm star-like triblock copolymers as nanoreactors (i.e., star-like P4VP-b-PtBA-b-PEO for PEO-capped MAPbBr3/SiO2 core/shell NCs, and star-like P4VP-b-PtBA-b-PS for PS-capped MAPbBr3/SiO2 core/shell NCs).
Rationally designed star-like P4VP-b-PtBA-b-PEO triblock copolymer was then used as a nanoreactor for crafting PEO-capped MAPbBr3/SiO2 core/shell NCs. Star-like P4VP-b-PtBA-b-PEO was dissolved in the mixed solvents of chloroform, isopropanol, and methanol (the volume ratio of chloroform/isopropanol/methanol = 20:5:3) (see Materials and Methods), forming a stable unimolecular micelle with fully expanded P4VP chains and collapsed PtBA and PEO chains. This heterogeneous swelling occurred because of differences in solubility: Chloroform is a good solvent for all P4VP, PtBA, and PEO, whereas isopropanol and methanol are poor solvents for both PtBA and PEO but good solvents for P4VP. Under argon protection, presynthesized lead oleate was introduced into the solution containing chloroform, isopropanol, methanol, and star-like P4VP-b-PtBA-b-PEO with vigorous stirring, and preferentially formed a complex with the inner P4VP blocks owing to strong coordination interaction between the pyridyl groups of P4VP and the metal moieties of precursors (33). Notably, no such interaction occurred between the lead oleate and PtBA or PEO blocks as neither block has any functional groups for coordination. Subsequent introduction of tetraoctylammonium bromide (TOABr; 1 M in chloroform) prompted the ionic metathesis reaction (7) in the space populated by the P4VP blocks, where lead oleate previously selectively partitioned in the P4VP blocks was converted into lead bromide NC (PbBr2 NC) capped by PtBA-b-PEO (lower third panel in Fig. 1).
The thermolysis of the PtBA block (33) into poly(acrylic acid) (PAA) yielded PAA-b-PEO–tethered PbBr2 NC. Note that one advantage of the synthesis of MAPbBr3 PNC using presynthesized PbBr2 NCs as template is that the thermolysis necessary to convert PtBA to PAA block has to be performed at high temperature (i.e., 200°C), which can decompose most organolead perovskite materials (fig. S3A). In contrast, PbBr2 NC was found to survive this harsh environment. Just as bulk organolead halide perovskite can easily be formed by reacting lead halide with the corresponding organoammonium halide, lead halide NCs are expected to be readily transformed into organolead halide PNCs. Hence, PAA-b-PEO–capped PbBr2 NCs (fig. S3B) were in situ turned into PAA-b-PEO–capped MAPbBr3 NCs by reacting with methylammonium bromide (MABr). Passivation of PNC with the SiO2 shell was then accomplished via in situ hydrolysis of tetramethyl orthosilicate (TMOS) in the compartment occupied by the PAA blocks (lower first panel in Fig. 1), whose chain length dictates the thickness of the SiO2 shell (16).
Figure 2A shows a representative transmission electron microscopy (TEM) image of PEO-capped MAPbBr3/SiO2 core/shell NCs with an average MAPbBr3 core diameter of 6.1 ± 0.3 nm and a SiO2 shell thickness of 8.9 ± 0.4 nm, crafted by exploiting star-like triblock copolymer P4VP-b-PtBA-b-PEO as nanoreactor (Sample A in Table 1). The core/shell nanostructure is evident owing to the differential electron scattering ability of MAPbBr3 and SiO2. We note that as a living free radical polymerization technique, ATRP affords excellent control over the molecular weight (MW) of polymer (34). Thus, it enables the synthesis of star-like triblock copolymers with tunable MWs and narrow MW distribution (i.e., low polydispersity index) of P4VP and PtBA blocks, which in turn yield uniform PEO-capped MAPbBr3/SiO2 core/shell NCs with the MAPbBr3 core diameter and SiO2 shell thickness readily tailored by varying the lengths of the P4VP and PtBA blocks, respectively. Figure S3C shows PEO-capped MAPbBr3/SiO2 core/shell NCs with increased dimension (MAPbBr3 core diameter = 8.0 ± 0.3 nm and SiO2 shell thickness = 9.8 ± 0.4 nm; Sample B in Table 1) as the MWs (i.e., chain length) of both P4VP and PtBA increased.
Fig. 2. TEM images of PEO-capped MAPbBr3/SiO2 core/shell NCs.
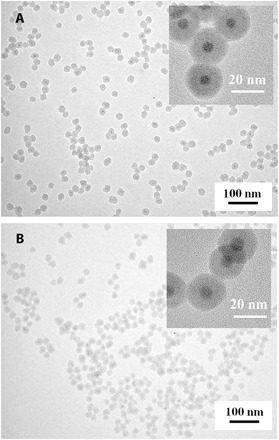
(A) MAPbBr3 core diameter of 6.1 ± 0.3 nm and SiO2 shell thickness of 8.9 ± 0.4 nm, crafted by capitalizing on star-like P4VP-b-PtBA-b-PEO (Sample A in Table 1) as nanoreactor, and (B) MAPbBr3 core diameter of 6.8 ± 0.3 nm and SiO2 shell thickness of 8.0 ± 0.3 nm, crafted by using star-like P4VP-b-PtBA-b-PS (Sample C in Table 1) as nanoreactor.
Table 1. Summary of MWs of amphiphilic star-like P4VP-b-PtBA-b-PEO and P4VP-b-PtBA-b-PS triblock copolymers and the corresponding dimension of polymer-capped MAPbBr3/SiO2 core/shell NCs.
| Samples |
Mn,P4VP* (kg/mol) |
Mn,PtBA† (kg/mol) |
Mn,PS‡ (kg/mol) |
Mn,PEO§ (kg/mol) | PDI§ |
Dimensions of MAPbBr3/SiO2 core/shell NCs (core diameter/shell thickness)║ (nm) |
| Sample A | 8000 | 14,600 | – | 5000 | 1.15 | 6.1 ± 0.3/8.9 ± 0.4 |
| Sample B | 9300 | 21,600 | – | 5000 | 1.18 | 8.0 ± 0.3/9.8 ± 0.4 |
| Sample C | 8500 | 13,800 | 5,000 | – | 1.19 | 6.8 ± 0.3/8.0 ± 0.3 |
| Sample D | 7800 | 15,600 | 10,000 | – | 1.19 | 6.0 ± 0.4/8.9 ± 0.5 |
*Number average MW of the inner P4VP block (Mn,P4VP) was calculated from 1H NMR data. The calculation method was adapted from a previous work (33).
†Number average MW, Mn of each PtBA block calculated on the basis of 1H NMR data from the MW difference between PtBA block and P4VP block. The intermediate PtBA block can be calculated by the following equation , where Mn,P4VP and M4VP are MWs of the inner P4VP block and the 4VP monomer, respectively. Ab and Aa are the integral area of the protons in the pyridine group of P4VP block and the integral area of methyl protons in the tert-butyl group of PtBA block, respectively. Mn,PtBA and Mn,P4VP are MWs of the intermediate PtBA block and the inner P4VP block, respectively. Ae and Ab are the integral area of methyl protons in the tert-butyl group of PtBA block and the integral area of protons in the pyridine group of P4VP block, respectively. MtBA and M4VP are MWs of the tBA monomer and the 4VP monomer, respectively.
‡Number average MW of the outer PS block was calculated from 1H NMR data based on the following equation , where Mn,PS, Mn,PtBA, and Mn,P4VP are the MWs of the outer PS block, intermediate PtBA block, and inner P4VP block, respectively. Aa and Ab are the integral area of methyl protons in the tert-butyl group of PtBA block and the integral area of protons in the phenyl group and the pyridine group of PS and P4VP blocks, respectively. MSt, MtBA, and MP4VP are MWs of St, tBA, and 4VP monomers, respectively.
§PDI was recorded by DMF GPC.
║The core diameter and shell thickness of core/shell NCs were determined by performing image analysis on TEM images of MAPbBr3/SiO2 core/shell NCs.
To demonstrate the importance of the unimolecular micellar nature of star-like P4VP-b-PtBA-b-PEO in the formation of MAPbBr3/SiO2 NCs, a control experiment using a linear triblock copolymer P4VP-b-PtBA-b-PEO counterpart to template the growth of MAPbBr3 PNCs was conducted. It is not unexpected that MAPbBr3 PNCs were not observed. Instead, large particles with poor colloidal stability were formed, due to the failure of forming a stable unimolecular nanoreactor in the mixed solvents upon the addition of perovskite precursors (fig. S3D).
Figure 3A compares the x-ray diffraction (XRD) profiles of MAPbBr3/SiO2 core/shell NCs capped with PEO, MAPbBr3 core NCs capped with PAA-b-PEO, and pure bulk SiO2 as control. Prior to the growth of the SiO2 shell, the position of the characteristic peaks of the XRD pattern of PAA-b-PEO–capped MAPbBr3 core NC is identical to the bulk MAPbBr3 perovskite (fig. S4A) (9, 16), indicating the successful synthesis of MAPbBr3 NCs. The pronounced broadening of the diffraction peak further confirms the nanocrystallinity of the as-synthesized MAPbBr3 core NC. After coating with the SiO2 shell, the diffraction profile clearly presents a superposition of diffraction patterns of MAPbBr3 NCs and SiO2, while the peak positions of the MAPbBr3 core remains unchanged, signifying that its structural integrity was preserved. This substantiates the success in synthesizing MAPbBr3/SiO2 core/shell NCs. The UV-visible spectrum of PEO-capped MAPbBr3/SiO2 core/shell NCs displays a clear absorption peak at 525 nm (fig. S4B). The PL spectra before and after SiO2 coating are shown in Fig. 3B, where the emission peak of PNCs is located at 529 nm, and more importantly, no appreciable peak shift after the SiO2 shell growth was seen. This further corroborates the retention of the structural integrity during the SiO2 passivation process via in situ rapid hydrolysis of silicon alkoxide precursors. The PL quantum yields before and after the SiO2 coating are 86 and 88%, which are comparable to previous studies (8). The average decay time of MAPbBr3 NCs is increased from 17.6 ns (for PEO-b-PtBA–capped MAPbBr3 NCs) to 19.4 ns (for PEO-capped MAPbBr3/SiO2 core/shell NCs) after the SiO2 coating (Fig. 3E), signifying that a portion of the surface defects on MAPbBr3 PNC is passivated by the SiO2 shell.
Fig. 3. Structure and optical characterization of PEO-capped MAPbBr3/SiO2 core/shell NCs.
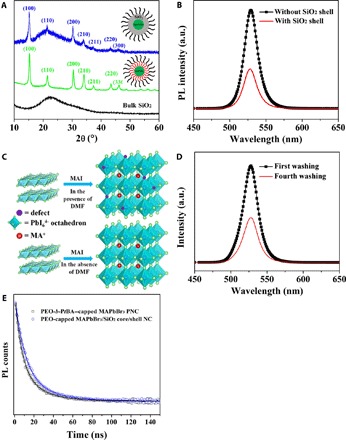
(A) XRD patterns of MAPbBr3/SiO2 core/shell NCs capped with PEO, MAPbBr3 core NCs capped with PAA-b-PEO, and pure bulk SiO2. (B) PL spectra of MAPbBr3 core NC before and after the SiO2 shell coating dispersed in toluene. a.u., arbitrary units. (C) Schematic illustration of direct conversion of PbI2 into MAPbI3. (D) Repeated washing of PEO-capped MAPbBr3/SiO2 core/shell NCs using n-hexane as a precipitant and toluene as a good solvent. The decrease in PL intensity in (B) and (D) is due to the change in the NC concentration. (E) Time-resolved PL decay traces of PEO-b-PtBA–capped MAPbBr3 NCs (τ1 = 7.6 ns, τ2 = 25.4 ns, and τave = 17.6 ns) and PEO-capped MAPbBr3/SiO2 core/shell NCs (τ1 = 9.8 ns, τ2 = 29.6 ns, and τave = 19.4 ns), where black and blue curves are the corresponding biexponential fits, respectively.
The success in the synthesis of MAPbBr3/SiO2 core/shell NCs provides inspiration for exploring the possibility of generalizing our amphiphilic star-like block copolymer strategy to craft iodide-containing PNCs, which are extremely sensitive to moisture and undergo rapid degradation even under ambient condition (24). For simplicity, star-like PAA-b-PEO was synthesized and used as a nanoreactor to direct the growth of methylammonium lead iodide (MAPbI3) PNCs. Similar to MAPbBr3, MAPbI3 PNCs were produced via in situ conversion of PEO-capped PbI2 NCs into the resulting MAPbI3 PNCs by mixing with methylammonium iodide. The XRD profile confirms the formation of MAPbI3 PNCs (fig. S4C, lower red profile). The TEM images of PEO-capped MAPbI3 PNCs and oleylamine– and oleic acid–cocapped MAPbI3 PNCs can be seen in fig. S4 (D and E), respectively. The stability of the as-synthesized PEO-capped MAPbI3 PNCs was largely improved (14 days; fig. S5A) in comparison with that of MAPbI3 PNCs prepared via the LARP route (8) in the glove box (36 hours; fig. S5B), where the latter experienced an even faster degradation in open air, resulting in an MAPbI3-H2O intermediate (white flocculent precipitates) (35) that displayed no characteristic absorption of MAPbI3 PNCs (fig. S5C). Although PbI2 has a good solubility in polar solvents, such as DMF, N-methyl-2-pyrrolidone (NMP), dimethyl sulfoxide (DMSO), and N,N-diethylformamide (DEF), it has a strong propensity to form PbI2-solvent intermediate in these polar solvents (fig. S6, A and B). The possible fast degradation can be attributed to the fact that MAPbI3 PNCs formed by the reaction of the PbI2-solvent intermediate with methylammonium iodide inevitably contain residual solvent molecules or intrinsic iodine vacancies in the crystal structure (28). These intrinsic halide vacancies, and vacancies generated due to the removal of solvent molecules during the process of formation of PNCs, could transform MAPbI3 PNCs into the MAPbI3-water complex in the presence of a trace amount of water from solvents or open air, leading to instantaneous degradation and the emission loss of PNCs (28). This accounts for the low stability of MAPbI3 PNCs prepared by the LARP method (8), which involves the use of DMF.
To probe the solvent effect on the stability of MAPbI3 PNCs, we investigated the influence of alcohols and common hydrocarbon solvents on the crystal structure of PbI2. Systematic XRD patterns confirmed that alcohols (e.g., methanol, ethanol, isopropanol) and common hydrocarbon solvents (e.g., toluene, n-hexane, heptane) do not have the ability to coordinate with PbI2 to form the intermediate; instead, PbI2 crystals are produced (i.e., the primary peak located at 12.7°; fig. S6, C to F). Hence, to decrease the defects in PNCs, it is desirable to avoid the use of solvents (e.g., DMF) that can coordinate with PbI2. Therefore, it is clear that the enhanced stability of as-synthesized MAPbI3 NCs via the star-like PAA-b-PEO diblock copolymer nanoreactor strategy at ambient conditions, which uses methanol as a solvent, is due partly to the fact that this strategy enables the crafting of PNCs in the absence of any organic solvents that can potentially form the PbI2-solvent intermediate, thereby eliminating the possible crystal defects (e.g., halide vacancies) responsible for the rapid degradation of PNCs. The direct transformation of PbI2 into MAPbI3 in our strategy without using DMF or other solvents that coordinate with PbI2 is illustrated in Fig. 3C.
Notably, star-like block copolymer (both diblock and triblock) nanoreactors offer not only a robust means of crafting organolead halide (e.g., MAPbBr3 and MAPbI3) PNCs but also a mild and nondestructive route to purifying them. It remains a grand challenge to translate the conventional NC purification method (e.g., using polar solvent as a precipitant and nonpolar solvent as a good solvent) to organolead halide PNCs synthesized by conventional methods. This is due to the highly unstable surfaces because of dynamic equilibrium of the association and dissociation of surface ligands and the strong decomposition tendency of organolead halide PNCs in several polar solvents owing to their ionic crystal nature (30). The polar solvent effect on the structural stability of MAPbBr3 and MAPbI3 PNCs is presented in fig. S7.
Recent density functional theory (DFT) calculations suggested that oxygen and water molecules, as well as OH−, have a strong tendency to bond to surface defects (e.g., halide vacancy and halide-ammonium vacancy) on the (010) plane of MAPbBr3 due to the formation of hydrogen bonds, which likely reduces the formation energy of other defects, thus accelerating the perovskite decomposition (36). On the basis of recent experimental results and DFT calculation (36), deliberate efforts to control the amount and type of alcohols should be taken during the purification of conventionally prepared PNCs to prevent their decomposition. In stark contrast, our star-like triblock copolymer nanoreactor strategy circumvents this issue by completely avoiding the use of any polar or protic antisolvents (i.e., acetone, methanol, ethanol, isopropanol, butanol, etc.) commonly used in conventional routes to PNCs (31). Because of a large unfavorable interaction between PEO and n-hexane (the PEO/n-hexane interaction parameter, χPEO/n-hexane = 1.7), n-hexane with dipole moment of 0.08 D and relative polarity of 0.0009 is an ideal destabilizing solvent for the PEO-capped PNC purification that preserves the structural integrity of PNCs (37).
The dipole moment and relative polarity of n-hexane are even lower than those of toluene (relative polarity = 0.099) (table S1), which is a widely used good solvent for PNCs synthesized using the conventional LARP method (8). Figure 3D presents the PL spectra of repeatedly washed, purified PEO-capped MAPbBr3/SiO2 core/shell NCs using toluene as a solvent and n-hexane as a precipitant. MAPbBr3/SiO2 NCs retain their inherent optical properties and display no appreciable emission wavelength shift, contrasting greatly to PNCs synthesized by conventional approaches, which show a large shift in emission wavelength (38).
Ligand exchange strategies have been widely used in metal chalcogenide quantum dots to tune the surface capping ligands. Unfortunately, these strategies cannot be extended to PNCs as ligand exchange involves the exposure of PNCs to harsh environments. For example, the use of protic solvents such as methanol can decompose PNCs because of the hydroxyl functional groups in protic solvents. Moreover, the ligand binding to the PNC surface is highly dynamic, which further makes surface functionalization of PNCs a daunting challenge (39). Our amphiphilic star-like block copolymer nanoreactor strategy renders the easy tailoring of surface chemistry of PNCs during the synthesis of star-like block copolymers. Specifically, hydrophobic PS-capped MAPbBr3/SiO2 core/shell NCs were crafted using star-like P4VP-b-PtBA-b-PS as nanoreactor prepared by three sequential ATRP as discussed below, and PEO-capped MAPbBr3/SiO2 core/shell NCs were created by using star-like P4VP-b-PtBA-b-PEO as nanoreactor synthesized by combining two consecutive ATRP with a click reaction, thereby dispensing with the need for ligand exchange and eliminating the associated decomposition issues noted above.
Star-like P4VP-b-PtBA-b-PS triblock copolymers were synthesized by consecutive ATRP of 4VP, tBA, and St monomers (middle fourth panel in Fig. 1). Monomodal GPC traces (fig. S1E) and DLS measurement (fig. S2E) substantiated the successful synthesis of star-like P4VP-b-PtBA-b-PS with well-defined MW and narrow MW distribution of each block. Star-like P4VP-b-PtBA-b-PS was then exploited as a nanoreactor to create PS-capped MAPbBr3/SiO2 core/shell NCs (all middle panels in Fig. 1) in a similar procedure to the preparation of PEO-capped MAPbBr3/SiO2 core/shell NCs. Figure 2B shows a representative TEM image of PS-capped MAPbBr3/SiO2 core/shell NCs with an MAPbBr3 core diameter of 6.8 ± 0.3 nm and an SiO2 shell thickness of 8.0 ± 0.3 nm (using Sample C in Table 1 as nanoreactor). Noticeably, the versatility of surface ligands tethered on the surface of PNCs by our nanoreactor strategy is virtually unlimited. We envision that any functional polymer (e.g., light responsive, thermal responsive, pH sensitive) can be grafted as the outer block of star-like block copolymers by ATRP, click reaction, or other polymerization techniques, for investigation into stimuli-responsive properties from surface ligands and the intrinsic optoelectronic properties of PNCs, as well as their synergistic or coupling properties. Moreover, these polymer-capped PNCs are likely to enable a good dispersion in the identical polymer matrix for a wide range of potential applications in optoelectronics, sensors, etc.
DISCUSSION
As our PNCs are dual-shelled, that is, passivated with a SiO2 shell and stably ligated with either PEO or PS chains, we now turn our attention to evaluate the stabilities of these polymer-capped core/shell NCs, including repeated solvent washing to represent the colloidal stability, mixing PNCs of different compositions to reflect the chemical composition stability, UV light irradiation to demonstrate the photostability, and exposing to moist air and immersing into water to manifest the moisture and water stability.
PS-capped MAPbBr3/SiO2 core/shell NCs were chosen to study the colloidal stability of PNCs by repetitive organic solvent washing. As discussed above, both organic-inorganic hybrid and all-inorganic PNCs prepared by conventional methods tend to lose surface capping ligands (e.g., oleic acid, oleylamine, or both) due to the dynamic association and dissociation of surface short-chain ligands (38), leading to the aggregation of PNCs, and thus the change in PL intensity and peak position. However, it is imperative to perform repetitive washing involving the use of good solvents and precipitants to purify as-synthesized PNCs. Hence, it requires much effort during the purification process to obtain the PNC colloidal solution while preventing the decomposition and aggregation of PNCs, particularly for iodide-containing PNCs. In marked contrast, our star-like P4VP-b-PtBA-b-PS triblock copolymer nanoreactor strategy affords the permanent ligating of PS chains, as discussed above, on the surface of MAPbBr3/SiO2 core/shell NCs, thereby conferring their great colloidal stability. Fig. 4A demonstrates that PS-capped MAPbBr3/SiO2 core/shell NCs did not exhibit appreciable change in the PL peak position even after four times of washing. The decreased PL intensity of the washed PS-capped MAPbBr3/SiO2 NCs is because of change of NC toluene concentration during the washing process. Oppositely, MAPbBr3 PNCs prepared by the LARP method had a pronounced red shift of PL peak due to their aggregation induced by the loss of surface capping ligands (i.e., octylamine and/or oleic acid) (fig. S8A).
Fig. 4. Investigation into an array of stabilities of PS-capped MAPbBr3/SiO2 core/shell NCs.
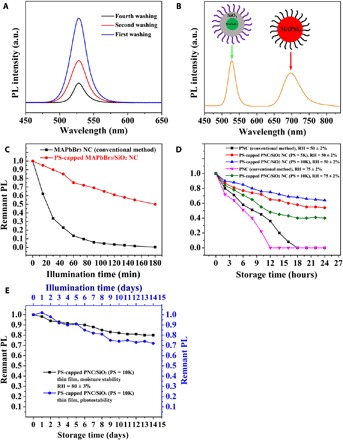
(A) Colloidal stability during repeated solvent washing using n-hexane as a precipitant and toluene as a solvent. The decrease in PL intensity is due to the change in the NC concentration. (B) Chemical composition stability of PS-capped MAPbBr3/SiO2 core/shell NCs (λPS-capped MAPbBr3/SiO2 = 529 nm) mixed with PEO-capped MAPbI3 NCs (λPEO-capped MAPbI3 = 697 nm) in toluene. (C) Photostability of PS-capped MAPbBr3/SiO2 core/shell NC toluene solution in response to continuous illumination of 365-nm UV light at RH of 55 ± 2%. (D) Moisture stability of PS-capped MAPbBr3/SiO2 core/shell NC toluene solution with varied PS chain lengths (i.e., MWPS = 5K and 10K) stored under different moisture conditions (i.e., RH = 50 and 75%). (E) Moisture stability (i.e., RH = 80%) and photostability (under 450-nm illumination) of 10K PS-capped MAPbBr3/SiO2 core/shell NC thin film stored without inert gas protection.
It is well known that the halide composition in PNCs can be reversibly tuned from chloride (Cl−), to bromide (Br−), to iodide (I−) while retaining the size and shape of as-prepared PNCs by simply mixing with compounds capable of delivering halide ions at room temperature (i.e., anion exchange), thereby effectively tuning PL across the whole visible range. However, fast anion exchange reactions occur when mixing PNCs of different halide compositions, which eventually leads to the compositional homogenization of PNCs (40). Figure 4B displays the PL spectrum of the mixed PS-capped MAPbBr3/SiO2 core/shell NCs and PEO-capped MAPbI3 PNCs. Two distinct PL peaks assigned to green-emitting MAPbBr3/SiO2 NCs and red-emitting PEO-capped MAPbI3 PNCs, respectively, are seen, manifesting the absence of the anion exchange process between Br− and I−. The appearance of two respective characteristic PL peaks, as opposed to a halide composition homogenization (fig. S8B), can be ascribed to the dual protection of MAPbBr3 PNCs with inorganic SiO2 shell and organic PS ligands. These inorganic and organic shells effectively function as barriers to the exchange of halide anions, demonstrating outstanding chemical composition stability.
Moreover, the photostability of PS-capped MAPbBr3/SiO2 core/shell NCs was assessed by continuous illumination with 365-nm UV light. The short ligand–capped MAPbBr3 PNCs prepared by the LARP method (see in the Supplementary Materials) were used as control and found to undergo a significant PL drop to nearly zero after 70 min due to the UV-induced decomposition of PNCs at relative humidity (RH) of 55 ± 2%. In sharp contrast, the PL intensity of PS-capped MAPbBr3/SiO2 core/shell NCs still retained 50% of their original value after 180-min illumination, reflecting excellent photostability of PS-capped PNCs (Fig. 4C).
Last, the moisture stability of PS-capped MAPbBr3/silica PNCs was evaluated under different RH conditions. Not unexpectedly, oleylamine- and oleic acid–cocapped MAPbBr3 PNCs prepared by the conventional approach experienced a significant PL intensity loss when exposed to humid air. The PL intensity dropped to nearly zero at RH of 75 ± 2% after 12 hours (Fig. 4D). Notably, the dual-shell architecture, that is, surface passivation of MAPbBr3 with SiO2, followed by hydrophobic PS having bulky pendent benzene groups affords an excellent diffusion barrier to moisture, thereby markedly increasing the stability of PNCs. Capping MAPbBr3/SiO2 NCs with longer PS chains (from MWPS = 5K to MWPS = 10K) was found to further improve their moisture stability at fixed RH (50 ± 2%). To further demonstrate the superior moisture stability and photostability of PS-capped MAPbBr3/SiO2 NCs, the thin film composed of core/shell NCs was prepared and exposed to moisture and continuous illumination. The thin film was able to retain an 80% remnant PL after a 14-day storage under an 80% humidity and a 72% remnant PL after a 14-day continuous illumination (λ = 470 nm). Notably, thin films composed of long PS-capped MAPbBr3/SiO2 NCs (MWPS = 10K), prepared by spin coating the NC toluene solution onto a glass substrate, still displayed highly bright green emission under 365-nm UV illumination after immersion into deionized (DI) water for 30 min (Fig. 5, A to C). Such superior water stability can also be ascribed to the dual shielding of SiO2 shell and PS chains. When in contact with water, the outer hydrophobic long PS chains capped on the surface of MAPbBr3/SiO2 NCs collapsed due to the unfavorable interaction between PS and water molecules diffused from intentionally added water, forming a dense PS shell situated on the SiO2 shell surface, and thus, further effectively blocking water penetration through the double shells, leading to enhanced stability against water, as illustrated in Fig. 5D. Moreover, the PS-capped MAPbBr3/SiO2 NC colloidal solution stored at ambient condition in a closed vial still exhibited high remnant PL after 18 months, while the remnant PL of conventionally prepared PNCs completely disappeared (fig. S8C). Moreover, the excellent water resistance of PS-capped MAPbBr3/SiO2 NCs colloidal solution was demonstrated by directly mixing a colloidal solution with water (fig. S9).
Fig. 5. Digital images of evolution of PS-capped MAPbBr3/SiO2 core/shell NC thin film spin coated on a glass substrate immersed into DI water.
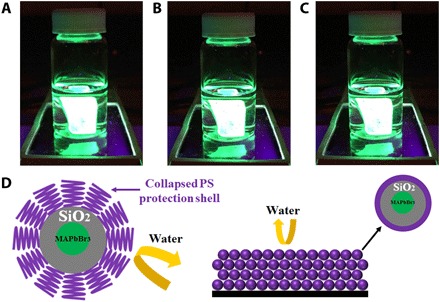
(A) 0 min, (B) 15 min, and (C) 30 min. (D) Schematic illustration of dual-shelled MAPbBr3 NC (i.e., PS-ligated MAPbBr3/SiO2 core/shell NC) in solution (left) and in thin film state (right) when in contact with water. The outer PS chains collapses, forming a dense PS shell situated on the SiO2 shell surface to further effectively distance water from the MAPbBr3 core and result in enhanced water stability.
In summary, we developed an unconventional, robust nanoreactor strategy based on judiciously designed amphiphilic star-like diblock and triblock copolymers with well-defined MW and narrow MW distribution of each block to craft intimate polymer-capped organic-inorganic perovskite NCs and organic-inorganic perovskite/SiO2 core/shell NCs, respectively. These strongly ligated NCs have precisely controlled dimensions, tunable surface chemistry, and an array of appealing stabilities. The dimension controllability of NCs is readily rendered by templating their growth in the compartment occupied by hydrophilic blocks with well-defined MW (i.e., length) of star-like block copolymers. For PEO-capped MAPbI3 NCs, the diameter of the MAPbI3 NC is dictated by the length of the inner PAA block of the star-like PAA-b-PEO diblock copolymer nanoreactor. For PEO-capped (or PS-capped) MAPbBr3/SiO2 core/shell NCs, the diameter of the MAPbBr3 core and the thickness of the SiO2 shell are governed by the length of the inner P4VP block and the intermediate PtBA block (hydrolyzed into PAA block for templating the SiO2 shell growth), respectively, of the star-like P4VP-b-PtBA-b-PEO (or P4VP-b-PtBA-b-PS) triblock copolymer nanoreactor. The surface chemistry tunability of organic-inorganic perovskite NCs is imparted by grafting the outer block of interest to star-like copolymers, that is, the PEO block by click reaction as in the star-like P4VP-b-PtBA-b-PEO nanoreactor or the PS block by ATRP as in the star-like P4VP-b-PtBA-b-PS nanoreactor. A set of markedly improved stabilities of organic-inorganic perovskite NCs are conveniently achieved, including colloidal stability, chemical composition stability, photostability, and moisture and water stability, as a direct consequence of the dual protection attributed to the SiO2 shell passivation followed by intimate and permanent capping of polymer ligands. We envision that our amphiphilic star-like block copolymer nanoreactor strategy may provide a versatile platform for crafting a diverse stable organic-inorganic nanohybrid, as well as all-inorganic perovskite/SiO2 core/shell NCs, stably ligated with a polymer of different functionalities (e.g., semiconducting, ferroelectric, photoresponsive, thermal responsive, or pH sensitive) for a spectrum of applications in bioimaging, biosensors, photonic materials and devices, perovskite-based LEDs, lasers, high-energy ionization radiation detections, multiphoton emission, and scintillators.
MATERIALS AND METHODS
Materials
Methanol (99.8%), ethanol (99.8), isopropyl alcohol (>98.0%), tert-butanol (>99.0%), toluene (99.5%), chloroform (99.8%), dichloromethane (>99.5%), tetrahydrofuran (GR ACS grade), anisole (99.0%), acetone (ACS reagent, >99.5%), γ-butyrolactone (GBL; >99.0%), DMSO (>99.0%), DEF (99.0%), anhydrous DMF (>99.8%), anhydrous N-methyl-2-pyrrolidinone (NMP; 99.5%), heptane (99.0%), anhydrous n-hexane (95.0%), anhydrous 2-propanol (99.5%), α-bromoisobutyryl bromide (99.0%), sodium azide (>99.5%), diphenyl ether (99.0%), methylamine solution [33 weight % (wt %) in absolute ethanol], hydrobromic acid (48 wt % in water; >99.99%), hydriodic acid (contains no stabilizer, distilled; 57 wt % in H2O; 99.99% trace metals basis), anhydrous diethyl ether (>99.0%), sodium bicarbonate (>99.7%), CuCl2 (99.999% trace metals basis), Me6TREN (97.0%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA; 99.0%), TMOS (98.0%), naphthalene (99.0%), sodium (chunk; 98.0%), potassium (chunk; 98.0%), diphenylmethane (99.0%), hydrochloric acid solution (volumetric, 0.1 M; endotoxin free), propargyl bromide solution (80 wt % in xylene), poly(ethylene glycol) methyl ether (MW = 5000 g/mol), trifluoroacetic acid (99.0%), TOABr (98.0%), lead acetate trihydrate [Pb(CH3CO2)2•3H2O, 99.999% trace metals basis], aluminum oxide powder (activated, neutral; Brockmann Activity I), ethyl α-bromoisobutyrate (98.0%), tetrabutylammonium iodide (regent grade; 98.0%), tetrabutylammonium bromide (98.0%), oleylamine (technical grade; 70.0%), and oleic acid (technical grade; 90.0%) were purchased from Sigma-Aldrich and used as received.
β-CD (>97.0%, Sigma-Aldrich) was recrystallized twice from water and dried under vacuum for 48 hours at 50°C. The traces of water were removed by azeotropic distillation in toluene for 2 hours at 110°C prior to the bromination reaction. CuBr (99.999% trace metals basis) and (99.995% trace metals basis) were stirred overnight in acetic acid, filtered, rinsed with ethanol and diethyl ether successively, and dried in vacuum overnight to remove any impurities prior to polymerization. Lead oleate was synthesized by reacting lead acetate with oleic acid under vacuum. St (Sigma-Aldrich) and tBA (Sigma-Aldrich) were washed with aqueous sodium hydroxide and DI water to remove inhibitors, followed by drying by MgSO4 and distilled over CaH2 under reduced pressure prior to use. 4-Vinylpyridine (4VP; Sigma-Aldrich) was distilled over CaH2 under reduced pressure. All three monomers were stored in sealed ampoules under nitrogen and kept in the dark at 0°C. All other reagents were purified by common purification procedures.
Synthesis of PbBr2 NCs capped by poly(tert-butyl acrylate)-block-PEO using star-like P4VP-b-PtBA-b-PEO as nanoreactor
Poly(tert-butyl acrylate)-block-PEO-capped PbBr2 NCs (denoted PtBA-b-PEO-capped PbBr2 NC) were prepared by using star-like P4VP-b-PtBA-b-PEO as nanoreactor. Typically, 10 mg of star-like P4VP-b-PtBA-b-PEO was dissolved in the mixture solution containing 20 ml of chloroform, 5 ml of isopropanol, and 3 ml of methanol, followed by the addition of 385.06 mg of Pb-oleate (0.5 mmol) (synthesized by reacting lead acetate with oleic acid) under argon protection. The system was continuously stirred for 2 hours before 546.8 mg of TOABr (1 mmol) was added into the reaction solution, yielding a light milky colloidal solution within 10 min. The solution was continuously stirred at ambient temperature for another 2 hours. The PtBA-b-PEO–capped PbBr2 NCs were purified by washing with methanol twice and centrifuged at 6000 rpm. PtBA-b-PS–capped PbBr2 NCs were obtained using star-like P4VP-b-PtBA-b-PS as nanoreactor. The formation mechanism is similar to that of PtBA-b-PEO–capped PbBr2 NCs.
Synthesis of PbBr2 NCs capped by PAA-block-PEO via in situ thermolysis of PtBA-b-PEO–capped PbBr2 NCs
PAA-block-PEO–capped PbBr2 NCs (PAA-b-PEO–capped PbBr2 NCs) were crafted by in situ thermolysis of surface tethering PtBA-b-PEO diblock copolymer ligand into PAA-b-PEO in high–boiling point organic solvent. Briefly, PtBA-b-PEO–capped PbBr2 NCs were dissolved in diphenyl ether under strong sonication for 4 hours to generate a homogeneous solution. The solution was transferred into a three-neck flask, and the system was purged by argon for 20 min. After that, the solution was gradually heated to 200°C in 1 hour and maintained at this temperature for 1 hour. The resulting PbBr2 NCs were extracted using a separatory funnel twice and washed by water. The water-soluble PAA-b-PEO–capped PbBr2 NCs were obtained by removing water using rotary evaporation, yielding a white powder-like product. To facilitate the formation of PNCs, the trace amount of water was further removed by a freeze dryer for 24 hours. Preparation of PAA-b-PS–capped PbBr2 NCs followed the same procedure as that of PAA-b-PEO–capped PbBr2 NCs.
Synthesis of MAPbBr3/SiO2 core/shell NCs capped by PEO as ligands using PAA-b-PEO–capped PbBr2 NCs as nanoreactor
MAPbBr3/SiO2 core/shell NCs were synthesized by in situ transformation of PbBr2 into MAPbBr3 in the presence of MABr, followed by hydrolysis of TMOS in the space occupied by PAA chains. In the typical synthesis of core/shell NCs, PAA-b-PEO–capped PbBr2 NCs were dissolved in a 20-ml mixture solution containing toluene and chloroform (the volume ratio of toluene/chloroform was 1:1) under argon protection. MABr (0.16 M) isopropanol solution was added dropwise into the solution noted above under vigorous stirring via a syringe pump for 1 hour. The solution was continuously stirred at 40°C for another 2 hours before the excess MABr was removed by centrifugation. Subsequently, 0.1 ml of TMOS was added into the supernatant under vigorous stirring, followed by purification using toluene as a solvent and hexane as a precipitator for three times. The purified MAPbBr3/SiO2 core/shell NCs were redissolved in toluene and remained stable for several weeks. Synthesis of PS-capped MAPbBr3/SiO2 core/shell NCs followed the same procedure as that of PEO-capped MAPbBr3/SiO2 core/shell NCs, except star-like P4VP-b-PtBA-b-PS was used as nanoreactor.
Synthesis of MAPbI3 PNCs using star-like PAA-b-PEO as nanoreactor
Preparation of PEO-capped MAPbI3 PNCs followed the same procedure as that of PtBA-b-PEO–capped MAPbBr3 PNCs. Briefly, 10 mg of star-like PAA-b-PEO was dissolved in 10 ml of methanol, followed by the addition of lead oleate (1 mmol) under argon protection. The system was continuously stirred for 2 hours before TOABr (1 mmol) was added into the reaction solution. The solution was continuously stirred at ambient temperature for another 2 hours, yielding a light yellow colloidal solution. The PEO-capped PbI2 NCs were purified by washing with methanol twice and centrifuged at 6000 rpm to discard any precipitants. The solvent was removed by rotary evaporation, and the resulting PbI2 NC solid was redissolved in toluene, followed by the addition of MAI isopropanol solution. The reaction solution was continuously stirred at ambient temperature for another 2 hours to enable the complete conversion of PEO-capped PbI2 NCs into PEO-capped MAPbI3 PNCs. The MAPbI3 PNCs were purified by centrifugation at 6000 rpm to remove the excess MAI. Further purification was conducted by using toluene as a solvent and hexane as a precipitator.
Supplementary Material
Acknowledgments
Funding: This work is supported by the AFOSR (FA9550-19-1-0317), NSF (CMMI 1727313 and 1914713, CBET 1803495), DTRA (HDTRA1-18-1-0004), DOE-STTR (DE-SC0018611), and the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering Award # DE-FG02-09ER46604 (spectral measurements). Author contributions: Z.L. and Y.H. initiated this work and designed the research program; Y.H., Y.J.Y., Y.W.H., G.V.B.-M., S.L., and C.H.L. performed the experiments; Z.L., Y.H., V.V.T., N.T., and Z.K. wrote the paper. All the authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax4424/DC1
Experimental Section
Table S1. Dipole moment and normalized polarity of common organic solvents.
Fig. S1. Characterization of macroinitiator and various 21-armed star-like homopolymer and copolymers.
Fig. S2. NMR spectra of polymers and DLS measurements of star-like P4VP-b-PtBA-b-PEO copolymer in different solvents.
Fig. S3. Structural characterization of PNCs prepared by conventional method and star-like block copolymer nanoreactor strategy, respectively.
Fig. S4. XRD patterns, UV-vis absorption and PL spectra, and TEM images of perovskites.
Fig. S5. UV absorption spectra of MAPbI3 PNCs prepared by different methods.
Fig. S6. XRD patterns of MAPbI3 prepared by reacting MAI with PbI2 dissolved in different solvents.
Fig. S7. Stability of MAPbX3 PNCs synthesized by conventional method (i.e., LARP) upon exposure to various common polar organic solvents by the addition of a designated volume of these solvents into the purified PNC toluene solution.
Fig. S8. MAPbBr3 PNCs prepared by conventional method when exposed to repeated solvent washing and mixing with PNCs of different compositions.
Fig. S9. Digital images of evolution of PS-capped MAPbBr3/SiO2 core/shell NCs dispersed in toluene with the addition of 20% water (by volume) under 365-nm UV light.
REFERENCES AND NOTES
- 1.Turren-Cruz S.-H., Hagfeldt A., Saliba M., Methylammonium-free, high-performance, and stable perovskite solar cells on a planar architecture. Science 362, 449–453 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Lin K., Xing J., Na Quan L., Pelayo García de Arquer F., Gong X., Lu J., Xie L., Zhao W., Zhang D., Yan C., Li W., Liu X., Lu Y., Kirman J., Sargent E. H., Xiong Q., Wei Z., Perovskite light-emitting diodes with external quantum efficiency exceeding 20 percent. Nature 562, 245–248 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Chen Q., Wu J., Ou X., Huang B., Almutlaq J., Zhumekenov A. A., Guan X., Han S., Liang L., Yi Z., Li J., Xie X., Wang Y., Li Y., Fan D., Teh D. B. L., All A. H., Mohammed O. F., Bakr O. M., Wu T., Bettinelli M., Yang H., Huang W., Liu X., All-inorganic perovskite nanocrystal scintillators. Nature 561, 88–93 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Akkerman Q. A., Rainò G., Kovalenko M. V., Manna L., Genesis, challenges and opportunities for colloidal lead halide perovskite nanocrystals. Nat. Mater. 17, 394–405 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Berhe T. A., Su W.-N., Chen C.-H., Pan C.-J., Cheng J.-H., Chen H.-M., Tsai M.-C., Chen L.-Y., Dubale A. A., Hwang B.-J., Organometal halide perovskite solar cells: degradation and stability. Energ. Environ. Sci. 9, 323–356 (2016). [Google Scholar]
- 6.Kovalenko M. V., Protesescu L., Bodnarchuk M. I., Properties and potential optoelectronic applications of lead halide perovskite nanocrystals. Science 358, 745–750 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Protesescu L., Yakunin S., Bodnarchuk M. I., Krieg F., Caputo R., Hendon C. H., Yang R. X., Walsh A., Kovalenko M. V., Nanocrystals of cesium lead halide perovskites (CsPbX3, X=Cl, Br, and I): Novel optoelectronic materials showing bright emission with wide color gamut. Nano Lett. 15, 3692–3696 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang F., Zhong H., Chen C., Wu X.-g., Hu X., Huang H., Han J., Zou B., Dong Y., Brightly luminescent and color-tunable colloidal CH3NH3PbX3 (X = Br, I, Cl) quantum dots: Potential alternatives for display technology. ACS Nano 9, 4533–4542 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Schmidt L. C., Pertegás A., Gozález-Carrero S., Malinkiewicz O., Agouram S., Mínguez Espallargas G., Bolink H. J., Galian R. E., Pérez-Prieto J., Nontemplate synthesis of CH3NH3PbBr3 perovskite nanoparticles. J. Am. Chem. Soc. 136, 850–853 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Bekenstein Y., Koscher B. A., Eaton S. W., Yang P. D., Alivisatos A. P., Highly luminescent colloidal nanoplates of perovskite cesium lead halide and their oriented assemblies. J. Am. Chem. Soc. 137, 16008–16011 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Malgras V., Henzie J., Takei T., Yamauchi Y., Stable blue luminescent CsPbBr3 perovskite nanocrystals confined in mesoporous thin films. Angew. Chem. Int. Ed. 57, 8881–8885 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Rainò G., Becker M. A., Bodnarchuk M. I., Mahrt R. F., Kovalenko M. V., Stöferle T., Superfluorescence from lead halide perovskite quantum dot superlattices. Nature 563, 671–675 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Swarnkar A., Marshall A. R., Sanehira E. M., Chernomordik B. D., Moore D. T., Christians J. A., Chakrabarti T., Luther J. M., Quantum dot–induced phase stabilization of α-CsPbI3 perovskite for high-efficiency photovoltaics. Science 354, 92–95 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Li G., Rivarola F. W., Davis N. J., Bai S., Jellicoe T. C., de la Peña F., Hou S., Ducati C., Gao F., Friend R. H., Greenham N. C., Tan Z. K., Highly efficient perovskite nanocrystal light-emitting diodes enabled by a universal crosslinking method. Adv. Mater. 28, 3528–3534 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Xu Y. Q., Chen Q., Zhang C., Wang R., Wu H., Zhang X., Xing G., Yu W. W., Wang X., Zhang Y., Xiao M., Two-photon-pumped perovskite semiconductor nanocrystal lasers. J. Am. Chem. Soc. 138, 3761–3768 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Huang S. Q., Li Z., Kong L., Zhu N., Shan A., Li L., Enhancing the stability of CH3NH3PbBr3 quantum dots by embedding in silica spheres derived from tetramethyl orthosilicate in “waterless” toluene. J. Am. Chem. Soc. 138, 5749–5752 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Li Z. C., Kong L., Huang S., Li L., Highly luminescent and ultrastable CsPbBr3 perovskite quantum dots incorporated into a silica/alumina monolith. Angew. Chem. Int. Ed. 56, 8134–8138 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Loiudice A., Saris S., Oveisi E., Alexander D. T. L., Buonsanti R., CsPbBr3 QD/AlOx inorganic nanocomposites with exceptional stability in water, light, and heat. Angew. Chem. Int. Ed. 56, 10696–10701 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Hu H., Wu L., Tan Y., Zhong Q., Chen M., Qiu Y., Yang D., Sun B., Zhang Q., Yin Y., Interfacial synthesis of highly stable CsPbX3/Oxide janus nanoparticles. J. Am. Chem. Soc. 140, 406–412 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Chang Y., Yoon Y. J., Li G., Xu E., Yu S., Lu C.-H., Wang Z., He Y., Lin C. H., Wagner B. K., Tsukruk V. V., Kang Z., Thadhani N., Jiang Y., Lin Z., All-inorganic perovskite nanocrystals with a stellar set of stabilities and their use in white light-emitting diodes. ACS Appl. Mater. Interfaces 10, 37267–37276 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Malgras V., Tominaka S., Ryan J. W., Henzie J., Takei T., Ohara K., Yamauchi Y., Observation of quantum confinement in monodisperse methylammonium lead halide perovskite nanocrystals embedded in mesoporous silica. J. Am. Chem. Soc. 138, 13874–13881 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Wang H. C., Lin S. Y., Tang A. C., Singh B. P., Tong H. C., Chen C. Y., Lee Y. C., Tsai T. L., Liu R. S., Mesoporous silica particles integrated with all-inorganic CsPbBr3 perovskite quantum-dot nanocomposites (MP-PQDs) with high stability and wide color gamut used for backlight display. Angew. Chem. Int. Ed. 55, 7924–7929 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Wang Y. N., He J., Chen H., Chen J., Zhu R., Ma P., Towers A., Lin Y., Gesquiere A. J., Wu S.-T., Dong Y., Ultrastable, highly luminescent organic-inorganic perovskite-polymer composite films. Adv. Mater. 28, 10710–10717 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Liu F., Zhang Y., Ding C., Kobayashi S., Izuishi T., Nakazawa N., Toyoda T., Ohta T., Hayase S., Minemoto T., Yoshino K., Dai S., Shen Q., Highly luminescent phase-stable CsPbl3 perovskite quantum dots achieving near 100% absolute photoluminescence quantum yield. ACS Nano 11, 10373–10383 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Imran M., Caligiuri V., Wang M., Goldoni L., Prato M., Krahne R., Trizio L., Manna L., Benzoyl halides as alternative precursors for the colloidal synthesis of lead-based halide perovskite nanocrystals. J. Am. Chem. Soc. 140, 2656–2664 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H., Chen B., Wang Z., Hung T., Susha A., Zhong H., Rogach A., Water resistant CsPbX3 nanocrystals coated with polyhedral oligomeric silsesquioxane and their use as solid state luminophores in all-perovskite white light-emitting devices. Chem. Sci. 7, 5699–5703 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luo B. B., Pu Y.-C., Lindley S., Yang Y., Lu L., Li Y., Li X., Organolead halide perovskite nanocrystals: Branched capping ligands control crystal size and stability. Angew. Chem. Int. Ed. 55, 8864–8868 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Zhang F., Huang S., Wang P., Chen X., Zhao S., Dong Y., Zhong H., Colloidal synthesis of air-stable CH3NH3PbI3 quantum dots by gaining chemical insight into the solvent effects. Chem. Mater. 29, 3793–3799 (2017). [Google Scholar]
- 29.Wang L. L., Williams N., Malachosky E., Otto J., Hayes D., Wood R., Guyot-Sionnest P., Engel G., Scalable ligand-mediated transport synthesis of organic-inorganic hybrid perovskite nanocrystals with resolved electronic structure and ultrafast dynamics. ACS Nano 11, 2689–2696 (2017). [DOI] [PubMed] [Google Scholar]
- 30.De Roo J., Ibáñez M., Geiregat P., Nedelcu G., Walravens W., Maes J., Martins J., Driessche I., Kovalenko M., Hens Z., Highly dynamic ligand binding and light absorption coefficient of cesium lead bromide perovskite nanocrystals. ACS Nano 10, 2071–2081 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Zhang Q., Yin Y. D., All-inorganic metal halide perovskite nanocrystals: Opportunities and challenges. ACS Cent. Sci. 4, 668–679 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Almeida G., Ashton O., Goldoni L., Maggioni D., Petralanda U., Mishra N., Akkerman Q., Infante I., Snaith H., Manna L., The phosphine oxide route toward lead halide perovskite nanocrystals. J. Am. Chem. Soc. 140, 14878–14886 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pang X. C., He Y. J., Jung J. H., Lin Z. Q., 1D nanocrystals with precisely controlled dimensions, compositions, and architectures. Science 353, 1268–1272 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Matyjaszewski K., Atom transfer radical polymerization (ATRP): Current status and future perspectives. Macromolecules 45, 4015–4039 (2012). [Google Scholar]
- 35.Zhu Z., Hadjiev V., Rong Y., Guo R., Cao B., Tang Z., Qin F., Li Y., Wang Y., Hao F., Venkatesan S., Li W., Baldelli S., Guloy A., Fang H., Hu Y., Yao Y., Wang Z., Bao J., Interaction of organic cation with water molecule in perovskite MAPbI3: From dynamic orientational disorder to hydrogen bonding. Chem. Mater. 28, 7385–7393 (2016). [Google Scholar]
- 36.Liu Y., Palotas K., Yuan X., Hou T., Lin H., Li Y., Lee S., Atomistic origins of surface defects in CH3NH3PbBr3 perovskite and their electronic structures. ACS Nano 11, 2060–2065 (2017). [DOI] [PubMed] [Google Scholar]
- 37.C. Reichardt, Solvents And Solvent Effects In Organic Chemistry (Wiley-VCH, ed. 3, 2003). [Google Scholar]
- 38.Kim Y., Yassitepe E., Voznyy O., Comin R., Walters G., Gong X., Kanjanaboos P., Nogueira A., Sargent E., Efficient luminescence from perovskite quantum dot solids. ACS Appl. Mater. Interfaces 7, 25007–25013 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Meyns M., Perálvarez M., Heuer-Jungemann A., Hertog W., Ibáñez M., Nafria R., Genc A., Arbiol J., Kovalenko M., Carreras J., Cabot A., Kanaras A., Polymer-enhanced stability of inorganic perovskite nanocrystals and their application in color conversion LEDs. ACS Appl. Mater. Interfaces 8, 19579–19586 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Akkerman Q. A., D’Innocenzo V., Accornero S., Scarpellini A., Petrazza A., Prato M., Manna L., Tuning the optical properties of cesium lead halide perovskite nanocrystals by anion exchange reactions. J. Am. Chem. Soc. 137, 10276–10281 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/11/eaax4424/DC1
Experimental Section
Table S1. Dipole moment and normalized polarity of common organic solvents.
Fig. S1. Characterization of macroinitiator and various 21-armed star-like homopolymer and copolymers.
Fig. S2. NMR spectra of polymers and DLS measurements of star-like P4VP-b-PtBA-b-PEO copolymer in different solvents.
Fig. S3. Structural characterization of PNCs prepared by conventional method and star-like block copolymer nanoreactor strategy, respectively.
Fig. S4. XRD patterns, UV-vis absorption and PL spectra, and TEM images of perovskites.
Fig. S5. UV absorption spectra of MAPbI3 PNCs prepared by different methods.
Fig. S6. XRD patterns of MAPbI3 prepared by reacting MAI with PbI2 dissolved in different solvents.
Fig. S7. Stability of MAPbX3 PNCs synthesized by conventional method (i.e., LARP) upon exposure to various common polar organic solvents by the addition of a designated volume of these solvents into the purified PNC toluene solution.
Fig. S8. MAPbBr3 PNCs prepared by conventional method when exposed to repeated solvent washing and mixing with PNCs of different compositions.
Fig. S9. Digital images of evolution of PS-capped MAPbBr3/SiO2 core/shell NCs dispersed in toluene with the addition of 20% water (by volume) under 365-nm UV light.