

Abstract
Infectious diseases remain a threat to critically ill patients, particularly with the rise of antibiotic-resistant bacteria. Septic shock carries a mortality of up to ~40% with no compelling evidence of promising therapy to reduce morbidity or mortality. Septic shock survivors are also prone to nosocomial infections. Treatment with toll-like receptor 4 (TLR4) agonists have demonstrated significant protection against common nosocomial pathogens in various clinically relevant models of infection and septic shock. TLR4 agonists are derived from a bacteria cell wall or synthesized de novo, and more recently novel small molecule TLR4 agonists have also been developed. TLR4 agonists augment innate immune functions including expansion and recruitment of innate leukocytes to the site of infection. Recent studies demonstrate TLR4-induced leukocyte metabolic reprogramming of cellular metabolism to improve antimicrobial function. Metabolic changes include sustained augmentation of macrophage glycolysis, mitochondrial function, and tricarboxylic acid cycle flux. These findings set the stage for the use of TLR4 agonists as standalone therapeutic agents or antimicrobial adjuncts in patient populations vulnerable to nosocomial infections.
Keywords: Toll-like receptor 4, lipopolysaccharide, monophosphoryl lipid A, phosphorylated hexaacyl disaccharides, aminoalkyl glucosamine 4-phosphate, neoseptins, ugi, innate immunity, endotoxin tolerance, metabolic reprogramming
Graphical Abstract
Introduction
Infection remains a serious healthcare problem globally, due to the rising threat of antimicrobial resistance and hospital-acquired infections [1, 2]. Sepsis and septic shock are among of the most common causes of death for patients in the intensive care unit, with a mortality ranging from 15–40% [3–5]. In 2016, the Third International Consensus Conference for Sepsis and Septic Shock (Sepsis-3) developed new terms and defined sepsis as “life-threatening organ dysfunction caused by a dysregulated host response to infection” [5]. Therapy for septic shock remains focused on early antimicrobial treatment and hemodynamic and organ function support [6]. Various therapeutic strategies ranging across in vitro human cell, live animal, and human trials have been attempted in order to target factors thought to drive sepsis pathobiology, including Toll-like receptor 4 (TLR4) antagonists [7, 8], anti-inflammatory and antioxidant agents [9–11], growth factors to augment leukocyte number and function [12–14], and even extracorporeal blood filtration technology [15–18]. While some of these experimental therapeutic interventions have demonstrated benefit in small pilot human subject studies, these have not been examined further in large clinical trials [12, 14, 15]. Thus, new approaches are needed to decrease the incidence and severity of sepsis in vulnerable populations. TLR4 agonists have demonstrated the ability to augment antimicrobial protection and enhance resistance to infection and sepsis [19–21]. In this narrative, we discuss the immunomodulatory properties of TLR4 agonists and future prospective applications for TLR4 agonists to aid in the management of complex infections, including severe sepsis and septic shock.
TLRs are a class of pattern recognition receptors expressed by leukocytes, endothelial cells, and parenchymal cells [22, 23]. They recognize distinct microbial ligands and play a critical role in initiation of innate immune responses to infection. Activation of TLRs induces robust cytokine expression, leukocyte activation, and co-stimulatory receptor expression. Due to their ability to induce potent inflammatory responses, TLR agonists have a profound impact on host immune function. The TLR4 agonist lipopolysaccharide (LPS), a structural component of the cell wall of Gram-negative bacteria, is a potent activator of immune responses. Considerable focus has been invested on the ability of LPS and other TLR4 agonists to modify adaptive immune responses. This has resulted in the commercial development of TLR4 agonists, particularly monophosphoryl lipid A (MPLA), as vaccine adjuvants. MPLA is a component of vaccine adjuvant formulations, such as AS04, that are currently used in commercially available vaccines against the human papilloma virus and shingles (Varicella zoster) [24]. The potency of MPLA as a vaccine adjuvant arises from its ability to activate the antigen presenting functions of dendritic cells and other antigen presenting cells. However, there has been less emphasis on how TLR4 agonists modify innate antimicrobial immune responses.
It was first observed in the 1950s that mice primed with LPS had improved resistance to infection with several Gram-negative pathogens [25]. However, due to its significant toxicity in humans, studies involving the therapeutic benefits of LPS on infection resistance were largely abandoned. Furthermore, it has long been recognized that LPS and other TLR4 agonists induce endotoxin tolerance, an attenuated inflammatory response to further LPS challenge [26, 27]. Some investigators have postulated that endotoxin tolerance represents a state of immune suppression based on observations that mice deficient in pro-inflammatory cytokine production are susceptible to otherwise sub-lethal bacterial infections [28, 29]. Moreover, clinical studies have associated the development of endotoxin tolerance with worsened patient outcomes and increased incidence of secondary nosocomial infections in critically ill patients [30–32]. However, a cause and effect relationship has not been identified.
A growing body of literature indicates that TLR4 agonists, including LPS and MPLA, both of which induce endotoxin tolerance, augment innate responses to infection including enhanced bacterial clearance, despite suppressed pro-inflammatory cytokine responses [33–38]. However, the specific mechanisms by which TLR4 agonists confer resistance to infection remain to be fully elucidated. Recent studies have revealed novel insights into the cellular and metabolic alterations by which TLR4 agonists augment innate host immunity against infection.
This review will focus on TLR4 agonists that have demonstrated potent innate immunomodulatory properties and the mechanisms by which these agonists modify host immune responses. The potential of utilizing these agonists as protective therapies for the prevention of microbial infections and sepsis in high-risk patients will be explored.
TLR4 Agonists Overview
Naturally Sourced Agonists
Lipopolysaccharide
LPS derived from a Gram-negative bacterial cell wall is composed of a hydrophilic polysaccharide backbone of repeating sugar units (O-antigen and Core antigen) linked to a lipid A moiety [39]. Lipid A is a disaccharide bearing variable numbers of acyl chains that interdigitate into the bacterial cell wall. The two glucosamine residues of native lipid A are each substituted with a phosphate group at positions 1 and 4, respectively (Figure 1A). The diphosphate conformation confers potent pro-inflammatory activity and in vivo toxicity [40, 41]. The interaction between the principal lipid A moiety of LPS and TLR4 receptor on innate immune cells leads to cellular activation and production of pro-inflammatory mediators such as cytokines and chemokines. LPS typically binds to the co-factors LPS-binding binding protein (LBP) and CD14 prior to engaging with the myeloid differentiation factor 2 (MD-2)/TLR4 ectodomain (Figure 2A) [42–44]. TLR4 and MD-2 must form a heterodimer that is capable of recognizing and binding LPS [41]. The LPS/CD14 complex is transferred to the MD-2/TLR4 complex for binding to the TLR4 ectodomain where five of the six acyl chains engage deep into the heterodimer pocket (Figure 2B) [45–49]. Binding of LPS with TLR4 leads to activation of the MyD88 adaptor-like (MAL)/myeloid differentiation primary response 88 (MyD88) pathway as well as endocytosis of the MD-2/TLR4 complex. This is followed by interaction with TRIF-related adaptor molecule (TRAM)/TIR-domain-containing adapter-inducing interferon-β (TRIF) proteins and activation of that pathway (Figure 3). TLR4-induced MyD88 and TRIF signaling is demonstrated by IκB kinase (IKK) and interferon regulatory factor 3 (IRF3) phosphorylation, respectively [50–53]. Activation of the TRIF-dependent pathway is associated with type I interferons and RANTES production, while production of pro-inflammatory cytokines such IL-6 are facilitated by activation of either the TRIF- and MyD88-pathways (Figure 3) [54, 55]. Specifically, after dimerization of TLR4, under the MyD88 pathway, the IL-1 receptor-associated kinase (IRAK) become active with phosphorylation of IRAK1 by IRAK4 [54]. Phosphorylated IRAK1 then activates tumor necrosis factor receptor-associated factor 6 (TRAF6) [55], leading to the activation of transforming growth factor-β-activated protein kinase 1 (TAK1) which engages with TGF-β-activated kinase 1/MAP3K7 binding protein 1 (TAB1), TAB2, and TAB 3 [56]. The TAK1/TAB1/TAB2/TAB3 complex then phosphorylates the IKK complex which has two catalytic subunits IKKα and IKKβ, and IKKγ/NEMO (NF-kB essential modulator) [57]. What follows is the degradation of inhibitor of κB (IκB) which leads to the development of NF-kB dimers that translocate to the nucleus [57]. TAK1 also activates MAP kinase kinase 3 (MKK3) and MKK6, leading to the activation of c-Jun N-terminal Kinase (JNK) and p38 [56]. P38 then activates the transcription factors CREB and JNK to induce AP-1 formation and nuclear translocation (Figure 3) [58–62]. P38 then activates the transcription factor CREB and JNK increase AP-1 transcription factor [58]. Regarding the alternative, TRIF pathway, cytosolic TRIF-related adaptor molecule (TRAM) recruits TIR-domain-containing adapter-inducing interferon-β (TRIF) to the TLR4 receptor. The LPS-induced activation leads to internalization of the TLR4/TRAM/TRIF complex via an endosome [59]. As well as signaling to through RIP1 to TRAF6, this leads to the recruitment of TNF-R-associated factor 3 (TRAF3) to TRIF, which initiates activation of IKKi and TANK-binding kinase 1(TBK1) [57, 60]. Interferon regulatory factor 3 (IRF3) is then translocated to the nucleus, leading to the production of inflammatory mediators and type 1 interferons (Figure 3) [61, 62].
Fig. 1. Chemical structures of naturally sourced toll-like receptor 4 (TLR4) agonists.

(A) lipopolysaccharide (LPS), and (B) monophosphoryl lipid A (MPLA).
Fig 2. Schematic of toll-like receptor 4 (TLR4) interactions with accessory proteins.

(A) Extracellular LPS binds LPS-binding protein (LBP). From there, LPS is transferred to CD14, which is associated with the ectodomain of TLR4. (B) CD14 facilitates recognition of LPS by transferring it to the complex of TLR4 and myeloid differentiation factor 2 (MD-2).
Fig 3. Toll-like receptor 4 (TLR4) signaling.

LPS binding induces TLR4 dimerization at the cell surface and activation of MAL and MyD88. This induces phosphorylation of IRAK, which signals through TRAF6 and TAK1, recruiting TAB proteins. This complex signals through MKKs to p38 and JNK to activate CREB and AP-1 transcription factors. The TAK/TAB complex also signals through the IKK complex to activate NFκB. These pathways lead to production of proinflammatory cytokines. LPS binding induces endocytosis of TLR4, activating signaling through TRAM and TRIF to TRAF3. This ultimately activates transcription factor IRF3 and produces type 1 interferons. TRIF also communicates with TRAF6 via RIP1, activating the MKK and NFκB pathways as well.
Two fundamental elements of lipid A contribute to its potential to induce an innate immune response: the number and length of acyl chains and the number of phosphate groups. Longer acyl chains and the presence of two phosphate groups independently provoke a more robust immune response, [63] while the presence of 6 acyl chains facilitates optimal TLR4 activation. Reducing the number of lipid A acyl chains to four renders the ligand a TLR4 antagonist [41].
Monophosphoryl Lipid A
Cleaving the C1 phosphate group from lipid A generates MPLA (Figure 1B). MPLA was originally derived from Salmonella minnesota strain 595, upon a series of acid- followed by base-hydrolysis reactions, which yielded a heterogeneous mixture of lipids with the primary lipid A species being 3-O-deacyl-4’-monophosphoryl lipid A [64, 65]. MPLA has 6 acyl chains and, like LPS, these acyl chains enter the convex domain formed by the MD-2/TLR4 complex. MPLA possesses similar immunomodulatory properties as LPS but is approximately 100-fold less toxic. The decreased toxicity is largely due to absence of the C1 phosphate group and a greatly reduced ability to induce pro-inflammatory cytokine production and deleterious leukocyte activation [41, 60]. One factor that appears to contribute to differences in pro-inflammatory activity among LPS and MPLA is the differential ability of the agents to activate the NLRP3 inflammasome. Recent papers indicate that LPS potently induces expression of NLRP3, procaspase 1 and pro-IL-1β and secretion of IL-1β whereas MPLA is a weak inducer of those factors [66, 67]. Further studies show that MPLA is a weak activator of mast cells, whereas LPS potently induces mast activation [68]. Unlike LPS, MPLA does not induce release of superoxide and myeloperoxidase from neutrophils and does not require LBP and CD 14 to engage with the TLR4 ectodomain [69, 70]. Furthermore, the two phosphate groups on LPS interact with the dimerization interface of TLR4, and the single phosphate in MPLA may influence the decreased TLR4 dimerization noted with MPLA binding [70, 71].
MPLA has been administered to humans by intravenous infusion at doses of up to 20 μg/kg. Astiz and colleagues reported that humans did not experience subjective side effects until doses of 10 μg/kg or higher were administered. At 20 μg/kg, humans experienced mild to moderate symptomatology in association with increases in heart rate, body temperature and elevated concentrations of TNF-α, IL-6 and IL-8 in plasma. Further studies showed that systemic administration of MPLA induces endotoxin tolerance in humans. Pretreatment with MPLA led to fewer symptoms and lower cytokine concentrations during endotoxin infusion compared to controls [72]. MPLA was previously identified as a TRIF-biased TLR4 agonist, however recent work demonstrated both pathways become activated, with MyD88 predominance [73, 74]. The potent immunomodulatory properties of MPLA accompanied by its low toxicity has led to its use as an adjunct in various vaccine adjuvant systems. Intradermal injection of MPLA in non-human primates leads to the recruitment of CD14+CD16− monocytes and myeloid dendritic cells into local lymph nodes, which facilitate antigen presentation and augment vaccine efficacy [75]. TLR4 agonists promote TH1 polarization which is characterized by an increased production of interferon- γ (IFN-γ) and opsonization of IgG1 and IgG3 immunoglobulins, whereas alum generates TH2 responses (74–76). MPLA also primes the T-cell populations for long-term survival and induces antigen specific T-cell clonal expansion [73, 76]. Adding MPLA to vaccine adjuvant systems increases antibody titers by up to 20-fold [77]. These vaccine adjuvant systems include AS01 which is used in the malaria and herpes zoster vaccines, and AS04 which is used in the human papillomavirus and hepatitis B vaccines [24].
Synthetic Agonists
Phosphorylated Hexaacyl Disaccharides (PHADs)
There are TLR4 agonists under development that are synthetized de novo. MPLA itself can be synthesized de novo and is currently available for non-clinical applications from commercial vendors. Phosphorylated hexaacyl disaccharides (PHADs) are another family of ultrapure, synthetic TLR4 agonists. Like LPS and MPLA, the acyl chains of PHADs activate both MyD88- and TRIF-pathways [21]. Similar to MPLA, PHADs have one phosphate group. Three different products, termed PHAD, 3-deacyl PHAD (3D PHAD) and 3D(6-Acyl)-PHAD, have been developed, each with varying 14 carbon acyl side chain conformations (Figure 4A–C). Differences among the three PHADs involve modifications of the location of the 5th acyl chain. 3D(6-Acyl)-PHAD has the strongest resemblance to MPLA, having the 5th acyl chain attached to the hydroxyl group of the 6th acyl chain. 3D PHAD lacks the 5th acyl chain, thus forming a pentaacyl agonist, as compared to the hexaacyl products PHAD and 3D(6-Acyl)-PHAD. The pentaacyl 3D PHAD retains the ability to activate TLR4 and retains immunoenhancing properties similar to the hexaacyl agonists MPLA, PHAD, and 3D(6-Acyl)-PHAD [21]. Thus, the presence of six acyl chains may be less important than the length of the acyl chains. In a study by Stover et al., modifications in the left secondary acyl chain of hexaacyl aminoalkyl glucosaminide phosphate (AGP) TLR4 agonists led to changes in TLR4 activity [78]. Changing the left secondary acyl chain from 6–14 carbons increased TLR4 activation, where decreasing acyl chain length to 8 carbons or less resulted in significantly lower TLR4 activation [78].
Fig. 4. Chemical structures of phosphoryl hexaacyl disaccharide (PHAD) derivatives and aminoalkyl glucosamine 4-phosphate molecules.

(A) 3D(6-acyl)-PHAD, (B) PHAD, (C) 3D-PHAD, (D) CRX-527, and (E) CRX-547.
Aminoalkyl glucosamine 4-phosphate
AGP is a synthetic lipid A TLR4 agonist. Within the AGP class, there are two structurally similar agonists, CRX-547 and CRX-527 [79]. CRX-547 is derived from CRX-527 upon modification of the L-seryl AGP to the D-isomer (Figure 4D–E). CRX-527 is a potent inducer of the MyD88- and TRIF-signaling pathways, as noted by increased concentrations of TNF-α, and CXCL-10 and RANTES, respectively [79]. However, CRX-547 has significantly reduced MyD88 signaling yet retains much of the TRIF-dependent signaling activity [79]. The TRIF selectivity may result from CRX-547 impeding homodimerization and other conformational changes in the MD-2/TLR4 complex necessary for MyD88 binding [80].
Synthetic Small Molecule Agonists
Neoseptins
Neoseptins were identified upon screening nearly 100,000 compounds using functional assays for induction of TNF-α from pretreated mouse macrophages.[81]. Specifically, neoseptin-3 is a potent TLR4 agonist when compared to LPS despite a lack of structural similarity (Figure 5A) [82]. Neoseptin-3 activates the MD-2/TLR4 complex, yet is independent of CD14. Neoseptin stimulation failed to induce TNF-α in TLR4- and MD-2-deficient mouse macrophages, whereas isolated CD14-deficient mice with intact MD-2/TLR4 complex demonstrated MyD88 and TRIF signaling activity upon neoseptin stimulation. Of note, neoseptin-3 failed to stimulate TLR4 in human THP-1 monocytes, thus functioning as a TLR4 agonist specific to mice and offering limited therapeutic potential [82].
Fig. 5. Chemical structures of synthetic small molecule toll-like receptor 4 (TLR4) agonists.

(A) Neoseptin-3, and (B) Ugi.
Ugi compounds
In a study by Marshall et al., high throughput screening of small molecules led to the identification of a novel class of TLR4 agonists called Ugi compounds (Figure 5B) [83]. These new TLR4 agonists have demonstrated potent TLR4 activation by measuring cytokine induction in TLR4-transfected human embryonic kidney cells (HEK), but not on TLR4 in mouse cells. However, inclusion of human MD-2 with mouse TLR4 led to partial activation. Ugi also failed to activate TLR4 in rat, rabbit, ferret and cotton rat cells. Prospects for Ugi include formulation for use as vaccine adjuvants with greater potency than MPLA based on greater TLR4 activity in HEK cells.
Augmentation of Innate Antimicrobial Responses by TLR4 Agonists
As noted above, much effort has been devoted to developing TLR4 agonists as vaccine adjuvants. The remainder of this paper will focus on reviewing the ability of TLR4 agonists to augment innate antimicrobial immunity.
Endotoxin tolerance
Defining the factors responsible for infection susceptibility is essential for understanding how to therapeutically modulate the immune response to infection. One of the most frequently used assessments of innate immune function in clinical research settings is the magnitude of the cytokine response induced by LPS. Numerous investigators have reported that an attenuated cytokine response to LPS (i.e. endotoxin tolerance) is associated with increased morbidity and mortality in infected critically ill patients [30–32]. However, the relationship between the magnitude of the cytokine response to LPS and infection susceptibility is merely correlative. Studies have failed to demonstrate that LPS-elicited cytokine responses provide an accurate assessment of the host response to infection. To directly address this issue, Fensterheim et al. hypothesized that innate antimicrobial efficiency, rather than the cytokine responses elicited by exposure to TLR agonists, determines infection outcomes [20]. The ability of TLR agonists to induce endotoxin tolerance or sensitization had no correlation with the ability of these agonists to enhance antimicrobial effector cell functions and improve pathogen clearance and resistance to infection. Specifically, treatment of mice with CpG-ODN, a TLR9 agonist, elicited endotoxin sensitization, as indicated by significant increases in plasma concentrations of IL-6, IFN-γ, and IL-1β in response to LPS challenge. In contrast, mice pretreated with the TLR4 agonists MPLA or LPS elicited endotoxin tolerance, as indicated by attenuation of the same cytokine levels. However, mice pretreated with the TLR agonists LPS, MPLA, or CpG demonstrated equal levels of protection from subsequent bacterial infection [20]. Antimicrobial protection was associated with an increased recruitment of phagocytes, such as neutrophils and macrophages, to the site of infection [20]. The alternate hypothesis that endotoxin tolerance is immunoprotective is challenged by evidence wherein pretreatment with CpG led to increased LPS-elicited cytokine responses in vivo, resulting in antimicrobial protection against bacterial infection similar to that elicited by treatment with LPS or MPLA [20]. Collectively, these findings suggest that TLR4 agonist-induced augmentation of the host response to infection via enhancement of antimicrobial functions such as leukocyte recruitment, phagocytosis and microbial killing will reduce systemic inflammation and facilitate resolution of infection.
Several further studies have shown that priming with TLR4 agonists will augment host resistance to infection despite inducing endotoxin tolerance. Murphey and colleagues primed mice with LPS from E. coli or Pseudomonas aeruginosa and observed suppressed IL-12 and IFN-γ production but greatly augmented bacterial clearance and survival in mice challenged with P. aeruginosa [37, 84, 85].
Cellular Responses
Earlier studies with LPS have correlated improved resistance to infection from a variety of pathogens with enhancements in phagocyte recruitment, improved bacterial clearance, and attenuation of pro-inflammatory cytokine production [20, 60, 86, 87]. More recent studies have demonstrated similar immunomodulatory properties of more clinically-applicable agonists such as MPLA and PHADs (Table 1) [19, 21, 33, 36]. These studies reveal insights into the cellular mechanisms by which TLR4 agonists mediate improved bacterial clearance, including expansion of innate immune cell precursors, enhanced leukocyte mobilization and recruitment to sites of infection, and augmented antimicrobial functions of innate immune cells, which together lead to more effective pathogen clearance [33, 34, 36].
Table 1.
TLR4 agonists that modify innate immune responses to infection
| TLR4 Agonist | Modified Innate Response in Models of Infection | Reference(s) |
|---|---|---|
| LPS | ↑ Macrophage-mediated bacterial clearance during polymicrobial sepsis | Deng et.al. J Immunol 2013 [86] |
| ↓ Bacterial burden & serum cytokines during P. aeruginosa & Salmonella enterica infection | Lehner et al. Infect Immun 2001; Murphey et al. Shock 2007 [35, 84] | |
| ↑ Survival & decreased serum cytokines during Cryptococcus neoformans infection | Rayhane et al. Infect Immun 2000 [87] | |
| ↑ Recruitment of phagocytes to the site of P. aeruginosa infection | Varma et al. Infect Immun 2005 [85] | |
| ↑ Leukocyte recruitment after S. aureus infection | Murphey et al. Shock 2008 [37] | |
| ↑ Neutrophil recruitment, antimicrobial function, bacterial clearance, and survival during fecal peritonitis | Wynn et al. Blood 2008 [89] | |
| MPLA | ↑ Recruitment of innate leukocytes to sites of P. aeruginosa & S. aureus infections | Bohannon et al. J Leukoc Biol 2015; Fensterheim et al. J Immunol 2018 [19, 36] |
| ↑ Expansion of innate leukocyte progenitors in the bone marrow | Bohannon et al. J Leukoc Biol 2015 [19] | |
| ↑ Survival during P. aeruginosa, S. aureus, & C. albicans infections | Bohannon et al. J Leukoc Biol 2015; Romero et al. Infect Immun 2011; Fensterheim et al. J Immunol 2018 [19, 33, 36] | |
| ↑ Bacterial clearance during P. aeruginosa | Bohannon et al. J Leukoc Biol 2015; Romero et al. Infect Immun 2011 [19, 33] | |
| ↑ Bacterial clearance during P. aeruginosa & S. aureus infections, which is preserved up to 10 days after treatment | Fensterheim et al. J Immunol 2017; Fensterheim et al. J Immunol 2018 [20, 36] | |
| ↑ Antimicrobial functions of innate phagocytes | Fensterheim et al. J Immunol 2018 [36] | |
| ↑ Macrophage metabolic phenotype characterized by sustained heightened glycolysis and oxidative phosphorylation | Fensterheim et al. J Immunol 2018 [36] | |
| ↓ Cytokines in the plasma & site of infection | Fukuda et al. Shock 2019 [38] | |
| ↑ Bacterial clearance & attenuated lung injury during P. aeruginosa pneumonia in a post-burn ovine model | Fukuda et al. Shock 2019 [38] | |
| ↑ Human neutrophil antimicrobial functions | Ruchaud-Sparagano et al. Immunol Cell Biol 2014 [69] | |
| PHADs | ↑ Recruitment of innate leukocytes to sites of P. aeruginosa & S. aureus infections | Hernandez et al. Crit Care Med 2019 [21] |
| ↑ Survival during P. aeruginosa & S. aureus infections | Hernandez et al. Crit Care Med 2019 [21] | |
| ↑ Bacterial clearance during P. aeruginosa & S. aureus infections, which is preserved up to 10 days after treatment | Hernandez et al. Crit Care Med 2019 [21] | |
| ↑ Antimicrobial functions of innate phagocytes | Hernandez et al. Crit Care Med 2019 [21] | |
| ↓ Cytokines in the plasma & site of infection | Hernandez et al. Crit Care Med 2019 [21] | |
Romero and colleagues demonstrated that treatment with MPLA greatly facilitated the recruitment of neutrophils and monocytes to the peritoneal cavity in response to cecal ligation and puncture [33]. Depletion of neutrophils ablated the ability of MPLA to augment resistance to infection. Similarly, Murphey et al., showed that innate leukocyte expansion and recruitment was crucial to LPS-induced resistance to Staphylococcus aureus infection [37]. Wynn et al., demonstrated pretreatment with LPS enhanced neutrophil recruitment with increased oxidative burst production, reduced bacteremia, and improved survival from fecal induced peritonitis [88]. Ruchaud-Sparagano and colleagues demonstrated the ability of MPLA to augment antimicrobial functions of human neutrophils. Treatment of human neutrophils with MPLA augmented neutrophil chemotaxis and bacterial killing without inducing extracellular release of cytotoxic mediators [69].
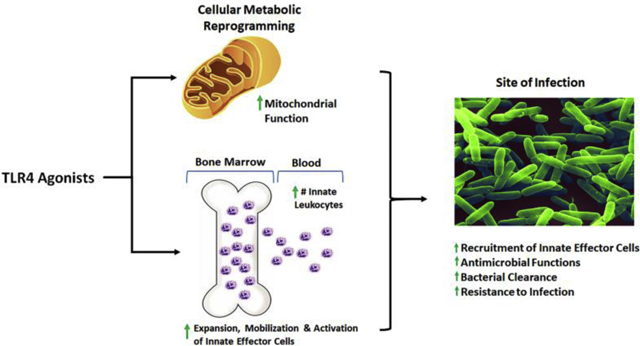
Bohannon and colleagues further elucidated potential cellular mechanisms for mediating the augmented innate leukocyte responses triggered by TLR4 priming. Induction of granulocyte colony stimulating factor (G-CSF) and the chemokines CXCL1 and CXCL2 by MPLA stimulated bone marrow progenitor expansion, as well as mobilization and recruitment of neutrophils, which are essential for mediating clearance of bacteria (Figure 6) [34, 36, 74]. MPLA treatment also caused neutrophil activation, as indicated by increased expression of CD11b and shedding of L-selectin [34, 74] (Figure 6). Direct antimicrobial functions including phagocytosis and respiratory burst functions of monocytes and neutrophils were also enhanced by TLR4 agonists [21, 36]. Of note, MPLA-induced protection against infection was independent of the adaptive immune system, as RAG2−/− mice deficient in T and B cells remained protected from infection [20]. Interestingly, when G-CSF was neutralized prior to MPLA treatment, MPLA-induced augmented resistance to infection and attenuation of pro-inflammatory cytokines were ablated. Rather, the treated mice showed high levels of serum cytokines after infection similar to control mice [34]. This observation suggests that the augmentation of microbial clearance is likely to be the primary mechanism conferring protection after TLR4 agonist treatment.
Fig. 6. Mechanism by which monophosphoryl lipid A (MPLA) increases bacterial clearance and resistance to infection.

MPLA agonism of TLR4 induces G-CSF production and decreases CXCR4 expression, leading to an increase in myeloid progenitors in bone marrow. Increased G-CSF, along with induction of CXCL1 and CXCL2 promote mobilization of neutrophils from the bone marrow into the blood, which are then recruited to the site of infection. MPLA induced activation of neutrophils, evidenced by increased CD11b with concurrent shedding of L-selectin. These activated and mobilized neutrophils display increased antimicrobial activity, including augmented phagocytosis and respiratory burst, effectively leading to increased bacterial clearance and resistance to infection.
Both MPLA and PHADs augment resistance to both Gram-negative P. aeruginosa and Gram-positive S. aureus infection in mice [20, 21, 33, 34, 36]. Further, MPLA was also shown to improve resistance against fungal Candida albicans infection [36]. When evaluated for antimicrobial effectiveness, all PHADs were equipotent immunomodulators when compared to MPLA. In murine models of infection with P. aeruginosa and S. aureus, all PHADs and MPLA demonstrated equipotent antimicrobial protection, lasting up to 10 days after treatment. Antimicrobial protection was associated with increased leukocyte recruitment to the site of infection and enhanced phagocytosis and respiratory burst functions in neutrophils, monocytes, and macrophages [21, 36]. Systemic infection with S. aureus in mice pretreated with PHADs and MPLA demonstrated lower bacterial burden in lung, kidney and spleen tissue [36].
MPLA priming has also been demonstrated to protect against infection in a large animal model following severe trauma. Fukuda et al., demonstrated that severely burn-injured sheep which received systemic infusion with MPLA exhibited attenuated acute lung injury, lower blood lactate levels, attenuated plasma cytokine production, less vascular leakage, improved hemodynamic parameters, and attenuated lung bacteria burden during subsequent P. aeruginosa pneumonia. Importantly, systemic MPLA infusion was well-tolerated by burned sheep, and induced only mild and transient hemodynamic alterations. These findings are important in that they demonstrate that MPLA can be administered safely in a model that closely mimics human critical illness and protects against subsequent infection and organ injury [38].
The finding that TLR4 agonists have the ability to mediate protection against a variety of pathogens, including Gram-negative and Gram-positive bacteria and fungi, is noteworthy because it demonstrates that even though these agonists are derived from Gram-negative organisms, they retain the ability to induce resistance to diverse pathogens. This is consistent with prior studies revealing that a myriad of products derived from various microbial organisms can induce broad resistance against pathogens [37, 84, 89, 90]. This raises the question of whether activation of innate antimicrobial responses by these agonists is mediated through a common molecular pathway. The discovery of this common pathway would pave the way for a multitude of potential therapeutics to decrease the incidence of nosocomial infections in vulnerable patients.
TLR4 agonist-induced reprogramming of cellular metabolism
The innate immune response to infection is metabolically taxing for both trafficking leukocytes and resident tissue cells [91]. Thus, the capacity for metabolic adaptation is essential to mount a successful defense against infection. Recent studies have demonstrated that TLR agonists induce dynamic alterations in substrate utilization and cellular metabolism to enhance adaptation and, in turn, antimicrobial activity [20]. Important roles for glycolytic metabolism, tricarboxylic acid (TCA) cycle intermediates, and oxidative phosphorylation (OXPHOS) machinery have been described. In this section, we will review the changes seen in each of these core metabolic pathways after TLR4 agonism.
TLR4 agonist-induced alterations in glycolysis
Aerobic metabolism, including consumption of pyruvate to feed the TCA cycle and OXPHOS, is often the predominant source of ATP for cells residing in an oxygen-rich environment. In contrast, leukocytes have adapted to not only survive in a hypoxic environment, but also to harness the intracellular hypoxic machinery to drive inflammatory processes [92]. Over fifty years ago, researchers observed that activated macrophages consume glucose at an elevated rate as compared to unstimulated cells [93]. Several decades later Krawczyk and colleagues observed that stimulation of dendritic cells with LPS triggered enhanced glycolysis despite the presence of oxygen, a process termed aerobic glycolysis [94]. This metabolic shift is mediated by signaling through the PI3K/Akt pathway and accompanied by a decrease in oxygen consumption. It was later shown that LPS specifically induces expression of pyruvate kinase isoenzyme M2 (PKM2), which not only augments glycolysis, but also promotes expression of hypoxia related genes [95]. Surprisingly, Haschemi et al., showed that instead of using glucose to fuel enhanced OXPHOS, LPS-stimulated macrophages enrich the pentose-phosphate pathway and excrete additional lactate, two hallmarks of metabolic reprogramming favoring glycolysis [96]. Additional studies found that hypoxia-inducible factor-1α (HIF-1 α) stabilization drove aerobic glycolysis in innate leukocytes [97, 98]. In a landmark discovery, Tannahill and colleagues showed that aerobic glycolysis was not simply a byproduct of immune activation; rather, they showed the deliberate stabilization of HIF-1α enhanced glycolysis and directly fueled inflammatory responses in macrophages [99].
Based on this work, pro-inflammatory, or “M1,” macrophages are characterized by an acute reliance on glycolysis. Several hypotheses exist as to why this is the case. First, while OXPHOS generates significantly more ATP than glycolysis on a per-glucose basis, an activated cell may successfully complete aerobic glycolysis at a rate several fold higher than that possible for OXPHOS. In turn, these cells may have higher production of ATP for a short period of time [100]. Additionally, glycolytic intermediates can serve as precursor substrates for protein and lipid synthesis, meaning that enhanced glycolysis may uniquely provide building blocks for mediators of inflammation and antimicrobial activity [100].
Augmentation of glycolysis after stimulation is not limited to LPS. Indeed, other TLR agonists including CpG and polyinosinic:polycytidylic acid (Poly I:C) trigger a similar effect [20]. Furthermore, these ligands do not simply induce transient changes. Studies with MPLA and CpG also demonstrated a durable elevation in glycolytic rate three days after the removal of the initial stimulus [20]. Less is known about the consequences of blocking glycolytic metabolism during TLR agonism. However, inhibition of HIF-1α during LPS stimulation inhibits macrophage recruitment and function [12]. Additionally, Fensterheim et al., demonstrated that inhibiting the activity of the mechanistic target of rapamycin (mTOR), which is upstream of HIF-1α prevents the protective phenotype generated by MPLA priming in vivo [36].
TLR4 agonist-induced alterations in mitochondrial metabolism
The field of immunomodulation is increasingly steering towards the discovery of novel immune related functions of the mitochondrial TCA cycle intermediates. There is a growing interest in evaluating the physiological consequences of the alterations of the levels of mitochondrial metabolites as a result of inflammatory stimulus [101, 102]. Our studies and others have shown that TLR4 agonist-induced stimulation of innate leukocytes such as macrophages and monocytes, leads to metabolic reprogramming of the mitochondrial TCA cycle resulting in intracellular accumulation of major TCA cycle intermediates such as citrate, itaconate, succinate, and malate (Table 2) [36, 99, 101, 103, 104]. Each of these metabolites play a unique role in regulating cellular functions, as described in the following sections.
Table 2.
Toll-like receptor 4 agonist mediated augmentations in mitochondrial metabolism and immunoregulation
| Metabolite | Immunomodulatory Effect | Reference(s) |
|---|---|---|
| MALATE (Via ↑ production and shuttling back into the mitochondria) | Associated with ↑ TCA flux cycle, ↑ oxygen consumption rate, & ↑ phagocytic capacity | Fensterheim et.al. J Immunol 2018 [36] |
| CITRATE (Via ↑ accumulation via downregulation of IDH) | ↑ Expression of mitochondrial citrate carrier, ↑ Export to cytosol, downstream inflammatory response | Infantino et. al. Biochem J 2011 [105] |
| ↑ Itaconate | Jha et. al. Immunity 2015 [104] | |
| ITACONATE (Via accumulation of citrate and ↑ Irg1) | Antiinflammatory | |
| ↑ Nrf2, heme oxygenase 1, glutathione | Mills et. al. Nature 2018 [111] | |
| ↓ IL-1β, IL-6 | Bambouskova et.al. Nature 2018 [112] | |
| ↓ SDH activity (↓ proinflammatory cytokines, ↓ ROS) | Lampropoulou et. al.Cell Metab 2016 [114] | |
| Antimicrobial | ||
| Inhibition of Microbial glyoxalate shunt | Cordes et. al. Annu Rev Nutr 2015 [115] | |
| Inhibits microbial growth | Michelucci et. al.PNAS 2013 [109] | |
| Sasikaran et. al.Nat Chem Biol 2014 [116] | ||
| SUCCINATE (Via downregulation of SDH and glutamine anapleurosis) | ↑ IL-1β, inflammation, | Tannahill et. al. Nature 2013 [99] |
| HIF-1α stabilization | Mills et. al. Trends Cell Biol 2014 [118] | |
| Succinylation of target proteins via ↓ SIRT5 | Tannahill et. al. Nature 2013 [99] | |
| ↑ mitochondrial ROS generation | Chouchani et. al. Cell Metab 2016 [121] | |
Jha et al., demonstrated that LPS-induced activation of macrophages leads to the induction of breakpoints in the TCA cycle via downregulation of isocitrate dehydrogenase (IDH) and succinate dehydrogenase (SDH) function, which contributes significantly towards accumulation of citrate and succinate [104]. Fensterheim et al., demonstrated that MPLA-activated macrophages actively export citrate out from mitochondria to the cytosol, where it is broken down into acetyl-CoA and oxaloacetate [36]. At 24 hours after MPLA stimulation, oxaloacetate is sequentially converted to malate and pyruvate. On the other hand, malate is shuttled back into the mitochondria to replenish the mitochondrial pool of oxaloacetate at 72 hours post-MPLA stimulation, leading to increased TCA cycle flux. This increased TCA flux is also temporally associated with a significant increase in mitochondrial content, oxygen consumption rate and increased phagocytic capacity. Another study by Infantino et al., showed that LPS stimulation of macrophages upregulates the expression of mitochondrial citrate carrier (CIC) protein levels and subsequent increase of cytosolic citrate levels. Citrate is required for the pro-inflammatory response of activated macrophages [105–108]. Therefore, we hypothesize that initial citrate accumulation upon TLR4 agonist-induced stimulation of innate leukocytes fuels the inflammatory response, which ultimately transitions over time to increase TCA cycle flux and enhance the phagocytic capacity and an anti-inflammatory resolution response.
Increased citrate is also diverted towards production of itaconate, as a result of inflammatory stimulus-induced upregulation of a unique mitochondrial enzyme, known as Immunoresponsive gene 1 (Irg1) [104, 109]. Itaconate is increasingly being recognized as one of the key players inducing metabolic reprogramming of leukocytes [110]. One of the first studies by Michelucci et al., showed that LPS-induced upregulation of Irg1 led to increased itaconate levels in mouse and human leukocytes [109]. A time course study by Zhu et al., demonstrated that relative itaconate levels peak at 8–24 hours and then gradually decline until 96 hours upon LPS stimulation of THP-1 monocytic cells [103]. Itaconate has been shown to exert an anti-inflammatory effect on macrophage function. Mills et al., demonstrated that treatment of macrophages with 4-octyl itaconate (cell permeable itaconate analogue) significantly increased LPS induced nuclear respiratory factor 2 (Nrf2) levels, which ultimately lead to a significant increase in anti-inflammatory genes including heme oxygenase 1 and glutathione synthesis [111]. Pretreatment with 4-octyl itaconate also decreased LPS-induced increase in the pro-inflammatory cytokines IL-1β and IL-6. Other studies using another itaconate analogue, dimethyl-itaconate, have also demonstrated similar findings of enhanced Nrf2-mediated reduction in the inflammatory response [112].
LPS stimulation of macrophages has also been shown to inhibit SDH activity leading to succinate accumulation [104, 113]. Lampropoulou et al., showed that itaconate inhibits SDH activity in LPS-stimulated macrophages leading to decreased succinate oxidation, which limits generation of excess reactive oxygen species (ROS) and pro-inflammatory cytokines [114]. Itaconate is also known to inhibit the microbial glyoxylate shunt which is critical for bacterial survival [115]. Numerous studies have now shown that itaconate inhibits the growth of a variety of microbes including Escherichia coli, Salmonella enteretica, Mycobacterium tuberculosis, S. aureus and others [109, 116]. LPS stimulation of macrophages has been shown to accumulate up to 8 mM levels of intracellular itaconate and activated macrophages actively transport itaconate to the extracellular milieu [115, 117]. Therefore, TLR4 agonist treatment-induced increase in itaconate not only acts as an antimicrobial metabolite but also limits inflammation. Our studies and others have shown that pretreatment with MPLA and low dose LPS significantly reduces infection-induced inflammation and provides prolonged protection against bacterial and fungal models of murine sepsis [20, 33, 34]. It is reasonable to hypothesize that increased itaconate levels upon pretreatment with microbial ligands such as MPLA and PHADs might be one of the major mechanisms responsible for strengthening host immunity leading to protection against deadly infections.
Inhibition of SDH activity by itaconate and glutamine anapleurosis leads to succinate accumulation upon LPS stimulation of macrophages [104]. LPS-induced increase in succinate accumulation leads to increased IL-1β production and inflammation [99]. Succinate is empirically viewed as a metabolite fueling inflammation. The pro-inflammatory effects of succinate have been shown to be mediated via enhanced mitochondrial ROS production, HIF-1α stabilization, signaling through its G-protein coupled receptor (SUCNR1) and post-translational modification of other proteins via succinylation [118]. LPS-induced increase in succinate in macrophages stabilizes HIF-1α protein under normoxic conditions and HIF-1α plays a critical role in inflammatory response of myeloid cells [99, 119, 120]. Along with an increase in succinate levels, an intact SDH activity is also important to sustain macrophage pro-inflammatory responses, as treatment with dimethyl malonate (SDH inhibitor) decreased HIF-1α expression and inflammatory gene expression [120]. Increasing oxidation of high levels of accumulated succinate by active SDH favors augmentation of mitochondrial ROS generation via reverse electron transport [121]. Mitochondrial ROS are critical for microbial clearance. Upon TLR stimulation, phagosomes recruit mitochondria and augmented mitochondrial ROS aids in clearance of phagocytosed microbes [122]. To implicate the role of SDH generated ROS in microbial clearance and immunomodulation, further in vivo studies are needed in animal models of infection. Succinate also alters immune cell function via succinylation of target proteins while the sirtuin enzyme, SIRT5, is responsible for desuccinylation [123]. LPS has been shown to reduce SIRT5 expression leading to increased succinylation of cellular proteins [99]. Upon SIRT5 knock down, increased succinylation has been shown to augment SDH activity, leading to increased mitochondrial respiration [124]. However, none of the studies have evaluated cytokines as a target for succinylation. Base on available literature, it is quite possible that TLR4 agonist-induced succinate accumulation and succinylation of target proteins might be one of the important mechanisms for metabolic reprogramming of leukocytes, which warrants further investigation. Overall, it is evident that TLR4 stimulation reprograms cellular metabolism of innate leukocytes leading to augmentation of mitochondrial function and TCA cycle flux which ultimately aid in microbial clearance and improved survival [20, 36].
Conclusions
Infections continue to be a significant problem in healthcare systems globally and with the emergence of antibiotic resistance, we need new strategies to aid in the prevention and treatment of infections. LPS is a potent TRL4 agonist, inducing a robust inflammatory response, and has been shown to prime the immune system and provide protection against infectious agent. However, due to its severe toxicity in humans it is not a therapeutic candidate. The vaccine adjuvant MPLA has similar immunomodulatory properties to LPS, demonstrating protection against Gram-negative and -positive infections and is well tolerated by humans. MPLA, with 1/100th the toxicity of LPS, provides antimicrobial resistance by augmenting the innate immune function via expansion and mobilization of leukocytes to the site of infection, enhancing phagocytic function and respiratory burst, and by reprograming leukocyte cellular metabolism. However, MPLA is only approved as a vaccine adjuvant for clinical use and is thus unavailable to use as an independent antimicrobial agent. This leaves a need for TLR4 agonists which possess similar immunomodulatory properties as MPLA that can function as antimicrobial adjuncts for clinical use, such as PHADs and other clinically relevant TLR4 agonists. The primary success of TLR4 agonists in clinically relevant models of infection are focused on pretreatment prior to infection or injury, thus making these agents prospects for perioperative or peri-trauma therapeutic agents. More studies are necessary to identify optimal dosing, safety, pharmacodynamic and pharmacokinetic profiles for PHADs and other TLR4 agonists. With the rising rate of antimicrobial resistance, TLR4 agonists may serve to function as an adjunct to limit the time necessary for antibiotic therapy and possibly reduce the rate of infections.
Acknowledgments
This manuscript was supported by U.S. National Institute of Health, Institute of General Medicine Grants K08 GM123345 to AH, R01 GM12171 to JKB, T32 grant, 5T32GM108554-05 to NKP, and R01 GM119197 to DLW and ERS.
Abbreviations
- AGP
aminoalkyl glucosaminide phosphate
- AP-1
activator protein 1
- AS04
adjuvant system 04
- CpG-ODN
cytosine phosphate guanine oligodeoxynucleotide
- CREB
cAMP response element-binding protein
- G-CSF
granulocyte-colony stimulating factor
- HIF-1α
hypoxia-inducible factor-1α
- IFN-γ
interferon-γ
- Irg1
immunoresponsive gene 1
- IκB
inhibitor of κB
- IKK
IκB kinase
- IRAK
IL-1 receptor-associated kinase
- IRF3
interferon regulatory factor 3
- JNK
c-Jun N-terminal kinase
- LPS
lipopolysaccharide
- MAL
MyD88 adapter-like
- MKK
MAP kinase kinase
- MD-2
myeloid differentiation factor 2
- MPLA
monophosphoryl lipid A
- mTOR
mammalian target of rapamycin
- MyD88
myeloid differentiation primary response 88
- NEMO
NF-κB essential modulator
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NLRP3
NLR family pyrin domain containing 3
- Nrf2
nuclear respiratory factor 2
- OXPHOS
oxidative phosphorylation
- PHAD
phosphorylated hexaacyl disaccharide
- PKM2
pyruvate kinase isoenzyme M2
- Poly I:C
polyinosinic:polycytidylic acid
- RANTES
regulated upon activation normal T cell expressed and secreted
- RIP1
receptor-interacting protein 1
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- SIRT5
sirtuin 5
- SUCNR1
succinate receptor
- TAB
TGF-β-activated kinase 1/MAP3K7 binding protein
- TAK1
transforming growth factor-β-activated protein kinase
- TBK1
TANK-binding protein 1
- TCA
tricarboxylic acid
- TLR
Toll-like receptor
- TRAM
TRIF-related adaptor molecule
- TRIF
TIRdomain-containing adapter-inducing interferon-β
Footnotes
Declaration of interests
☒ The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Chemical compounds studied in this article: 3D(6-acyl)-PHAD® (PubChem CID 136212447); 3D-PHAD® (PubChem CID 136212443); CRX-527 (PubChem CID 9877226); Glycopyranoside Lipid A (PubChem CID 131846120); Hexaacyl monophosphoryl lipid A (PubChem CID 10877033); Lipopolysaccharide (PubChem CID: 11970143); Neoseptin 3 (PubChem CID 77461013)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hampton T, Report reveals scope of US antibiotic resistance threat, JAMA 310(16) (2013) 1661–3. [DOI] [PubMed] [Google Scholar]
- [2].Marston HD, Dixon DM, Knisely JM, Palmore TN, Fauci AS, Antimicrobial Resistance, JAMA 316(11) (2016) 1193–1204. [DOI] [PubMed] [Google Scholar]
- [3].Hatfield KM, Dantes RB, Baggs J, Sapiano MRP, Fiore AE, Jernigan JA, Epstein L, Assessing Variability in Hospital-Level Mortality Among U.S. Medicare Beneficiaries With Hospitalizations for Severe Sepsis and Septic Shock, Crit Care Med 46(11) (2018) 1753–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, Jernigan JA, Martin GS, Septimus E, Warren DK, Karcz A, Chan C, Menchaca JT, Wang R, Gruber S, Klompas M, Program CDCPE, Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009–2014, JAMA 318(13) (2017) 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC, The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3), JAMA 315(8) (2016) 801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rello J, Valenzuela-Sanchez F, Ruiz-Rodriguez M, Moyano S, Sepsis: A Review of Advances in Management, Adv Ther 34(11) (2017) 2393–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Opal SM, Laterre PF, Francois B, LaRosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Perrotin D, Tidswell M, Jauregui L, Krell K, Pachl J, Takahashi T, Peckelsen C, Cordasco E, Chang CS, Oeyen S, Aikawa N, Maruyama T, Schein R, Kalil AC, Van Nuffelen M, Lynn M, Rossignol DP, Gogate J, Roberts MB, Wheeler JL, Vincent JL, Group AS, Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial, JAMA 309(11) (2013) 1154–62. [DOI] [PubMed] [Google Scholar]
- [8].Rice TW, Wheeler AP, Bernard GR, Vincent JL, Angus DC, Aikawa N, Demeyer I, Sainati S, Amlot N, Cao C, Ii M, Matsuda H, Mouri K, Cohen J, A randomized, double-blind, placebo-controlled trial of TAK-242 for the treatment of severe sepsis, Crit Care Med 38(8) (2010) 1685–94. [DOI] [PubMed] [Google Scholar]
- [9].Desaki M, Takizawa H, Ohtoshi T, Kasama T, Kobayashi K, Sunazuka T, Omura S, Yamamoto K, Ito K, Erythromycin suppresses nuclear factor-kappaB and activator protein-1 activation in human bronchial epithelial cells, Biochem Biophys Res Commun 267(1) (2000) 124–8. [DOI] [PubMed] [Google Scholar]
- [10].Kikuchi T, Hagiwara K, Honda Y, Gomi K, Kobayashi T, Takahashi H, Tokue Y, Watanabe A, Nukiwa T, Clarithromycin suppresses lipopolysaccharide-induced interleukin-8 production by human monocytes through AP-1 and NF-kappa B transcription factors, J Antimicrob Chemother 49(5) (2002) 745–55. [DOI] [PubMed] [Google Scholar]
- [11].Hsu BG, Lee RP, Yang FL, Harn HJ, Chen HI, Post-treatment with N-acetylcysteine ameliorates endotoxin shock-induced organ damage in conscious rats, Life Sci 79(21) (2006) 2010–6. [DOI] [PubMed] [Google Scholar]
- [12].Docke WD, Randow F, Syrbe U, Krausch D, Asadullah K, Reinke P, Volk HD, Kox W, Monocyte deactivation in septic patients: restoration by IFN-gamma treatment, Nat Med 3(6) (1997) 678–81. [DOI] [PubMed] [Google Scholar]
- [13].Presneill JJ, Harris T, Stewart AG, Cade JF, Wilson JW, A randomized phase II trial of granulocyte-macrophage colony-stimulating factor therapy in severe sepsis with respiratory dysfunction, Am J Respir Crit Care Med 166(2) (2002) 138–43. [DOI] [PubMed] [Google Scholar]
- [14].Meisel C, Schefold JC, Pschowski R, Baumann T, Hetzger K, Gregor J, Weber-Carstens S, Hasper D, Keh D, Zuckermann H, Reinke P, Volk HD, Granulocyte-macrophage colony-stimulating factor to reverse sepsis-associated immunosuppression: a double-blind, randomized, placebo-controlled multicenter trial, Am J Respir Crit Care Med 180(7) (2009) 640–8. [DOI] [PubMed] [Google Scholar]
- [15].Cruz DN, Antonelli M, Fumagalli R, Foltran F, Brienza N, Donati A, Malcangi V, Petrini F, Volta G, Bobbio Pallavicini FM, Rottoli F, Giunta F, Ronco C, Early use of polymyxin B hemoperfusion in abdominal septic shock: the EUPHAS randomized controlled trial, JAMA 301(23) (2009) 2445–52. [DOI] [PubMed] [Google Scholar]
- [16].Schadler D, Pausch C, Heise D, Meier-Hellmann A, Brederlau J, Weiler N, Marx G, Putensen C, Spies C, Jorres A, Quintel M, Engel C, Kellum JA, Kuhlmann MK, The effect of a novel extracorporeal cytokine hemoadsorption device on IL-6 elimination in septic patients: A randomized controlled trial, PLoS One 12(10) (2017) e0187015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Honore PM, Jacobs R, Joannes-Boyau O, De Regt J, De Waele E, van Gorp V, Boer W, Verfaillie L, Spapen HD, Newly designed CRRT membranes for sepsis and SIRS--a pragmatic approach for bedside intensivists summarizing the more recent advances: a systematic structured review, ASAIO J 59(2) (2013) 99–106. [DOI] [PubMed] [Google Scholar]
- [18].Pickkers P, Payen D, What’s new in the extracorporeal treatment of sepsis?, Intensive Care Med 43(10) (2017) 1498–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bohannon JK, Luan L, Hernandez A, Afzal A, Guo Y, Patil NK, Fensterheim B, Sherwood ER, Role of G-CSF in monophosphoryl lipid A-mediated augmentation of neutrophil functions after burn injury, J Leukoc Biol (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Fensterheim BA, Guo Y, Sherwood ER, Bohannon JK, The Cytokine Response to Lipopolysaccharide Does Not Predict the Host Response to Infection, J Immunol 198(8) (2017) 3264–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hernandez A, Luan L, Stothers CL, Patil NK, Fults JB, Fensterheim BA, Guo Y, Wang J, Sherwood ER, Bohannon JK, Phosphorylated Hexaacyl Disaccharides Augment Host Resistance Against Common Nosocomial Pathogens, Crit Care Med (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Namas R, Zamora R, Namas R, An G, Doyle J, Dick TE, Jacono FJ, Androulakis IP, Nieman GF, Chang S, Billiar TR, Kellum JA, Angus DC, Vodovotz Y, Sepsis: Something old, something new, and a systems view, J Crit Care 27(3) (2012) 314 e1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Vandenbon A, Teraguchi S, Akira S, Takeda K, Standley DM, Systems biology approaches to toll-like receptor signaling, Wiley Interdiscip Rev Syst Biol Med 4(5) (2012) 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Garcon N, Di Pasquale A, From discovery to licensure, the Adjuvant System story, Human vaccines & immunotherapeutics 13(1) (2017) 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Landy M, Pillemer L, Increased resistance to infection and accompanying alteration in properidin levels following administration of bacterial lipopolysaccharides, J Exp Med 104(3) (1956) 383–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Biswas SK, Lopez-Collazo E, Endotoxin tolerance: new mechanisms, molecules and clinical significance, Trends Immunol 30(10) (2009) 475–87. [DOI] [PubMed] [Google Scholar]
- [27].Wolk K, Docke WD, von Baehr V, Volk HD, Sabat R, Impaired antigen presentation by human monocytes during endotoxin tolerance, Blood 96(1) (2000) 218–23. [PubMed] [Google Scholar]
- [28].O’Brien DP, Briles DE, Szalai AJ, Tu AH, Sanz I, Nahm MH, Tumor necrosis factor alpha receptor I is important for survival from Streptococcus pneumoniae infections, Infect Immun 67(2) (1999) 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dai WJ, Bartens W, Kohler G, Hufnagel M, Kopf M, Brombacher F, Impaired macrophage listericidal and cytokine activities are responsible for the rapid death of Listeria monocytogenes-infected IFN-gamma receptor-deficient mice, J Immunol 158(11) (1997) 5297–304. [PubMed] [Google Scholar]
- [30].Pena OM, Hancock DG, Lyle NH, Linder A, Russell JA, Xia J, Fjell CD, Boyd JH, Hancock RE, An Endotoxin Tolerance Signature Predicts Sepsis and Organ Dysfunction at Initial Clinical Presentation, EBioMedicine 1(1) (2014) 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, Rautanen A, Gordon AC, Garrard C, Hill AV, Hinds CJ, Knight JC, Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study, Lancet Respir Med 4(4) (2016) 259–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Hotchkiss RS, Monneret G, Payen D, Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy, Nat Rev Immunol 13(12) (2013) 862–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Romero CD, Varma TK, Hobbs JB, Reyes A, Driver B, Sherwood ER, The Toll-like receptor 4 agonist monophosphoryl lipid a augments innate host resistance to systemic bacterial infection, Infect Immun 79(9) (2011) 3576–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bohannon JK, Luan L, Hernandez A, Afzal A, Guo Y, Patil NK, Fensterheim B, Sherwood ER, Role of G-CSF in monophosphoryl lipid A-mediated augmentation of neutrophil functions after burn injury, Journal of leukocyte biology 99(4) (2016) 629–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lehner MD, Ittner J, Bundschuh DS, van Rooijen N, Wendel A, Hartung T, Improved innate immunity of endotoxin-tolerant mice increases resistance to Salmonella enterica serovar typhimurium infection despite attenuated cytokine response, Infect Immun 69(1) (2001) 463–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fensterheim BA, Young JD, Luan L, Kleinbard RR, Stothers CL, Patil NK, McAtee-Pereira AG, Guo Y, Trenary I, Hernandez A, Fults JB, Williams DL, Sherwood ER, Bohannon JK, The TLR4 Agonist Monophosphoryl Lipid A Drives Broad Resistance to Infection via Dynamic Reprogramming of Macrophage Metabolism, J Immunol 200(11) (2018) 3777–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Murphey ED, Fang G, Sherwood ER, Endotoxin pretreatment improves bacterial clearance and decreases mortality in mice challenged with Staphylococcus aureus, Shock 29(4) (2008) 512–8. [DOI] [PubMed] [Google Scholar]
- [38].Fukuda S, Ihara K, Bohannon JK, Hernandez A, Patil NK, Luan L, Stothers C, Stark R, Prough DS, Herndon DN, Sherwood ER, Enkhbaatar P, Monophosphoryl Lipid A Attenuates Multiorgan Dysfunction During Post-Burn Pseudomonas Aeruginosa Pneumonia In Sheep, Shock (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shands JW, The Morphologic Structure of Isolated Bacterial Lipopolysaccharide, J. Mol. Biol. 25(1) (1966) 15–21. [DOI] [PubMed] [Google Scholar]
- [40].Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zahringer U, Seydel U, Di Padova F, et al. , Bacterial endotoxin: molecular relationships of structure to activity and function, FASEB J 8(2) (1994) 217–25. [DOI] [PubMed] [Google Scholar]
- [41].Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO, The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex, Nature 458(7242) (2009) 1191–5. [DOI] [PubMed] [Google Scholar]
- [42].Pugin J, Schurer-Maly CC, Leturcq D, Moriarty A, Ulevitch RJ, Tobias PS, Lipopolysaccharide activation of human endothelial and epithelial cells is mediated by lipopolysaccharide-binding protein and soluble CD14, Proc Natl Acad Sci U S A 90(7) (1993) 2744–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC, CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein, Science 249(4975) (1990) 1431–3. [DOI] [PubMed] [Google Scholar]
- [44].Ulevitch RJ, Recognition of bacterial endotoxins by receptor-dependent mechanisms, Adv Immunol 53 (1993) 267–89. [DOI] [PubMed] [Google Scholar]
- [45].Bingle CD, Craven CJ, Meet the relatives: a family of BPI- and LBP-related proteins, Trends Immunol 25(2) (2004) 53–5. [DOI] [PubMed] [Google Scholar]
- [46].Schumann RR, Old and new findings on lipopolysaccharide-binding protein: a soluble pattern-recognition molecule, Biochem Soc Trans 39(4) (2011) 989–93. [DOI] [PubMed] [Google Scholar]
- [47].Miyake K, Roles for accessory molecules in microbial recognition by Toll-like receptors, J Endotoxin Res 12(4) (2006) 195–204. [DOI] [PubMed] [Google Scholar]
- [48].Tobias PS, Soldau K, Gegner JA, Mintz D, Ulevitch RJ, Lipopolysaccharide binding proteinmediated complexation of lipopolysaccharide with soluble CD14, J Biol Chem 270(18) (1995) 10482–8. [DOI] [PubMed] [Google Scholar]
- [49].Tobias PS, Soldau K, Kline L, Lee JD, Kato K, Martin TP, Ulevitch RJ, Cross-linking of lipopolysaccharide (LPS) to CD14 on THP-1 cells mediated by LPS-binding protein, J Immunol 150(7) (1993) 3011–21. [PubMed] [Google Scholar]
- [50].Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R, TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-beta, Nature immunology 9(4) (2008) 361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Oshiumi H, Sasai M, Shida K, Fujita T, Matsumoto M, Seya T, TIR-containing adapter molecule (TICAM)-2, a bridging adapter recruiting to Toll-like receptor 4 TICAM-1 that induces interferon-beta, Journal of Biological Chemistry 278(50) (2003) 49751–49762. [DOI] [PubMed] [Google Scholar]
- [52].Rowe DC, McGettrick AF, Latz E, Monks BG, Gay NJ, Yamamoto M, Akira S, O’Neill LA, Fitzgerald KA, Golenbock DT, The myristoylation of TRIF-related adaptor molecule is essential for Toll-like receptor 4 signal transduction, P Natl Acad Sci USA 103(16) (2006) 6299–6304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Kagan JC, Medzhitov R, Phosphoinositide-mediated adaptor recruitment controls toll-like receptor signaling, Cell 125(5) (2006) 943–955. [DOI] [PubMed] [Google Scholar]
- [54].Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S, Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway, Science 301(5633) (2003) 640–3. [DOI] [PubMed] [Google Scholar]
- [55].Kolanowski ST, Dieker MC, Lissenberg-Thunnissen SN, van Schijndel GM, van Ham SM, ten Brinke A, TLR4-mediated pro-inflammatory dendritic cell differentiation in humans requires the combined action of MyD88 and TRIF, Innate immunity 20(4) (2014) 423–30. [DOI] [PubMed] [Google Scholar]
- [56].Picard C, Casanova JL, Puel A, Infectious diseases in patients with IRAK-4, MyD88, NEMO, or IkappaBalpha deficiency, Clin Microbiol Rev 24(3) (2011) 490–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yesudhas D, Gosu V, Anwar MA, Choi S, Multiple roles of toll-like receptor 4 in colorectal cancer, Front Immunol 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Butler MP, Hanly JA, Moynagh PN, Pellino3 is a novel upstream regulator of p38 MAPK and activates CREB in a p38-dependent manner, J Biol Chem 280(30) (2005) 27759–68. [DOI] [PubMed] [Google Scholar]
- [59].Chessler AD, Ferreira LR, Chang TH, Fitzgerald KA, Burleigh BA, A novel IFN regulatory factor 3-dependent pathway activated by trypanosomes triggers IFN-beta in macrophages and fibroblasts, J Immunol 181(11) (2008) 7917–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bohannon JK, Hernandez A, Enkhbaatar P, Adams WL, Sherwood ER, The immunobiology of toll-like receptor 4 agonists: from endotoxin tolerance to immunoadjuvants, Shock 40(6) (2013) 451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tanimura N, Saitoh S, Matsumoto F, Akashi-Takamura S, Miyake K, Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling, Biochem Biophys Res Commun 368(1) (2008) 94–9. [DOI] [PubMed] [Google Scholar]
- [62].Zhang Y, Liang C, Innate recognition of microbial-derived signals in immunity and inflammation, Sci China Life Sci 59(12) (2016) 1210–1217. [DOI] [PubMed] [Google Scholar]
- [63].Shah NR, AlBitar-Nehme S, Kim E, Marr N, Novikov A, Caroff M, Fernandez RC, Minor Modifications to the Phosphate Groups and the C3 ‘ Acyl Chain Length of Lipid A in Two Bordetella pertussis Strains, BP338 and 18–323, Independently Affect Toll-like Receptor 4 Protein Activation, Journal of Biological Chemistry 288(17) (2013) 11751–11760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Needham BD, Carroll SM, Giles DK, Georgiou G, Whiteley M, Trent MS, Modulating the innate immune response by combinatorial engineering of endotoxin, Proc Natl Acad Sci U S A 110(4) (2013) 1464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Garcon N, Preclinical development of AS04, Methods Mol Biol 626 (2010) 15–27. [DOI] [PubMed] [Google Scholar]
- [66].Luo M, Hu L, Li D, Wang Y, He Y, Zhu L, Ren W, MD-2 regulates LPS-induced NLRP3 inflammasome activation and IL-1beta secretion by a MyD88/NF-kappaB-dependent pathway in alveolar macrophages cell line, Mol Immunol 90 (2017) 1–10. [DOI] [PubMed] [Google Scholar]
- [67].Dolunay A, Senol SP, Temiz-Resitoglu M, Guden DS, Sari AN, Sahan-Firat S, Tunctan B, Inhibition of NLRP3 Inflammasome Prevents LPS-Induced Inflammatory Hyperalgesia in Mice: Contribution of NF-kappaB, Caspase-1/11, ASC, NOX, and NOS Isoforms, Inflammation 40(2) (2017) 366–386. [DOI] [PubMed] [Google Scholar]
- [68].Schulke S, Flaczyk A, Vogel L, Gaudenzio N, Angers I, Loschner B, Wolfheimer S, Spreitzer I, Qureshi S, Tsai M, Galli S, Vieths S, Scheurer S, MPLA shows attenuated pro-inflammatory properties and diminished capacity to activate mast cells in comparison with LPS, Allergy 70(10) (2015) 1259–68. [DOI] [PubMed] [Google Scholar]
- [69].Ruchaud-Sparagano MH, Mills R, Scott J, Simpson AJ, MPLA inhibits release of cytotoxic mediators from human neutrophils while preserving efficient bacterial killing, Immunol Cell Biol 92(9) (2014) 799–809. [DOI] [PubMed] [Google Scholar]
- [70].Kolanowski ST, Lissenberg-Thunnissen SN, Emal D, van Ham SM, Ten Brinke A, Monophosphoryl lipid A-induced pro-inflammatory cytokine expression does not require CD14 in primary human dendritic cells, Inflamm Res 65(6) (2016) 449–58. [DOI] [PubMed] [Google Scholar]
- [71].Ohto U, Fukase K, Miyake K, Satow Y, Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa, Science 316(5831) (2007) 1632–4. [DOI] [PubMed] [Google Scholar]
- [72].Astiz ME, Rackow EC, Still JG, Howell ST, Cato A, Voneschen KB, Ulrich JT, Rudbach JA, Mcmahon G, Vargas R, Stern W, Pretreatment of Normal Humans with Monophosphoryl Lipid-a Induces Tolerance to Endotoxin - a Prospective, Double-Blind, Randomized, Controlled Trial, Crit Care Med 23(1) (1995) 9–17. [DOI] [PubMed] [Google Scholar]
- [73].Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC, The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4, Science 316(5831) (2007) 1628–32. [DOI] [PubMed] [Google Scholar]
- [74].Hernandez A, Bohannon JK, Luan L, Fensterheim BA, Guo Y, Patil NK, McAdams C, Wang J, Sherwood ER, The role of MyD88- and TRIF-dependent signaling in monophosphoryl lipid A-induced expansion and recruitment of innate immunocytes, J Leukoc Biol 100(6) (2016) 1311–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Kwissa M, Nakaya HI, Oluoch H, Pulendran B, Distinct TLR adjuvants differentially stimulate systemic and local innate immune responses in nonhuman primates, Blood 119(9) (2012) 2044–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].McAleer JP, Vella AT, Understanding how lipopolysaccharide impacts CD4 T-cell immunity, Crit Rev Immunol 28(4) (2008) 281–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Baldridge JR, McGowan P, Evans JT, Cluff C, Mossman S, Johnson D, Persing D, Taking a Toll on human disease: Toll-like receptor 4 agonists as vaccine adjuvants and monotherapeutic agents, Expert opinion on biological therapy 4(7) (2004) 1129–38. [DOI] [PubMed] [Google Scholar]
- [78].Stover AG, Da Silva Correia J, Evans JT, Cluff CW, Elliott MW, Jeffery EW, Johnson DA, Lacy MJ, Baldridge JR, Probst P, Ulevitch RJ, Persing DH, Hershberg RM, Structure-activity relationship of synthetic toll-like receptor 4 agonists, J Biol Chem 279(6) (2004) 4440–9. [DOI] [PubMed] [Google Scholar]
- [79].Bowen WS, Minns LA, Johnson DA, Mitchell TC, Hutton MM, Evans JT, Selective TRIFDependent Signaling by a Synthetic Toll-Like Receptor 4 Agonist, Sci Signal 5(211) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Khalaf JK, Bowen WS, Bazin HG, Ryter KT, Livesay MT, Ward JR, Evans JT, Johnson DA, Characterization of TRIF selectivity in the AGP class of lipid A mimetics: role of secondary lipid chains, Bioorg Med Chem Lett 25(3) (2015) 547–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Whitby LR, Boger DL, Comprehensive Peptidomimetic Libraries Targeting Protein-Protein Interactions, Accounts Chem Res 45(10) (2012) 1698–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Morin MD, Wang Y, Jones BT, Su LJ, Surakattula MMRP, Berger M, Huang H, Beutler EK, Zhang H, Beutler B, Boger DL, Discovery and Structure-Activity Relationships of the Neoseptins: A New Class of Toll-like Receptor-4 (TLR4) Agonists, J Med Chem 59(10) (2016) 4812–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Marshall JD, Heeke DS, Rao E, Maynard SK, Hornigold D, McCrae C, Fraser N, Tovchigrechko A, Yu L, Williams N, King S, Cooper ME, Hajjar AM, Woo JC, A Novel Class of Small Molecule Agonists with Preference for Human over Mouse TLR4 Activation, Plos One 11(10) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Murphey ED, Fang G, Varma TK, Sherwood ER, Improved bacterial clearance and decreased mortality can be induced by LPS tolerance and is not dependent upon IFN-gamma, Shock 27(3) (2007) 289–95. [DOI] [PubMed] [Google Scholar]
- [85].Varma TK, Durham M, Murphey ED, Cui W, Huang Z, Lin CY, Toliver-Kinsky T, Sherwood ER, Endotoxin priming improves clearance of Pseudomonas aeruginosa in wild-type and interleukin-10 knockout mice, Infect Immun 73(11) (2005) 7340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Deng M, Scott MJ, Loughran P, Gibson G, Sodhi C, Watkins S, Hackam D, Billiar TR, Lipopolysaccharide clearance, bacterial clearance, and systemic inflammatory responses are regulated by cell type-specific functions of TLR4 during sepsis, J Immunol 190(10) (2013) 5152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Rayhane N, Fitting C, Lortholary O, Dromer F, Cavaillon JM, Administration of endotoxin associated with lipopolysaccharide tolerance protects mice against fungal infection, Infect Immun 68(6) (2000) 3748–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Wynn JL, Scumpia PO, Winfield RD, Delano MJ, Kelly-Scumpia K, Barker T, Ungaro R, Levy O, Moldawer LL, Defective innate immunity predisposes murine neonates to poor sepsis outcome but is reversed by TLR agonists, Blood 112(5) (2008) 1750–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Williams DL, Sherwood ER, Browder IW, McNamee RB, Jones EL, Rakinic J, Di Luzio NR, Effect of glucan on neutrophil dynamics and immune function in Escherichia coli peritonitis, J Surg Res 44(1) (1988) 54–61. [DOI] [PubMed] [Google Scholar]
- [90].Tzianabos AO, Cisneros RL, Prophylaxis with the immunomodulator PGG glucan enhances antibiotic efficacy in rats infected with antibiotic-resistant bacteria, Ann N Y Acad Sci 797 (1996) 285–7. [DOI] [PubMed] [Google Scholar]
- [91].Taylor CT, Colgan SP, Regulation of immunity and inflammation by hypoxia in immunological niches, Nat Rev Immunol 17(12) (2017) 774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Nizet V, Johnson RS, Interdependence of hypoxic and innate immune responses, Nat Rev Immunol 9(9) (2009) 609–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Hard GC, Some biochemical aspects of the immune macrophage, Br J Exp Pathol 51(1) (1970) 97–105. [PMC free article] [PubMed] [Google Scholar]
- [94].Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ, Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation, Blood 115(23) (2010) 4742–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, van den Bosch MWM, Quinn SR, Domingo-Fernandez R, Johnston DGW, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vander Heiden M, Xavier RJ, O’Neill LAJ, Pyruvate Kinase M2 Regulates Hif-1alpha Activity and IL-1beta Induction and Is a Critical Determinant of the Warburg Effect in LPSActivated Macrophages, Cell Metab 21(2) (2015) 347. [DOI] [PubMed] [Google Scholar]
- [96].Haschemi A, Kosma P, Gille L, Evans CR, Burant CF, Starkl P, Knapp B, Haas R, Schmid JA, Jandl C, Amir S, Lubec G, Park J, Esterbauer H, Bilban M, Brizuela L, Pospisilik JA, Otterbein LE, Wagner O, The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism, Cell Metab 15(6) (2012) 813–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Frede S, Stockmann C, Freitag P, Fandrey J, Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB, Biochem J 396(3) (2006) 517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Blouin CC, Page EL, Soucy GM, Richard DE, Hypoxic gene activation by lipopolysaccharide in macrophages: implication of hypoxia-inducible factor 1alpha, Blood 103(3) (2004) 1124–30. [DOI] [PubMed] [Google Scholar]
- [99].Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LA, Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha, Nature 496(7444) (2013) 238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].O’Neill LA, Kishton RJ, Rathmell J, A guide to immunometabolism for immunologists, Nat Rev Immunol 16(9) (2016) 553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Murphy MP, O’Neill LAJ, Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers, Cell 174(4) (2018) 780–784. [DOI] [PubMed] [Google Scholar]
- [102].Patil NK, Bohannon JK, Hernandez A, Patil TK, Sherwood ER, Regulation of leukocyte function by citric acid cycle intermediates, J Leukoc Biol 106(1) (2019) 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Zhu X, Meyers A, Long D, Ingram B, Liu T, Yoza BK, Vachharajani V, McCall CE, Frontline Science: Monocytes sequentially rewire metabolism and bioenergetics during an acute inflammatory response, Journal of leukocyte biology 105(2) (2019) 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, Pearce EJ, Driggers EM, Artyomov MN, Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization, Immunity 42(3) (2015) 419–30. [DOI] [PubMed] [Google Scholar]
- [105].Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, Palmieri F, Iacobazzi V, The mitochondrial citrate carrier: a new player in inflammation, Biochem J 438(3) (2011) 433–6. [DOI] [PubMed] [Google Scholar]
- [106].Infantino V, Iacobazzi V, Menga A, Avantaggiati ML, Palmieri F, A key role of the mitochondrial citrate carrier (SLC25A1) in TNFalpha- and IFNgamma-triggered inflammation, Biochim Biophys Acta 1839(11) (2014) 1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Infantino V, Iacobazzi V, Palmieri F, Menga A, ATP-citrate lyase is essential for macrophage inflammatory response, Biochem Biophys Res Commun 440(1) (2013) 105–11. [DOI] [PubMed] [Google Scholar]
- [108].Infantino V, Pierri CL, Iacobazzi V, Metabolic routes in inflammation: the citrate pathway and its potential as therapeutic target, Curr Med Chem (2018). [DOI] [PubMed] [Google Scholar]
- [109].Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, Buttini M, Linster CL, Medina E, Balling R, Hiller K, Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production, Proceedings of the National Academy of Sciences of the United States of America 110(19) (2013) 7820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Hooftman A, O’Neill LAJ, The Immunomodulatory Potential of the Metabolite Itaconate, Trends in immunology 40(8) (2019) 687–698. [DOI] [PubMed] [Google Scholar]
- [111].Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, Szpyt J, Runtsch MC, King MS, McGouran JF, Fischer R, Kessler BM, McGettrick AF, Hughes MM, Carroll RG, Booty LM, Knatko EV, Meakin PJ, Ashford MLJ, Modis LK, Brunori G, Sevin DC, Fallon PG, Caldwell ST, Kunji ERS, Chouchani ET, Frezza C, DinkovaKostova AT, Hartley RC, Murphy MP, O’Neill LA, Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1, Nature 556(7699) (2018) 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, Korenfeld D, Mathyer ME, Kim H, Huang LH, Duncan D, Bregman H, Keskin A, Santeford A, Apte RS, Sehgal R, Johnson B, Amarasinghe GK, Soares MP, Satoh T, Akira S, Hai T, de Guzman Strong C, Auclair K, Roddy TP, Biller SA, Jovanovic M, Klechevsky E, Stewart KM, Randolph GJ, Artyomov MN, Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta-ATF3 inflammatory axis, Nature 556(7702) (2018) 501–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, Koseki H, Cabrales P, Murphy AN, Hiller K, Metallo CM, Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels, J Biol Chem 291(27) (2016) 14274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC, Griss T, Weinheimer CJ, Khader S, Randolph GJ, Pearce EJ, Jones RG, Diwan A, Diamond MS, Artyomov MN, Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation, Cell Metab 24(1) (2016) 158–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Cordes T, Michelucci A, Hiller K, Itaconic Acid: The Surprising Role of an Industrial Compound as a Mammalian Antimicrobial Metabolite, Annu Rev Nutr 35 (2015) 451–73. [DOI] [PubMed] [Google Scholar]
- [116].Sasikaran J, Ziemski M, Zadora PK, Fleig A, Berg IA, Bacterial itaconate degradation promotes pathogenicity, Nat Chem Biol 10(5) (2014) 371–7. [DOI] [PubMed] [Google Scholar]
- [117].Sugimoto M, Sakagami H, Yokote Y, Onuma H, Kaneko M, Mori M, Sakaguchi Y, Soga T, Tomita M, Non-targeted metabolite profiling in activated macrophage secretion, Metabolomics 8(4) (2012) 624–633. [Google Scholar]
- [118].Mills E, O’Neill LA, Succinate: a metabolic signal in inflammation, Trends Cell Biol 24(5) (2014) 313–20. [DOI] [PubMed] [Google Scholar]
- [119].Hollander AP, Corke KP, Freemont AJ, Lewis CE, Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint, Arthritis Rheum 44(7) (2001) 1540–4. [DOI] [PubMed] [Google Scholar]
- [120].Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA, Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages, Cell 167(2) (2016) 457–470 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP, A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury, Cell Metab 23(2) (2016) 254–63. [DOI] [PubMed] [Google Scholar]
- [122].West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S, TLR signalling augments macrophage bactericidal activity through mitochondrial ROS, Nature 472(7344) (2011) 476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, Xu Y, Zhou L, Chu Y, Zhang C, Qin X, Yang P, Yu H, SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1beta Production and to Prevent DSS-Induced Colitis in Mice, Cell Rep 19(11) (2017) 2331–2344. [DOI] [PubMed] [Google Scholar]
- [124].Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, Lombard DB, Zhao Y, SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways, Molecular cell 50(6) (2013) 919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]