

Abstract
Background
Long noncoding RNAs (lncRNAs) play an important role in diverse biological processes and have been widely studied in recent years. However, the roles of lncRNAs in leaf pigment formation in ginkgo (Ginkgo biloba L.) remain poorly understood.
Results
In this study, lncRNA libraries for mutant yellow-leaf and normal green-leaf ginkgo trees were constructed via high-throughput sequencing. A total of 2044 lncRNAs were obtained with an average length of 702 nt and typically harbored 2 exons. We identified 238 differentially expressed lncRNAs (DELs), 32 DELs and 49 differentially expressed mRNAs (DEGs) that constituted coexpression networks. We also found that 48 cis-acting DELs regulated 72 target genes, and 31 trans-acting DELs regulated 31 different target genes, which provides a new perspective for the regulation of the leaf-color mutation. Due to the crucial regulatory roles of lncRNAs in a wide range of biological processes, we conducted in-depth studies on the DELs and their targets and found that the chloroplast thylakoid membrane subcategory and the photosynthesis pathways (ko00195) were most enriched, suggesting their potential roles in leaf coloration mechanisms. In addition, our correlation analysis indicates that eight DELs and 68 transcription factors (TFs) might be involved in interaction networks.
Conclusions
This study has enriched the knowledge concerning lncRNAs and provides new insights into the function of lncRNAs in leaf-color mutations, which will benefit future selective breeding of ginkgo.
Keywords: Leaf-color mutation, Differentially expressed lncRNAs, Target genes, Functional analysis
Background
Only 1 to 2% of the total RNAs produced by eukaryotic cells during transcription are encoded to produce proteins, and the remaining RNAs are called noncoding RNAs (ncRNAs). ncRNAs play important roles in cells, such as rRNAs and tRNAs in protein synthesis, snRNAs in the splicing of nascent RNA, and microRNAs, siRNAs and piRNAs in inhibiting gene expression [1]. Among ncRNAs, there is a widely distributed class of ncRNA transcripts with lengths greater than 200 nucleotides and no protein-encoding function, named long noncoding RNAs (lncRNAs) [2–5]. Most lncRNAs are transcribed by RNA polymerase II and have a structure similar to that of mRNA, such as 5′ caps and 3′ poly (A) tails [6, 7]. According to the genomic location of lncRNAs relative to neighboring genes, lncRNAs can be divided into five classes: sense lncRNA, antisense lncRNA, intergenic lncRNA (lincRNA), intronic lncRNA, and bidirectional lncRNA [8]. Moreover, lncRNAs can be classified into signals, decoys, guides and scaffolds based on molecular function [3, 9].
LncRNAs can be dynamically expressed during differentiation, and different mature lncRNAs can be formed by polyadenylation and different alternative splicing events, allowing the same gene to form different lncRNA transcripts [10]. LncRNA is universally transcribed in eukaryotic cells and distributed in the cytoplasm, organelles and nucleus but mainly in the nucleus. LncRNA was first identified in a sequencing analysis of mice in 2002 [11]. Currently, the functional mechanisms of lncRNA in humans and animals have been studied in great depth, especially in terms of diseases. In recent years, with the continuous improvement of bioinformatics technology, including high-throughput sequencing technology and other biological technologies, research on plant lncRNAs has developed rapidly and received increasing attention. Currently, lncRNAs have been widely identified in plants such as Arabidopsis thaliana [12], Zea mays [13], Salvia miltiorrhiza [14], and Populus [15]. LncRNAs can affect a series of biological processes, such as epigenetic regulation, cell cycle regulation, cell differentiation regulation and secondary metabolite synthesis, by regulating the level of target genes [16–18].
Ginkgo (Ginkgo biloba L.) is a well-known relict plant that originates from China and has been described as a “living fossil” [19]. As a multifunctional tree species, ginkgo has important economic and medicinal values [20] and has attracted researchers’ attention with many studies have been reported on the origin and evolution, cytology, molecular biology, tree breeding and medicinal value of ginkgo [19, 21–25]. Ginkgo is also a popular ornamental species and widely cultivated worldwide [24]. However, there are few studies on its ornamental characteristics [26]. Leaf color is an important trait of ginkgo as a landscape plant. The most attractive ornamental feature of ginkgo is its golden leaves in autumn [26]. The yellow color mutant identified in ginkgo showed the phenotypic trait of yellow leaves for the entire leaf development period and had a longer foliage period than common ginkgo [27]. Thus, the mutant not only possesses an excellent ornamental value but also provides an ideal material to study the genetic control of the leaf pigment synthesis.
Previous studies have provided an understanding of the protein-coding genes involved in leaf-color mutation [26, 27], but the role of lncRNAs in the yellow-leaf mutation has rarely been reported. In this study, normal green leaves and mutant yellow leaves of ginkgo were used as research materials to investigate their regulatory mechanism of lncRNAs in leaf-color mutation. Our objectives are to (1) establish lncRNA libraries, identify and characterize the putative lncRNAs expressed; (2) construct a coexpression network for differentially expressed lncRNAs (DELs) and differentially expressed mRNAs (DEGs); (3) predict the target genes of cis- and trans-acting lncRNAs and their functions; and (4) perform correlation analysis between lncRNAs and transcription factors (TFs) in ginkgo leaves. These findings will provide a scientific foundation for further research on the potential function of leaf-color mutations and benefit the future selective breeding and cultivation of ginkgo.
Results
RNA sequencing and identification of lncRNAs in ginkgo leaves
To obtain comprehensive lncRNA transcripts (Additional file 3: data S1), we used six (three yellow-colored leaves (YL) and three normal green-colored leaves (GL)) cDNA libraries by RNA sequencing (RNA-seq) on the Illumina HiSeq X platform. After paired-end sequencing, there was a total of 493.71 M raw reads generated from GL and 484.42 M from YL (Table 1). The raw sequencing data were submitted to the Short Reads Archive (SRA) database under the accession number SRP182122. After the low-quality sequences were filtered out, approximately 931.52 M clean reads were generated from the six libraries with an average GC content of 42.89%. Among them, more than 93% of the sequences were located on the reference ginkgo genome.
Table 1.
Quality of the sequencing data
| Sample | Raw reads | Raw bases | Clean reads | Clean bases | Valid bases | Q30 | GC |
|---|---|---|---|---|---|---|---|
| Gb_GL1 | 165.33 M | 24.80 G | 155.75 M | 22.39 G | 90.30% | 92.07% | 43.17% |
| Gb_GL2 | 164.41 M | 24.66 G | 154.79 M | 22.27 G | 90.32% | 92.47% | 43.22% |
| Gb_GL3 | 163.97 M | 24.60 G | 154.84 M | 22.41 G | 91.11% | 92.79% | 42.97% |
| Gb_YL1 | 165.47 M | 24.82 G | 156.14 M | 22.38 G | 90.16% | 92.13% | 42.69% |
| Gb_YL2 | 165.07 M | 24.76 G | 156.08 M | 22.58 G | 91.19% | 92.43% | 42.55% |
| Gb_YL3 | 163.88 M | 24.58 G | 153.92 M | 22.03 G | 89.61% | 91.69% | 42.74% |
LncRNA is a class of RNA molecules that have lengths over 200 bp and no protein-coding ability. Therefore, the coding potential of transcripts is predicted to determine whether the transcripts are lncRNAs. The candidate lncRNAs were further screened by using Coding Potential Calculator (CPC) analysis, Coding-Non-Coding Index (CNCI) analysis, Protein Families (Pfam) protein domain analysis and predictor of long noncoding RNAs and messenger RNAs based on an improved k-mer scheme (PLEK) analysis methods (Fig. 1a). After the candidate lncRNAs were screened, 2044 lncRNA sequences were finally predicted (≥ 200 bp). The average length of the lncRNA sequences was 702, the N50 was 840, and the total length of all lncRNA sequences was 1,436,133. The newly predicted lncRNA length distribution indicates that the number of lncRNAs between 201 and 300 bp was at most 314, and the number of lncRNAs between 1801 and 1900 bp was only 10 (Fig. 1b). Moreover, lncRNAs can be classified into four types according to their location in relationship with known protein-coding transcripts: intergenic lncRNA (lincRNA, U), intronic lncRNA (I), antisense lncRNA (X), and sense-overlapping lncRNA (O); the results show that the number of U-type lncRNAs was the highest (1074), nearly 6.5 times that of I-type lncRNAs (Fig. 1c). The number of exons predicted in lncRNAs is shown in Fig. 1d, indicating that lncRNAs primarily harbored two exons. In addition, since some lncRNAs can act as precursors to known miRNAs in plants, we aligned miRNA precursors with the lncRNA sequences we identified. Six novel lncRNAs were identified as precursors to known miRNAs. The results showed that TCONS_00021802 had the most miRNA precursors, while TCONS_00016273 had the fewest miRNA precursors.
Fig. 1.

Summary of the characteristic long noncoding RNAs (lncRNAs) in ginkgo. a Venn diagram showing the numbers of candidate lncRNAs in ginkgo according to the Coding Potential Calculator (CPC) and Coding-Non-Coding Index (CNCI) as well the Protein Families (Pfam) and predictor of long noncoding RNAs and messenger RNAs based on an improved k-mer scheme (PLEK) databases. b LncRNA sequence length distribution. The lncRNA number is on the vertical axis, and the lncRNA length range is on the horizontal axis. c Statistical chart of lncRNA types. Types of lncRNAs on the vertical axis and number of lncRNAs on the horizontal axis. U: intergenic lncRNA; I: intronic lncRNA; X: antisense lncRNA; and O: sense-overlapping lncRNA. d Statistical graph of exon number of lncRNAs. The number of lncRNAs is on the vertical axis, and the number of exon(s) contained in lncRNAs is on the horizontal axis
LncRNAs expression level analysis
The expression levels of 2044 lncRNA transcripts were estimated via their fragments per kilobase per million reads (FPKM, Additional file 4: data S2). The results showed that these lncRNA expression levels were different, as shown in a boxplot (Fig. 2a). The average lncRNA expression levels of the YL group are significantly lower than those of the GL group (F = 34.180, p = 0.004). The results of interval expression values showed that the transcript number and distribution of gene expression values were different between YL and GL groups (Fig. 2b). Most transcripts had FPKM values greater than or equal to 10, whereas fewest number of transcripts had FPKM values between 0.5 and 1. The significance analysis results indicated that the YL group had more numbers of transcripts in FPKM 0–0.5 (F = 29.252, p = 0.006) and FPKM 0.5–1 (F = 33.155, p = 0.005), and less numbers in FPKM ≥10 (F = 23.957, p = 0.008) than the counterpart.
Fig. 2.

Long noncoding RNAs (lncRNAs) expression level analysis between GL and YL group in ginkgo. a Boxplot of fragments per kilobase per million reads (FPKM) values; b Transcript expression within each sample. The different colors of the FPKM expression distribution map represent different ranges of FPKM values
Identification and validation of DELs between GL and YL
A total of 238 DELs were screened according to the results of significance test (Additional file 5: data S3) between the GL and the YL groups. Among these DELs, 135 were significantly upregulated and 103 were significantly downregulated in the YL compared with GL group (Fig. 3). Moreover, there were more DEGs (1361, Additional file 6: data S4) than DELs. The overall distribution of DELs is depicted in a volcano plot. The red color represents the significantly upregulated lncRNAs, and the green color indicates significantly downregulated lncRNAs in the YL group (Fig. 3a). It can be seen from the heat map clustering that the same kind of samples appear in the same cluster by clustering analysis, and the genes in the same cluster may have similar biological functions (Fig. 3b).
Fig. 3.

Differentially expressed lncRNAs (DELs) in ginkgo (Gb_GL and Gb_YL). a represents the differential volcano plot, the x-axis represents log2 fold change, and the y-axis represents log10 p-value. P-value < 0.05 and |log2 fold change| > 1; b Heat map clustering results of different groups. Red indicates high long noncoding RNA (lncRNA) expression, and blue indicates low lncRNA expression
To further verify the transcriptional patterns of DELs from the RNA-seq analysis, ten DELs were randomly selected and examined using quantitative real-time PCR (qRT-PCR) at the expression level. Although the expression multiples of several DELs verified by qRT-PCR were not completely the same as those of the FPKM values, the expression levels of these 10 DELs screened from the qRT-PCR analysis were consistent with those deduced from the FPKM values (Additional file 2: Figure S1, Additional file 5: data S3). Hence, these results indicated that the transcriptomic analysis results were reproducible and reliable, and would be useful for further studies of the lncRNAs functions (especially DELs) in GL and YL of ginkgo.
DEL and DEG coexpression analysis
To further explore the functional relationship between DELs and DEGs, we constructed a coexpression network for DELs and DEGs in Fig. 4. The results showed that a total of 32 DELs and 49 DEGs constituted coexpression networks. The networks provide candidate DELs related to pigment function. Among them, TCONS_00014724 and six DEGs have a coexpression network. TCONS_00018140 and four DEGs (Gb_32501, Gb_08822, Gb_29710 and Gb_06804) exhibited coexpression patterns. The Gb_11361 gene was involved in the ko00195 photosynthesis pathway and was designated as photosystem I P700 chlorophyll a apoprotein A1 according to SwissProt. Gb_11361 was coexpressed with two DELs (TCONS_00020469 and TCONS_00023547). Most DELs and DEGs might have one-to-one coexpression patterns.
Fig. 4.

The coexpression network diagram of differentially expressed lncRNAs (DELs) and differentially expressed mRNAs (DEGs). The red arrow node represents DELs, and the green circular node represents DEGs
Functional analysis of DEL target genes
To investigate the potential function of DELs, we predicted the candidate targets of cis- and trans-acting lncRNA. For the cis-acting lncRNAs, we searched for all coding genes in the upstream and downstream 100 kb range of DELs. These genes and lncRNAs with significant coexpression intersected. The results showed that 48 DELs may regulate 72 target genes within a range of 100 kb. These genes that are genomically neighboring and coexpressed in expression patterns are likely to be regulated by lncRNAs. For trans-acting lncRNAs (Fig. 5), the results of the lncRNA and mRNA analyses indicated that there were 31 different DELs that regulated 31 different target genes. Interestingly, four different lncRNAs (TCONS_00019467, TCONS_00019468, TCONS_00019469, and TCONS_00019471) have the same target gene Gb_31006. Another trans-acting lncRNA called TCONS_00018671 has seven target genes (Fig. 5). Nine DELs demonstrated one-to-one associations with nine DEGs.
Fig. 5.

Analytical network diagram (Top50) of trans-acting differentially expressed lncRNAs (DELs) and differentially expressed mRNAs (DEGs). The red arrow node represents DELs, and the green circular node represents DEGs
To further explore the potential biological function of lncRNAs, we performed Gene Ontology (GO) annotation analysis on cis- and trans-targets of lncRNAs (Fig. 6a). GO analysis results revealed that lncRNA target genes were categorized according to biological process (BP), cellular component (CC), and molecular function (MF). The protein-chromophore linkage and ATP synthesis coupled electron transport subcategories were enriched in the BP category. In the CC category, the chloroplast thylakoid membrane subcategory was the most enriched, followed by photosystem I. The oxidoreductase activity, chlorophyll binding and quinone binding were enriched in the MF category. We also analyzed the predicted target genes of lncRNAs using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database (Fig. 6b). The most highly represented pathways included photosynthesis (ko00195) and oxidative phosphorylation (ko00190).
Fig. 6.

Enrichment analysis of lncRNA targets. a Gene Ontology (GO) enrichment analysis of long noncoding RNA (lncRNA) targets. b Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis of lncRNA targets
Correlation analysis of lncRNAs and TFs
Because TFs play important roles in the network of leaf-color mutation genes, we identified TFs including MYB-related, bHLH, HSF, and others through a correlation analysis of lncRNAs and TFs. To further reveal the potential functions of DELs, we observed multiple pairs of lncRNA-TF relationships for each lncRNA. Each lncRNA-TF relationship is the result of the enrichment of multiple mRNAs. The results showed that the whole network constituted by these connections was divided into three clusters, including one large network and two relatively small networks (Fig. 7a). In the present study, we determined that TCONS_00007638 (lncRNA) interacts with ONIVA02G03520.1 (MYB-related TF) and five other mRNAs. Seven DELs, ten mRNAs and one TF have a relatively complex interaction network (Fig. 7a). Moreover, we also focused on the TFs targeted by lncRNAs in ginkgo. A total of 8 DELs and 68 TFs have interaction networks (Fig. 7b). ONIVA02G03520.1 (MYB-related TF) and 29 different lncRNAs have an interaction network. Itr_sc000748.1_g00024.1 (MYB-related TF) and TCONS_00011938, RrC14701_p1 (HSF TF) and TCONS_00002911, and SMil_00011498-RA_Salv (bHLH TF) and TCONS_00012957 have interaction networks.
Fig. 7.

Interaction network diagrams. a Interaction networks of lncRNAs, mRNAs and transcription factors (TFs). b Interaction networks of lncRNAs and TFs. Red nodes represent TFs, green nodes represent lncRNAs, and blue nodes represent mRNAs
Discussion
LncRNAs play an important role in diverse biological processes and have been widely studied in recent years [12, 15, 28, 29]. Nevertheless, lncRNAs remain poorly understood in the context of leaf-color mutation and pigment formation in ginkgo. Leaf-color mutation is different pigment formation result characterized by a series of transitions that are coordinated by a network of interacting genes and pathways. Herein, we have constructed coexpression networks for DELs and DEGs, predicted the target genes of lncRNAs, and performed correlation functional research in leaf-color mutation leaves. Our study not only enriched the knowledge of lncRNAs but also provided new insights into the potential functions of lncRNAs in plants. These RNA-seq data might provide molecular targets that assist in the selective breeding and production of yellow-leaf ginkgo.
Bioinformatics technology for transcriptome analyses has been rapidly improved, and many lncRNAs have been identified in plants. For example, 9686 lncRNAs were found in Norway spruce [30]. Several studies have shown that lncRNAs are similar to mRNA-encoding proteins, but the number of lncRNA transcripts is lower than that of mRNA transcripts, and lncRNAs usually exist in the nucleus [31, 32]. Our study has confirmed Cui et al. [33] finding that the lncRNA expression levels in ginkgo leaves are lower and shorter in length than those of the coding genes. Similar findings were reported in previous studies [31, 32], and this conclusion was universal in plants [33]. These common factors may indicate the essential regulation of lncRNAs during growth, development and evolution [28]. In addition, ginkgo lncRNAs contained fewer exons (mostly two exons) than the coding genes, which may be responsible for the differences in their evolution and function. This result is similar to the angiosperm poplar and gymnosperm Norway spruce [30, 34]. LncRNAs are typically greater than 200 nucleotides in length, whereas only 3% of lncRNA were > 1 kb in length in this ginkgo study. A similar condition was observed in the study of lncRNAs in maize [35]. This indicates that a small number of long lncRNAs exist in plants.
RNA plays not only an auxiliary role as an intermediate carrier of genetic information but also a role in a variety of regulatory functions. LncRNA is essentially RNA, a long chain composed of nucleotides, which can affect the biological activities of eukaryotes through various mechanisms of action [5, 7]. Since the functions of lncRNAs are highly complex and diverse, unlike the mRNA sequences that can provide potential functional information, the sequence motifs of lncRNAs generally do not provide information to predict lncRNA functions [9, 32]. However, lncRNAs may regulate gene expression either in a cis- or trans-acting manner [36]. The regulatory roles of lncRNAs in gene expression were achieved through acting on the adjacent target genes, and this was known as the cis-acting process of lncRNAs [37–39]. Transposable elements can regulate adjacent gene expression as a cis-element [40]. To further analyze the lncRNA function, we have obtained 48 DELs with 72 target genes within a 100 kb range; therefore, these 72 target genes may be regulated by DELs. Because the trans-acting lncRNAs regulate gene expression at independent loci [41], 31 DELs regulated 31 different target genes in this study, which are involved in pigment formation process and may be an important reason for leaf-color mutation. In addition, several studies have shown that lncRNAs coordinate miRNAs, forming multiple feedforward pathways to regulate a range of target genes [42, 43]. For example, some lncRNAs can act as precursors for miRNAs [44]. Some studies have proposed that lncRNAs may also function as miRNA primary transcripts, targets, or target mimics, providing a new mechanism for the regulation of miRNA activity [45–47]. Thus, the identification and analysis of the correlation between lncRNAs and miRNA precursors will help elucidate regulatory processes [43]. These results will also help explore the functions of the corresponding lncRNAs [48]. We detected six novel lncRNAs as precursors to known miRNAs that were identified in this study, which lays a foundation for the subsequent study of leaf-color mutations.
LncRNA can participate in the regulation of gene expression through various mechanisms [49–51]. Several lncRNAs can regulate mRNAs by binding or interacting with their targets [28, 52]. This could be the result of the direct regulation of lncRNAs with the promoter region or other cis-regulated elements of its coexpressed protein-coding genes [53]. To investigate whether ginkgo lncRNAs have the potential to interact with sequences of their targets, we performed GO and KEGG analysis on cis- and trans-targets of lncRNAs. The results showed that the chloroplast thylakoid membrane subcategory was most enriched in the CC category and that photosynthesis pathways (ko00195) were most enriched in the KEGG pathway analysis. The chloroplast thylakoid membrane subcategory was highly enriched, which may result in a green-deficient leaf color or lead to an abnormal leaf color [54]. Chlorophyll is an important pigment related to photosynthesis, and leaf-color variations are closely related to pigment synthesis [26, 48]. This suggests that the complex mechanism of leaf-color variation requires a coordinated regulatory network of posttranscriptional gene expression [55]. Although pigment synthesis may be a multifactorial phenotypic trait, only a few pathways regulating pigment synthesis have been validated [28, 56], and our understanding of the role of lncRNA in pigment synthesis is very limited [57]. Hence, we established a substantial number of coexpression modules to reveal the regulatory relationships and functions of leaf-color mutation or pigment synthesis. These coexpression analyses implied the functional correlation of lncRNA and protein coding, especially for TFs (MYB-related and bHLH). LncRNAs interact with a myriad of genes encoding TFs [43]. A previous study indicated that the trans-acting lncRNA HID1 associates with the chromatin of the TF gene PIF3 and can repress its transcription in Arabidopsis [58]. Several studies have shown that lncRNAs can regulate the activity of TFs [59–61]. Studies have indicated that the regulatory factors involved in pigment synthesis include MYB, bHLH and WD40, among which MYB plays the most important role in regulating anthocyanin synthesis, which has been proven in apple and grape species [62, 63]. These results provides a useful source for further research on pigment formation in plants.
Conclusions
In this study, we obtained 2044 lncRNAs including 238 DELs involved in the ginkgo leaf-color mutation through high-throughput sequencing. The results showed that 48 cis-acting DELs might regulate 72 target genes, and 31 trans-acting DELs might regulate 31 different target genes. The chloroplast thylakoid membrane subcategory and photosynthesis pathways (ko00195) were most enriched in GO and KEGG analyses. In addition, 32 DELs and 49 DEGs constituted coexpression networks, and eight DELs and 68 TFs had interaction networks. This study will provide a basis for subsequent studies on the molecular biology of the leaf color of ginkgo and will also provide a reference for the study of other plants in related fields.
Methods
Plant materials and RNA sequencing
Since ginkgo is listed as “Endangered” on the red list, we first obtained permission to collect ginkgo leaves and branches. Plant materials were collected from about 150-year-old ginkgo tree in Jiujiang city, Jiangxi Province, China (29°49′ N, 116°40’E). The ginkgo leaves exhibited GL and YL phenotypes on a main branch of the tree. And YL phenotype was identified as a xantha mutant (Ginkgo biloba “Wannianjin”) by Professor Fuliang Cao. Phenotypes of GL and YL mutant also exhibited in the Additional file 2: Figure S1 of our previous research [27]. In addition, several GL and YL scions were grafted onto rootstocks in the ginkgo germplasm nursery at Nanjing Forestry University Base. These samples were fully expanded mature leaves (free from pests and diseases). Three leaves were sampled per replication with three replicates for each group in the same period. The total RNA from these leaves (GL and YL) was extracted and purified as previously described [23] with a slight modification in the Illumina sequencing platform (HiSeq™ X) used for reference transcriptome sequencing (Ginkgo genome: http://gigadb.org/dataset/100209, [64]).
Identification of lncRNAs
First, each of the reconstructed transcripts was pooled to generate a collection of transcripts representative of transcription using StringTie software [65]. Then, based on the characteristics of lncRNA, we used a strict four-step screening method to obtain candidate lncRNAs. Step 1: Cuffcompare software was used to compare the merged transcripts with reference transcripts one-by-one to clarify the location of the remaining transcripts. Transcripts with the words “I”, “U”, “X”, and “O” were retained by screening the candidate lncRNA transcripts. Step 2: Transcripts greater than 200 bp in length and harboring 2 or more exons were screened and retained. Step 3: The transcripts screened in the second step were predicted and analyzed for their coding capacity, and these transcripts with coding potential were removed. The software used for the characterization of the obtained lncRNA sequences included CPC [66] and CNCI [67] as well as the Pfam [68] and PLEK databases [69]. Step 4: For species with known lncRNAs, the lncRNA sequences obtained in the third step were aligned with the known lncRNA using BLASTN to remove the duplicate sequences. After the known lncRNA sequences were combined, quantitative analysis was performed. Otherwise, the lncRNA sequences obtained in the third step were directly used for quantitative analysis. Furthermore, the transcripts obtained for each sample were merged and compared to known transcripts using Cuffcompare. Finally, the abovementioned candidate lncRNAs were further screened by using the most widely used coding potential analysis methods, including CPC analysis, CNCI analysis, Pfam protein domain analysis, and PLEK analysis. The final lncRNA sequence statistics were obtained (Fig. 8). Moreover, lncRNAs were aligned to the Rfam database using INFERNAL, and the family of lncRNAs was annotated. BLAST was used to map lncRNAs to miRBase for potential miRNA precursors, and lncRNAs with miRNA precursor coverage greater than 90% were selected.
Fig. 8.
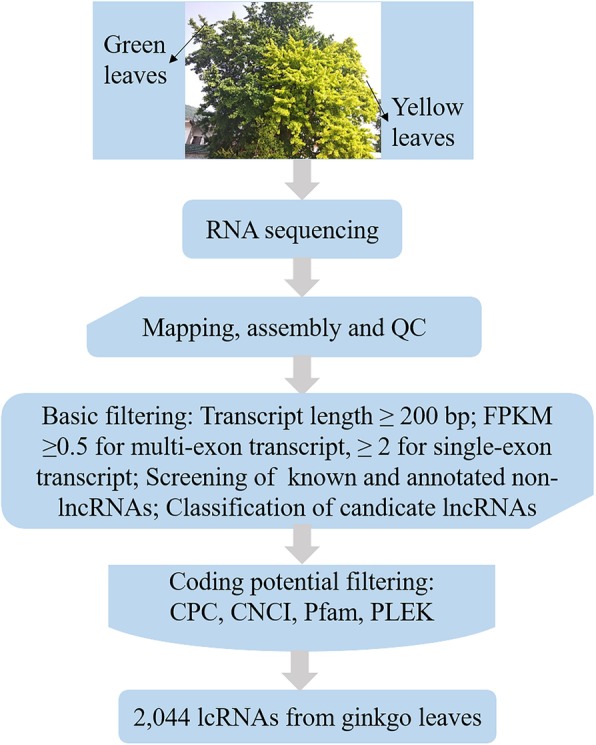
Identification of ginkgo long noncoding RNAs (lncRNAs)
Expression level annotation and DEL screening
We used a reference transcript as a library, and the abundance of the expression of each transcript in each sample was determined by the method of sequence similarity alignment. Bowtie 2 [70] and expression analysis [71] were used. The transcript expression quantity was calculated using the FPKM method [72]. The number of counts of each sample lncRNA was normalized by DESeq software [73], and the difference multiple was calculated. The difference in the number of reads was tested by negative binomial distribution test. Finally, the DELs were screened according to the different multiple and differential significance test results.
DEL and DEG coexpression analysis
Transcriptome sequencing assembly and functional annotations were performed according to the methods of Wu et al. (2018) [23]. To identify the DEGs, the two different groups were statistically compared via DESeq software [73]. Specifically, the differential expression were tested via a negative binomial distribution and a shrinkage estimator for the variance of the distribution. The false discovery rate was used as a threshold for the p-value for multiple tests to judge the significance of gene expression differences. A Pearson correlation test was used to determine the correlation between the expression data for the DELs and DEGs. The relationship pairs with a correlation coefficient greater than 0.8 and a p-value less than or equal to 0.05 were considered to have a coexpression relationship. Top50 was used to construct the coexpression network.
Target gene prediction
Because lncRNAs mainly have a cis- or trans-acting function on target genes, lncRNAs were divided into two cases to predict target genes. On the one hand, all protein-coding genes near the lncRNAs in the upstream and downstream 100 kb and significantly coexpressed with the lncRNA were screened as target genes. On the other hand, the target genes of trans-acting lncRNAs were identified by the correlation of expression levels rather than the positional relationships. These lncRNA target genes were functionally annotated using the GO (http://geneontology.org/) and KEGG (http://www.genome.jp/kegg/) databases. Based on the results of the differentially expressed coexpression analysis, lncRNA and mRNA with different chromosomes were selected as candidate targets to extract candidate sequences. The RNA interaction software RIsearch-v2.0 was used to predict the binding of candidate lncRNAs and mRNAs at the nucleic acid level. According to the screening condition, the number of bases between two nucleic acid molecules directly interacting with each other was no less than 10, and the free energy of base binding was no more than 50; the screened lncRNA and mRNA may have direct regulation.
Correlation analysis of lncRNAs and TFs
For each DEL, the coexpressed coding genes were calculated, and the significance of differential mRNA enrichment in each TF entry was calculated using the hypergeometric distribution test method. The result of the calculation returned a p-value that was significant for the enrichment. Then, the intersection of the lncRNA coexpression coding gene set and TF set was calculated. The hypergeometric distribution was used to calculate the enrichment degree of the intersection, and the TFs significantly related to lncRNAs were obtained, thereby identifying the TFs that may play a regulatory role in combination with lncRNAs.
Real-time quantitative PCR validation
We randomly selected 10 DELs from the results of the transcriptional analysis and confirmed them by qRT-PCR. All qRT-PCR experiments were performed on an ABI ViiA 7 Real-time PCR platform (Applied Biosystems, Carlsbad, CA, USA). All reactions were performed in triplicate. The PCR program was performed according to Xu et al. [74], and the glyceraldehyde-3-phosphate dehydrogenase gene (forward primer [5′-3′]: GGTGCCAAAAAGGTGGTCAT; reverse primer [5′-3′]: CAACAACGAACATGGGAGCAT) was used as a reference gene [23]. All primers for lncRNAs were designed with Oligo v6.0 software and are listed in Additional file 1: Table S1. We normalized the relative expression of the genes with the 2−ΔΔCt method [75].
Supplementary information
Additional file 1: Table S1. Primer pairs for quantitative real-time PCR.
Additional file 2: Figure S1. The expression patterns of ten lncRNAs in the leaves of Ginkgo. LncRNAs expression were analysed by quantitative real-time PCR (qRT-PCR) (a) and by their values of fragments per kilobase per million reads (FPKM) (b). The error bars represent the standard errors of the means of three independent replicates.
Additional file 3: Data S1. Full-length sequences of lncRNAs.
Additional file 4: Data S2. Gene expression.
Additional file 5: Data S3. Differentially expressed lncRNAs.
Additional file 6: Data S4. Differentially expressed mRNAs.
Acknowledgments
Not applicable.
Abbreviations
- BP
Biological process
- CC
Cellular component
- CNCI
Coding-Non-Coding Index
- CPC
Coding Potential Calculator
- DEGs
Differentially expressed mRNAs
- DELs
Differentially expressed lncRNAs
- FPKM
Fragments per kilobase per million reads
- Ginkgo
Ginkgo biloba L
- GL
Normal green-colored leaves
- GO
Gene Ontology
- I
Intronic lncRNA
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- lincRNA, U
Intergenic lncRNA
- lncRNA
long noncoding RNA
- MF
Molecular function
- ncRNAs
noncoding RNAs
- O
Sense-overlapping lncRNA
- Pfam
Protein Families
- PLEK
Predictor of long noncoding RNAs and messenger RNAs based on an improved k-mer scheme
- qRT-PCR
quantitative real-time PCR
- RNA-seq
RNA sequencing
- SRA
Short Reads Archive
- TFs
Transcription factors
- X
Antisense lncRNA
- YL
Yellow-colored leaves
Authors’ contributions
GW and FC conceived and designed the project. YW and JG participated in the data analysis. YW drafted the manuscript. TW and GW modified manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by the Special Fund for Forest Scientific Research in the Public Welfare (201504105), the Agricultural Science and Technology Independent Innovation Funds of Jiangsu Province (CX(16)1005), the National Key Research and Development Program of China (2017YFD0600700), the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX18_0954), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). The funding bodies provided the financial support to the research projects (experimental costs and publication fees), but funder didn’t involve in the experiment design, data analysis or preparation of manuscript.
Availability of data and materials
All raw data from Illumina sequencing including lncRNAs and RNA-seq data have been submitted to the Short Reads Archive database under the accession number SRP182122.
Ethics approval and consent to participate
Plant materials were collected from ginkgo tree in Jiujiang city, Jiangxi Province, China. Sampling was permitted by the Ginkgo Engineering Technology Research Center of the State Forestry Administration.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yaqiong Wu, Email: ya_qiong@126.com.
Jing Guo, Email: guojingg0921@163.com.
Tongli Wang, Email: tongli.wang@ubc.ca.
Fuliang Cao, Email: caofuliangnjfu@163.com.
Guibin Wang, Email: gbwang@njfu.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12870-019-2141-z.
References
- 1.Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73:2491–2509. doi: 10.1007/s00018-016-2174-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11:59. doi: 10.1186/1741-7007-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.St. Laurent G, Wahlestedt C, Kapranov P. The landscape of long noncoding RNA classification. Trends Genet. 2015;31:239–251. doi: 10.1016/j.tig.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamaguchi A, Abe M. Regulation of reproductive development by non-coding RNA in Arabidopsis: to flower or not to flower. J Plant Res. 2012;125:693–704. doi: 10.1007/s10265-012-0513-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu J, Fu H, Wu Y, Zheng X. Function of lncRNAs and approaches to lncRNA-protein interactions. Sci China Life Sci. 2013;56:876–885. doi: 10.1007/s11427-013-4553-6. [DOI] [PubMed] [Google Scholar]
- 6.Gibb EA, Brown CJ, Lam WL. The functional role of long non-coding RNA in human carcinomas. Mol Cancer. 2011;10:38. doi: 10.1186/1476-4598-10-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marchese FP, Raimondi I, Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017;18:206. doi: 10.1186/s13059-017-1348-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma L, Bajic VB, Zhang Z. On the classification of long non-coding RNAs. RNA Biol. 2013;10:924–933. doi: 10.4161/rna.24604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Yang L, Chen L-L. Life without a tail: new formats of long noncoding RNAs. Int J Biochem Cell Biol. 2014;54:338–349. doi: 10.1016/j.biocel.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, Jung C, Xu J, Wang H, Deng S, Bernad L, et al. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell. 2012;24:4333–4345. doi: 10.1105/tpc.112.102855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lv Y, Liang Z, Ge M, Qi W, Zhang T, Lin F, et al. Genome-wide identification and functional prediction of nitrogen-responsive intergenic and intronic long non-coding RNAs in maize (Zea mays L.) BMC Genomics. 2016;17:350. doi: 10.1186/s12864-016-2650-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li D, Shao F, Lu S. Identification and characterization of mRNA-like noncoding RNAs in Salvia miltiorrhiza. Planta. 2015;241:1131–1143. doi: 10.1007/s00425-015-2246-z. [DOI] [PubMed] [Google Scholar]
- 15.Liu S, Sun Z, Xu M. Identification and characterization of long non-coding RNAs involved in the formation and development of poplar adventitious roots. Ind Crops Products. 2018;118:334–346. doi: 10.1016/j.indcrop.2018.03.071. [DOI] [Google Scholar]
- 16.Huanca-Mamani W, Arias-Carrasco R, Cárdenas-Ninasivincha S, Rojas-Herrera M, Sepúlveda-Hermosilla G, Caris-Maldonado J, et al. Long non-coding RNAs responsive to salt and boron stress in the hyper-arid lluteño maize from Atacama desert. Genes. 2018;9:E170. doi: 10.3390/genes9030170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang T-H, Polacek N, Zywicki M, Huber H, Brugger K, Garrett R, et al. Identification of novel non-coding RNAs as potential antisense regulators in the archaeon Sulfolobus solfataricus. Mol Microbiol. 2005;55:469–481. doi: 10.1111/j.1365-2958.2004.04428.x. [DOI] [PubMed] [Google Scholar]
- 19.Zhou Z, Zheng S. The missing link in Ginkgo evolution. Nature. 2003;423:821–822. doi: 10.1038/423821a. [DOI] [PubMed] [Google Scholar]
- 20.Guo J, Wu Y, Wang B, Lu Y, Cao F, Wang G. The effects of fertilization on the growth and physiological characteristics of Ginkgo biloba L. Forests. 2016;7:293. doi: 10.3390/f7120293. [DOI] [Google Scholar]
- 21.Chassagne F, Huang X, Lyles JT, Quave CL. Validation of a 16th century traditional chinese medicine use of Ginkgo biloba as a topical antimicrobial. Front Microbiol. 2019;10:775. doi: 10.3389/fmicb.2019.00775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng J, Wang B, He G, Wang Y, Tang X, Wang S, et al. Metabolomics integrated with transcriptomics reveals redirection of the phenylpropanoids metabolic flux in Ginkgo biloba. J Agric Food Chem. 2019;67:3284–3291. doi: 10.1021/acs.jafc.8b06355. [DOI] [PubMed] [Google Scholar]
- 23.Wu Y, Guo J, Zhou Q, Xin Y, Wang G, Xu L-A. De novo transcriptome analysis revealed genes involved in flavonoid biosynthesis, transport and regulation in Ginkgo biloba. Ind Crops Products. 2018;124:226–235. doi: 10.1016/j.indcrop.2018.07.060. [DOI] [Google Scholar]
- 24.Wu Y, Zhou Q, Huang S, Wang G, Xu L-A. SNP development and diversity analysis for Ginkgo biloba based on transcriptome sequencing. Trees. 2019;33:587–597. doi: 10.1007/s00468-018-1803-z. [DOI] [Google Scholar]
- 25.Zhou Q, Mu K, Xu M, Ma X, Ni Z, Wang J, et al. Variation in the concentrations of major secondary metabolites in Ginkgo leaves from different geographical populations. Forests. 2017;8:266. doi: 10.3390/f8080266. [DOI] [Google Scholar]
- 26.Li W-X, Yang S-B, Lu Z, He Z-C, Ye Y-L, Zhao B-B, et al. Cytological, physiological, and transcriptomic analyses of golden leaf coloration in Ginkgo biloba L. Hortic Res. 2018;5:18. doi: 10.1038/s41438-018-0025-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Yu W, Wang G, Cao F, Cai J, Wang H. Comparative proteomic and physiological analysis reveals the variation mechanisms of leaf coloration and carbon fixation in a Xantha mutant of Ginkgo biloba L. Int J Mol Sci. 2016;17:E1794. doi: 10.3390/ijms17111794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feng D, Li Q, Yu H, Kong L, Du S. Transcriptional profiling of long non-coding RNAs in mantle of Crassostrea gigas and their association with shell pigmentation. Sci Rep. 2018;8:1436. doi: 10.1038/s41598-018-19950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Ai G, Zhang C, Cui L, Wang J, Li H, et al. Expression and diversification analysis reveals transposable elements play important roles in the origin of Lycopersicon-specific lncRNAs in tomato. New Phytol. 2016;209:1442–1455. doi: 10.1111/nph.13718. [DOI] [PubMed] [Google Scholar]
- 30.Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin Y-C, Scofield DG, et al. The Norway spruce genome sequence and conifer genome evolution. Nature. 2013;497:579–584. doi: 10.1038/nature12211. [DOI] [PubMed] [Google Scholar]
- 31.Kour S, Rath PC. Long noncoding RNAs in aging and age-related diseases. Ageing Res Rev. 2016;26:1–21. doi: 10.1016/j.arr.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 33.Cui J, Zhao J, Zhao J, Xu H, Wang L, Jin B. Cytological and miRNA expression changes during the vascular cambial transition from the dormant stage to the active stage in Ginkgo biloba L. Trees. 2016;30:2177–2188. doi: 10.1007/s00468-016-1443-0. [DOI] [Google Scholar]
- 34.Tian J, Song Y, Du Q, Yang X, Ci D, Chen J, et al. Population genomic analysis of gibberellin-responsive long non-coding RNAs in Populus. J Exp Bot. 2016;67:2467–2482. doi: 10.1093/jxb/erw057. [DOI] [PubMed] [Google Scholar]
- 35.Li L, Eichten SR, Shimizu R, Petsch K, Yeh C-T, Wu W, et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014;15:R40. doi: 10.1186/gb-2014-15-2-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- 37.Marquardt S, Raitskin O, Wu Z, Liu F, Sun Q, Dean C. Functional consequences of splicing of the antisense transcript COOLAIR on FLC transcription. Mol Cell. 2014;54:156–165. doi: 10.1016/j.molcel.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu X, Shi T, Iqbal S, Zhang Y, Liu L, Gao Z. Genome-wide discovery and characterization of flower development related long non-coding RNAs in Prunus mume. BMC Plant Biol. 2019;19:64. doi: 10.1186/s12870-019-1672-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L, Xia X, Jiang H, Lu Z, Cui J, Cao F, et al. Genome-wide identification and characterization of novel lncRNAs in Ginkgo biloba. Trees. 2018;32:1429–1442. doi: 10.1007/s00468-018-1724-x. [DOI] [Google Scholar]
- 40.Kunarso G, Chia N-Y, Jeyakani J, Hwang C, Lu X, Chan Y-S, et al. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet. 2010;42:631–634. doi: 10.1038/ng.600. [DOI] [PubMed] [Google Scholar]
- 41.Li S, Yu X, Lei N, Cheng Z, Zhao P, He Y, et al. Genome-wide identification and functional prediction of cold and/or drought-responsive lncRNAs in Cassava. Sci Rep. 2017;7:45981. doi: 10.1038/srep45981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Braconi C, Kogure T, Valeri N, Huang N, Nuovo G, Costinean S, et al. microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene. 2011;30:4750–4756. doi: 10.1038/onc.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Feng SJ, Zhang XD, Liu XS, Tan SK, Chu SS, Meng JG, et al. Characterization of long non-coding RNAs involved in cadmium toxic response in Brassica napus. RSC Adv. 2016;6:82157–82173. doi: 10.1039/C6RA05459E. [DOI] [Google Scholar]
- 44.Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, et al. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet. 2007;39:1033–1037. doi: 10.1038/ng2079. [DOI] [PubMed] [Google Scholar]
- 45.Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14:699–712. doi: 10.1038/nrm3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y-C, Chen Y-Q. Long noncoding RNAs: new regulators in plant development. Biochem Biophys Res Commun. 2013;436:111–114. doi: 10.1016/j.bbrc.2013.05.086. [DOI] [PubMed] [Google Scholar]
- 47.Chen M, Wang C, Bao H, Chen H, Wang Y. Genome-wide identification and characterization of novel lncRNAs in populus under nitrogen deficiency. Mol Gen Genomics. 2016;291:1663–1680. doi: 10.1007/s00438-016-1210-3. [DOI] [PubMed] [Google Scholar]
- 48.Zhang G, Chen D, Zhang T, Duan A, Zhang J, He C. Transcriptomic and functional analyses unveil the role of long non-coding RNAs in anthocyanin biosynthesis during sea buckthorn fruit ripening. DNA Res. 2018;25:465–476. doi: 10.1093/dnares/dsy017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adelman K, Egan E. More uses for genomic junk. Nature. 2017;543:183–185. doi: 10.1038/543183a. [DOI] [PubMed] [Google Scholar]
- 50.Kung JTY, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193:651–669. doi: 10.1534/genetics.112.146704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun M, Kraus WL. From discovery to function: the expanding roles of long noncoding RNAs in physiology and disease. Endocr Rev. 2015;36:25–64. doi: 10.1210/er.2014-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu J, Wang H, Chua N-H. Long noncoding RNA transcriptome of plants. Plant Biotechnol J. 2015;13:319–328. doi: 10.1111/pbi.12336. [DOI] [PubMed] [Google Scholar]
- 53.Deforges J, Reis RS, Jacquet P, Sheppard S, Gadekar VP, Hart-Smith G, et al. Control of cognate sense mRNA translation by cis-natural antisense RNAs. Plant Physiol. 2019;180:305–322. doi: 10.1104/pp.19.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li Y, Zhang Z, Wang P, Wang SA, Ma L, Li L, et al. Comprehensive transcriptome analysis discovers novel candidate genes related to leaf color in a Lagerstroemia indica yellow leaf mutant. Genes Genom. 2015;37:851–863. doi: 10.1007/s13258-015-0317-y. [DOI] [Google Scholar]
- 55.Chen L-L, Carmichael GG. Decoding the function of nuclear long non-coding RNAs. Curr Opin Cell Biol. 2010;22:357–364. doi: 10.1016/j.ceb.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ren H, Wang G, Chen L, Jiang J, Liu L, Li N, et al. Genome-wide analysis of long non-coding RNAs at early stage of skin pigmentation in goats (Capra hircus) BMC Genomics. 2016;17:67. doi: 10.1186/s12864-016-2365-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koch L. Screening for lncRNA function. Nat Rev Genet. 2017;18:70. doi: 10.1038/nrg.2016.168. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, Fan X, Lin F, He G, Terzaghi W, Zhu D, et al. Arabidopsis noncoding RNA mediates control of photomorphogenesis by red light. Proc Natl Acad Sci U S A. 2014;111:10359–10364. doi: 10.1073/pnas.1409457111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Martianov I, Ramadass A, Barros AS, Chow N, Akoulitchev A. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature. 2007;445:666–670. doi: 10.1038/nature05519. [DOI] [PubMed] [Google Scholar]
- 60.Shamovsky I, Ivannikov M, Kandel ES, Gershon D, Nudler E. RNA-mediated response to heat shock in mammalian cells. Nature. 2006;440:556–560. doi: 10.1038/nature04518. [DOI] [PubMed] [Google Scholar]
- 61.Willingham AT, Orth AP, Batalov S, Peters EC, Wen BG, Aza-Blanc P, et al. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science. 2005;309:1570–1573. doi: 10.1126/science.1115901. [DOI] [PubMed] [Google Scholar]
- 62.Chagne D, Lin-Wang K, Espley RV, Volz RK, How NM, Rouse S, et al. An ancient duplication of apple MYB transcription factors is responsible for novel red fruit-flesh phenotypes. Plant Physiol. 2013;161:225–239. doi: 10.1104/pp.112.206771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deluc L, Bogs J, Walker AR, Ferrier T, Decendit A, Merillon JM, et al. The transcription factor VvMYB5b contributes to the regulation of anthocyanin and proanthocyanidin biosynthesis in developing grape berries. Plant Physiol. 2008;147:2041–2053. doi: 10.1104/pp.108.118919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guan R, Zhao Y, Zhang H, Fan G, Liu X, Zhou W, et al. Draft genome of the living fossil Ginkgo biloba. GigaScience. 2016;5:49. doi: 10.1186/s13742-016-0154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33:290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang Y-J, Yang D-C, Kong L, Hou M, Meng Y-Q, Wei L, et al. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017;45:W12–W16. doi: 10.1093/nar/gkx428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map (SAM) format and SAMtools. Transplant Proc. 2009;19:1653–1654. [Google Scholar]
- 68.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nat Methods. 2013;10:71–73. doi: 10.1038/nmeth.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12:R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anders S, Huber W. Differential expression of RNA-Seq data at the gene level–the DESeq package. European Molecular Biology Laboratory: Heidelberg, Germany; 2012. [Google Scholar]
- 74.Xu M, Chen C, Cai H, Wu L. Overexpression of PeHKT1;1 improves salt tolerance in populus. Genes. 2018;9:475. doi: 10.3390/genes9100475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Primer pairs for quantitative real-time PCR.
Additional file 2: Figure S1. The expression patterns of ten lncRNAs in the leaves of Ginkgo. LncRNAs expression were analysed by quantitative real-time PCR (qRT-PCR) (a) and by their values of fragments per kilobase per million reads (FPKM) (b). The error bars represent the standard errors of the means of three independent replicates.
Additional file 3: Data S1. Full-length sequences of lncRNAs.
Additional file 4: Data S2. Gene expression.
Additional file 5: Data S3. Differentially expressed lncRNAs.
Additional file 6: Data S4. Differentially expressed mRNAs.
Data Availability Statement
All raw data from Illumina sequencing including lncRNAs and RNA-seq data have been submitted to the Short Reads Archive database under the accession number SRP182122.