

Abstract
Background
Glioblastoma is the most frequent and aggressive brain tumour in humans with median survival from 12 to 15 months after the diagnosis. This is mostly due to therapy resistant glioblastoma stem cells in addition to intertumour heterogeneity that is due to infiltration of a plethora of host cells. Besides endothelial cells, mesenchymal stem cells and their differentiated progenies, immune cells of various differentiation states, including monocytes, comprise resident, brain tumour microenvironment. There are compelling evidence for CCL5/CCR5 in the invasive and metastatic behaviour of many cancer types. CCR5, a G-protein coupled receptor, known to function as an essential co-receptor for HIV entry, is now known to participate in driving tumour heterogeneity, the formation of cancer stem cells and the promotion of cancer invasion and metastasis. Clinical trials have recently opened targeting CCR5 using a humanized monoclonal antibody (leronlimab) for metastatic triple negative breast cancer (TNBC) or a small molecule inhibitor (maraviroc) for metastatic colon cancer. There are important CCL5 and CCR5 structure and signalling mechanisms in glioblastoma. In addition, the CCL5/CCR5 axis directs infiltration and interactions with monocytes/macrophages and mesenchymal stem cells, comprising glioblastoma stem cell niches.
Conclusions
CCR5 is highly expressed in glioblastoma and is associated with poor prognosis of patients. CCL5/CCR5 is suggested to be an excellent new target for glioblastoma therapy. The molecular mechanisms, by which chemoattractant and receptor respond within the complex tissue microenvironment to promote cancer stem cells and tumour heterogeneity, should be considered in forthcoming studies.
Key words: cytokines, CCL5-RANTES, glioblastoma, tumour microenvironment, mesenchymal stem cells, signalling
Introduction
Brain tumours originate from various types of cells, of which gliomas are most frequent. Recent epidemiological data in UK confirmed that glioblastoma (GB) is also the most common among glial tumours with 5–7 cases per 100.000 individuals yearly, and represents 50% of all gliomas.1 The World Health Organisation (WHO) distinguishes four grades of glioma, of which GB is the most aggressive, invasive, and lethal among all types of brain tumours. According to the standard histological classification, GB originates from neoplastic glial cells, also called astrocytes, either via the de novo pathway without clinical or histologic evidence of a less malignant precursor lesions (primary glioblastoma) or via the progressive pathway through development from a low-grade astrocytoma, progressing to anaplastic astrocytoma into diffuse glioblastoma (secondary glioblastoma). The major marker of secondary glioblastoma is mutated isocitrate dehydrogenase (IDH1)2, although also expressed in the proneural subtype of primary glioblastoma. Regardless of the origin, GB is characterized by histological features, such as necrosis, vascular proliferation and pleomorphism.3 Contrary to most tumour types, irradiation and chemotherapy have proven to be ineffective to impair GB progression in longer term, demonstrating its remarkable therapeutic resistance.4 Commonly used chemotherapeutic is temozolomide (TMZ), showing the highest effectiveness in GB.3, 5 However, only in about 55–60% of patients with methylated, i.e. silenced gene coding for O6-methylguanine deoxyribonucleic acid (DNA) methyltransferase (MGMT), thereby lowering the enzyme expression, the responsiveness to TMZ is more effective.6 There is thus continuous search for new, adjuvant therapeutics, including kinase inhibitors, anti-angiogenic agents and recently also immunotherapeutics, to increase average survival of glioblastoma patients.
There are other reasons for GB therapy resistance, i.e. the presence of glioblastoma stem cells (GSCs), mostly due to their high DNA repair mechanisms expression.7 The heterogeneity and plasticity of these cells that carry the genetic fingerprint of the developing glioma, has been recognised as one of the additional obstacles for their resistance. GSCs, similar to normal neural cells, which are precursors of glial and neural cells, express the characteristic stemness genes (e.g. CD133, Sox2, Nanog, Olig 4, Notch, etc.), in addition to selected oncogenes and tumour suppressor genes.8 The initial transcriptome analyses by Philips et al 9 and Verhaak et al.10, set the basis for the Cancer Genome Atlas (TCGA), defining four different glioblastoma subtypes: the proneural (PN), mesenchymal (MES), neural (N), and classical (CL) by their major genomic characteristics, which are: PDGFRA/IDH1 (in PN), NF1/TP53 (in MES) and epidermal growth factor receptor EGFR abnormalities i.e. amplification of the epidermal growth factor (EGF) (in N) and mutation in EGFRvIII/PTEN (in CL). These GB subtypes differ significantly in survival rate, being shorter in MES subtype. However, mixed subtypes are observed in the single tumour, giving rise to intra-tumour heterogeneity.11
Tumour microenvironment
Solid tumour progression is not only relaying on the genetic and epigenetic variations of cancerous cells acquired during their evolution, but also on how their homotypic and heterotypic interactions with the stromal cells of associated microenvironment are. The “tumour microenvironment” (TME) consists not only of local, resident cells being invaded by the tumour cells, but also of infiltrating host cells, e.g. bone-marrow and blood-derived mesenchymal stem cells (MSC) and haematopoietic stem cells (HSC) and their progenitors, e.g. mature lymphocytes, macrophages, etc. Very recently, Salmon et al.12 reviewed specific determinants that might influence tumours development and argue that unrevealing these selective interactions, mediating for example tumour immunity should facilitate development of immunotherapeutic precision strategies for patients with cancer.
In glioblastoma in addition to their autonomous (inter-tumour) heterogeneity11, the increasing attention is paid to their non-autonomous heterogeneity (intra-tumour heterogeneity), that is presenting an obstacle to a more informed treatment, as we still do not understand the ability of GB cells to manipulate and exploit these non-cancerous cells collectively termed “tumour stroma”. As stated by Broekman et al.13, almost all cell types in the GB stroma are affected: the tumour is able to stimulate angiogenesis and co-opt existing vasculature, suppress immune cell functions, disarm microglia and macrophages that should recognize and fight these “foreign elements” in the brain and coerce astrocytes into supporting tumour modification extracellular matrix (ECM) to facilitate invasion. GB cells recruit innate immune cells and change their phenotype to support tumour growth and suppress adaptive immune responses.14 The increasing understanding of how T cells access the brain and how the tumour tricks the immune response, offers new strategies for mobilizing an anti-immune tumour response. For example, GB cells extensive cross-talk with microglia and infiltrating macrophages, through the release of cytokines (see below), extracellular vesicles exchange and connecting nanotubes and microtubules, results in their support to malignancy, as reviewed by Matias et al.15 Among these cells, MSC homing to GB, have crucial effects on the microenvironment, either by their de-differentiation to other stromal cells, via paracrine effectors such as immunomodulatory cytokines16, or by direct interactions with GB cells.17
Thus, GB cells spreading to the brain involve multiple modes of communication with stromal cells as extensively reviewed by Matias et al.15 The aim of this review is to reveal the complex interactions of one of the most important cytokines, affecting glioblastoma progression i.e., chemokine (C-C motif) ligand 5 (CCL5) and its receptors.
Inflammatory chemokines&cytokines in cancer
As reviewed by Balkwill18, chemokines are chemotactic cytokines that cause directed migration of stromal cells, such as leukocytes, that are induced by inflammatory cytokines. Chemokine signalling results in transcription of target genes that are involved in cell invasion, motility, interactions with the extracellular matrix (ECM) and cell survival. Chemokine signalling can coordinate cell movement during inflammation, as well as the homeostatic transport of HSCs, MSCs, myeloids cells lymphocytes, dendritic cells and neutrophils, as well as cancer-associated fibroblasts (CAFs).19 Directed migration of cells that express the appropriate chemokine receptor, occurs along a chemical gradient of ligand - known as the chemokinereceptor axis — allowing cells to move towards high local concentrations of chemokines. More than 50 human chemokines and 20 chemokine receptors have been discovered so far. Cytokines, as pro-inflammatory mediators, when excessive, also play a role in causing chronic inflammation, for example induced by bacterial (e.g. by H. pylori) or after viral (Hepatitis B) infections, inducing immune suppression.20 Various cancers have a specific complex chemokine network that influences the immune-cell recruitment of inflammatory cells to the tumour milieu, but being neutralised by cancer immune suppression, and providing an immunological privilege that enables neoplasia, i.e. tumour cell growth, survival and migration, angiogenesis and metastasis.19,21
CCL5 - RANTES and its receptors
The cytokine CCL5, also classified as C-C motif chemokine 5, has been initially termed RANTES (“Regulated upon Activation Normal T cell Expressed and Secreted”), and as a potent chemokine, attracting leukocytes.22 Later, CCL5 was recognised as a versatile inflammatory mediator, expressed by breast cancer cells (BC), where along with CCL2 it promoted pro-malignant activities by attracting macrophages, T-cells and granulocytes, as well as mesenchymal stem cells and enhancing angiogenesis.23 CCL5 has been suggested as potential therapeutic target to impair the disease progression. In immune cells, CCL5 was reported initially as a HIV-suppressive factor and expressed mainly by CD8+ T cells.24 It binds to its cognate receptor C-C chemokine receptor type 5 (CCR5), which is (alongside C-X-C chemokine receptor type 4 (CXCR4)), an HIV entry co-receptor into CD4+ T cells.25 The Food and Drug Administration approved the first CCR5-based entry inhibitor, now called maraviroc (MRV) in 2009, and based on this, new drugs that promote CCR5 and CXCR4 internalization, independent of canonical cellular signalling, provided clinical benefits for HIV patients with minor side effects.
The mode of CCL5 action thus comprises binding not only to its cognate and the most studied interacting partner, the CCR5 membrane receptor, but also to other members of the G-protein coupled receptors (GPCR), such as C-C chemokine receptor type 1 (CCR1) and C-C chemokine receptor type 3 (CCR3). In addition to that, non-conventional or auxiliary receptors of CCL5 are CD4426 and GPR75 (Figure 1A).27 This is not unusual, as many chemokine receptors display promiscuous ligand binding, meaning they have more than one high-affinity ligand.28 Such variety of CCL5 interactions causes the activation of multiple pathways and gives the ligand a diverse range of not only physiological, but also pathological functions, including in cancer.29 In this vein, CCL5 has been shown to be highly expressed in a plethora of cancer types, such as colorectal30, lung31, prostate32, breast28,33,34 and cervical cancer.35 In addition, tissue or plasma CCL5 also serve as a marker of poor prognosis, as it is the case in cervical35, prostate32, gastric36, breast37, pancreatic cancer38, as well as in glioblastoma.39 The cells that express and secrete CCL5 in cancer, can either be cancerous cells themselves or stromal cells.40
Figure 1.

Chemokine CCL5 promiscuous binding to receptors. A variety of chemokine-receptors are interacting with CCL5/CCR5 in the signalling axis: (A) in addition to the cognate receptor CCR5, CCL5 binds also to CCR3 and CCR1. Non-conventional receptors are GPR75 and CD44. (B) The chemokines which bind to CCR5 are also CCL5, CCL4 and CCL3.
The receptor CCR5, classified as C-C chemokine receptor 5, alternatively termed also CD195, is the main receptor through which CCL5 transduces signalisation.41,42 Structurally, CCR5 is a GPCR, as are many other chemokine receptors.41 This means that it has a N-terminal extracellular tail responsible for ligand binding, seven hydrophobic transmembrane regions, the six loops that connect them, and a C-terminal cytosolic tail. This is crucial for transducing the signal caused by ligand binding after its heterotrimeric G-proteins binding or through G-protein independent pathways.41
Similar to other chemokine receptors, CCR5 also acts redundantly for signalling pathways.28 High affinity ligands that bind to CCR5 are CCL5, as well as chemokine (C-C motif) ligand 3 (CCL3) (also known as MIP-1α) and chemokine (C-C motif) ligand 4 (CCL4) (also known as MIP-1β) (Figure 1B).42,43 As already mentioned, substantial research has been dedicated to the role of CCR5 in HIV infection; M-tropic or macrophage strain HIV-1 uses CD4 as its main receptor to bind to and enter CD4+ T cells, but for this it also needs co-receptors, CCR5 and CXCR4 acting as such. Small molecular inhibitors (maraviroc [MRV]) and the humanized monoclonal antibody (leronlimab)44,45 are CCR5 antagonists that inhibit HIV-1 virus from entering the T-cells.43 MRV binds to CCR5 and acts as an allosteric inverse agonist, locking CCR5 in an inactive conformation.42 However, recent research has focused more on cancer, as similarly to CCL5, CCR5 is overexpressed in many types of cancers, including breast28,33,34,46, prostate32 and in particular glioblastoma.47,48 Both maraviroc49 and leronlimab46 have been shown to potently block cancer metastasis in murine xenografts.
The impact of CCL5/CCR5 signalling in glioblastoma
The canonical (conventional) way of signalling is through a hepta-helical chemokine receptor and adjacent G proteins, more specifically their Gα subunit and Gβγ dimer.41 Upon ligand binding, CCR5 activates Gαβγ trimer by causing guanosine exchange (GDP à GTP) and the dissociation of the membrane-bound Gα subunit from the Gβγ dimer.50 Activated Gα affects adenylyl cyclase, and subsequently cellular cyclic adenosine monophosphate (cAMP) levels that activates cytosolic protein kinase A (PKA). Gα together with Gβγ affect various targets (e.g. PLCβ), resulting in the production of secondary messengers, such as inositol-1,4,5-triphosphate (IP3), diacylglycerol (DAG) and increased cytosolic calcium concentration. Both Gα and Gβγ trigger calcium influx, therefore the direct interaction of chemokine and its receptor can be confirmed by a calcium mobilisation assay.38 Influx of calcium activates calcium-dependant pathways (NF-κB), as well pathways independent of G-proteins, all favouring malignancy in one way or the other.41
The CCL5/CCR5 axis has been recently reported as a mechanism of tumour progression in pancreatic38, gastric20 and breast cancer.33, 44 Noteworthy, in cancer, the CCL5-receptors signalling can favour cancer progression directly by affecting proliferation, migration and cell survival of cancer cells, or indirectly, by affecting tumour microenvironment, i.e. by recruiting pro-tumour and/or anti-inflammatory effector cells.20, 51 The state of the art in affecting the key hallmarks of glioblastoma progression, first described by Kouno et al.47 and recent reports on MES-GB subtypes29 and glioblastoma stem cells52, 53 will be discussed below.
Direct impact on glioblastoma cells
Cancer cell proliferation is regulated by many pathways, one of them, most commonly mediated by CCL5/CCR5 signalling, is mammalian target of rapamycin mTOR) pathway. This pathway is convergent with the Phosphatidylinositol 3-kinase (PI3K) pathway, as they both activate the Akt (protein kinase B) pathway (Figure 2).54
Figure 2.
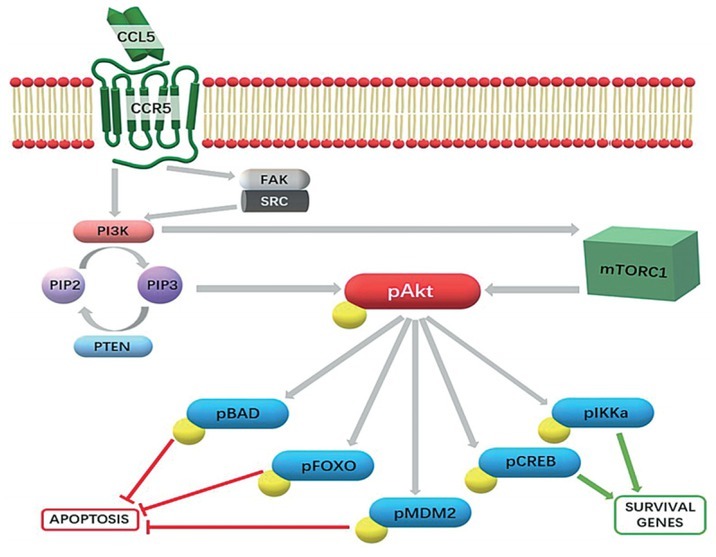
Phosphatidylinositol 3-kinase (PI3K)/pAkt-kinase pathway as a central CCL5/CCR5 signalling cascade in cancer cells. Upon CCL5 binding to its cognate receptor CCR5, primarily the PI3K/Akt pathway is triggered. This favours the phosphorylation of PIP2 to PIP3, a secondary messenger responsible for the activation of the Akt kinase, which in turn phosphorylates several downstream effectors. This causes the inhibition of pro-apoptotic effectors and the upregulation of survival genes. Another target of PI3K is the mammalian target of rapamycin complex 1 (mTORC1), which also activates the Akt kinase. However, it has also been shown that a secondary intracellular target, Focal Adhesion kinase (FAK) binding-SRC kinase complex can be activated, resulting in additional PI3K activation (in prostate cancer62).
The “mTOR is a kinase, encoded by mTOR gene MTOR and is the member of phosphatidylinositol 3 kinase (PI3K)-related kinase family that plays an important role in transcriptional activation, as it regulates the eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1). The role of 4E-BP1 is to sequester the eukaryotic translation initiation factor 4E (eIF4E), inhibiting translation. By inducing hyper phosphorylation of 4E-BP1, mTOR complex 1 (mTORC1) disables its eIF4E binding, enhancing the rate of translation.51 Binding to CCR5, CCL5 has been proven to activate the mTOR/4E-BP1 pathway, inducing the translation of a specific subset of mRNAs that have a long and highly structured 5’-UTR region, coding for cell survival and growth related onco-proteins, such as cyclin D1, c-Myc, and Dad-1.20 Indeed, CCL5/CCR5 signalling stimulated survival and proliferation of MCF-7 breast cancer cells through the mTOR/4E-BP1 pathway.55 Although similar has not directly been shown in glioblastoma, there are numerous reports on the role of the mTOR pathway in GB.56 Moreover, Khan et al.52 have shown that inhibition of mTOR complexes, mTORC1 and mTORC2, significantly increased the in vitro and in vivo sensitivity of glioblastoma stem cells to radiotherapy. The relevance of mTOR in GSCs, has been recently demonstrated by Mecca et al.54, by inhibition of mTORC1/2 in glioblastoma, causing persistent and dramatic reduction in p-Akt levels, which inhibited GSCs’ proliferation. Along these lines, Pan et al.29, demonstrated that CCL5/CCL5-receptor signalling in GB cells created an autocrine signalling circuit, important for high-grade glioma growth. Interestingly, they found that increased CCL5 expression was restricted to both human and mouse MES-GB, subtype characterized by NF1 protein (neurofibromin). This protein negatively regulated CCL5 expression through suppression of AKT/mTOR signalling. Zhao et al.56 also reported that GB cell proliferation is mediated through CCR5 signalling. Using BrdU incorporation in vitro in the GB cells U87 and U251, they determined that CCL5 stimulation significantly enhanced proliferation. In their experiments, CCL5/CCR5 axis activation triggered the PI3K/Akt pathway to promote proliferation, whereas PI3K inhibitors decreased Akt phosphorylation, which in turn decreased proliferation. However, both mTOR and PI3K are known to activate the Akt pathway, but the mutual relation of these two pathways is not clear. Further studies in GB are urgently needed due to the notions that most aggressive MES-GB and GSCs are affected by CCL5/CCR5 mediated treatment.
Migration and invasion
Cell migration along or through 3D extracellular matrix (ECM) is fundamental to normal tissue formation and regeneration, stem cells and immune cells trafficking, and cancer cell invasion and metastasis.57 As in various cancer cell types, migratory glioblastoma cell acquire mesenchymal type of movement58, where invasion rates are governed by the capacity of cells to induce a proteolytic cascade. This includes metalloproteases (MMPs), plasminogen and its activators as well as cathepsins59 and integrin- actomyosin mediated mechano-coupling. The process starts with cell polarisation of the actin cytoskeleton, enabling directional movement of the migrating cell. By forming frontal protrusions that activate integrin receptors, the cells are attached to the ECM integrins. Intracellularly, this triggers activity of small cytosolic GTPase proteins, RhoG, Cdc42 and Rac, which are essential in coordinating these processes58 and thereby metastasis in vivo.60 Monomeric G-actin polymerises into F-actin filaments, resulting in actomyosin contraction and subsequently migration, which is linked to FAK intracellular signalling and subsequent activation of PI3K at the leading edge.
Early studies showed the importance of CCR5 in the invasion of breast and prostate cancer cells.61 In human lung cancer cells, CCL5/CCR5 activation augments the migratory ability by increasing their surface expression of αvβ3 integrin31 and phosphorylation of PI3K/Akt kinases. Further, the authors have shown that by PI3K/Akt inhibitors or transfection of the CCL5 treated cells by mutant PI3K and Akt, lead to a decrease in αvβ3 integrin expression and migration. CCL5/CCR5 activation of PI3K/Akt signalling also activated IKKα/β and NF-κB, again enhancing αvβ3 integrin expression and migration.31 Presumably, the major mechanisms of CCL5 and other chemokines activation involves the PI3K-γ isoform through the Gβγ dimer of the G-protein, which is coupled with the CCR5 receptor.51
Actin polymerisation as a result of CCL5 activation of CCR5 was observed also in pancreatic cancer38 and recently in breast cancer epithelial cell lines34, enhancing migration by a PI3K/Akt pathway. In GB cell lines U87 and U251, CCL5 stimulation enhanced their migratory ability.56 After treatment of U87 cells with a PI3K inhibitor and CCR5 siRNA, inhibition of Akt phosphorylation was demonstrated in CCL5-treated cells and significant inhibition of growth was observed in U87 glioma xenografts in mouse model. Finally, high CCR5 expression in MES- GB was correlated with high p-Akt expression in patients’ samples. We have shown that GB cell invasion is triggered by intracellular cathepsin B62, followed by its activation of plasminogen system63 and finally activating executive metalloproteases of which MMP-9 is directly degrading EMC, and the latter was also downregulated by downregulated CCR5-PI3K/ Akt signalisation.56 Wang et al.64 reported that hypoxia, frequently found in GB, also induced CCR5 expression in U87 cells in vitro. This lead to an increase in matrix metalloproteinase-9 (MMP-9) expression and secretion and enhanced GB cancer cell invasion. The activation of CCR5 by hypoxia is undoubtedly one of the important mechanisms for enhancing cell migration via CCR5, but not the only one, as hypoxia simultaneously activates numerous signalling pathways.
Cell survival
Tumour’s maintenance of cancer cell survival is a necessity for its progression. This is achieved by overexpression of DNA repair and/or by increasing the apoptotic threshold to avoid cancer cell death. CCR5 signalling promotes breast cancer cell survival in both ways34, but in glioblastoma the CCL5/CCR5 activation mostly affects apoptosis. In human breast cancer, high CCR5 expression correlates with poor outcome, as recently reported by Jiao et al.34 In vitro, reintroduction of CCR5 expression into CCR5-negative breast carcinoma BCa SUM-159 cells that were irradiated, lower level of DNA damage marker γH2AX was demonstrated, indicating an increased DNA repair vs. CCR5 negative cells. CCR5-expressing BCa raised more metastases in animal model. Single cell analysis revealed that CCR5 governs PI3K/Akt and cell survival signalling. The drug maraviroc dramatically enhanced cell killing effect, mediated by DNA-damaging chemotherapeutic agent doxorubicin. As CCR5 augments DNA repair, the CCR5 inhibitors may enhance the tumour-specific treatments, allowing for lower doses of standard chemotherapy and radiation.
In GB, Pan et al.29 reported that GB cell survival also involves CCL5 signalling, although interestingly not by binding to CCR5, but to an auxiliary receptor CD44, and in an autocrine manner triggers signalisation that inhibits apoptosis. In the MES-GB subtype, CCL5 expression was shown to be increased and consistently with its role as a GB growth regulator, CCL5 knockdown in MES-GB cells reduced their survival in vitro, and increased mouse GB survival in vivo. Noteworthy, these authors demonstrated that CCL5 operates via CD44, activating the effector caspase-3 to inhibit apoptosis of MES-GB cells.
Impact on tumour microenvironment
Immunosuppression
Tumour-induced immunosuppression involves recruitment of different cells forming tumour microenvironment, such as tumour infiltrating lymphocytes (TIL), myeloid-derived suppressor cells (MDSCs), innate lymphoid cells, mesenchymal stem cells (MSC), immature dendritic cells (IDC) and tumour-associated macrophages (TAM), many of these cells expressing CCR5 and its ligand CCL5.46,48,65 TAMs actually comprise as two ontogenetically distinct subsets, microglia and glioblastoma infiltrated macrophages (MDMs) derived from monocyte are representing about 30% of all cells in glioblastoma.66,67 The difference between MDMs and microglia is also reflected in cytokine gene expression.65 Microglia mediated immuno-suppression dwells also on the CCL5/CCR5 and effect of CCR5 signalling on TAM activation (polarization) and GB progression has been investigated by Laudati et al.48 The main finding reveals that the functional relationship exists between the chemokine-CCR5 system and microglia polarization. Overall, the pharmacological blockade of CCR5 prevents the occurrence of a M2 anti-inflammatory microglia state (Figure 3).
Figure 3.
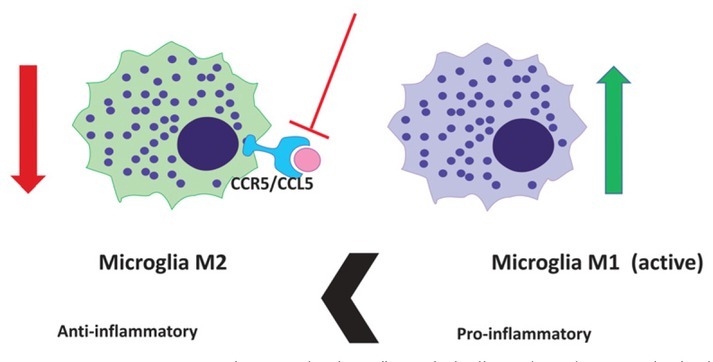
CCL5-CCR5 system and microglia polarization. The pharmacological blockade of CCR5 with maraviroc prevents the activity of glioblastoma-associated anti-inflammatory microglia M2 phenotype in and induces (green arrow) the conversion to prevailing pro-inflammatory M1 microglia phenotype.
Furthermore, under conditions mimicking the late stage of glioma pathology, CCR5 blockade thus induces a prevailing M1 pro-inflammatory state. Such changes in microglia polarization profile are potentially associated with cytotoxic and anti-tumour properties, which leads to a potential reduction in tumour growth (Figure 3), as emphasised by Jiao et al.46 Taken together, these changes suggest a possible clinical exploitation of CCR5 antagonists in the treatment of human GB. However, it has been shown in vivo, that the nature of microglia and TAM is not as simple binary as the M1/ M2 paradigm predicts. TAM phenotypes are much more complex to distinguish in the context of human pathology, compared to in vitro experiments by immunosuppressive myeloid cells (IMC) of both monocytic and granulocytic lineages.68 Ban et al.69 demonstrated that the absence of the autocrine CCL5 abrogated the generation of granulocytic myeloid-derived suppressor cells and tumorassociated macrophages. In parallel, enhanced maturation of intra-tumoral neutrophils and macrophages occurred in spite of tumor-derived CCL5. Maraviroc was used to block the CCL5/ CCR5 causing a reprograming of MDMs: initially they expressed anti-inflammatory effectors, but after maraviroc treatment they also underwent repolarisation and expressed significantly more pro-inflammatory mediators. Targeting the host CCL5 in bone marrow via nanoparticle-delivered expression silencing in combination with araviroc, resulted in robust immunities, suggesting that the myeloid CCL5/CCR5 axis is an excellent target for cancer immunotherapy.69
The concept of hierarchical tumour evolution from cancer stem cells is now widely accepted and proven also in GB.7 GSCs and their progenies, represent the final obstacles for therapy failure, as these represent the most resistant cell phenotype in the GB. These cells, although a minority of total cancer cell populations, define the functional progression of GB by expressing a panel of stemless markers and cell damage resistance genes. Besides molecular set up of these cells’ subpopulation, also the micro environmental cues contribute to their resistance by stromal cells’ protection in the so-called tumour tissue niches. We have recently shown the cellular and functional features of GB niches around a fraction of arterioles by immuno-histochemistry.70 Besides the crucial endothelial-GSCs paracrine interactions, maintained mainly by CCL12-CXCR4 axis, other interactions with resident mesenchymal stem cells (MSCs) are plausible, and may be maintained by CCL5/CCR5 axis as shown on Figure 4 (unpublished data).
Figure 4.

CCL5 and CCR5 in Glioblastoma microenvironment. (A) Fluorescence immunohistochemical labelling of CCR5 in glioblastoma associated macrophages. Macrophages in the tumour section were immuno-fluorescently labelled to detect antigen CD68 (marker for macrophages), as well as CCR5 expressions in glioblastoma tissue samples of glioblastoma patient. Nuclei were stained with DAPI (blue), CD68 with Alexa Fluor 546 (red) and CCR5 with Alexa Fluor 488 (green) dye. CCR5 is expressed in macrophages, shown in yellow in merged pictures. 40x magnification was used. (B) CCL5 is expressed in proximity to MSC on glioblastoma tissue slide. Immunofluorescence images of GB tissue slides, labelled for CD105 marker of Mesenchymal stem cells (green positive cells) and CCL5 (red). We observe selective localisation of CCL5 in glioblastoma cells and colonalization in the fraction of mesenchymal stem cells, expressing CCL5. This indicates the involvement of MSCs in the CCL5/CCR5 signalling in glioblastoma. CCL5 was also expressed in glioblastoma cells.
CCL5/CCR5 mediated cell-cross talk in glioblastoma
The basic question when investigating chemokine paracrine signalling is what attracts what, or when considering autocrine CCL5/CCR5 loop also, what activates what. Typical examples of the dilemma are numerous heterotypic interactions among heterogeneous glioblastoma cell subtypes, and stromal cells as listed above, microglia, infiltrating macrophages, lymphocytes, neutrophils, MSCs, neurons and neural stem cells, endothelial cells, etc. By categorically studying bilateral ligand and receptor expression by cell types, the mechanisms of individual cell types in CCL5/CCR5 signalling in GB may be elucidated. We are still far from being able to interpret complex multiple interactions under in vivo conditions. There are three types of situations, considering the trigger of signalling activation is the ligand CCL5.
External activation of receptor CCR5 expressing glioblastoma cells
The most commonly observed situation in CCL5/CCR5 signalling is activating host CCR5, expressed by differentiated glioblastoma cells, by stromal cells such as TAM, as discussed above and by Wang et al.64 As GB cells highly express CCR5, being activated by adding macrophage conditioned media, containing CCL5, which was even over-expressed under hypoxic conditions65, resulted in enhanced GB cell invasion. Another study71 observed microglia specific activation of growth of Neurofibromatosis 1 glioma cells, presumably (MES-GB) expressing CCR5. RNA-sequencing of microglia cells revealed CCL5 to be highly expressed. Its functionality was determined by CCL5 neutralising antibodies that also reduced glioma growth in in vivo murine model. Taken together, stromal cells activation via tumour CCR5 is a common functional mode of the CCL5/CCR5 axis in GB and presents a potential therapy target.
Activating stroma by ligand CCL5 expressing glioblastoma cells
Another way of heterotypic cellular cross-talk in glioblastoma is via secreted CCL5 by GB cells. This ligand affects infiltrating or stromal cells that express CCR5, thus affecting their intracellular signalisation that results for example in the immunosuppression of the GB microenvironment, as has been discussed above in chapter Immunosuppression. Besides modifying macrophages, T-reg lymphocytes, expressing CCR5, are recruited effectors of GB, known to be important players in immunosuppression.72 However, T-reg recruitment in GB in relation to CCL5/CCR5 signalling has been poorly studied so far. Similarly, as discussed above48, host microglia express CCR5 and rely on tumour (GB) derived CCL5 to maintain anti-inflammatory properties and migrate to attracting GB tissues. These results confirm the hypothesis of Kouno J. et al. that CCR5 ligands are overexpressed in GB in order to attract effector cells that modulate local immunity.47
Autocrine activation of glioblastoma cells, expressing both CCL5 and CCR5
Another hypothesis by Kouno et al. was that the upregulation of CCR5 and its ligand in glioblastoma serves to facilitate an autocrine system that enhances GB proliferation.47 This means that GB cells express both ligand and receptor, and thus activate the pathways downstream of CCR5 in a cell autonomous manner, as also suggested by Pan et al.29 This cross-talk via CCL5/CCR5 in cancer is also known for other cancers, indeed expressing both, the receptor and the ligand. In osteosarcoma cells, Wang et al.73, have shown that cells expressing both, CCL5/CCR5, regulate the VEGF expression, as a result of the autocrine CCL5/CCR5 activation attracts endothelial progenitor cells (EPC), contributing to tumour angiogenesis and subsequently, malignancy.
In MES-GB subtype, the autocrine CCL5/CCR5 activation loop has been examined as well by adding CCL5. Because no significant difference between wild-type tumours and those with additional stromal CCL5 was noticed, they concluded that CCL5 promotes survival and proliferation of the cells in a cell-autonomous and autocrine manner. Low grade gliomas seem to rely on stromal chemokine stimulation, whereas high grade gliomas (GB), establish autocrine chemokine stimulation. An interesting interpretation of this is that the ability of autocrine activation grants gliomas for relative stromal independency and this in turn causes the stromal cells to have less control over the regulation of the tumour leading in tumour malignancy.29
Acknowledgements
This work was supported by Slovenian Research Agency Programme P1-0245 (to T.T.L.) and by the European Program of Cross-Border Cooperation for Slovenia-Italy Interreg TRANS-GLIOMA (T.T.L.).
References
- 1.Philips A, Henshaw DL, Lamburn G, O’Carroll MJ. Brain tumours: rise in glioblastoma multiforme incidence in England 1995-2015 suggests an adverse environmental or lifestyle factor. J Environ Public Health. 2018:2170208. doi: 10.1155/2018/7910754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molenaar RJ, Maciejewski JP, Wilmink JW, Van Noorden CJF. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene. 2018;37:1949–60. doi: 10.1038/s41388-017-0077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20. doi: 10.1007/s00401-016-1545-1. et al. [DOI] [PubMed] [Google Scholar]
- 4.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. et al. [DOI] [PubMed] [Google Scholar]
- 5.Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1100–8. doi: 10.1016/S1470-2045(14)70379-1. et al. [DOI] [PubMed] [Google Scholar]
- 6.Hegi ME, Genbrugge E, Gorlia T, Stupp R, Gilbert MR, Chinot OL. MGMT promoter methylation cutoff with safety margin for selecting glioblastoma patients into trials omitting temozolomide: a pooled analysis of four clinical trials. Clin Cancer Res. 2018;25:1809–16. doi: 10.1158/1078-0432.ccr-18-3181. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–17. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin. 2010;60:166–93. doi: 10.3322/caac.20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–73. doi: 10.1016/j.ccr.2006.02.019. et al. [DOI] [PubMed] [Google Scholar]
- 10.Verhaak RGW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teng J, da Hora CC, Kantar RS, Nakano I, Wakimoto H, Batchelor TT. Dissecting inherent intratumor heterogeneity in patient-derived glioblastoma culture models. Neuro Oncol. 2017;19:820–32. doi: 10.1093/neuonc/now253. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salmon H, Remark R, Gnjatic S, Merad M. Host tissue determinants of tumour immunity. Nat Rev Cancer. 2019;19:215–27. doi: 10.1038/s41568-019-0125-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broekman ML, Maas SLN, Abels ER, Mempel TR, Krichevsky AM, Breakefield XO. Multidimensional communication in the microenvirons of glioblastoma. Nat Rev Neurol. 2018;14:482–95. doi: 10.1038/s41582-018-0025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH. The immune landscape of cancer. Immunity. 2018;48:812–30. doi: 10.1016/j.immuni.2018.03.023. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matias D, Balça-Silva J, da Graça GC, Wanjiru CM, Macharia LW, Nascimento CP. Microglia/astrocytes-glioblastoma crosstalk: crucial molecular mechanisms and microenvironmental factors. Front Cell Neurosci. 2018;12:1–22. doi: 10.3389/fncel.2018.00235. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Motaln H, Koren A, Gruden K, Ramšak Ž, Schichor C, Lah TT. Heterogeneous glioblastoma cell cross-talk promotes phenotype alterations and enhanced drug resistance. Oncotarget. 2015;6:40998–1017. doi: 10.18632/oncotarget.5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oliveira MN, Pillat MM, Motaln H, Ulrich H, Lah TT. Kinin-B1 receptor stimulation promotes invasion and is involved in cell-cell interaction of co-cultured glioblastoma and mesenchymal stem cells. Sci Rep. 2018;8:1299. doi: 10.1038/s41598-018-19359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:240–50. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 19.Lazennec G, Richmond A. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol Med. 2010;16:133–44. doi: 10.1016/j.molmed.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aldinucci D, Casagrande N. Inhibition of the CCL5/CCR5 axis against the progression of gastric cancer. Int J Mol Sci. 2018;19:1477. doi: 10.3390/ijms19051477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ben-Baruch A.. Inflammation-associated immune suppression in cancer: the roles played by cytokines, chemokines and additional mediators. Sem Cancer Biology. 2006;16:38–52. doi: 10.1016/j.semcancer.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 22.Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347:669–71. doi: 10.1038/347669a0. [DOI] [PubMed] [Google Scholar]
- 23.Soria G, Ben-Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008;267:271–85. doi: 10.1016/j.canlet.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 24.Cocchi F, Tresoldi E, Björndal A, Fredriksson R, Colognesi C, Deng HK. Identification of RANTES, MIP-1α, and MIP-1β as the major HIV-suppressive factors produced by CD8+T cells. Science. 1995;270:1811–5. doi: 10.1126/science.270.5243.1811. et al. [DOI] [PubMed] [Google Scholar]
- 25.Alkhatib G. The biology of CCR5 and CXCR4. Curr Opin HIV AIDS. 2009;4:96–103. doi: 10.1097/COH.0b013e328324bbec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roscic-Mrkic B, Fischer M, Leemann C, Manrique A, Gordon CJ, Moore JP. RANTES (CCL5) uses the proteoglycan CD44 as an auxiliary receptor to mediate cellular activation signals and HIV-1 enhancement. Blood. 2003;102:1169–77. doi: 10.1182/blood-2003-02-0488. et al. [DOI] [PubMed] [Google Scholar]
- 27.Liu B, Hassan Z, Amisten S, King AJ, Bowe JE, Huang GC. The novel chemokine receptor, G-protein-coupled receptor 75, is expressed by islets and is coupled to stimulation of insulin secretion and improved glucose homeostasis. Diabetologia. 2013;56:2467–76. doi: 10.1007/s00125-013-3022-x. et al. [DOI] [PubMed] [Google Scholar]
- 28.Velasco-Velazquez M, Xolalpa W, Pestell RG. The potential to target CCL5/CCR5 in breast cancer. Expert Opin Ther Targets. 2014;18:1–11. doi: 10.1517/14728222.2014.949238. [DOI] [PubMed] [Google Scholar]
- 29.Pan Y, Smithson LJ, Ma Y, Hambardzumyan D, Gutmann DH. Ccl5 establishes an autocrine high-grade glioma growth regulatory circuit critical for mesenchymal glioblastoma survival. Oncotarget. 2017;8:32977–89. doi: 10.18632/oncotarget.16516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cambien B, Richard-Fiardo P, Karimdjee BF, Martini V, Ferrua B, Pitard B. CCL5 neutralization restricts cancer growth and potentiates the targeting of PDGFRβ in colorectal carcinoma. PLoS One. 2011;6:e28842. doi: 10.1371/journal.pone.0028842. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang CY, Fong YC, Lee CY, Chen MY, Tsai HC, Hsu HC. CCL5 increases lung cancer migration via PI3K, Akt and NF-κB pathways. Biochem Pharmacol. 2009;77:794–803. doi: 10.1016/j.bcp.2008.11.014. et al. [DOI] [PubMed] [Google Scholar]
- 32.Vaday GG, Peehl DM, Kadam PA, Lawrence DM. Expression of CCL5 (RANTES) and CCR5 in prostate cancer. Prostate. 2006;66:124–34. doi: 10.1002/pros.20306. [DOI] [PubMed] [Google Scholar]
- 33.Pervaiz A, Zepp M, Mahmood S, Ali DM, Berger MR, Adwan H. CCR5 blockage by maraviroc: a potential therapeutic option for metastatic breast cancer. Cellular Oncology. 2018;42:93–106. doi: 10.1007/s13402-018-0415-3. [DOI] [PubMed] [Google Scholar]
- 34.Jiao X, Velasco-Velázquez MA, Wang M, Li Z, Rui H, Peck AR. CCR5 Governs DNA damage repair and breast cancer stem cell expansion. Cancer Res. 2018;78:1657–71. doi: 10.1158/0008-5472.CAN-17-0915. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niwa Y, Akamatsu H, Niwa H, Sumi H, Ozaki Y, Abe A. Correlation of tissue and plasma RANTES levels with disease course in patients with breast or cervical cancer. Clin Cancer Res. 2001;7:285–9. doi: 10.1158/1078-0432.ccr-06-0074. [DOI] [PubMed] [Google Scholar]
- 36.Sugasawa H, Ichikura T, Kinoshita M, Ono S, Majima T, Tsujimoto H. Gastric cancer cells exploit CD4+ cell-derived CCL5 for their growth and prevention of CD8+ cell-involved tumor elimination. Int J Cancer. 2008;122:2535–41. doi: 10.1002/ijc.23401. doi: 10.1002/ijc.23401. et al. [DOI] [PubMed] [Google Scholar]
- 37.Yaal-Hahoshen N, Shina S, Leider-Trejo L, Barnea I, Shabtai EL, Azenshtein E. The chemokine CCL5 as a potential prognostic factor predicting disease progression in stage II breast cancer patients. Clinical Cancer Res. 2006;12:4474–80. doi: 10.1158/1078-0432.CCR-06-0074. et al. [DOI] [PubMed] [Google Scholar]
- 38.Sushil KS, Mishra MK. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci Rep. 2018;8:1323. doi: 10.1038/s41598-018-19643-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pham K, Luo D, Liu C, Harrison JK. CCL5, CCR1 and CCR5 in murine glioblastoma: Immune cell infiltration and survival rates are not dependent on individual expression of either CCR1 or CCR5. J Neuroimmunol. 2012;246:10–7. doi: 10.1016/j.jneuroim.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borsig L, Wolf MJ, Roblek M, Lorentzen A, Heikenwalder M. Inflammatory chemokines and metastasis-tracing the accessory. Brit Dental J. 2014;33:3217–24. doi: 10.1038/onc.2013.272. [DOI] [PubMed] [Google Scholar]
- 41.Oppermann M. Chemokine receptor CCR5: Insights into structure, function, and regulation. Cellular Signalling. 2004;16:1201–10. doi: 10.1016/j.cellsig.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 42.Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–63. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: Positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- 44.Kaplon H, Reichert JM. Antibodies to watch in 2019. MAbs. 2019;11:219–38. doi: 10.1080/19420862.2018.1556465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dhody K, Pourhassan N, Kazempour K, Green D, Badri S, Mekonnen H. PRO 140, a monoclonal antibody targeting CCR5, as a long-acting, single-agent maintenance therapy for HIV-1 infection. HIV Clin Trials. 2018;19:85–93. doi: 10.1080/15284336.2018.1452842. et al. [DOI] [PubMed] [Google Scholar]
- 46.Jiao X, Nawab O, Patel T, Kossenkov AV, Halama N, Jaeger D. Recent advances targeting CCR5 for cancer and its role in immuno-oncology. Cancer Res Cancers. 2019;179:4801–7. doi: 10.1158/0008-5472.CAN-19-1167. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kouno J, Nagai H, Nagahata T, Onda M, Yamaguchi H, Adachi K. Up-regulation of CC chemokine, CCL3L1, and receptors, CCR3, CCR5 in human glioblastoma that promotes cell growth. J Neurooncol. 2004;70:301–7. doi: 10.1007/s11060-004-9165-3. et al. [DOI] [PubMed] [Google Scholar]
- 48.Laudati E, Currò D, Navarra P, Lisi L. Blockade of CCR5 receptor prevents M2 microglia phenotype in a microglia-glioma paradigm. Neurochem Int. 2017;108:100–8. doi: 10.1016/j.neuint.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 49.Velasco-Velazquez M, Jiao X, De La Fuente M, Pestell TG, Ertel A, Lisanti MP. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 2012;72:3839–50. doi: 10.1158/0008-5472.CAN-11-3917. et al. [DOI] [PubMed] [Google Scholar]
- 50.Peng WT, Sun WY, Li XR, Sun JC, Du JJ, Wei W. Emerging roles of G protein-coupled receptors in hepatocellular carcinoma. Int J Mol Sci. 2018;19 doi: 10.3390/ijms19051366. pii: E1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murooka TT, Rahbar R, Platanias LC, Fish EN. CCL5-mediated T-cell chemotaxis involves the initiation of mRNA translation through mTOR/4E-BPl. Blood. 2008;111:4892–901. doi: 10.1182/blood-2007-11-125039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kahn J, Hayman TJ, Jamal M, Rath BH, Kramp T, Camphausen K. The mTORC1/mTORC2 inhibitor AZD2014 enhances the radiosensitivity of glioblastoma stem-like cells. Neuro Oncol. 2014;16:29–37. doi: 10.1093/neuonc/not139. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mecca C, Giambanco I, Bruscoli S, Bereshchenko O, Fioretti B, Riccardi C. PP242 counteracts glioblastoma cell proliferation, migration, invasiveness and stemness properties by inhibiting mTORC2/AKT. Front Cell Neurosci. 2018;12:99. doi: 10.3389/fncel.2018.00099. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mecca C, Giambanco I, Donato R, Arcuri C. Targeting mTOR in glioblastoma: rationale and preclinical/clinical evidence. Dis Markers. 2018;18:1–10. doi: 10.1155/2018/9230479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Murooka TT, Rahbar R, Fish EN. CCL5 promotes proliferation of MCF-7 cells through mTOR-dependent mRNA translation. Biochem Biophys Res Commun. 2009;387:381–6. doi: 10.1016/j.bbrc.2009.07.035. [DOI] [PubMed] [Google Scholar]
- 56.Zhao L, Wang Y, Xue Y, Lv W, Zhang Y, He S. Critical roles of chemokine receptor CCR5 in regulating glioblastoma proliferation and invasion. Acta Biochim Biophys Sin. 2015;47:890–8. doi: 10.1093/abbs/gmv095. [DOI] [PubMed] [Google Scholar]
- 57.Wolf K, Friedl P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 2011;21:746–8. doi: 10.1016/j.tcb.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 58.Friedl P, Wolf K. Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–74. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 59.Lah TT, Duran Alonso MB, Van Noorden CJF. Antiprotease therapy in cancer: hot or not? Expert Opin Biol Ther. 2006;6:257–79. doi: 10.1517/14712598.6.3.257. [DOI] [PubMed] [Google Scholar]
- 60.Bouzahzah B, Albanese C, Ahmed F, Pixley F, Lisanti MP, Segall JD. Rho family GTPases regulate mammary epithelium cell growth and metastasis through distinguishable pathways. Mol Med. 2001;7:816–30. et al. [PMC free article] [PubMed] [Google Scholar]
- 61.Sicoli D, Jiao X, Ju X, Velasco-Velazquez M, Ertel A, Addya S. CCR5 receptor antagonists block metastasis to bone of v-Src oncogene-transformed metastatic prostate cancer cell lines. Cancer Res. 2014;74:7103–14. doi: 10.1158/0008-5472.CAN-14-0612. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gole B, Huszthy PC, Popović M, Jeruc J, Ardebili YS, Bjerkvig R. The regulation of cysteine cathepsins and cystatins in human gliomas. Int J Cancer. 2012;131:1779–89. doi: 10.1002/ijc.27453. et al. [DOI] [PubMed] [Google Scholar]
- 63.Colin C, Voutsinos-Porche B, Nanni I, Fina F, Metellus P, Intagliata D. High expression of cathepsin B and plasminogen activator inhibitor type-1 are strong predictors of survival in glioblastomas. Acta Neuropathol. 2009;118:745–54. doi: 10.1007/s00401-009-0592-2. et al. [DOI] [PubMed] [Google Scholar]
- 64.Wang Y, Liu T, Yang N Xu S, Li X, Wang D. Hypoxia and macrophages, promote glioblastoma invasion by the CCL4-CCR5 axis. Oncol Rep. 2016;36:3522–8. doi: 10.3892/or.2016.5171. et al. [DOI] [PubMed] [Google Scholar]
- 65.Müller S, Kohanbash G, Liu SJ, Alvarado B, Carrera D, Bhaduri A. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017;18:234. doi: 10.1186/s13059-017-1362-4. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matias D, Balça-Silva J, da Graça GC, Wanjiru CM, Macharia LW, Nascimento CP. Microglia/astrocytes-glioblastoma crosstalk: crucial molecular mechanisms and microenvironmental factors. Front Cell Neurosci. 2018;12:235. doi: 10.3389/fncel.2018.00235. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morisse MC, Jouannet S, Dominguez-Villar M, Sanson M, Idbaih A. Interactions between tumor-associated macrophages and tumor cells in glioblastoma: unraveling promising targeted therapies. Expert Rev Neurother. 2018;18:729–37. doi: 10.1080/14737175.2018.1510321. [DOI] [PubMed] [Google Scholar]
- 68.Ransohoff RM. A polarizing question: Do M1 and M2 microglia exist. Nature Neuroscience. 2016;19:987–91. doi: 10.1038/nn.4338. [DOI] [PubMed] [Google Scholar]
- 69.Ban Y, Mai J, Li X, Mitchell-Flack M, Zhang T, Zhang L. Targeting autocrine CCL5-CCR5 axis reprograms immunosuppressive myeloid cells and reinvigorates antitumor immunity. Cancer Res. 2017;77:2857–68. doi: 10.1158/0008-5472.CAN-16-2913. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hira VVV, Aderetti DA, van Noorden CJF. Glioma stem cell nichesn in human glioblastoma are periarteriolar. J Histochem Cytochem. 2018;66:349–58. doi: 10.1369/0022155417752676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Solga AC, Pong WW, Kim KY, Cimino PJ, Toonen JA, Walker J. RNA sequencing of tumor-associated microglia reveals Ccl5 as a stromal chemokine critical for neurofibromatosis-1 glioma growth. Neoplasia. 2015;17:776–88. doi: 10.1016/j.neo.2015.10.002. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chakraborty R, Rooney C, Dotti G, Savoldo B. Changes in chemokine receptor expression of regulatory T cells after ex vivo culture. J Immunother. 2012;35:329–36. doi: 10.1097/CJI.0b013e318255adcc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang SW, Liu SC, Sun HL, Huang TY. CCL5/CCR5 axis induces vascular endothelial growth factor-mediated tumor angiogenesis in human osteosarcoma microenvironment. Carcinogenesis. 2014;36:104–14. doi: 10.1093/carcin/bgu218. [DOI] [PubMed] [Google Scholar]