

Abstract
B-cell precursor (BCP) ALL carry a variety of classical V(D)J rearrangements as well as genomic fusions and translocations. Here, we assessed the value of genomic capture high-throughput sequencing (gc-HTS) in BCP ALL (n = 183) for the identification and implementation of targets for minimal residual disease (MRD) testing. For TRδ, a total of 300 clonal rearrangements were detected in 158 of 183 samples (86%). Beside clonal Vδ2-Dδ3, Dδ2-Dδ3, and Vδ2-Jα we identified a novel group of recurrent Dδ-Jα rearrangements, comprising Dδ2 or Dδ3 segments fused predominantly to Jα29. For IGH-JH, 329 clonal rearrangements were identified in 172 of 183 samples (94%) including novel types of V(D)J joining. Oligoclonality was found in ~1/3 (n = 57/183) of ALL samples. Genomic breakpoints were identified in 71 BCP-ALL. A distinct MRD high-risk subgroup of IGH-V(D)J-germline ALL revealed frequent deletions of IKZF1 (n = 7/11) and the presence of genomic fusions (n = 10/11). Quantitative measurement using genomic fusion breakpoints achieved equivalent results compared to conventional V(D)J-based MRD testing and could be advantageous upon persistence of a leukemic clone. Taken together, selective gc-HTS expands the spectrum of suitable MRD targets and allows for the identification of genomic fusions relevant to risk and treatment stratification in childhood ALL.
Subject terms: Cancer genetics, Cancer, Cancer genetics, Cancer
Introduction
Precise monitoring of minimal residual disease (MRD) during the first weeks of treatment supports decisions on escalation or de-escalation of therapy in patients with acute lymphoblastic leukemia (ALL)1,2. The treatment response measured by sensitive MRD techniques at the end of induction therapy (EOI) has been shown to be one of the strongest parameters for risk stratification. Initially, Southern blot and semiquantitative PCR-based methodologies were applied which demonstrated frequent clonal rearrangements of IGH and TR-δ segments in BCP-ALL3. Current technological approaches to the measurement of MRD comprise quantitative real-time PCR or multicolor flow cytometry allowing for reproducible quantification of residual leukemic cells with a sensitivity of one leukemic cell among 10,000 normal cells (1 × 10−4)4,5. In order to trace leukemic cells in a clinical setting, the rapid identification of suitable markers is essential for their subsequent quantification. A PCR-based screening method is currently implemented as the standard procedure for the detection of patient-specific V(D)J rearrangements6. With the establishment of recent, PCR-based high-throughput sequencing techniques (HTS) the analytical process of identification and characterization of patient-specific V(D)J rearrangements largely relies on bioinformatics7–12. As an alternative to PCR-based amplification of target sequences, HTS of directly captured genomic fragments has recently been employed for the identification of rearrangements13,14. The advantage of the latter approach is the potential identification of unknown fusion sequences, i.e., rare or unusual V(D)J rearrangements or chromosomal translocations not confined to IG or TR genes which could be used for MRD diagnostics and/or targeted therapy of actionable fusion proteins, such as ABL or JAK2 class fusions15,16.
Here we show that genomic capture high-throughput sequencing (gc-HTS) identified a novel type of recurrent clonal V(D)J TRδ Dδ-Jα rearrangements as well as diverse genomic fusion breakpoints which can be used as sensitive and specific markers in clinical diagnostics of MRD for risk-adapted treatment stratification and targeted intervention. ALL samples devoid of detectable IGH-V(D)J rearrangements were associated with high levels of MRD at EOI and often featured genomic fusion events, such as DUX4-IGH and EPOR-IGH.
Patients, material, and methods
Patients involved in this study were enrolled in COALL03-07 and COALL08-09 multicenter trials (www.clinicaltrials.gov: GPOH-COALL08-09 EU-21076/NCT01228331) except for BCR-ABL1 positive ALL patients who were treated according to the international EsPhALL trial (EudraCT 2004-001647-30 and clinicaltrials.gov Identifier: 00287105)17. Patient samples were obtained after written informed consent of the patients’ parents or legal guardians and with approval by institutional ethics boards (PVN3409; EudraCT-Nr: 2009-012758-18).
All gc-HTS analysed cases (n = 183) were diagnosed as B-precursor ALL devoid of KMT2A and ETV6-RUNX1 rearrangements including a subgroup of n = 19 BCP-ALL carrying a known BCR-ABL rearrangement as a positive control for precise breakpoint characterization. For identification of IKZF1 and P2RY8-CRLF2 (PAR1) deletions multiplex ligation-dependent probe amplification (Salsa MLPA kit P335; (MRC Holland)) was used. MRD-negative or weakly positive ALL (minimum quantitative threshold < 1 × 10−4) exhibiting genetic low-risk features were excluded from this study except n = 7 subsequently relapsing ALL (n = 3 MRD-negative and n = 4 weakly positive cases). Selected ALL samples were grouped into three different quantitative categories based on their EOI MRD levels (≥1 × 10−2 [MRDvery_high], ≥1 × 10−3 [MRDhigh], and <1 × 10−3 [MRDmoderate]) as indicated in Fig. 1.
Fig. 1. Experimental flow diagram.
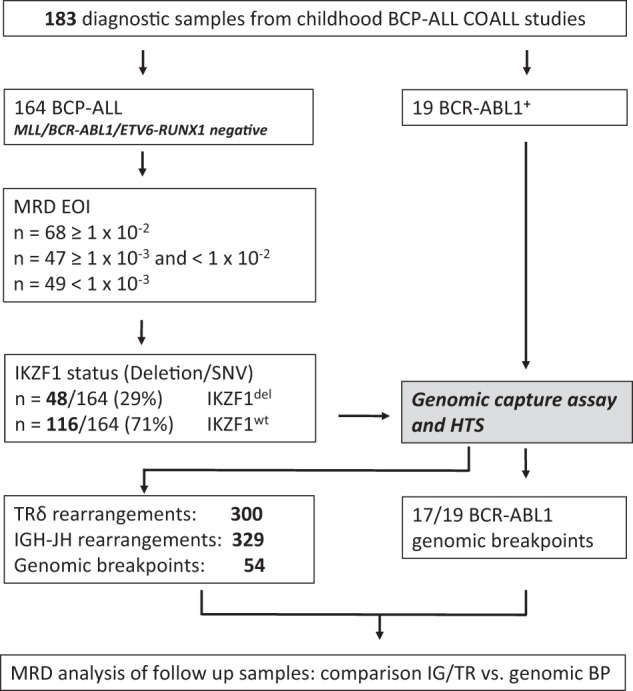
A total number of 183 BCP-ALL cases were analyzed with the gc-HTS approach. Nineteen of them had a known BCR-ABL1 fusion identified by either FISH or molecular genetics. The 164 remaining cases were further divided in three subgroups according to the MRD status at EOI therapy. IKZF1 deletions/mutations were detected by MLPA or Sanger sequencing. Genomic fusion breakpoints were used for quantification of follow-up samples by RQ-PCR.
Diagnostic samples were analyzed by gc-HTS to detect clonal TRδ or IGH-JH rearrangements at diagnosis of ALL in comparison to a standard PCR-based approach. In parallel, we analyzed gene-fusion events targeting cytokine receptors or kinases as observed in Ph-like ALL15,16. Methodological details and the open source software Segemehl for detection of gene fusions have been previously described13,18. Supplemental Table 1 defines the exact chromosomal coordinates of all captured genomic regions. Novel gene fusions detected by gc-HTS were confirmed by PCR and Sanger sequencing. Quantification of classical V(D)J rearrangements as well as for non V(D)J rearrangements/genomic fusion breakpoints (that include large intra-chromosomal deletions like EBF1-PDGFRß or chromosomal translocation breakpoints) followed EUROMRD guidelines4. Sequence data have been submitted to the European Nucleotide Archive (ENA) and they are publicly available at: http:/www.ebi.ac.uk/ena/data/view/PRJEB35051. Sample identifier are available upon request.
Results
In this study we sought to explore the potential of gc-HTS to concurrently identify clonal rearrangements and genomic breakpoints for the sensitive and specific detection of minimal residual disease in B-cell precursor ALL (Fig. 1).
TRδ clonality
First, we examined the TRδ gene locus on chromosome 14p12 using 196 capture probes that cover a 42 kb region between Vδ2 and Vδ3 (Fig. 2a and Supplemental Fig. 1). This region is commonly involved in clonal rearrangements in ALL19,20. We identified clonal TRδ rearrangements in 158 out of 183 BCP-ALL samples (86%), the majority of which involved Vδ2 joined to Dδ3 or to Jα as shown in Table 1. The conception of the applied gc-HTS approach and bioinformatics methodologies allowed for the identification of previously unrecognized Jα-rearrangements fused either to Dδ2- or to Dδ3-segments. Beside well-characterized clonal Vδ2-Jα joinings, we identified 63 rearrangements of Jα segments to Dδ2 (n = 39) or Dδ3 (n = 24) in 53 of 183 samples (29%), with Jα29 being the most common single TRJα fragment (n = 25). The frequency of these rearrangements substantially exceeded the previously reported prevalence in gene-specific PCR-based analyses20,21. To revalidate NGS-derived TRδ rearrangements by Sanger sequencing, amplicons were generated by a PCR strategy positioning forward primers in intronic Dδ2 and Dδ3 regions combined with a Jα29 specific reverse primer (Fig. 2b). Since the predictive value of MRD testing is dependent on the (sub)clonality of targeted rearranged sequences, we determined the number of TRδ rearrangements per sample. TRδ oligoclonality defined as greater than two sequences per ALL sample was observed in 34 of 158 positive specimens, the frequency of which is in accordance with previous reports based on Southern Blot or PCR-based methods19. Several ALL cases demonstrated clonally related TRδ-rearrangements with regard to their N-region insertions, probably due to ongoing recombination from Dδ2- Dδ3 to Dδ2-Jα segments (Fig. 2c)22. The knowledge of these additional clones is of importance for subsequent MRD testing as oligoclonal TRδ targets generally have a lower stability compared to clonal rearrangements.
Fig. 2. Identification of TRδ clonality.

a Schematic view of the captured TRδ region. Novel incomplete TRδ rearrangements were detectable fused to one of the 61 Jα segments. b Validation of either Dδ2 or Dδ3–Jα29 rearrangement in selected cases by PCR and subsequent Sanger sequencing. c Several cases showed either oligoclonality with >2 sequences or ongoing rearrangements within the TRδ locus. A number before/after brackets describe the number of nucleotides deleted within the corresponding germline segment. d Real-time quantitative PCR was performed in a selected ALL sample with Dδ3-Jα29 rearrangement; quantitative range (QR) 1 × 10−4.
Table 1.
Frequency of clonal IGH/TRδ rearrangements.
| Type of rearrangement | number of positive rearrangements | number of positive samples (Σ 183) |
|---|---|---|
| TRδ | 300 | 158 (86%) |
| Vδ2-Dδ3 | 118 | 88 (48%) |
| Dδ2-Dδ3 | 34 | 29 (16%) |
| Vδ2-Jα* | 85 | 71 (39%) |
| Dδ - Jα* | 63 | 53 (29%) |
| • Dδ2- Jα* | 39 | |
| • Dδ3- Jα* | 24 | |
| Vδ-Jδ | 2 | 2 (1%) |
| TRδ oligoclonality (>2) | 111 | 34 (19%) |
| IGH | 329 | 172 (94%) |
| V(D)J | 254 | 157 (86%) |
| DJ | 75 | 56 (31%) |
| IGH oligoclonality (>2) | 66 | 19 (10%) |
Jα*—one of the Jα01 to Jα61 segments, with the majority of cases positive for Jα29
IGH clonality
In BCP-ALL, clonal IGH rearrangements are very common and the most suitable and sensitive marker for MRD detection3,6. To examine the IGH-JH locus on chromosome 14q32 by gc-HTS we selected 184 capture probes comprising 22 kb of genomic sequence (Fig. 3a). IGH-V(D)J-joining was identified in 172 of the 183 samples (94%) with 329 clonal sequences including complete V(D)J-rearrangements as well as incomplete DJ-joining. Hence, gc-HTS identified n = 11 ALL samples truly devoid of IGH-V(D)J rearrangements (Table 2 and Table 3). Oligoclonal IGH-V(D)J rearrangements were identified in n = 19 cases (10%).
Fig. 3. Schematic view of the captured IGH-JH area on chromosome 14q.

a At least one clonal DH-JH or VH(DH)JH rearrangement was identified in 172/183 cases indicative of an IGH germline status in the remaining 11 cases. b A FOXI3-JH6 genomic fusion breakpoint was identified in case ALL UPN22. Fifty eight breakpoint spanning reads covered the breakpoint and for the same sample VIDJIL identified two different clonal rearrangements with 66 and 72 reads, respectively. c For UPN30 two genomic fusion breakpoints (BCL7C-Dδ3 and Dδ2-BCL7C) were sequenced. The different breakpoints were located 11.5 Kb downstream of the BCL7C stop codon.
Table 2.
Frequency of genomic rearrangements within the three different MRD groups.
| MRD value EOI/Genomic rearrangement | ≥1 × 10−2 | <1 × 10−2 | <1 × 10−3 |
|---|---|---|---|
| (n = 68) | (n = 47) | (n = 49)a | |
| No rearrangement | 37 | 39 | 36 |
| DUX4-IGH | 6 | 3 | 1 |
| CRLF2-IGH | 6 | – | – |
| EBF1-PDGFR-ß | 5 | – | – |
| EPOR-IGH | 5 | – | 1b |
| PAR1-Del | 4 | 3 | 3 (2c) |
| other | 6 | 2 | 8 |
| 31 (46%) | 8 (17%) | 13 (26%) |
EOI end of induction treatment
aThis group included seven selected relapse cases with weakly positive or negative MRD
bPatient had a relapse and was initially grouped as MRD-EOI “positive, not quantifiable”, but new MRD marker would re-assign this patient to the second group
cTwo out of the three cases were included because of a relapse and “positive, not quantifiable” MRD
Table 3.
Basic clinical data of samples without any IGH_V(D)J rearrangement.
| Age at Dx (y) | MRD_EOI | IGH_V(D)J | IKZF1 | UPN | Fusion | Response EOI | Therapy |
|---|---|---|---|---|---|---|---|
| 17 | 9.00E-03 | – | del 1–8 | 37 | BCR-ABL1 | yes | Tx |
| 17.5 | 9.00E-02 | – | del 4–7 | 21 | CRLF2-IGH | LateResp | Tx |
| 11 | 9.00E-02 | – | del 2–8 | 1 | DUX4-IGH | LateResp | Tx |
| 16 | 2.00E-02 | – | wt | 10 | DUX4-IGH | pesistent MRD | Tx |
| 5 | 2.00E-03 | – | del 4–7 | 8 | DUX4-IGH | Yes | Remission |
| 10 | 7.00E-02 | – | wt | 61 | EBF-PDGFRB | LateResp | /COALLa |
| 7.5 | 4.00E-01 | – | del 2–7 | 12 | EPOR-IGH | LateResp | TRM |
| 13 | 2.00E-01 | – | del 4–8 | 16 | EPOR-IGH | LateResp | Tx |
| 17.5 | 9.00E-02 | – | del 2–3 | 13 | EPOR-IGH | LateResp | Tx |
| 16 | 5.00E-02 | – | del 2–8 | 69 | FOXP1-ABL1 | LateResp | Tx |
| 12 | 2.00E-02 | – | wt | 72 | n.d. | LateResp | Rel/Tx 2.rem |
n.d. not detectable, Rel relapse, Rem remission, Tx Hematopoietic Stem Cell Transplantation
EOI end of induction therapy, TRM treatment related mortality
aoff protocol treatment intensification
Gene fusions and identification of genomic breakpoints
To advance the clinical assessment of MRD further beyond TR/IG V(D)J rearrangements we explored the potential of gc-HTS to identify the genomic breakpoints of leukemia-specific gene fusions as diagnostic targets. As a positive control, we first examined breakpoint-flanking sequences in a subgroup of BCR-ABL1 positive ALL (n = 19) by gc-HTS. ABL1-directed capturing and subsequent paired-end sequencing identified the genomic breakpoint in 17 of 19 samples at bp resolution indicating a high sensitivity and specificity of our analytical approach. As a next step TRδ, IGH, kinase and cytokine receptor genes as outlined in Supplemental Table 1 were analyzed with regard to genomic fusion breakpoints. Overall, among n = 183 BCP-ALL we identified n = 71 non-V(D)J gene-fusion events and deciphered their genomic fusion breakpoints. These mutations recurrently involved ABL1 (n = 17 BCR-ABL1; n = 1 FOXP1-ABL1; n = 1 RCSD1-ABL1), CRLF2 (n = 11 PAR1 deletions; n = 5 CRLF2-IGH), DUX4 (n = 10 DUX4-IGH), EPOR (n = 6 EPOR-IGH), PDGFRB (n = 5 EBF1-PDGFRB), JAK2 (n = 1 ETV6-JAK2; n = 1 BCR-JAK2; n = 1 PAX5-JAK2) and less frequently mostly transcription-factor associated genes (Table 4 and Table 5). Among the latter group, FOXI3 was identified as a novel, previously unrecognized fusion partner of IGH. The FOXI3 gene on chromosome 2 belongs to the large family of forkhead box transcription-factor genes consisting of two exons, which encode for a 422-aa protein with activities in embryogenesis, bone modeling and potentially in carcinogenesis23. Recently, functional mapping of FOXI3 has revealed a nuclear localization sequence (NLS) and a C-terminal transactivation domain (TAD)24. We mapped the genomic breakpoint to a region 3859 bp upstream of the ATG start codon, which was fused to IGH-JH6 suggesting an aberrant expression driven by the IGH-enhancer (Fig. 3b). In accordance, FOXI3 transcripts were found to be abundantly expressed in the FOXI3-JH6 rearranged ALL in contrast to a barely discernible or absent expression in 25 randomly selected primary BCP-ALL samples (Supplemental Fig. 2).
Table 4.
Basic clinical and molecular data of IGH and TRδ associated gene fusions.

Table 5.
Basic clinical and molecular data of non-IGH/TRδ associated gene fusions.

Additional chromosomal rearrangements into IGH-JH involved CEBPD, CTNNA3, AHDC1, MCCC2, and ELK2AP (Table 4 and Table 6). We identified CEBPD-IGH fusions (n = 3) in two ALL samples from patients with Down syndrome (DS)25,26. Breakpoints were located in different introns of the SPIDR gene displaying 53 kb and 7.8 kb distance to the 3’-end of CEBPD at primary diagnosis (UPN23 and UPN24). The latter patient developed relapsing disease 10 years after the initial diagnosis, albeit displaying a different breakpoint 257 kb downstream of the 3’-end of CEBPD indicative of a different sub-clone or potentially an independent secondary leukemia. Furthermore, in UPN27 a genomic fusion of IGH-JH4 into the MCCC2 gene comprising 17 coding exons on chromosome 5q13 was found with the breakpoint located in intron 16. As a consequence, MCCC2 exon 17 and a part of intron 16 were fused to the IGH-JH4 region suggesting that the translocation event is non-functional or that an alternative gene further downstream is involved. Finally, in UPN29 we identified a genomic breakpoint 8994 bp 5’ of the first exon of ELK2AP, a processed pseudogene on chromosome 14q3227,28. The functional consequences of the ELK2AP-JH1 fusion that was likely caused by an inversion affecting the IGH locus are unknown29. The exact genome coordinates of all novel genomic breakpoints identified here were confirmed by PCR and Sanger sequencing (Table 6).
Table 6.
Genomic coordiates of novel genomic breakpoints.
| UPN | Fusion | Gene_A | hg38 | Gene_B | hg38 | FSR | PCR/Seq | Comment |
|---|---|---|---|---|---|---|---|---|
| 22 | FOXI3_IGH | FOXI3_5’ | chr2: 88456394 | IGH_JH6 | chr14:105863240 | 58 (H) | Yes | Dx |
| 23 | CEBPD_IGH | CEBPD_3’ (53Kb) | chr8:47684174 | IGH_JH4 | chr14:105864260 | 13 (M) | Yes | Dx |
| 24 | CEBPD_IGH | CEBPD_3’ (7Kb) | chr8:47729469 | IGH_JH5-JH6 | chr14:105863653 | 6(M) | Yes | Dx |
| 24 | CEBPD_IGH | CEBPD_3’ (257Kb) | chr8:47479748 | IGH_JH5 | chr14:105863864 | 4(M) | Yes | Relapse |
| 25 | AHDC1_IGH | AHDC1_3' | chr1:27547201 | IGH_JH4 | chr14:105864248 | 15 (H) | Yes | Dx |
| 26 | CTNNA3_IGH | CTNNA3 intron 6 | chr10:67402606 | IGH_JH4 | chr14:105864251 | 22(M) | Yes | Dx |
| 27 | MCCC2_IGH | MCCC2_intron 16 | chr5:71655478 | IGH_JH4 | chr14:105864257 | 73(M) | Yes | Dx |
| 29 | ELK2AP-IGH | ELK2AP_5' | chr14_105681797 | IGH_JH1 | chr14:105865454 | 168(H) | Yes | Dx (possible inversion) |
| 29 | ELK2AP-IGH | id | id | id | id | n.t. | Yes | Relapse |
| 30 | BCL7C-TRδ | BCL7C_3' | chr16:30876320 | TRδ_Dδ2 | chr14:22439011 | 99 (H) | Yes | Dx (rev) |
| 30 | BCL7C-TRδ | BCL7C_3' | chr16:30876329 | TRδ_Dδ3 | chr14:22449125 | 4(H) | Yes | Dx (forw) |
| 31 | ACIN-TRδ | ACIN1_5' | chr14:23095837 | TRδ_Dδ2 | chr14_22439016 | 44(H) | Yes | Dx |
| 65 | CSF1R-? | CSF1R_intron 11 | chr5:150067115 | ? | chr5:92365792 | 36(M) | Yes | Dx (forw) |
| 65 | CSF1R-? | CSF1R_intron 11 | chr5:150067680 | CCNJL_intron 4 | chr5:160261418 | 10(M) | Yes | Dx (rev) |
All novel genomic breakpoints detected by gc-HTS were confirmed by PCR and Sanger Sequencing. Comment: Fusion detected at diagnosis (dx) and/or relapse
FSR fusion spanning reads; (H) HiSeq, (M) Miseq, forw forward orientation, rev reverse orientation
Taken together, IGH-associated gene fusions were detected in 31 samples, with DUX4 rearrangements as the most frequent partner gene identified in 10 ALL cases. Genomic breakpoints were distributed either within a region 5’ upstream of DUX4 or within the 3’-coding region in exon 1 fused to IGH-JH or IGH-DH as previously reported (Supplemental Fig. 3)30–32.
With regard to TRδ, we identified two previously unrecognized rearrangements into the BCL7C and ACIN1 gene loci, respectively. This type of fusion, which likely places the target gene under the control of the TRδ enhancer/promoter, has been previously described in T-ALL33–35. The BCL7C gene on chromosome 12q24 was identified as a novel fusion partner of TRδ (Fig. 3c). Split-read and subsequent PCR analyses revealed that the TRδ-Dδ2 segment was fused to the untranslated 3’-region of BCL7C, whereas TRδ-Dδ3 is linked to a region 3’ of BCL7C exon 6 comprising the complete coding region of this gene (UPN30; Fig. 3c). Both breakpoints showed variable insertions of random nucleotides similar to a previously described inversion that joins TRδ-Dδ3 and BCL11B34. BCL7C belongs to the small group of BCL7 genes, among which BCL7A has previously been detected in high-grade B-cell lymphoma as a partner gene in a complex chromosomal translocation t(8;14;12) that involves c-MYC as well as IGH-VH336. The second rearrangement involved the TRδ-Dδ2 segment fused to a region immediately 5’ of the start codon of ACIN1 located on chromosome 14q11 indicative of a non-functional break on the reverse strand (Supplemental Fig. 4; UPN 31). Of note, ACIN1 has recently been described as a fusion partner gene of NUTM1 in ALL37. The TRδ-Dδ3 related breakpoint site, which normally activates the corresponding target gene through strong TRδ enhancer elements, could not be identified. The most adjacent gene on the reverse strand belongs to the CEBP family (CEBPE), which is associated with translocations affecting the IGH locus on chromosome 14q3225.
With regard to kinase coding genes we detected single genomic breakpoints of previously described gene fusions that involve RCSD1-ABL1, FOXP1-ABL1, BCR-JAK2, PAX5-JAK2, and ETV6-JAK215,16,38. We identified a novel breakpoint in CSF1R intron 11 fused to a yet undefined intergenic region on chromosome 5. This genomic fusion contains a random seven-nucleotide insertion that was not discernible in previously analyzed ALL cases indicating a specific breakpoint with yet unknown functional relevance. The reverse breakpoint was located in intron 10 fused to intron 4 of the CCNJL gene (Table 6; UPN65)
Very high MRD is associated with non-V(D)J genomic rearrangements
A high burden of MRD after induction therapy (EOI) is generally associated with a poor prognosis1,2,39. To investigate type and prevalence of genomic rearrangements in MRD-defined very high-risk ALL we categorized n = 164 ALL samples into three subgroups according to their quantitative MRD EOI levels as outlined in Materials and Methods. In the MRDvery_high subgroup, 31 of 68 ALL samples (46%) carried genomic rearrangements compared to 17 and 26% fusion events in ALL exhibiting a moderate or high MRD burden, respectively (Table 2). All EBF1-PDGFR-ß and CRLF2-IGH rearrangement positive cases as well as five of six ALL harboring EPOR-IGH fusions were found in the MRD very high-risk group. Strikingly, six out of 10 DUX4-rearranged cases exhibited very high MRD levels at EOI (≥1 × 10−2) that led to hematopoietic stem cell transplantation in first remission. Another DUX4rearr ALL (UPN5) with an MRD burden of 9 × 10−3 at EOI developed two relapses, finally exhibiting a phenotypical switch to the myeloid lineage but stable clonal V(D)J-rearrangements used as molecular MRD markers40. ALL harboring DUX4-rearrangements have previously been described as a prognostically favorable ALL-subgroup often containing concomitant intragenic ERGdel or a deregulation of ERG by expression of an alternatively spliced, dominant-negative ERG isoform (ERGalt)31. In our DUX4rearr ALL cohort, ERGdel were detected in 2/10 cases by MLPA analysis, whereas ERGalt was identified in 4 of 5 available DUX4rearr ALL samples by isoform-specific RT-PCR in accordance with previously reported data31.
Among the small group of primary ALL (n = 7) relapsing later on with weakly positive or negative MRD at EOI analyzed in this study, a single case harbored an EPOR-IGH rearrangement (UPN11). Intriguingly, re-analysis of MRD in this case utilizing the EPOR-IGH breakpoint and a clonal Vδ2-Jα22 rearrangement as additional markers identified by gc-HTS would have resulted in an assignment to a higher risk group and more intensified treatment (Table 4).
A germline V(D)J constitution at the IGH locus as observed in 11 out of 183 analyzed ALL (6%) indicates a leukemic transformation at a very immature, early stage of B-cell development3,9. Germline IGH-V(D)J ALL were associated with poor treatment response, frequent IKZF1 deletions and genomic rearrangements such as DUX4-IGH, EPOR-IGH, BCR-ABL1, FOXP1-ABL1, EBF1-PDGFR-ß, or a CRLF2-IGH (Table 3).
Novel V(D)J and non-V(D)J genomic rearrangements are useful markers in MRD diagnostics
As outlined above we identified several novel TRδ-Jα clonal rearrangements which we sought to evaluate as optional markers for MRD diagnostics. Moreover, we established individual, patient-specific RQ-PCR assays based on genomic breakpoint sequences of non-V(D)J rearrangements. In regard to novel TRδ Dδ2- and Dδ3-specific rearrangements, we designed two RQ-PCR assays based on their germline sequences to allow for a Jα-independent quantification (Fig. 2d). In comparison, conventional and novel MRD markers showed similar sensitivities in the detection of EOI MRD presenting values deviating less than 0.5 log from each other as evaluated according to the EURO-MRD guidelines for quantification. We identified novel TRδ/α markers in almost a quarter of ALL, which could improve clinical diagnostics of MRD because of their specific amplification with a superior signal to noise ratio and a quantitative range of 1 × 10−4. These features render TRδ markers more suitable than other targets such as TRγ due to a low or absent background amplification.
In line with this notion, gc-HTS identified three additional MRD markers in a relapsed ALL (UPN29; Table 2 and Supplemental Table 2), which revealed slightly higher MRD values targeting the new rearrangements (Dδ2-Jα09; 5’-VH4-JH6 and ELK2AP-JH1) compared to routinely assessed TRß and a concomitant loss of two TRγ markers. Targeting the IGH locus in this sample, gc-HTS identified two unusual IGH-JH associated fusions not detectable by a PCR-based approach, both of which were discernible at relapse. Firstly, an IGH-JH6 segment was rearranged into an intronic region between two IGH-VH segments (7365 bp 5’ of VH7*34). Secondly, a somatic fusion event of the ETS-family pseudogene ELK2AP with an IGH-JH1 segment was deciphered at primary diagnosis and at relapse.
Beside novel TR and IGH rearrangements, a total of n = 25 non-V(D)J rearrangements including their respective breakpoints were evaluated as diagnostic targets for MRD analysis. In contrast to the BCR-ABL1-positive ALL-subgroup which clearly exhibited breakpoint-specific signals upon remission likely originating from a rearrangement persisting in the hematopoietic stem cell population, non-V(D)J rearrangements in general revealed MRD kinetics that were very similar to conventional IG/TR-V(D)J rearrangements (Fig. 4 and Table 3)41.
Fig. 4. RQ-PCR assays using the genomic fusion breakpoint.

a In sample UPN 15 conventional PCR screening identified three common TRγ/ß clonal marker used for MRD quantification; gc-HTS identified an additional EPOR-JH4 breakpoint with a RQ-PCR QR of 1 × 10−4. Because of a poor response towards frontline treatment as reflected by substantial molecular residual disease the patient was transplanted in first remission, but developed a relapse one year later. Both TR- and genomic breakpoint directed analytical approaches confirmed long term molecular remission after second transplantation (>3.5 years). b MRD follow-up for a BCR-ABL1-positive patient (UPN47). The patient relapsed 10 years after first diagnosis. In contrast to IG/TR targeted MRD testing showing a rapid early response, BCR-ABL1 genomic fusion breakpoint based RQ-PCR revealed persistently high MRD levels during the first year of treatment. At relapse, only the genomic fusion breakpoint-specific PCR showed a specific signal with loss of all previously identified IG/TR related markers.
Overall, gc-HTS alleviates the identification and implementation of genomic breakpoints and fusions as targets for MRD analysis, which might improve the monitoring of patients with a slow molecular response to chemotherapy and/or immunotherapy requiring sensitive surveillance of subclonal disease.
Discussion
The analysis of MRD by real-time quantitative genomic PCR or multicolor flow cytometry has been firmly established as an integral part of clinical diagnostics of acute lymphoblastic leukemia4,5. Both methodological approaches complement each other and allow for treatment stratification in the vast majority of patients. Nevertheless, a small but significant number of patients (~5–10%) lack leukemia-specific conventional genomic V(D)J rearrangements at TR/IG loci or unambiguous cell surface epitopes preventing a reliable and sensitive measurement of MRD42. In addition, RQ-PCR and flow cytometry are limited in the sensitive detection of subclonal disease, which is often driven by non-V(D)J rearrangements or gene fusions not detected by conventional PCR-based MRD analyses. In line with this notion, it is generally appealing to implement predisposing or causative genetic lesions as diagnostic targets for the measurement of MRD as established for various MLL-gene fusions in infant ALL or, as recently published, for ETV6-RUNX1 breakpoints43–45. High-throughput genome, exome and transcriptome sequencing has recently unfolded a variety of previously unrecognized genetic subtypes of B-cell precursor acute lymphoblastic leukemia46. Here, we present gc-HTS as an analytical approach to the combined identification of clonality and individual genomic breakpoints in B-cell precursor ALL. In accordance with PCR-based analyses, we identified classical V(D)J rearrangements at IGH and TRδ gene loci in 95% and 84% of ALL samples, respectively6. As an advancement, our PCR-independent approach allows for the detection of V(D)J recombinations and non-V(D)J rearrangements not discernible by PCR using pre-selected primer sets. Hence, gc-HTS substantially expands the spectrum of individual markers per ALL sample including distinct genomic aberrations, the latter of which are less prone to clonal evolution and early loss of specificity. In contrast, secondary mutational events or targets with an oligoclonal appearance such as PAR1- or ERG- deletions are less suitable for a reliable and robust MRD analysis.
In comparison, PCR-based HTS has a greater capability to detect minor V(D)J clones at diagnosis7,47. In a recently published study, paired analysis of diagnostic/relapse ALL samples by PCR-based HTS revealed that initial (“major”) clones with a frequency >5% or an absolute number of reads (ARC) >10,000 showed the highest stability at relapse48. gc-HTS does not achieve a read-depth comparable with PCR-based HTS but our preliminary data indicate a detection limit of genomic capturing for rearrangements in heterogeneous cell populations with a blast fraction of ~20%, which is sufficient for most diagnostic settings. To date, clinically relevant RQ-PCR-based MRD testing is confined to initially selected major clones. Currently, most NGS-based approaches use a nested PCR workflow for clonality detection, a strategy that requires rigorous precautions under strict GLP conditions7,10–12,47. By contrast, gc-HTS affixes sample-specific barcodes to the genomic fragments during the initial steps of library preparation, minimizing the risk of false-positive target assignment and contamination. In effect, the lower read-depth of gc-HTS might be compensated by the direct sequencing of enriched genomic fragments that enables us to detect rearrangements not detectable by PCR-based methods.
Beyond conventional rearrangements, previously unrecognized IG and recurrent TRδ rearrangements were deciphered, which increase the spectrum of suitable targets for clinical diagnostics of MRD. By identification of the Dδ-Jα subgroup additional targets are available for MRD testing. Nevertheless, based on these results TRδ-related oligoclonality or ongoing rearrangement patterns should be handled with caution as they can potentially result in divergent MRD values in follow-up samples. Importantly, novel previously unrecognized fusion partner genes of IGH were detected the function of which has yet to be defined. In line with earlier observations, we identified a subgroup of children with ALL displaying a germline IGH that was associated with a poor response to induction treatment and accordingly high levels of MRD3,9. Strikingly, this IGH germline ALL group exhibited a high frequency of IKZF1 deletions and non-V(D)J rearrangements including CRLF2-IGH, EPOR-IGH, and EBF1-PDGFRß that can be exploited as leukemia-specific targets in clinical diagnostics of MRD. Early identification of this high-risk subgroup is a prerequisite for treatment stratification or timely intervention. Unexpectedly, the IGH germline ALL-subgroup also included DUX4-rearranged ALL lacking concurrent ERG-deletions, but some expressed the dominant-negative ERGalt isoform49. In contrast to the previously reported favorable outcome of DUX4rearr-ALL we observed a remarkably poor treatment response of DUX4rearr ALL mostly leading to hematopoietic stem cell transplantation in first or second remission30–32. In line with this notion, Zaliova and colleagues have recently described a DUX4 overexpressing subgroup that showed a poor molecular response to therapy50. Larger prospective studies are needed to determine the prognostic impact of DUX4rearr/ERGalt in ALL.
From a technological perspective, gc-HTS might advance the clinical diagnostics of MRD by precise identification of genomic breakpoints and non-captured genomic partner genes such as RCSD1 or FOXP1, which is necessary for treatment stratification for instance of ABL-class (genomic) fusion-carrying ALL51,52. Using gc-HTS in the BCR-ABL1-positive ALL group we observed rare failures in the identification of genomic breakpoints that were potentially due to AT-rich- or repetitive sequences at probe hybridization sites or gaps in probe design targeting intron 1 of ABL1. Improved probe design with greater coverage at these sites may eliminate such drawbacks in the future.
In summary, the PCR-independent genomic approach presented here constitutes a very sensitive and robust method that enables laboratories to detect a broad spectrum of markers relevant for MRD diagnostics and also for targeted therapeutic intervention for instance in Ph-like ALL. A bespoke selection of genomic targets, a flexible workflow and the requirement for only very small amounts of diagnostic material should make this method widely applicable.
Supplementary information
Acknowledgements
We are very grateful to all members of the Pediatric Hematology Laboratory at the University Medical Center Hamburg who supported this study.
Author contributions
U.z.S. planned the study and performed bioinformatic analysis of the samples, M.Ad. prepared samples for NGS and performed quantification of follow-up samples; G.E. provided patient data and selected samples; D.I. performed NGS and M.Al. established and performed bioinformatics analyses; M.A.H. planned, conceived, and supervised the study; and U.z.S. and M.A.H. wrote the manuscript. All authors revised and approved the final version of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41408-019-0257-x).
References
- 1.Cave H, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. European Organization for Research and Treatment of Cancer-Childhood Leukemia Cooperative Group. N. Engl. J. Med. 1998;339:591–598. doi: 10.1056/NEJM199808273390904. [DOI] [PubMed] [Google Scholar]
- 2.van Dongen JJ, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352:1731–1738. doi: 10.1016/S0140-6736(98)04058-6. [DOI] [PubMed] [Google Scholar]
- 3.Felix CA, et al. Characterization of immunoglobulin and T-cell receptor gene patterns in B-cell precursor acute lymphoblastic leukemia of childhood. J. Clin. Oncol. 1990;8:431–442. doi: 10.1200/JCO.1990.8.3.431. [DOI] [PubMed] [Google Scholar]
- 4.van der Velden VH, et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia. 2007;21:604–611. doi: 10.1038/sj.leu.2404586. [DOI] [PubMed] [Google Scholar]
- 5.Theunissen P, et al. Standardized flow cytometry for highly sensitive MRD measurements in B-cell acute lymphoblastic leukemia. Blood. 2017;129:347–357. doi: 10.1182/blood-2016-07-726307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Dongen JJ, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17:2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 7.Faham M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120:5173–5180. doi: 10.1182/blood-2012-07-444042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giraud M, et al. Fast multiclonal clusterization of V(D)J recombinations from high-throughput sequencing. BMC Genomics. 2014;15:409. doi: 10.1186/1471-2164-15-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wood B, et al. Measurable residual disease detection by high-throughput sequencing improves risk stratification for pediatric B-ALL. Blood. 2018;131:1350–1359. doi: 10.1182/blood-2017-09-806521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheijen Blanca, Meijers Ruud W. J., Rijntjes Jos, van der Klift Michèle Y., Möbs Markus, Steinhilber Julia, Reigl Tomas, van den Brand Michiel, Kotrová Michaela, Ritter Julia-Marie, Catherwood Mark A., Stamatopoulos Kostas, Brüggemann Monika, Davi Frédéric, Darzentas Nikos, Pott Christiane, Fend Falko, Hummel Michael, Langerak Anton W., Groenen Patricia J. T. A. Next-generation sequencing of immunoglobulin gene rearrangements for clonality assessment: a technical feasibility study by EuroClonality-NGS. Leukemia. 2019;33(9):2227–2240. doi: 10.1038/s41375-019-0508-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knecht Henrik, Reigl Tomas, Kotrová Michaela, Appelt Franziska, Stewart Peter, Bystry Vojtech, Krejci Adam, Grioni Andrea, Pal Karol, Stranska Kamila, Plevova Karla, Rijntjes Jos, Songia Simona, Svatoň Michael, Froňková Eva, Bartram Jack, Scheijen Blanca, Herrmann Dietrich, García-Sanz Ramón, Hancock Jeremy, Moppett John, van Dongen Jacques J. M., Cazzaniga Giovanni, Davi Frédéric, Groenen Patricia J. T. A., Hummel Michael, Macintyre Elizabeth A., Stamatopoulos Kostas, Trka Jan, Langerak Anton W., Gonzalez David, Pott Christiane, Brüggemann Monika, Darzentas Nikos. Quality control and quantification in IG/TR next-generation sequencing marker identification: protocols and bioinformatic functionalities by EuroClonality-NGS. Leukemia. 2019;33(9):2254–2265. doi: 10.1038/s41375-019-0499-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brüggemann Monika, Kotrová Michaela, Knecht Henrik, Bartram Jack, Boudjogrha Myriam, Bystry Vojtech, Fazio Grazia, Froňková Eva, Giraud Mathieu, Grioni Andrea, Hancock Jeremy, Herrmann Dietrich, Jiménez Cristina, Krejci Adam, Moppett John, Reigl Tomas, Salson Mikael, Scheijen Blanca, Schwarz Martin, Songia Simona, Svaton Michael, van Dongen Jacques J. M., Villarese Patrick, Wakeman Stephanie, Wright Gary, Cazzaniga Giovanni, Davi Frédéric, García-Sanz Ramón, Gonzalez David, Groenen Patricia J. T. A., Hummel Michael, Macintyre Elizabeth A., Stamatopoulos Kostas, Pott Christiane, Trka Jan, Darzentas Nikos, Langerak Anton W. Standardized next-generation sequencing of immunoglobulin and T-cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia; a EuroClonality-NGS validation study. Leukemia. 2019;33(9):2241–2253. doi: 10.1038/s41375-019-0496-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stadt UZ, et al. Rapid capture next-generation sequencing in clinical diagnostics of kinase pathway aberrations in B-cell precursor ALL. Pediatr. Blood Cancer. 2016;63:1283–1286. doi: 10.1002/pbc.25975. [DOI] [PubMed] [Google Scholar]
- 14.Wren D, et al. Comprehensive translocation and clonality detection in lymphoproliferative disorders by next-generation sequencing. Haematologica. 2017;102:e57–e60. doi: 10.3324/haematol.2016.155424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roberts KG, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014;371:1005–1015. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts KG, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012;22:153–166. doi: 10.1016/j.ccr.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biondi A, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): a randomised, open-label, intergroup study. Lancet Oncol. 2012;13:936–945. doi: 10.1016/S1470-2045(12)70377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoffmann S, et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol. 2014;15:R34. doi: 10.1186/gb-2014-15-2-r34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szczepanski T, et al. Cross-lineage T cell receptor gene rearrangements occur in more than ninety percent of childhood precursor-B acute lymphoblastic leukemias: alternative PCR targets for detection of minimal residual disease. Leukemia. 1999;13:196–205. doi: 10.1038/sj.leu.2401277. [DOI] [PubMed] [Google Scholar]
- 20.Szczepanski T, et al. Vdelta2-Jalpha rearrangements are frequent in precursor-B-acute lymphoblastic leukemia but rare in normal lymphoid cells. Blood. 2004;103:3798–3804. doi: 10.1182/blood-2003-08-2952. [DOI] [PubMed] [Google Scholar]
- 21.Ferret Y, et al. Multi-loci diagnosis of acute lymphoblastic leukaemia with high-throughput sequencing and bioinformatics analysis. Br. J. Haematol. 2016;173:413–420. doi: 10.1111/bjh.13981. [DOI] [PubMed] [Google Scholar]
- 22.Steenbergen EJ, et al. Frequent ongoing T-cell receptor rearrangements in childhood B-precursor acute lymphoblastic leukemia: implications for monitoring minimal residual disease. Blood. 1995;86:692–702. doi: 10.1182/blood.V86.2.692.bloodjournal862692. [DOI] [PubMed] [Google Scholar]
- 23.Mukherjee A, et al. A Review of FOXI3 regulation of development and possible roles in cancer progression and metastasis. Front. Cell Dev. Biol. 2018;6:69. doi: 10.3389/fcell.2018.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh S, Jangid RK, Crowder A, Groves AK. Foxi3 transcription factor activity is mediated by a C-terminal transactivation domain and regulated by the Protein Phosphatase 2A (PP2A) complex. Sci. Rep. 2018;8:17249. doi: 10.1038/s41598-018-35390-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Akasaka T, et al. Five members of the CEBP transcription factor family are targeted by recurrent IGH translocations in B-cell precursor acute lymphoblastic leukemia (BCP-ALL) Blood. 2007;109:3451–3461. doi: 10.1182/blood-2006-08-041012. [DOI] [PubMed] [Google Scholar]
- 26.Lundin C, et al. B-cell precursor t(8;14)(q11;q32)-positive acute lymphoblastic leukemia in children is strongly associated with Down syndrome or with a concomitant Philadelphia chromosome. Eur. J. Haematol. 2009;82:46–53. doi: 10.1111/j.1600-0609.2008.01166.x. [DOI] [PubMed] [Google Scholar]
- 27.Rao VN, et al. elk, tissue-specific ets-related genes on chromosomes X and 14 near translocation breakpoints. Science. 1989;244:66–70. doi: 10.1126/science.2539641. [DOI] [PubMed] [Google Scholar]
- 28.Yamauchi T, et al. Structural organization of the human Elk1 gene and its processed pseudogene Elk2. DNA Res. 1999;6:21–27. doi: 10.1093/dnares/6.1.21. [DOI] [PubMed] [Google Scholar]
- 29.Harindranath N, et al. The human elk-1 gene family: the functional gene and two processed pseudogenes embedded in the IgH locus. Gene. 1998;221:215–224. doi: 10.1016/S0378-1119(98)00448-X. [DOI] [PubMed] [Google Scholar]
- 30.Lilljebjorn H, et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat. Commun. 2016;7:11790. doi: 10.1038/ncomms11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat. Genet. 2016;48:1481–1489. doi: 10.1038/ng.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yasuda T, et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Nat. Genet. 2016;48:569–574. doi: 10.1038/ng.3535. [DOI] [PubMed] [Google Scholar]
- 33.Przybylski GK, et al. The effect of a novel recombination between the homeobox gene NKX2-5 and the TRD locus in T-cell acute lymphoblastic leukemia on activation of the NKX2-5 gene. Haematologica. 2006;91:317–321. [PubMed] [Google Scholar]
- 34.Przybylski GK, et al. Disruption of the BCL11B gene through inv(14)(q11.2q32.31) results in the expression of BCL11B-TRDC fusion transcripts and is associated with the absence of wild-type BCL11B transcripts in T-ALL. Leukemia. 2005;19:201–208. doi: 10.1038/sj.leu.2403619. [DOI] [PubMed] [Google Scholar]
- 35.Przybylski GK, et al. Molecular characterization of a novel chromosomal translocation t(12;14)(q23;q11.2) in T-lymphoblastic lymphoma between the T-cell receptor delta-deleting elements (TRDREC and TRAJ61) and the hypothetical gene C12orf42. Eur. J. Haematol. 2010;85:452–456. doi: 10.1111/j.1600-0609.2010.01508.x. [DOI] [PubMed] [Google Scholar]
- 36.Zani VJ, et al. Molecular cloning of complex chromosomal translocation t(8;14;12)(q24.1;q32.3;q24.1) in a Burkitt lymphoma cell line defines a new gene (BCL7A) with homology to caldesmon. Blood. 1996;87:3124–3134. doi: 10.1182/blood.V87.8.3124.bloodjournal8783124. [DOI] [PubMed] [Google Scholar]
- 37.Li Jian-Feng, Dai Yu-Ting, Lilljebjörn Henrik, Shen Shu-Hong, Cui Bo-Wen, Bai Ling, Liu Yuan-Fang, Qian Mao-Xiang, Kubota Yasuo, Kiyoi Hitoshi, Matsumura Itaru, Miyazaki Yasushi, Olsson Linda, Tan Ah Moy, Ariffin Hany, Chen Jing, Takita Junko, Yasuda Takahiko, Mano Hiroyuki, Johansson Bertil, Yang Jun J., Yeoh Allen Eng-Juh, Hayakawa Fumihiko, Chen Zhu, Pui Ching-Hon, Fioretos Thoas, Chen Sai-Juan, Huang Jin-Yan. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proceedings of the National Academy of Sciences. 2018;115(50):E11711–E11720. doi: 10.1073/pnas.1814397115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ernst T, et al. Identification of FOXP1 and SNX2 as novel ABL1 fusion partners in acute lymphoblastic leukaemia. Br. J. Haematol. 2011;153:43–46. doi: 10.1111/j.1365-2141.2010.08457.x. [DOI] [PubMed] [Google Scholar]
- 39.O’Connor D, et al. Use of minimal residual disease assessment to redefine induction failure in pediatric acute lymphoblastic leukemia. J. Clin. Oncol. 2017;35:660–667. doi: 10.1200/JCO.2016.69.6278. [DOI] [PubMed] [Google Scholar]
- 40.Zoghbi Annabelle, zur Stadt Udo, Winkler Beate, Müller Ingo, Escherich Gabriele. Lineage switch under blinatumomab treatment of relapsed common acute lymphoblastic leukemia without MLL rearrangement. Pediatric Blood & Cancer. 2017;64(11):e26594. doi: 10.1002/pbc.26594. [DOI] [PubMed] [Google Scholar]
- 41.Hovorkova L, et al. Monitoring of childhood ALL using BCR-ABL1 genomic breakpoints identifies a subgroup with CML-like biology. Blood. 2017;129:2771–2781. doi: 10.1182/blood-2016-11-749978. [DOI] [PubMed] [Google Scholar]
- 42.van Dongen JJ, van der Velden VH, Bruggemann M, Orfao A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. 2015;125:3996–4009. doi: 10.1182/blood-2015-03-580027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer C, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273–284. doi: 10.1038/leu.2017.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van der Velden VH, et al. Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-99 protocol. Leukemia. 2009;23:1073–1079. doi: 10.1038/leu.2009.17. [DOI] [PubMed] [Google Scholar]
- 45.Hoffmann, J. et al. High sensitivity and clonal stability of the genomic fusion as single marker for response monitoring in ETV6-RUNX1-positive acute lymphoblastic leukemia. Pediatr. Blood Cancer66, e27780 (2019). [DOI] [PubMed]
- 46.Lilljebjorn H, Fioretos T. New oncogenic subtypes in pediatric B-cell precursor acute lymphoblastic leukemia. Blood. 2017;130:1395–1401. doi: 10.1182/blood-2017-05-742643. [DOI] [PubMed] [Google Scholar]
- 47.Gawad C, et al. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120:4407–4417. doi: 10.1182/blood-2012-05-429811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Theunissen Prisca M.J., de Bie Maaike, van Zessen David, de Haas Valerie, Stubbs Andrew P., van der Velden Vincent H.J. Next-generation antigen receptor sequencing of paired diagnosis and relapse samples of B-cell acute lymphoblastic leukemia: Clonal evolution and implications for minimal residual disease target selection. Leukemia Research. 2019;76:98–104. doi: 10.1016/j.leukres.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 49.Clappier E, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. 2014;28:70–77. doi: 10.1038/leu.2013.277. [DOI] [PubMed] [Google Scholar]
- 50.Zaliova Marketa, Stuchly Jan, Winkowska Lucie, Musilova Alena, Fiser Karel, Slamova Martina, Starkova Julia, Vaskova Martina, Hrusak Ondrej, Sramkova Lucie, Stary Jan, Zuna Jan, Trka Jan. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica. 2019;104(7):1396–1406. doi: 10.3324/haematol.2018.204974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boer JM, den Boer ML. BCR-ABL1-like acute lymphoblastic leukaemia: from bench to bedside. Eur. J. Cancer. 2017;82:203–218. doi: 10.1016/j.ejca.2017.06.012. [DOI] [PubMed] [Google Scholar]
- 52.Boer JM, et al. Tyrosine kinase fusion genes in pediatric BCR-ABL1-like acute lymphoblastic leukemia. Oncotarget. 2017;8:4618–4628. doi: 10.18632/oncotarget.13492. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.