

Abstract
Chromosomal translocation leads to the juxtaposition of two otherwise separate DNA loci, which could result in gene fusion. These rearrangements at the DNA level are catastrophic events and often have causal roles in tumorigenesis. The oncogenic DNA messages are transferred to RNA molecules, which are in most cases translated into cancerous fusion proteins. Gene expression programs and signaling pathways are altered in these cytogenetically abnormal contexts. Notably, non-coding RNAs have attracted increasing attention and are believed to be tightly associated with chromosome-rearranged cancers. These RNAs not only function as modulators in downstream pathways but also directly affect chromosomal translocation or the associated products. This review summarizes recent research advances on the relationship between non-coding RNAs and chromosomal translocations and on diverse functions of non-coding RNAs in cancers with chromosomal rearrangements.
Keywords: chromosomal translocation, non-coding RNA, fusion protein, gene regulation, non-coding fusion transcript
Introduction
Cancer is, in essence, a genetic disease at the cellular level (Vogelstein and Kinzler, 2004). However, this consensus was not reached until the groundbreaking discovery of a chromosomal translocation designated the Philadelphia chromosome (Koretzky, 2007). Chromosomal translocation is a unique genetic aberration that can lead to tumorigenesis (Mitelman et al., 2007). This aberration is a characteristic feature of neoplasia in which a chromosome breaks and a portion is transferred to a different locus. Consequently, the formation of fusion genes, gene disruption or changes in regulatory elements may contribute to the dysregulation or malfunction of the corresponding genes (Roukos and Misteli, 2014). Specific chromosomal translocations are associated with distinct subtypes of diseases and have prognostic value. The most extensively studied consequence of rearrangement is the production of oncogenic fusion proteins, such as recurrent mixed lineage leukemia (MLL) fusions with t(11q23), BCR–ABL with t(9;22)(q34;q11), AML1–ETO with t(8;21)(q22;q22) and PML–RARA with t(15;17)(q22;q21) in hematological disorders and EWSR1–FLI1 with t(11;22)(q24;q12), EVT6–NTRK3 with t(12;15)(p13;q25), and EML4–ALK with inv(2)(p21p23) in sarcomas and carcinomas (Chen et al., 2010; Parker and Zhang 2013). By dysregulating gene expression programs or related signaling pathways, cancerous fusion proteins are causally involved in tumorigenesis. Thus, elucidation of the molecular networks governed by fusion proteins is important to determine the basis of cancer development and to lay the foundation for targeted therapy. One prominent example is the utilization of the kinase inhibitor, imatinib, which specifically inhibits the activity of the recurrent fusion protein BCR–ABL in chronic myeloid leukemia (CML) and changes the natural history of this disease (Druker, 2008; Rossari et al., 2018).
Due to advances in our knowledge of the complexity of the genome, we now know that non-coding RNAs (ncRNAs), which are transcribed from regions previously known as ‘junk’ DNA, are functional (Cech and Steitz, 2014; Morris and Mattick, 2014). The non-coding molecules, which include microRNAs (miRNAs), small nucleolar RNAs (snoRNAs), long ncRNAs (lncRNAs), and circular RNAs (circRNAs), have attracted increased attention in cancer research (Esteller, 2011; Anastasiadou et al., 2018). Studies in the last decade have uncovered the strong association between chromosome translocation-driven cancer and ncRNA. Although the most extensively studied ncRNAs in chromosome translocation-associated malignancies are miRNAs, researchers have also begun to focus on other ncRNAs. The ncRNA has been shown to be a critical and indispensable component in fusion protein-driven tumorigenesis. Moreover, ncRNAs might also be directly involved in chromosomal instability by shaping chromosomal translocation or redefining the definition of recurrent fusion transcripts. Thus, the following questions arise: what are the expression signatures of ncRNAs in cancers with chromosomal translocations? What are the causes of these changes? Are these changes irrelevant epiphenomena or are they functionally relevant? What is the relationship between ncRNAs and chromosomal rearrangements? In this review, we will discuss the new advances regarding these questions and their implications for the remaining important questions related to chromosome translocation and ncRNAs. Given that most of the studies on chromosomal rearrangements were related with hematological disorders, ncRNAs in blood malignancies will be the primary focus of this review, but chromosomal rearrangements in solid tumors will be extensively covered.
ncRNA dysregulation is linked to chromosomal translocation
The importance of the regulatory roles of ncRNA has been shown, and ncRNA expression profiling can provide classification, prognostic, and diagnostic information in cancer. Due to the complexity and heterogeneity of various malignancies, miRNAs were proposed as biomarkers a decade ago (Lu et al., 2005) and have shown close correlation with specific cytogenetic abnormalities (Dixon-McIver et al., 2008; Garzon et al., 2008; Li et al., 2008a). For example, miR-196 was downregulated in patients harboring 11q23 translocations in leukemia, and miR-21 exhibited higher expression in t(6;11) than in t(9;11) in blood malignancy (Garzon et al., 2008). The potential value of miRNAs as diagnostic marker for cytogenetically abnormal acute myeloid leukemia (AML) was further exploited in patient samples with translocations including t(8;21)/AML–ETO, inv(16)/CBFB–MYH1, t(15;17)/PML–RARA, and MLL rearrangements (MLL-r). Expression signatures involving as few as two (miR-126/miR-126*), three (miR-224, miR-368, and miR-382), and seven (miR-17-5p and miR-20a combined with five miRNAs above) were sufficient to distinguish core-binding factor (CBF), PML–RARA, and MLL-r leukemias, respectively (Li et al., 2008a). A recent study also showed that miRNAs could distinguish sub-entities such as MLL-related t(11;19)(q23;p13.1) and t(11;19)(q23;p13.3) (Bhatnagar et al., 2016). Further elucidation of the differences between closely related subtypes will be beneficial for diagnosis and application of targeted therapy.
Systematic expression profiling has also been used to analyze lncRNAs in recent years. We and others reported that lncRNAs were differentially expressed in MLL-r and non-MLL leukemias or other common rearrangements (Fang et al., 2014; Schwarzer et al., 2017). The specific profiles of subtypes of MLL-r, as well as those of infants and elderly patients, were further identified (Fang et al., 2014). Similar results were also found in B-cell acute lymphoblastic leukemia (B-ALL), prostate cancer, and Ewing sarcoma (Ren et al., 2012; Marques et al., 2014; Fernando et al., 2015; Ghazavi et al., 2016). A large cohort study of MLL-r validated its unique transcriptional landscape (Lavallee et al., 2015). Notably, the most differentially expressed gene, LOC100289656, is a pseudogene, from which an lncRNA is most likely transcribed. The high expression of LOC100289656 helped identify several cryptic MLL fusions that were previously undetected by standard cytogenetic analyses (Lavallee et al., 2015). These findings indicate that lncRNAs are a potentially powerful tool to predict minimal residual disease. Notably, LOC100289656 displayed specific expression patterns in MLL-r whereas it displayed no expression or very restricted expression in non-MLL leukemia or normal bone marrow and cord and peripheral blood cells. These findings suggest that some lncRNAs may function only in selective genetic contexts, such as chromosome rearrangements.
In addition, snoRNAs and their host genes have attracted renewed attention and have been recognized as new players in cancers (Williams and Farzaneh, 2012; McMahon et al., 2015). Several recent studies reported the general downregulation of a number of snoRNAs in normal hematopoiesis and leukemia (Valleron et al., 2012; Ronchetti et al., 2013; Warner et al., 2018). However, AML1–ETO, AML1–ETO9a, and MLL–AF9 enhanced snoRNA expression, and similar results were obtained in t(8;21) patient samples compared with purified CD34+ progenitor cells or normal bone marrow (Zhou et al., 2017). These findings indicate the unique genetic signatures of malignancies with chromosomal rearrangements. Interestingly, overexpression of the SNOR112–114 cluster marks acute promyelocytic leukemia (APL) (Valleron et al., 2012; Liuksiala et al., 2014). Overexpression of the 14q32 snoRNA transcripts located at the DLK1-DIO3 locus is believed to be related to fusion transcripts harboring the RARA gene (Cohen et al., 2012). Neither host gene expression nor alternative splicing and mutations accounted for the aberrant expression of snoRNAs (Valleron et al., 2012; Warner et al., 2018). Further analyses of the underlying mechanisms are needed. Interestingly, this locus also contains several lncRNAs and 54 miRNAs in addition to the snoRNA cluster. These ncRNAs are upregulated in megakaryopoiesis, and integrated microarray-based analysis suggested that these RNAs might be positive regulators of this process (Schwarzer et al., 2017).
Causes of abnormal expression of ncRNAs in cancers with chromosomal rearrangements
The altered expression patterns of ncRNAs raised the question of how the ncRNAs are activated or silenced in cytogenetically abnormal cancers and whether their dysregulation is the direct result of chromosomal rearrangements or secondary mutations in cooperation with fusion genes. The expression of ncRNAs is altered in a complex, incompletely understood manner. miRNAs have been shown to be affected by multiple factors, such as chromosomal abnormalities, epigenomics and processing (Calin and Croce, 2007). Previous studies have also demonstrated the direct involvement of ncRNAs in chromosome breakpoint regions. In this section, we will discuss the underlying mechanisms of the fusion protein-driven and non-fusion protein-driven dysregulation of ncRNAs in cancers with chromosomal rearrangements.
Fusion protein and epigenetic factor-driven transcriptional dysregulation
The recurring fusion genes in blood malignancies have been identified as transcriptional regulators in many cases (Chen et al., 2010). In these cases, a dysregulated transcriptional program strongly contributes to tumorigenesis (Chen et al., 2010). The activation or silencing of ncRNAs is believed to be directly controlled by fusion proteins bound to their respective regulatory elements (Figure 1A). For example, in the t(11;16) translocations, MLL fuses in-frame with > 60 partners, which leads to the production of functional fusion proteins (Krivtsov and Armstrong, 2007). The N-terminal MLL retains its chromosome-binding domain, while the most common C-terminal partners are nuclear proteins such as AF4, AF9, ENL, and ELL, which are involved in transcriptional activation or elongation (Dou and Hess, 2008). Thus, the ncRNAs regulated by MLL fusions should be overexpressed. Indeed, a large-scale genome-wide microarray analysis showed that, among 48 miRNAs that were significantly differentially expressed between MLL and non-MLL-rearranged AML patient samples, 47 of them showed increased expression levels (Li et al., 2008a), suggesting that MLL fusion proteins could promote the transcription of their downstream targets. The upregulated miRNAs between MLL and non-MLL leukemias are likely directly controlled by MLL chimeras. For example, MLL–AF9 binds to the promoter of miR-9 and promotes its expression through recruiting the histone H3 lysine 79 (H3K79) methylase DOT1L (Chen et al., 2013). The recruitment of RNA polymerase II and DOT1L-mediated H3K79 methylation is essential for MLL chimeras (Milne et al., 2005a,b; Krivtsov et al., 2008; Erfurth et al., 2008). The transduction of fusion proteins alone was sufficient to significantly upregulate these ncRNAs (Popovic et al., 2009; Mi et al., 2010; Li et al., 2012; Chen et al., 2013). Consistent with this mechanism, H3K79 methylation was found to be enriched on the promoters of a group of overexpressed lncRNAs in MLL leukemia, implying that their activation is directly dependent on fusion proteins (Fang et al., 2014). Similar results were found for other fusion proteins such as AML1–ETO, TEL–AML1, and PML–RARA (Fazi et al., 2007; Careccia et al., 2009; Saumet et al., 2009; Brauer-Hartmann et al., 2015; Tran et al., 2016). All these proteins exert repressive transcriptional functions by recruiting epigenetic modification enzymes, such as histone deacetylase (HDAC), nuclear receptor co-repressor 1 (NCOR1), nuclear receptor co-repressor 2 (NCOR2), DNA methyltransferase 1 (DNMT1), and DNA methyltransferase 3A (DNMT3A) (Chen et al., 2010; Gutierrez and Romero-Oliva, 2013). In addition to binding to promoters, fusion proteins could also bind distant regulatory elements such as enhancers and regulate the lncRNAs near these regions (Teppo et al., 2016) (Figure 1A).
Figure 1.
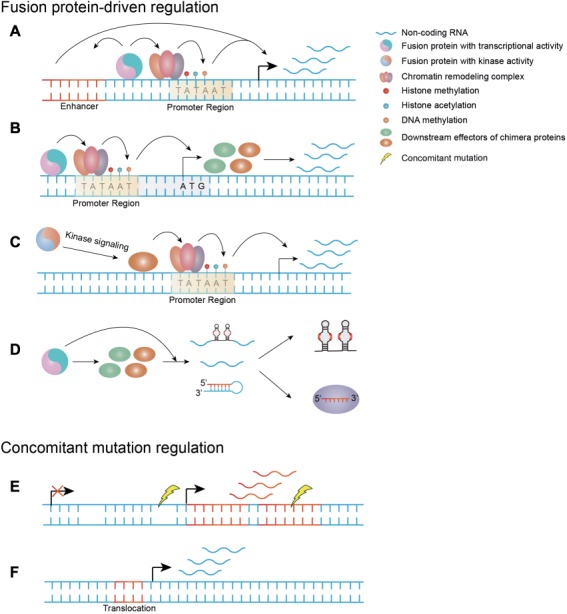
The causes of the dysregulation of ncRNAs. (A) Fusion proteins with transcriptional activity alter the expressions of ncRNAs by directly binding to their regulatory elements and recruiting epigenetic modification enzymes. (B and C) The dysregulation of ncRNAs is mediated by the downstream effectors of fusion proteins with transcriptional activity or kinase activity. (D) The dysregulation of ncRNAs is due to the disturbance of RNA processing/formation process by fusion proteins. (E) The loss or amplification of ncRNAs as concomitant mutations. (F) The activation or silencing of ncRNA expressions is directly triggered by chromosomal translocation.
Some targets of fusion proteins can also be controlled by the corresponding wild-type counterparts (Popovic et al., 2009; Saumet et al., 2009; Mi et al., 2010). For example, both wild-type MLL and MLL fusions bound the promoters of miR-17-92, and they could reinduce miR-196b expression in MLL knockout cells, particularly MLL fusion proteins (Popovic et al., 2009; Mi et al., 2010). However, fusion proteins bind a wider range of DNA-target sequences than their wild-type counterpart in some cases (May et al., 1993; Saumet et al., 2009; Linka et al., 2013). A study focusing on TEL–AML1 found that the majority of promoter regions were specific for fusion proteins and not bound by TEL or AML1 (Linka et al., 2013), indicating that this transcriptional program is unique to the cytogenetically abnormal context. While MLL and MLL fusions have uniformly activating functions (Popovic et al., 2009; Mi et al., 2010), fusion proteins and their wild-type counterparts, such as AML1–ETO and AML1, could elicit opposing effects (Bakshi et al., 2008).
Downstream effectors of fusion proteins control ncRNA dysregulation
Although fusion proteins with transcriptional activity could directly alter the expression of their targets, some of these proteins might be regulated by the downstream effectors of chimera proteins (Schotte et al., 2010; Velu et al., 2014; Huang et al., 2016; Mohr et al., 2017) (Figure 1B). For example, miR-196b is controlled by HOXA9 (Schotte et al., 2010), a pivotal downstream effector of MLL fusion proteins. HOXA9 competes with its antagonist GIF1 for binding sites on miR-196b and miR-21 (Velu et al., 2014). Thus, the activation of miR-196b and miR-21 is dependent on HOXA9 overexpression. This finding also partly explains why the alterations of some ncRNAs are not consistent with the regulatory properties of fusion proteins. For example, miR-26a was found to be significantly downregulated in the presence of transcriptional activating oncoproteins, such as MLL fusions (Huang et al., 2016). In fact, miR-26a is repressed by MYC, which is promoted by MLL chimeras (Huang et al., 2016). Although the transcriptional promotion of lncRNAs such as EWSAT1 is dependent on the positive transcriptional regulator EWS–FLI1 fusion in Ewing sarcoma, proximal promoter or more distal enhancer elements binding with fusion protein are not the causes for EWSAT1 overexpression (Marques et al., 2014). Based on the dependence on chimera proteins, this phenomenon could be due to the dysregulation of downstream targets of fusion proteins, or undetermined properties of oncogenic fusions. This hypothesis was further validated by the enhancement of rRNA transcription by AML1–ETO, which has transcriptionally repressing activity (Bakshi et al., 2008). By direct binding to rDNA repeats, AML1–ETO activates rRNA transcription instead of silencing this process (Bakshi et al., 2008). The activation may be due to the enrichment of the hypomethylation marker on the rDNA repeat-binding region of AML1–ETO (Bakshi et al., 2008). Although MLL fusions are activators, global hypermethylation was observed on the promoters in MLL-r infant ALL (Schafer et al., 2010), suggesting the involvement of cooperative events. The fusion protein-driven program involves multiple cooperative factors. Thus, the relationships among chimera proteins, epigenetics and ncRNAs remain unclear.
Kinase fusion protein-driven ncRNA dysregulation
Another major group of fusion proteins are chimeras with ‘always on’ kinase activity (Stransky et al., 2014; Nelson et al., 2017; Kim et al., 2018). The first chromosomal translocation product ever characterized was BCR–ABL in CML (Mitelman et al., 2007). New fusion proteins with constitutive kinase activity are continually being discovered (Yde et al., 2016; Hicks et al., 2018). The molecular dissection of BCR–ABL could serve as a paradigm for other fusions with kinase activity. The dysregulation of ncRNA expression was shown to be dependent on BCR–ABL (Venturini et al., 2007; Bueno et al., 2008; Guo et al., 2014a, 2015; Xu et al., 2014; Choi et al., 2016; Zhou et al., 2018). BCR–ABL knockdown or treatment with specific ABL kinase inhibitor, imatinib, reversed the aberrant expression of ncRNAs in a BCR–ABL kinase activity-dependent manner (Venturini et al., 2007; Xu et al., 2014; Guo et al., 2014a). BCR–ABL cannot directly induce the expression of ncRNAs, and instead exploits downstream signaling pathways or DNA methylation status to manipulate their expression (Figure 1C). The upregulation of miR-17-92 and lncRNA H19 was activated by c-MYC, which is a critical downstream target of BCR–ABL signaling in CML (Venturini et al., 2007; Guo et al., 2014a). Strikingly, miR-17-92 was downregulated rather than upregulated by BCR–ABL in B-ALL, suggesting various pathways of fusion proteins in different cellular contexts (Scherr et al., 2014). Only treatment with inhibitors of BCR–ABL-associated pathways changes the expression of fusion protein-dependent ncRNAs, such as miR-139-5p (Choi et al., 2016). A similar phenomenon was observed in other cancers harboring kinase fusion BCR–FGFR1 or ETV6–NTRK3 (Chen et al., 2018; Hu et al., 2018). The DNA methylation status in the upstream regions also contributes to the altered expression patterns of miR-139-5p, miR-203, and H19 (Bueno et al., 2008; Choi et al., 2016; Zhou et al., 2018), which may also be BCR–ABL kinase-dependent. Moreover, fusion proteins as transcriptional regulators might exploit kinase signaling to alter the expression of ncRNAs. For example, the addiction to highly phosphorylated SYK leads to the activation of c-MYC, which in turn stimulates the transcription of lncRNA MALAT1 in EWS–FLI1-driven multiple myeloma (Sun et al., 2017).
Disturbance of RNA processing machinery by fusion proteins
Sporadic reports have suggested that the disturbance of the RNA processing machinery alters ncRNA expression in diseases with chromosomal rearrangements. On the one hand, fusion proteins can block RNA processing (Figure 1D). One study showed that both the MLL fusion and the downstream target MYC directly bound and activated the pri-miR-150 promoter. However, the expression of mature miR-150 was specifically downregulated in MLL-r leukemia (Jiang et al., 2012). This downregulation is due to the overexpression of Lin28 by MYC, as Lin28 interferes with the miRNA maturation process (Viswanathan et al., 2008; Jiang et al., 2012). On the other hand, fusion proteins can also enhance non-coding ribonucleoprotein (RNP) formation (Figure 1D). As self-renewal is required for leukemogenesis, MLL–AF9 and AML1–ETO enhanced C/D box snoRNA expression (Zhou et al., 2017). However, these were not caused by transcriptional activation but by the promotion of small nucleolar ribonucleoprotein (snoRNP) formation (Zhou et al., 2017). Suppression of the key formation factor AES led to decreased snoRNA expression (Zhou et al., 2017).
Concomitant mutation-induced ncRNA dysregulation
In addition to fusion protein-driven change in ncRNA expression, other co-occurring events facilitate the expression alterations (Figure 1E). More than half of miRNAs are located at minimal regions of loss of heterozygosity (LOHs) and minimal regions of amplification in various cancers (Calin et al., 2004). A 7-Mb fragile region encoding 12% of all miRNAs is frequently lost in specific hematopoietic malignancies (Bueno et al., 2008). In addition to direct upregulation by fusion proteins, miR-17-92 has more than 2-fold amplification at corresponding DNA locus (Mi et al., 2010). This DNA increase could also be found at the regulatory region of ncRNAs. miR-142 is overexpressed in a subtype of B-cell tumors with t(8;17)(q24;q22) translocation (Kuriyama et al., 2018). Although the translocation involved pri-miR-142, it does not result in upregulation as the pri-miR-142 locus was truncated in the affected allele. Instead, a germline band with increased intensity suggests a gain in the upstream region of pri-miR-142 (Kuriyama et al., 2018). In addition, gain of DNA copies was reported on lncRNAs, such as PVT1 in PML–RARA-driven APL (Zeng et al., 2015).
Notably, some ncRNAs could be directly altered by chromosome rearrangements (Bousquet et al., 2008; Guastadisegni et al., 2008; Schneider et al., 2008; Chu et al., 2012) (Figure 1F). These changes might be due to the exchange of upstream regulatory elements of ncRNAs. First, chromosome translocation could activate the expression of ncRNAs. For example, in AML and myelodysplastic syndrome (MDS) cases with t(2;11)(p21;q23), no genes were mapped in this area. Elaborate analyses near the genomic locus revealed the overexpression of miR-125b-1, which is sufficient to interfere with primary human CD34+ cell differentiation (Bousquet et al., 2008). In addition to the activation of ncRNAs, silencing upon chromosomal translocation was observed in some regions, such as miR-29, which is located near the breakpoint region (Schneider et al., 2008).
The multifaceted functions of ncRNAs in fusion protein-driven cancers
The comprehensive and specific dysregulation of ncRNAs in fusion protein-driven malignancies raises the question whether ncRNAs play indispensable roles or are mere by-products of the genetic alterations. The most extensively studied ncRNAs are miRNAs. miRNAs target RNAs post-transcriptionally by fine-tuning the expression of mRNAs or repressing their translation (Lim et al., 2005). The mode of action for miRNA appears ‘simple’, as they function in a ‘teeterboard’ manner: high expression of oncogenic miRNA causes downregulation of tumor suppressors, and low expression of anticancer miRNAs results in upregulation of oncogenic genes (Figure 2A). However, in certain scenarios, miRNAs function in a much more complicated manner. Below are some examples of how miRNA functions in cytogenetically abnormal contexts, which can serve as a paradigm in fusion protein-driven cancers.
Figure 2.
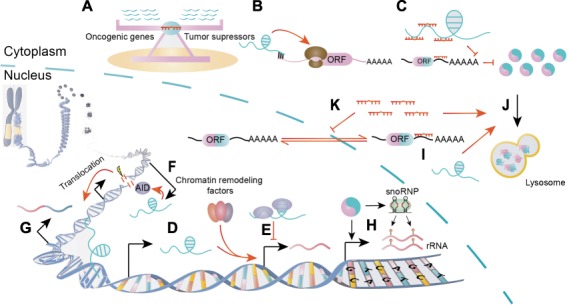
The various modes of actions of ncRNAs in fusion protein-driven cancers. (A) The miRNAs function in a teeterboard manner. (B–E) The lncRNAs play their roles by regulating translation (B), competing for miRNA binding (C), recruiting chromatin-remodeling complex (D), and binding proteins to regulate gene expressions (E). (F and G) The lncRNAs possibly facilitate AID-dependent gene translocation by transcription opposite to the sense strands (F) or bring two translocation partners into close proximity by directing interacting with their DNA loci (G). (H) Fusion proteins deregulate snoRNA and/or rRNA to exert procancer functions. (I–K) The ncRNAs can directly target fusion transcripts (I), regulate fusion protein degradation (J), or the nucleoplasmic transport of fusion mRNA pathways (K).
The functional complexity of miRNAs in a cytogenetically abnormal context
As miRNAs usually have multiple targets, defining a certain miRNA as oncogenic or as a tumor suppressor can be difficult in some cases. In fact, miRNAs may target oncogenes and tumor suppressors at the same time. For example, miR-196b was shown to delay MLL fusion-driven leukemogenesis in primary bone marrow transplantation (BMT), while accelerating this process in secondary transplantation (Li et al., 2012). Mechanically, it was found that both oncogenic HOXA9/MEIS1 and anticancer FAS are validated targets of miR-196b (Li et al., 2012). The forced expression of miR-196b retarded MLL leukemia that is addicted to HOXA9/MEIS1 signaling, while repression of Fas led to increased leukemia stem/initiating cells and aggressive disease (Li et al., 2012; Liu et al., 2013). These results reveal the complexity of miRNA networks in cytogenetically abnormal contexts. This is further supported by another example of miR-126. While expression and knockdown of miR-126 exerted opposing effects in cell lines, both overexpression and knockout of miR-126 accelerate AML1–ETO leukemogenesis in mouse model. These results may be because overexpression and depletion of miR-126 have preferential functions in distinct populations of cells in vivo. Ectopic expression of miR-126 induced genes highly expressed in leukemia stem/progenitor cells by silencing ERRFI1 and SPRED1, while miR-126 depletion activated genes highly expressed in committed progenitor cells by inducing FZD7 (Li et al., 2015). Compared with in vitro experiments, an in vivo study is more likely to provide a full view of the functions of ncRNAs. The discovery of multiple targets of miRNA raises the question of which ones are direct targets of a certain miRNA, especially in regard to non-canonical binding in specific cellular contexts, such as chromosomal translocation. By combining miRNA mimic pull-down and shRNA functional screening, a recent study claimed that Cdkn1b (p27Kip1) was a genuine target of miR-196 in the specific MLL-rearranged context (Meyer et al., 2018). In addition to their cell-intrinsic functions, miRNAs exert their roles in a non-cell intrinsic manner; for example, miR-125b induces VEGFA production in MLL–AF9-driven leukemia (Liu et al., 2017). Given that the same family of miRNAs often functions as a cluster, the disruption of multiple homologous miRNAs is sometimes required for effective targeted therapy (Mian and Zeleznik-Le, 2016). All the examples mentioned above demonstrate that miRNAs actually function in a more complex manner than previously believed.
The diverse functions of lncRNAs and snoRNAs
Unlike the ‘teeter board’ rules that guide miRNAs, other ncRNAs such as lncRNAs have diverse mechanisms in fusion protein-driven cancer. First, lncRNAs could regulate their adjacent genes by recruiting chromatin-remodeling factors, such as lncRNA CASC15 in RUNX1-rearranged leukemia (Fernando et al., 2017) (Figure 2D). Some natural antisense transcripts regulate their sense counterpart via overlapping regions (Figure 2B). For example, AS-RBM15 enhances the translation of RBM15, a regulator of megakaryocyte differentiation, and both of them are repressed in AML1–ETO leukemia (Tran et al., 2016). In addition to their cis-acting effects, lncRNAs could act in trans as competing RNAs for miRNAs or binding proteins to regulate gene expression, such as lncRNA-BGL3 and EWSAT1 in BCR–ABL and EWS–FLI1-mediated transformations, respectively (Marques et al., 2014; Guo et al., 2015) (Figure 2C and E). Notably, lncRNAs might have direct roles in shaping chromosomal translocations. First, the convergent transcription of the antisense non-coding region and the sense coding region can promote AID-dependent translocation (Qian et al., 2014; Meng et al., 2014; Lu et al., 2015) (Figure 2F). Furthermore, lncRNAs might even demarcate the major breakpoint region as exemplified by lnc-RP11-211G3.3.1-1 in BCL-6 breakage (Lu et al., 2015). This lncRNA precisely matches the boundary of the BCL6 translocation zone (Lu et al., 2015). Another possibility is that lncRNAs might bring two otherwise separate fusion partners into close proximity by mediating long-range intra/interchromosomal interactions (Wang et al., 2014) (Figure 2G). The relationship between lncRNAs and DNA translocation has yet to be validated.
The roles of snoRNAs in cancer have recently been revisited. In cancers with chromosomal rearrangements, snoRNAs function in both canonical and non-canonical manners (Figure 2H). rRNAs are essential for cancer cell proliferation, and studies in AML1–ETO-driven leukemia demonstrated that fusion proteins can either directly activate rRNA transcription or enhance snoRNP formation to catalyze the site-specific 2′-O-methylation of rRNAs (Bakshi et al., 2008; Zhou et al., 2017). The canonical function of C/D box snoRNA in rRNA methylation is required for leukemia self-renewal (Zhou et al., 2017). However, snoRNA can also act in a non-canonical manner. For example, ACA11, a H/ACA box snoRNA, was shown to regulate ribosomal protein genes by binding a novel non-canonical protein complex in multiple myeloma (Chu et al., 2012). Therefore, the roles of snoRNAs may be more diverse than previously predicted.
circRNAs emerge as new players
Although circRNAs were discovered over two decades ago, the development of next-generation RNA sequencing led to the rediscovery of thousands of circRNAs in eukaryotes (Salzman et al., 2012; Memczak et al., 2013; Jeck et al., 2013; Guo et al., 2014b). Importantly, circRNAs are emerging as key regulators in diseases (Qu et al., 2018; Liu et al., 2019). It was reported that circRNAs could be used as biomarkers to distinguish PML–RARA-driven APL with other subtypes of AML (You and Conrad, 2016; Li et al., 2018a; Zhang et al., 2014, 2016). The differential expression patterns suggest that circRNAs might be functional in malignancy with chromosome rearrangements. The proposed roles of circRNAs in cancer include mainly serving as miRNA sponges, interacting with proteins and cis-/trans-regulation of gene expressions (Bonizzato et al., 2016). However, some reports argued that the majority of circRNAs do not function as miRNA sponges (Guo et al., 2014b; Conn et al., 2015). Currently, our understanding on the functions of circRNAs in cancers with chromosome rearrangements is very limited. One experimentally validated example is the formation of oncogenic fusion circRNAs (f-circRNAs) upon chromosome translocation (Guarnerio et al., 2016). Both PML–RARA and MLL–AF9 fusions give rise to more than one f-circRNAs. Although f-circRNAs alone were not sufficient to trigger leukemia, the non-coding f-circRNAs contributed to disease progression in vivo when coupled with fusion proteins (Guarnerio et al., 2016). Nevertheless, the mechanism underlying the pathogenetic effects of f-circRNA remains to be uncovered. However, another group failed to detect these f-circRNAs with their home-developed software (You and Conrad, 2016). The low abundance of these f-circRNAs and insufficient sequencing depth might result in the failure in computational detection. More efforts are needed to shed light on the roles of circRNAs in cancers with chromosome translocation.
Direct fusion products targeted by ncRNAs
ncRNAs have also been shown to directly regulate fusion products by chromosomal translocation at both RNA and protein levels. First, a group of miRNAs, including miR-203, miR-23a, miR-196b, miR-30e, miR-320a, and miR-138, has been reported to directly target BCR–ABL in CML (Bueno et al., 2008; Liu et al., 2013; Xu et al., 2014; Xishan et al., 2014, 2015; Hershkovitz-Rokah et al., 2015) (Figure 2I). Their expression levels are inversely correlated with that of BCR–ABL, and their binding sites are on the coding region or 3′UTR of ABL (Bueno et al., 2008; Liu et al., 2013; Xu et al., 2014; Xishan et al., 2014, 2015; Hershkovitz-Rokah et al., 2015). Second, the fusion protein products could be regulated by finely controlling their degradation or the nucleocytoplasmic ratio. For example, we showed that miR-125b and HOTAIRM1 target PML–RARA by regulating its autophagic degradation (Chen et al., 2017; Zeng et al., 2014) (Figure 2J). Additionally, the forced expression of miR-1301 trapped BCR–ABL mRNA in the nucleus by targeting RanGAP1, which is responsible for nuclear protein export (Lin et al., 2016) (Figure 2K).
Fusion proteins can ‘connect’, ‘amplify’, and ‘switch’ ncRNA functions
In the specific situation of newly formed fusion proteins as the driving force in cancer with chromosomal rearrangements, several unique properties of ncRNA regulation and function are observed. First, fusion products can function as a ‘connector’ and thus confer context-specific regulation on ncRNAs. As an example, MALAT1 was downregulated only by MAPK/PI3K inhibitors in EVT6–NTRK3 kinase fusion-positive cell lines (Chen et al., 2018). This finding indicates that the fusion protein could rewire signaling pathways to exploit the functionally relevant lncRNAs. Next, fusion proteins can act as an ‘amplifier’ to strengthen the functions of ncRNAs. A subtype of aberrantly expressed ncRNAs has no or neglectable effects in normal tissues. However, these ncRNAs could enhance the oncogenic role of fusion proteins or could be essential for fusion protein-driven cancers (Mi et al., 2010; Jiang et al., 2012; Chen et al., 2013; Marques et al., 2014). Disruption or overexpression of these ncRNAs exhibits strong phenotypes only in the fusion protein-positive context. Thus, targeting these ncRNAs will not have adverse effects and should be investigated. Finally, the fusion proteins might serve as a ‘switcher’ to change the pro- or anticancer role of ncRNAs. For example, miR-9 plays a tumor suppressor role in EVI1-induced leukemia as it is repressed by the oncogene EVI1 (Senyuk et al., 2013). However, miR-9 is upregulated in MLL leukemia as MLL fusions override the repressive effects of EVI1 and consequently switch its role to a procancer one (Chen et al., 2013). In conclusion, the role of ncRNAs might be entirely different in fusion-protein driven cancer. Thus, the function of ncRNAs should be revisited in these specific contexts.
Non-coding RNAs are directly involved in chromosomal rearrangements
The discovery of chromosomal aberrations has furthered our understanding of tumorigenesis and provided reliable markers for diagnostic and therapeutic purposes. In the traditional view, the primary oncogenic entities in cancers with chromosome rearrangements are fusion proteins. Analysis of the coding products of fusion transcripts remains the focus of research. The universally used criteria for identifying clinically important fusion genes are that the capacity for coding proteins should be retained and the fusion transcripts are not spurious. Since our understanding of the functional genome has expanded to the non-coding genome, our view of fusion genes should be updated to include the non-coding dimension as well. For example, a recent study showed that the overwhelming majority of newly discovered fusion genes are out of frame in non-translocation-related sarcomas (Delespaul et al., 2017). Thus, the following questions need to be answered: (i) how does chromosomal translocation affect ncRNAs? (ii) Are the coding/non-coding fusion transcripts per se functionally relevant? Recently, several studies have begun to address these questions.
The ncRNA-convergent fusions
The direct interplay between chromosomal translocation and ncRNAs could be roughly summarized in four scenarios. First, more than a decade ago, miRNAs were shown to reside in hot spots for chromosomal abnormalities (Calin et al., 2004). Thus, chromosomal translocation might directly disturb the miRNA locus. The exchange of upstream regions could dysregulate miRNAs without disruption on their sequences (Figure 3A). A prominent example is miR-125b, which is frequently fused with the immunoglobulin heavy chain (IGH) locus in ALL (Sonoki et al., 2005; Chapiro et al., 2010; Tassano et al., 2010). The insertion of the IGH segment around the miRNA loci activates their expression. Incidental findings also indicate that miR-29 is deregulated by chromosomal translocation that involves its parental gene FRA7H (Schneider et al., 2008; Feldman et al., 2011). miR-29 is located in the intron of FRA7H and is thus deregulated together with FRA7H upon chromosomal translocation. In fact, dysregulation of intron-located small regulatory RNAs might be a paradigm for ncRNAs directly involved in chromosome rearrangements. As the traditional pipeline for discovering fusion genes filters out non-coding and non-recurrent fusion transcripts, this intriguing phenomenon remains unclear. However, a recent study shed light on this issue by searching for both out-of-frame and spurious fusion genes in breast cancer (Persson et al., 2017). The results were enriched for intron-encoded miRNAs, and the change in their upstream elements upregulated the miRNAs regardless of production of functional proteins of the coding regions (Persson et al., 2017; Rovira, 2018). This phenomenon is not restricted to miRNAs as another class of ncRNA, snoRNAs, is processed from the introns of paternal genes in eukaryotes as well (Dupuis-Sandoval et al., 2015) (Figure 3A). In multiple myeloma, WHSC1 is fused with IGH, which leads to the overexpression of the former gene. Notably, H/ACA box snoRNA ACA11 is located in the intron of WHSC1, and thus co-expressed with this gene (Chu et al., 2012). The protein product of WHSC1 is unable to transform wild-type or tumor-prone primary hematopoietic cells, while ACA11 exerts critical oncogenic role in multiple myeloma cells (Chu et al., 2012). The recurrent inclusion of specific ncRNAs with various 5′ partners redefines the meaning of ‘recurrence’ of fusion genes. Thus, the functional outcomes may be more important than the structural features of the fusions. These so-called ‘ncRNA-convergent fusions’ might substantially increase the number of oncogenic fusions.
Figure 3.
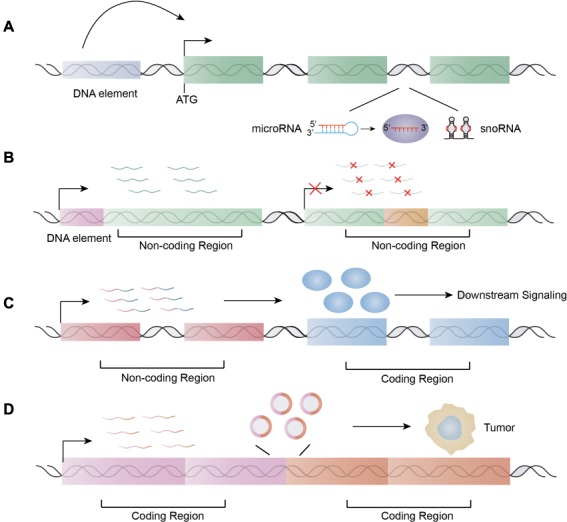
ncRNAs are directly involved in chromosome translocation. (A) ncRNA-convergent fusions. (B) The chromosomal translocation sites reside in the ncRNA regions: production of new ncRNAs by juxtaposition of regulatory elements upstream of non-coding regions or disruption of original ncRNA. (C) The fusions of non-coding regions with coding regions lead to the dysregulations of coding products and subsequent alterations of related signaling pathways. (D) The coding fusion transcripts or the derived non-coding isoforms, such as circRNAs by back-splicing, function as regulatory RNAs.
The disruption and generation of ncRNAs in breakpoint regions
Although the relationship between other ncRNAs and chromosome fragile sites has not been thoroughly studied, sporadic reports have indicated the close association of lncRNAs with chromosome breakpoint regions. A study detecting fusion transcripts by RNA-Seq in a pan-cancer panel of head and neck squamous cell carcinoma (HNSCC) identified only mRNA- and lncRNA-containing fusions (Bossi et al., 2017). This result suggests that the lncRNA locus might contain at least a proportion of chromosome breakpoint regions. Interestingly, a very recent paper stated that lncRNA FAM230C and its related genes carry a DNA translocation breakpoint element in their sequences (Delihas, 2018). One explanation for this is that the genes provide stability for these elements; however, this increases the possibility of DNA breakage and translocation within these lncRNAs. In fact, the second scenario involves the direct disruption of ncRNAs upon chromosomal translocation (Figure 3B). For example, in an orphan disease, MOMO syndrome, the balanced reciprocal translocation found in the patients disrupted a novel lncRNA gene named LINC00237 (Vu et al., 2012). The patients with MOMO syndrome failed to express this lncRNA. The specific expression pattern of this lncRNA and its strong relationship with MOMO syndrome indicate that it might be functionally relevant in the disease pathogenesis (Vu et al., 2012). These findings can also be extended to snoRNAs as a chromosome break within the U76 sequence that has been reported in GAS5–BCL6 fusions (Pickard and Williams, 2015). Whether snoRNA mediates double-stranded DNA break and recombination events is unclear. Furthermore, one study showed that chromosomal translocation could generate new ncRNAs that do not exist in normal tissues (Guastadisegni et al., 2008). These results suggest that ‘DNA reshuffling’ by chromosomal rearrangements can produce novel translocation-specific ncRNAs, which might be filtered out by the current bioinformatics pipeline for fusion protein identification. Although only incidental findings have been reported to date, we believe that future studies will uncover more examples and confirm that the disruptions or generations of ncRNAs upon chromosomal translocations are functionally important events in tumorigenesis.
Non-coding regions partner with coding transcripts
The third scenario involves the juxtaposition of non-coding regions with protein-coding regions, which change the expression of the protein-coding transcripts instead of resulting in the dysregulation of ncRNAs mentioned above (Figure 3C). This change does not result in a fusion protein because one of the fusion partners is non-coding. Instead, the protein-coding transcripts are dysregulated, which in turn lead to aberrant alterations of downstream signaling (Figure 3C). One such repeatedly reported fusion gene involves MALAT1 and GLI1 in epithelioid neoplasm, plexiform fibromyxoma, and gastroblastoma (Spans et al., 2016; Graham et al., 2017; Antonescu et al., 2018). The high expression of MALAT1 results in constant activation of GLI1, which in turn activates the sonic hedgehog signaling pathway (Spans et al., 2016; Graham et al., 2017). Similar results are also found on double minutes which are small fragments of extrachromosomal DNA observed in many human tumors. Fusion transcripts involving lncRNA PVT1 and the protein-coding gene NSMCE2 were identified on double minute chromosomes in AML patients and cell lines (Chinen et al., 2014).
The coding/non-coding bifunctional fusion transcripts
Along with the increasing investigations of the function of ncRNAs, the distinction between coding and non-coding genes has been slowly blurred. The RNA functions are believed to be Janus-faced because some annotated lncRNAs have been shown to encode small peptides, and some coding RNAs have regulatory functions independent of the proteins they encode (Kumari and Sampath, 2015; Nam et al., 2016; Dhamija and Menon, 2018). To further complicate this matter, protein-coding genes can also express lncRNA forms by alternative splicing or generate circRNA by back-splicing. Kumari and Sampath (2015) designated these bifunctional RNAs as ‘cncRNAs’ (coding and ncRNA). This finding raises the question of whether the protein-coding fusion transcripts per se have any regulatory functions. Using public t(8;21) AML RNA-Seq and miRNA-Seq data, a study predicted that the recurrent fusion partner RUNXT1 has a protein-coding-independent function and suggested that this molecule acts as a ceRNA in AML formation (Junge et al., 2017). Moreover, the primary role of fusion cncRNAs might be related to non-coding functions rather than coding functions in some cases. For example, the coding product of the fusion transcript SLC45A3–ELK4 is unlikely to perturb the protein pool of wild-type ELK4 in prostate cancer, as the abundance of fusion RNA is insufficient (Qin et al., 2017). In fact, SLC45A3–ELK4 functions as an lncRNA and non-coding mutant transcripts are sufficient to regulate cancer cell proliferation (Qin et al., 2017). In addition to the fusion transcript itself, other fusion ncRNAs, such as circRNA, are derived from the chimeric transcripts upon chromosome translocation. circRNAs are generated by a proactive back-splicing event in which the 3′ tail of one exon is joined to the 5′ head of an upstream exon (Jeck et al., 2013). The juxtaposition of otherwise separate genes upon chromosomal translocation results in the close proximity of distant complementary repetitive intronic sequence and thus favors new back-splicing events. This phenomenon is exemplified by the generation of oncogenic fusion circRNA from fusion protein-encoding transcripts both in leukemia and solid tumors (Guarnerio et al., 2016; Tan et al., 2018a,b). Functional analysis revealed that at least some of them are essential for oncogenesis, such as F-circEA/F-circEA-2a, F-circM9, and F-circPR in non-small cell lung cancer, MLL leukemia, and APL, respectively (Guarnerio et al., 2016; Tan et al., 2018a,b). These findings have challenged the view that the functions of coding transcripts resulting from chromosomal translocations are protein-dependent and add another non-coding layer to their functional complexity (Figure 3D).
Non-coding fusion transcripts as biomarkers
Although the functions of non-coding fusion transcripts remain largely unknown, they have been suggested as diagnostic and prognostic biomarkers. In some scenarios, lncRNA-containing fusions might show a closer relationship to the disease outcome than mRNA fusions, as reported in head and neck cancer (Bossi et al., 2017). In contrast to their parental linear fusion RNAs, circRNAs have a circular covalent bond structure that endows them with high resistance to exonuclease digestion (Li et al., 2018b). As circRNAs are enriched and stable in the exosomes of the peripheral blood, several fusion circRNAs have been proposed as liquid pathognomonic markers (Guarnerio et al., 2016; Tan et al., 2018b). These molecules may be a good choice when biopsies are difficult to obtain. Notably, chimeric RNAs are not exclusive to cancer, and some fusion RNAs exist in normal conditions because cis-splicing or trans-splicing at the RNA level can generate fusion transcripts (Jia et al., 2016; Elfman and Li, 2018). Some fusion RNAs resulting from cis-/trans-splicing are even identical in malignant and normal cells (Li et al., 2008b; Nacu et al., 2011; Ren et al., 2012; Yuan et al., 2013). Thus, researchers should be cautious when considering fusion RNAs as biomarkers or drug targets for cancer. Intriguingly, the chimeric transcripts generated at the RNA level might mediate DNA rearrangements, since this is a process that occurs in lower species (Li et al., 2008b; Zaphiropoulos, 2011; Jia et al., 2016). Chimeric RNA might serve as template for double-stranded DNA breakage repair or as a scaffold to bring two genomic loci into proximity to induce chromosomal rearrangements, thereby producing DNA translocation-triggered fusion transcripts (Jia et al., 2016; Elfman and Li, 2018). Alternatively, there might be no causal relationship between chimeric RNA generated by trans-splicing and chromosomal translocation. Instead, same factors might contribute to both trans-spliced RNA chimeras and DNA rearrangements, which could bring the separate gene loci into close proximity.
Perspective
Researches in the past decade suggested a direct link between chromosome translocation-driven cancer and ncRNAs. Although miRNAs are the best-studied ncRNAs in cytogenetically abnormal contexts, research focus has started to move to other ncRNAs, such as lncRNAs, circRNAs, and snoRNAs. However, the expression patterns and functions of these novel molecules in distinct subtypes of cancers with chromosomal rearrangements, especially solid tumors, are unclear. Hence, more efforts are needed to determine the profiles of these ncRNAs. Notably, several ncRNAs function only in malignancies with chromosomal rearrangements and are dispensable in normal conditions; thus, these molecules should be assessed as potential therapeutic targets. Given that direct targeting of ncRNAs is not applicable in the clinic, identification of targets in upstream pathways that control the expression of ncRNAs is needed. Thus, further elucidation of the complicated mechanisms underlying ncRNA regulation in cancers with chromosomal rearrangements is important.
In addition, the direct involvement of ncRNAs in the formation of fusion transcripts and regulation of chromosomal translocation requires further analyses. Although several studies have reported the existence of non-coding fusion transcripts, whether they are functional remains largely elusive. Determination of whether ncRNAs influence the formation of chromosomal rearrangements and the underlying mechanisms is also necessary. Based on the current findings, the transcription of ncRNAs or ncRNAs themselves should be taken into account when addressing how DNA translocations are formed. However, more experimental evidence is needed to validate this concept. Further analysis of ncRNAs may help determine the preferential regions of chromosomal translocation and provide explanations for this process.
Funding
This research was supported by the National Key R&D Program of China (2017YFA0504400) and the National Natural Science Foundation of China (81770174 and 31870818).
Conflict of interest: none declared.
References
- Anastasiadou E., Jacob L.S., and Slack F.J. (2018). Non-coding RNA networks in cancer. Nat. Rev. Cancer 18, 5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonescu C.R., Agaram N.P., Sung Y.S., et al. (2018). A distinct malignant epithelioid neoplasm with GLI1 gene rearrangements, frequent S100 protein expression, and metastatic potential: expanding the spectrum of pathologic entities with ACTB/MALAT1/PTCH1–GLI1 fusions. Am. J. Surg. Pathol. 42, 553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakshi R., Zaidi S.K., Pande S., et al. (2008). The leukemogenic t(8;21) fusion protein AML1–ETO controls rRNA genes and associates with nucleolar-organizing regions at mitotic chromosomes. J. Cell. Sci. 121, 3981–3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar B., Blachly J.S., Kohlschmidt J., et al. (2016). Clinical features and gene- and microRNA-expression patterns in adult acute leukemia patients with t(11;19)(q23;p13.1) and t(11;19)(q23;p13.3). Leukemia 30, 1586–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzato A., Gaffo E., Te K.G., et al. (2016). CircRNAs in hematopoiesis and hematological malignancies. Blood Cancer J. 6, e483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi P., Siano M., Bergamini C., et al. (2017). Are fusion transcripts in relapsed/metastatic head and neck cancer patients predictive of response to anti-EGFR therapies? Dis. Markers 2017, 6870614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet M., Quelen C., Rosati R., et al. (2008). Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. J. Exp. Med. 205, 2499–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer-Hartmann D., Hartmann J.U., Wurm A.A., et al. (2015). PML/RARα-regulated miR-181a/b cluster targets the tumor suppressor RASSF1A in acute promyelocytic leukemia. Cancer Res. 75, 3411–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno M.J., Perez D.C.I., Gomez D.C.M., et al. (2008). Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR–ABL1 oncogene expression. Cancer Cell 13, 496–506. [DOI] [PubMed] [Google Scholar]
- Calin G.A., and Croce C.M. (2007). Chromosomal rearrangements and microRNAs: a new cancer link with clinical implications. J. Clin. Invest. 117, 2059–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin G.A., Sevignani C., Dumitru C.D., et al. (2004). Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl Acad. Sci. USA 101, 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Careccia S., Mainardi S., Pelosi A., et al. (2009). A restricted signature of miRNAs distinguishes APL blasts from normal promyelocytes. Oncogene 28, 4034–4040. [DOI] [PubMed] [Google Scholar]
- Cech T.R., and Steitz J.A. (2014). The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94. [DOI] [PubMed] [Google Scholar]
- Chapiro E., Russell L.J., Struski S., et al. (2010). A new recurrent translocation t(11;14)(q24;q32) involving IGH@ and miR-125b-1 in B-cell progenitor acute lymphoblastic leukemia. Leukemia 24, 1362–1364. [DOI] [PubMed] [Google Scholar]
- Chen J., Odenike O., and Rowley J.D. (2010). Leukaemogenesis: more than mutant genes. Nat. Rev. Cancer 10, 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Price C., Li Z., et al. (2013). miR-9 is an essential oncogenic microRNA specifically overexpressed in mixed lineage leukemia-rearranged leukemia. Proc. Natl Acad. Sci. USA 110, 11511–11516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Nagel S., Schneider B., et al. (2018). A new ETV6–NTRK3 cell line model reveals MALAT1 as a novel therapeutic target—a short report. Cell Oncol. 41, 93–101. [DOI] [PubMed] [Google Scholar]
- Chen Z.H., Wang W.T., Huang W., et al. (2017). The lncRNA HOTAIRM1 regulates the degradation of PML–RARA oncoprotein and myeloid cell differentiation by enhancing the autophagy pathway. Cell Death Differ. 24, 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinen Y., Sakamoto N., Nagoshi H., et al. (2014). 8q24 amplified segments involve novel fusion genes between NSMCE2 and long noncoding RNAs in acute myelogenous leukemia. J. Hematol. Oncol. 7, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Kim Y.K., Park K., et al. (2016). MicroRNA-139-5p regulates proliferation of hematopoietic progenitors and is repressed during BCR–ABL-mediated leukemogenesis. Blood 128, 2117–2129. [DOI] [PubMed] [Google Scholar]
- Chu L., Su M.Y., Maggi L.J., et al. (2012). Multiple myeloma-associated chromosomal translocation activates orphan snoRNA ACA11 to suppress oxidative stress. J. Clin. Invest. 122, 2793–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Y., Hertzog K., Reish O., et al. (2012). The increased expression of 14q32 small nucleolar RNA transcripts in promyelocytic leukemia cells is not dependent on PML–RARA fusion gene. Blood Cancer J. 2, e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn S.J., Pillman K.A., Toubia J., et al. (2015). The RNA binding protein quaking regulates formation of circRNAs. Cell 160, 1125–1134. [DOI] [PubMed] [Google Scholar]
- Delespaul L., Lesluyes T., Perot G., et al. (2017). Recurrent TRIO fusion in nontranslocation-related sarcomas. Clin. Cancer Res. 23, 857–867. [DOI] [PubMed] [Google Scholar]
- Delihas N. (2018). A family of long intergenic non-coding RNA genes in human chromosomal region 22q11.2 carry a DNA translocation breakpoint/AT-rich sequence. PLoS One 13, e0195702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhamija S., and Menon M.B. (2018). Non-coding transcript variants of protein-coding genes—what are they good for? RNA Biol. 15, 1025–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon-McIver A., East P., Mein C.A., et al. (2008). Distinctive patterns of microRNA expression associated with karyotype in acute myeloid leukaemia. PLoS One 3, e2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y., and Hess J.L. (2008). Mechanisms of transcriptional regulation by MLL and its disruption in acute leukemia. Int. J. Hematol. 87, 10–18. [DOI] [PubMed] [Google Scholar]
- Druker B.J. (2008). Translation of the Philadelphia chromosome into therapy for CML. Blood 112, 4808–4817. [DOI] [PubMed] [Google Scholar]
- Dupuis-Sandoval F., Poirier M., and Scott M.S. (2015). The emerging landscape of small nucleolar RNAs in cell biology. Wiley Interdiscip. Rev. RNA. 6, 381–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfman J., and Li H. (2018). Chimeric RNA in cancer and stem cell differentiation. Stem Cells Int. 2018, 3178789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erfurth F.E., Popovic R., Grembecka J., et al. (2008). MLL protects CpG clusters from methylation within the Hoxa9 gene, maintaining transcript expression. Proc. Natl Acad. Sci. USA 105, 7517–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. (2011). Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861–874. [DOI] [PubMed] [Google Scholar]
- Fang K., Han B.W., Chen Z.H., et al. (2014). A distinct set of long non-coding RNAs in childhood MLL-rearranged acute lymphoblastic leukemia: biology and epigenetic target. Hum. Mol. Genet. 23, 3278–3288. [DOI] [PubMed] [Google Scholar]
- Fazi F., Racanicchi S., Zardo G., et al. (2007). Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 12, 457–466. [DOI] [PubMed] [Google Scholar]
- Feldman A.L., Dogan A., Smith D.I., et al. (2011). Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood 117, 915–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando T.R., Contreras J.R., Zampini M., et al. (2017). The lncRNA CASC15 regulates SOX4 expression in RUNX1-rearranged acute leukemia. Mol. Cancer 16, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando T.R., Rodriguez-Malave N.I., Waters E.V., et al. (2015). LncRNA expression discriminates karyotype and predicts survival in B-lymphoblastic leukemia. Mol. Cancer Res. 13, 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzon R., Volinia S., Liu C.G., et al. (2008). MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood 111, 3183–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazavi F., De Moerloose B., Van Loocke W., et al. (2016). Unique long non-coding RNA expression signature in ETV6/RUNX1-driven B-cell precursor acute lymphoblastic leukemia. Oncotarget 7, 73769–73780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham R.P., Nair A.A., Davila J.I., et al. (2017). Gastroblastoma harbors a recurrent somatic MALAT1–GLI1 fusion gene. Mod. Pathol. 30, 1443–1452. [DOI] [PubMed] [Google Scholar]
- Guarnerio J., Bezzi M., Jeong J.C., et al. (2016). Oncogenic role of fusion-circRNAs derived from cancer-associated chromosomal translocations. Cell 166, 1055–1056. [DOI] [PubMed] [Google Scholar]
- Guastadisegni M.C., Lonoce A., Impera L., et al. (2008). Bone marrow ectopic expression of a non-coding RNA in childhood T-cell acute lymphoblastic leukemia with a novel t(2;11)(q11.2;p15.1) translocation. Mol. Cancer 7, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G., Kang Q., Chen Q., et al. (2014a). High expression of long non-coding RNA H19 is required for efficient tumorigenesis induced by Bcr–Abl oncogene. FEBS Lett. 588, 1780–1786. [DOI] [PubMed] [Google Scholar]
- Guo G., Kang Q., Zhu X., et al. (2015). A long noncoding RNA critically regulates Bcr–Abl-mediated cellular transformation by acting as a competitive endogenous RNA. Oncogene 34, 1768–1779. [DOI] [PubMed] [Google Scholar]
- Guo J.U., Agarwal V., Guo H., et al. (2014b). Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 15, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez S.E., and Romero-Oliva F.A. (2013). Epigenetic changes: a common theme in acute myelogenous leukemogenesis. J. Hematol. Oncol. 6, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershkovitz-Rokah O., Modai S., Pasmanik-Chor M., et al. (2015). MiR-30e induces apoptosis and sensitizes K562 cells to imatinib treatment via regulation of the BCR–ABL protein. Cancer Lett. 356(2 Pt B), 597–605. [DOI] [PubMed] [Google Scholar]
- Hicks J.K., Henderson-Jackson E., Duggan J., et al. (2018). Identification of a novel MTAP–RAF1 fusion in a soft tissue sarcoma. Diagn. Pathol. 13, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T., Chong Y., Qin H., et al. (2018). The miR-17/92 cluster is involved in the molecular etiology of the SCLL syndrome driven by the BCR–FGFR1 chimeric kinase. Oncogene 37, 1926–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H., Jiang X., Wang J., et al. (2016). Identification of MLL-fusion/MYC ⊣miR-26 ⊣TET1 signaling circuit in MLL-rearranged leukemia. Cancer Lett. 372, 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeck W.R., Sorrentino J.A., Wang K., et al. (2013). Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19, 141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y., Xie Z., and Li H. (2016). Intergenically spliced chimeric RNAs in cancer. Trends Cancer 2, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Huang H., Li Z., et al. (2012). Blockade of miR-150 maturation by MLL-fusion/MYC/LIN-28 is required for MLL-associated leukemia. Cancer Cell 22, 524–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge A., Zandi R., Havgaard J.H., et al. (2017). Assessing the miRNA sponge potential of RUNX1T1 in t(8;21) acute myeloid leukemia. Gene 615, 35–40. [DOI] [PubMed] [Google Scholar]
- Kim P., Jia P., and Zhao Z. (2018). Kinase impact assessment in the landscape of fusion genes that retain kinase domains: a pan-cancer study. Brief. Bioinform. 19, 450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koretzky G.A. (2007). The legacy of the Philadelphia chromosome. J. Clin. Invest. 117, 2030–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov A.V., and Armstrong S.A. (2007). MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 7, 823–833. [DOI] [PubMed] [Google Scholar]
- Krivtsov A.V., Feng Z., Lemieux M.E., et al. (2008). H3K79 methylation profiles define murine and human MLL–AF4 leukemias. Cancer Cell 14, 355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari P., and Sampath K. (2015). cncRNAs: bi-functional RNAs with protein coding and non-coding functions. Semin. Cell Dev. Biol 47–48, 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama K., Enomoto Y., Suzuki R., et al. (2018). Enforced expression of MIR142, a target of chromosome translocation in human B-cell tumors, results in B-cell depletion. Int. J. Hematol. 107, 345–354. [DOI] [PubMed] [Google Scholar]
- Lavallee V.P., Baccelli I., Krosl J., et al. (2015). The transcriptomic landscape and directed chemical interrogation of MLL-rearranged acute myeloid leukemias. Nat. Genet. 9, 1030–1037. [DOI] [PubMed] [Google Scholar]
- Li H., Wang J., Mor G., et al. (2008b). A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science 321, 1357–1361. [DOI] [PubMed] [Google Scholar]
- Li S., Ma Y., Tan Y., et al. (2018a). Profiling and functional analysis of circular RNAs in acute promyelocytic leukemia and their dynamic regulation during all-trans retinoic acid treatment. Cell Death Dis. 9, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Yang L., and Chen L.L. (2018b). The biogenesis, functions, and challenges of circular RNAs. Mol. Cell 71, 428–442. [DOI] [PubMed] [Google Scholar]
- Li Z., Chen P., Su R., et al. (2015). Overexpression and knockout of miR-126 both promote leukemogenesis. Blood 126, 2005–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Huang H., Chen P., et al. (2012). miR-196b directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat. Commun. 3, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Lu J., Sun M., et al. (2008a). Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proc. Natl Acad. Sci. USA 105, 15535–15540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L.P., Lau N.C., Garrett-Engele P., et al. (2005). Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433, 769–773. [DOI] [PubMed] [Google Scholar]
- Lin T.Y., Chen K.C., Liu H.J., et al. (2016). MicroRNA-1301-mediated RanGAP1 downregulation induces BCR–ABL nuclear entrapment to enhance imatinib efficacy in chronic myeloid leukemia cells. PLoS One 11, e0156260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linka Y., Ginzel S., Kruger M., et al. (2013). The impact of TEL–AML1 (ETV6–RUNX1) expression in precursor B cells and implications for leukaemia using three different genome-wide screening methods. Blood Cancer J. 3, e151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C.X., Li X., Nan F., et al. (2019). Structure and degradation of circular RNAs regulate PKR activation in innate immunity. Cell 177, 865–880.e21. [DOI] [PubMed] [Google Scholar]
- Liu J., Guo B., Chen Z., et al. (2017). miR-125b promotes MLL–AF9-driven murine acute myeloid leukemia involving a VEGFA-mediated non-cell-intrinsic mechanism. Blood 129, 1491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Zheng W., Song Y., et al. (2013). Low expression of miR-196b enhances the expression of BCR–ABL1 and HOXA9 oncogenes in chronic myeloid leukemogenesis. PLoS One 8, e68442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liuksiala T., Teittinen K.J., Granberg K., et al. (2014). Overexpression of SNORD114-3 marks acute promyelocytic leukemia. Leukemia 28, 233–236. [DOI] [PubMed] [Google Scholar]
- Lu J., Getz G., Miska E.A., et al. (2005). MicroRNA expression profiles classify human cancers. Nature 435, 834–838. [DOI] [PubMed] [Google Scholar]
- Lu Z., Pannunzio N.R., Greisman H.A., et al. (2015). Convergent BCL6 and lncRNA promoters demarcate the major breakpoint region for BCL6 translocations. Blood 126, 1730–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques H.M., Simpson D., Ngok S.P., et al. (2014). Long noncoding RNA EWSAT1-mediated gene repression facilitates Ewing sarcoma oncogenesis. J. Clin. Invest. 124, 5275–5290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May W.A., Lessnick S.L., Braun B.S., et al. (1993). The Ewing’s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol. Cell. Biol. 13, 7393–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon M., Contreras A., and Ruggero D. (2015). Small RNAs with big implications: new insights into H/ACA snoRNA function and their role in human disease. Wiley Interdiscip. Rev. RNA 6, 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S., Jens M., Elefsinioti A., et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338. [DOI] [PubMed] [Google Scholar]
- Meng F.L., Du Z., Federation A., et al. (2014). Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell 159, 1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer S.E., Muench D.E., Rogers A.M., et al. (2018). miR-196b target screen reveals mechanisms maintaining leukemia stemness with therapeutic potential. J. Exp. Med. 215, 2115–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S., Li Z., Chen P., et al. (2010). Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc. Natl Acad. Sci. USA 107, 3710–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian Y.A., and Zeleznik-Le N.J. (2016). The miR-17 approximately 92 cluster contributes to MLL leukemia through the repression of MEIS1 competitor PKNOX1. Leuk. Res. 46, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne T.A., Dou Y., Martin M.E., et al. (2005a). MLL associates specifically with a subset of transcriptionally active target genes. Proc. Natl Acad. Sci. USA 102, 14765–14770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne T.A., Martin M.E., Brock H.W., et al. (2005b). Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res. 65, 11367–11374. [DOI] [PubMed] [Google Scholar]
- Mitelman F., Johansson B., and Mertens F. (2007). The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 7, 233–245. [DOI] [PubMed] [Google Scholar]
- Mohr S., Doebele C., Comoglio F., et al. (2017). Hoxa9 and Meis1 cooperatively induce addiction to Syk signaling by suppressing miR-146a in acute myeloid leukemia. Cancer Cell 31, 549–562.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris K.V., and Mattick J.S. (2014). The rise of regulatory RNA. Nat. Rev. Genet. 15, 423–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacu S., Yuan W., Kan Z., et al. (2011). Deep RNA sequencing analysis of readthrough gene fusions in human prostate adenocarcinoma and reference samples. BMC Med. Genomics 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J.W., Choi S.W., and You B.H. (2016). Incredible RNA: dual functions of coding and noncoding. Mol. Cells 39, 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K.N., Peiris M.N., Meyer A.N., et al. (2017). Receptor tyrosine kinases: translocation partners in hematopoietic disorders. Trends Mol. Med. 23, 59–79. [DOI] [PubMed] [Google Scholar]
- Parker B.C., and Zhang W. (2013). Fusion genes in solid tumors: an emerging target for cancer diagnosis and treatment. Chin. J. Cancer 32, 594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson H., Sokilde R., Hakkinen J., et al. (2017). Frequent miRNA-convergent fusion gene events in breast cancer. Nat. Commun. 8, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard M.R., and Williams G.T. (2015). Molecular and cellular mechanisms of action of tumour suppressor GAS5 LncRNA. Genes 6, 484–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovic R., Riesbeck L.E., Velu C.S., et al. (2009). Regulation of mir-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood 113, 3314–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J., Wang Q., Dose M., et al. (2014). B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 159, 1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F., Zhang Y., Liu J., et al. (2017). SLC45A3–ELK4 functions as a long non-coding chimeric RNA. Cancer Lett. 404, 53–61. [DOI] [PubMed] [Google Scholar]
- Qu S., Liu Z., Yang X., et al. (2018). The emerging functions and roles of circular RNAs in cancer. Cancer Lett. 414, 301–309. [DOI] [PubMed] [Google Scholar]
- Ren S., Peng Z., Mao J.H., et al. (2012). RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res. 22, 806–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchetti D., Mosca L., Cutrona G., et al. (2013). Small nucleolar RNAs as new biomarkers in chronic lymphocytic leukemia. BMC Med. Genomics 6, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossari F., Minutolo F., and Orciuolo E. (2018). Past, present, and future of Bcr–Abl inhibitors: from chemical development to clinical efficacy. J. Hematol. Oncol. 11, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roukos V., and Misteli T. (2014). The biogenesis of chromosome translocations. Nat. Cell Biol. 16, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira C. (2018). miRNA-convergent gene fusions. Mol. Cell. Oncol. 5, e1406433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman J., Gawad C., Wang P.L., et al. (2012). Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One 7, e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saumet A., Vetter G., Bouttier M., et al. (2009). Transcriptional repression of microRNA genes by PML–RARA increases expression of key cancer proteins in acute promyelocytic leukemia. Blood 113, 412–421. [DOI] [PubMed] [Google Scholar]
- Schafer E., Irizarry R., Negi S., et al. (2010). Promoter hypermethylation in MLL-r infant acute lymphoblastic leukemia: biology and therapeutic targeting. Blood 115, 4798–4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherr M., Elder A., Battmer K., et al. (2014). Differential expression of miR-17~92 identifies BCL2 as a therapeutic target in BCR–ABL-positive B-lineage acute lymphoblastic leukemia. Leukemia 28, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider B., Nagel S., Kaufmann M., et al. (2008). T(3;7)(q27;q32) fuses BCL6 to a non-coding region at FRA7H near miR-29. Leukemia 22, 1262–1266. [DOI] [PubMed] [Google Scholar]
- Schotte D., Lange-Turenhout E.A., Stumpel D.J., et al. (2010). Expression of miR-196b is not exclusively MLL-driven but is especially linked to activation of HOXA genes in pediatric acute lymphoblastic leukemia. Haematologica 95, 1675–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzer A., Emmrich S., Schmidt F., et al. (2017). The non-coding RNA landscape of human hematopoiesis and leukemia. Nat. Commun. 8, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senyuk V., Zhang Y., Liu Y., et al. (2013). Critical role of miR-9 in myelopoiesis and EVI1-induced leukemogenesis. Proc. Natl Acad. Sci. USA 110, 5594–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoki T., Iwanaga E., Mitsuya H., et al. (2005). Insertion of microRNA-125b-1, a human homologue of lin-4, into a rearranged immunoglobulin heavy chain gene locus in a patient with precursor B-cell acute lymphoblastic leukemia. Leukemia 19, 2009–2010. [DOI] [PubMed] [Google Scholar]
- Spans L., Fletcher C.D., Antonescu C.R., et al. (2016). Recurrent MALAT1–GLI1 oncogenic fusion and GLI1 up-regulation define a subset of plexiform fibromyxoma. J. Pathol. 239, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky N., Cerami E., Schalm S., et al. (2014). The landscape of kinase fusions in cancer. Nat. Commun. 5, 4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H., Lin D.C., Cao Q., et al. (2017). Identification of a novel SYK/c-MYC/MALAT1 signaling pathway and its potential therapeutic value in Ewing sarcoma. Clin. Cancer Res. 23, 4376–4387. [DOI] [PubMed] [Google Scholar]
- Tan S., Gou Q., Pu W., et al. (2018b). Circular RNA F-circEA produced from EML4–ALK fusion gene as a novel liquid biopsy biomarker for non-small cell lung cancer. Cell Res. 28, 693–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S., Sun D., Pu W., et al. (2018a). Circular RNA F-circEA-2a derived from EML4–ALK fusion gene promotes cell migration and invasion in non-small cell lung cancer. Mol. Cancer 17, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassano E., Acquila M., Tavella E., et al. (2010). MicroRNA-125b-1 and BLID upregulation resulting from a novel IGH translocation in childhood B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer 49, 682–687. [DOI] [PubMed] [Google Scholar]
- Teppo S., Laukkanen S., Liuksiala T., et al. (2016). Genome-wide repression of eRNA and target gene loci by the ETV6–RUNX1 fusion in acute leukemia. Genome Res. 26, 1468–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran N.T., Su H., Khodadadi-Jamayran A., et al. (2016). The AS-RBM15 lncRNA enhances RBM15 protein translation during megakaryocyte differentiation. EMBO Rep. 17, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valleron W., Laprevotte E., Gautier E.F., et al. (2012). Specific small nucleolar RNA expression profiles in acute leukemia. Leukemia 26, 2052–2060. [DOI] [PubMed] [Google Scholar]
- Velu C.S., Chaubey A., Phelan J.D., et al. (2014). Therapeutic antagonists of microRNAs deplete leukemia-initiating cell activity. J. Clin. Invest. 124, 222–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturini L., Battmer K., Castoldi M., et al. (2007). Expression of the miR-17-92 polycistron in chronic myeloid leukemia (CML) CD34+ cells. Blood 109, 4399–4405. [DOI] [PubMed] [Google Scholar]
- Viswanathan S.R., Daley G.Q., and Gregory R.I. (2008). Selective blockade of microRNA processing by Lin28. Science 320, 97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B., and Kinzler K.W. (2004). Cancer genes and the pathways they control. Nat. Med. 10, 789–799. [DOI] [PubMed] [Google Scholar]
- Vu P.Y., Toutain J., Cappellen D., et al. (2012). A homozygous balanced reciprocal translocation suggests LINC00237 as a candidate gene for MOMO (macrosomia, obesity, macrocephaly, and ocular abnormalities) syndrome. Am. J. Med. Genet. A 158A, 2849–2856. [DOI] [PubMed] [Google Scholar]
- Wang H., Li W., Guo R., et al. (2014). An intragenic long noncoding RNA interacts epigenetically with the RUNX1 promoter and enhancer chromatin DNA in hematopoietic malignancies. Int. J. Cancer 135, 2783–2794. [DOI] [PubMed] [Google Scholar]
- Warner W.A., Spencer D.H., Trissal M., et al. (2018). Expression profiling of snoRNAs in normal hematopoiesis and AML. Blood Adv. 2, 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G.T., and Farzaneh F. (2012). Are snoRNAs and snoRNA host genes new players in cancer? Nat. Rev. Cancer 12, 84–88. [DOI] [PubMed] [Google Scholar]
- Xishan Z., Xianjun L., Ziying L., et al. (2014). The malignancy suppression role of miR-23a by targeting the BCR/ABL oncogene in chromic myeloid leukemia. Cancer Gene Ther. 21, 397–404. [DOI] [PubMed] [Google Scholar]
- Xishan Z., Ziying L., Jing D., et al. (2015). MicroRNA-320a acts as a tumor suppressor by targeting BCR/ABL oncogene in chronic myeloid leukemia. Sci. Rep. 5, 12460. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xu C., Fu H., Gao L., et al. (2014). BCR–ABL/GATA1/miR-138 mini circuitry contributes to the leukemogenesis of chronic myeloid leukemia. Oncogene 33, 44–54. [DOI] [PubMed] [Google Scholar]
- Yde C.W., Sehested A., Mateu-Regue A., et al. (2016). A new NFIA:RAF1 fusion activating the MAPK pathway in pilocytic astrocytoma. Cancer Genet. 209, 440–444. [DOI] [PubMed] [Google Scholar]
- You X., and Conrad T.O. (2016). Acfs: accurate circRNA identification and quantification from RNA-Seq data. Sci. Rep. 6, 38820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H., Qin F., Movassagh M., et al. (2013). A chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis process. Cancer Discov. 3, 1394–1403. [DOI] [PubMed] [Google Scholar]
- Zaphiropoulos P.G. (2011). Trans-splicing in higher eukaryotes: implications for cancer development? Front. Genet. 2, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C., Yu X., Lai J., et al. (2015). Overexpression of the long non-coding RNA PVT1 is correlated with leukemic cell proliferation in acute promyelocytic leukemia. J. Hematol. Oncol. 8, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng C.W., Chen Z.H., Zhang X.J., et al. (2014). MIR125B1 represses the degradation of the PML–RARA oncoprotein by an autophagy-lysosomal pathway in acute promyelocytic leukemia. Autophagy 10, 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.O., Dong R., Zhang Y., et al. (2016). Diverse alternative back-splicing and alternative splicing landscape of circular RNAs. Genome Res. 26, 1277–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.O., Wang H.B., Zhang Y., et al. (2014). Complementary sequence-mediated exon circularization. Cell 159, 134–147. [DOI] [PubMed] [Google Scholar]
- Zhou F., Liu Y., Rohde C., et al. (2017). AML1–ETO requires enhanced C/D box snoRNA/RNP formation to induce self-renewal and leukaemia. Nat. Cell Biol. 19, 844–855. [DOI] [PubMed] [Google Scholar]
- Zhou J.D., Lin J., Zhang T.J., et al. (2018). Hypomethylation-mediated H19 overexpression increases the risk of disease evolution through the association with BCR–ABL transcript in chronic myeloid leukemia. J. Cell. Physiol. 233, 2444–2450. [DOI] [PubMed] [Google Scholar]