

Abstract
The processing of peptide tandem mass spectrometry data involves matching observed spectra against a sequence database. The ranking and calibration of these peptide-spectrum matches can be improved substantially by using a machine learning post-processor. Here, we describe our efforts to speed up one widely used post-processor, Percolator. The improved software is dramatically faster than the previous version of Percolator, even when using relatively few processors. We tested the new version of Percolator on a data set containing over 215 million spectra and recorded an overall reduction to 23% of the running-time as compared to the unoptimized code. We also show that the memory footprint required by these speedups is modest relative to that of the original version of Percolator.
Keywords: tandem mass spectrometry, machine learning, support vector machine, SVM, percolator
Graphical Abstract
1. Introduction
The field of proteomics is growing rapidly, driven in large part by advances in our ability to identify and quantify proteins of complex biological samples in a high throughput fashion. In particular, technological advances in tandem mass spectrometry have made full proteome measurements much easier to carry out, with the result that large data sets must be analyzed in more labs and with greater frequency than in the past. These analysis workflows typically consist of a detection phase, wherein observed spectra are assigned peptide sequences and the resulting peptide-spectrum matches are combined to draw inferences about the detection of peptides and proteins, and a subsequent quantitation phase, wherein the abundances of peptides and proteins are inferred. This work focuses on speeding up a particular algorithm, Percolator, which is used during the detection phase of tandem mass spectrometry analysis.12
1.1. Percolator
The first step in this phase of the analysis is to attempt to infer the identity of the peptide species primarily responsible for generating each observed spectrum. This task is most commonly carried out by a database search algorithm, which compares an observed spectrum to a series of theoretical spectra derived from peptides in a given database. The first database search algorithm was published in 1994,6 and dozens have been produced in the ensuing two decades. However, by the early 2000s it was becoming clear that interpreting the scores produced by any given search engine was challenging. At that time, state-of-the-art methods for attempting to separate correct from incorrect peptide-spectrum matches (PSMs) involved applying hand-selected, charge-state specific thresholds30 or using probabilistic reasoning to interpret the scores.18 Concurrently, two research groups developed machine learning methods for addressing this interpretation problem, one based on linear discriminant analysis14 and one based on support vector machines (SVMs).1 These methods provided a powerful new way to combine multiple PSM features into a single, interpretable quality measure.
Percolator, which was described several years later, is thus a second-generation method. Its primary innovation was to use semi-supervised learning to improve the generalization ability of the learning system (Figure 1). Early machine learning methods for PSM scoring suffered from poor generalizability because they were trained using hand-curated data sets, and this manual curation had to be repeated for each new experimental setting. Percolator avoids the need for manual curation by training a classifier to distinguish between real PSMs (called “target PSMs”) and decoy PSMs (i.e., shuffled or reversed peptide sequences). This task is semi-supervised because the decoy PSMs have labels (“incorrect”) but the target PSMs are an unlabeled mixture of correct and incorrect PSMs. Percolator uses an iterative semi-supervised machine learning algorithm in which the inner loop is an SVM classifier. The method is fast because Percolator makes use of an optimization algorithm specific to linear SVMs.23 A cross-validation scheme is employed to allow the target PSMs to also be used for statistical confidence estimation.9 Subsequently, Percolator was improved via the inclusion of sophisticated statistical confidence estimation procedures13 and the incorporation of a Bayesian protein inference algorithm.20
Figure 1: Overview of the Percolator algorithm.
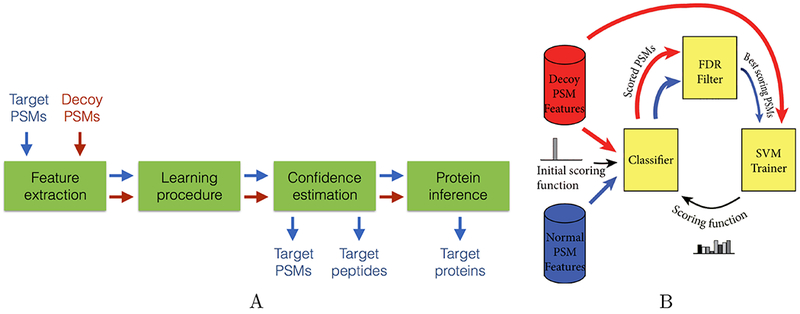
(A) Components of the software. Output PSMs, peptides and proteins are associated with q-values (a measure of false discovery rate) and posterior error probabilities. (B) The core learning algorithm. The input is a set of feature vectors, labeled as targets and decoys. Targets and decoys are ranked according to a selected feature, and target PSMs with 1% false discovery rate (FDR) are identified. An SVM is trained to discriminate between the selected targets and the full set of decoys. The SVM induces a new ranking, and the procedure iterates until the ranking stops changing.
Percolator has been broadly adopted and adapted by the computational mass spectrometry community. The initial publication demonstrated the utility of Percolator in conjunction with SEQUEST6 and Inspect.26 Subsequently, variants of Percolator were developed for use with Mascot,2 OMSSA,32 X!Tandem,35 and MS-GF+.8 Percolator has also been improved by use of a self-boosting machine learning scheme36 and for use with electron transfer dissociation data.34 Percolator is dis tributed via Github and is also distributed by Matrix Science and ThermoFisher as part of Mascot and Proteome Discoverer, respectively.
Since the initial description of Percolator, several competing algorithms have been published.5, 7, 37 For example, the widely used PeptideProphet algorithm was modified in 2008 to use the semi-supervised target-decoy approach pioneered by Percolator.3 Recently, an independent research group published a comparison of three different search engines in combination with five postprocessing methods.29 The study involved eight data sets produced from different proteomes and on a variety of mass spectrometry platforms. The primary conclusion was that “combinations involving Percolator achieved markedly more peptide and protein identifications at the same FDR level than the other 12 combinations for all data sets.” Also, for the three tested search engines, Percolator detected between 27.9% to 154% more PSMs at a 1% FDR compared to using the raw search engine scores.
1.2. Motivation
In this work, we describe our recent, successful efforts to dramatically speed up the Percolator software. Our motivation for carrying out this work was three-fold.
First, in general, faster software is better than slower software. Even when the input to Percolator is relatively small and the analysis only takes a few minutes, we believe that users will appreciate even faster execution. In many cases, the speedups reported here will enable interactive use of Percolator, since the improved software delivers results much more quickly. Particularly for large data sets, proper FDR control is critical to appropriately interpret the data. In such cases, long running times may deter researchers from running Percolator and hence may lead to invalid conclusions due to poorly calibrated or improperly interpreted search scores.
Second, faster software is particularly important for the analysis of large data sets. In analyses involving millions of spectra, the previous version of Percolator can take many hours or even days to run. Previously, we have argued that an effective speed-up can be obtained simply by downsampling (i.e., randomly discarding a portion of the input data).28 However, the downsampling approach can be problematic when the input data contains relatively few high-quality PSMs. Furthermore, from a practical perspective, downsampling introduces an additional user-level parameter—the downsampling rate—that may be difficult to set a priori.
Third, speeding up the Percolator algorithm has the potential to enable research into methods that use more complex sets of features. Currently, the standard version of Percolator uses a small set of features that varies slightly depending on the search engine. More complex feature sets include, for example, subscores corresponding to different ion types,25 scores from multiple search engines,31 similarity scores from fragment intensity prediction21 and probabilistic gradient information of PSMs.10 Such sets have the potential to improve Percolator’s performance but lead to a corresponding increase in running time. Percolator has also been adopted for applications that it was not originally intended for. For example, it was adapted for use for the annotaion of metabolites in imaging mass spectrometry,22 grouping of MS1-features in label-free quantification27 and with the OpenSWATH pipeline for analysis of data generated using data independent acquisition. In this setting, the number of features associated with each datapoint can be quite large. Furthermore, a side benefit of our speed-up efforts is that Percolator’s inference engine can now support, in principle, the use of a non-linear SVM classifier. This feature may be particularly useful in the context of research into more complex feature spaces, where non-linearity may be necessary to achieve good discrimination performance.
1.3. Contributions
In what follows, we describe how we sped up Percolator and provide empirical evidence that the new version is significantly faster. The speedups combat two major computational bottlenecks, which arise particularly in the analysis of massive proteomics data sets. The first such bottleneck concerns data input and output. In pursuing these speedups, we discovered, to our surprise, that a substantial portion of Percolator’s running time was dominated by reading and writing data. Accordingly, we show that overall runtime may be appreciably improved by speeding up input/output (I/O) tasks. The second bottleneck is the execution time required to learn SVM parameters. Recent work11 has tackled this bottleneck through software optimizations to Percolator’s SVM learning engine, and our efforts complement and further improve upon these optimizations.
On a massive data set containing over 215 million PSMs, the new version of Percolator achieves an overall speedup of 439% (81.4 hours down to 18.6 hours). We show that the improved software efficiently uses multiple processors, quickly approaching optimal performance using relatively few such processors. Furthermore, we show that the memory footprint required by these speedups is modest relative to that of the original version of Percolator. The improved version of Percolator is freely available with an Apache license at https://github.com/percolator/percolator/tree/pthread-speedup.
2. Methods
2.1. Software optimization
Percolator trains a series of SVM classifiers in an iterative, semi-supervised learning scheme. The program receives as input a collection of target and decoy PSMs, each one summarized using a fixed-length vector of features. A linear SVM is trained to distinguish high-scoring target PSMs from decoy PSMs. To prevent overfitting, this training is done using a nested cross-validation scheme.9 Thus, a separate SVM is learned using the PSMs within each fold, and the PSM scores are then merged. This training procedure repeats for a user specified number of iterations (ten, by default), updating the set of high-scoring target PSMs at each iteration. In the end, input PSMs are scored by the final learned SVM.
We started with Percolator v3.02, which uses a conjugate gradient least squares (CGLS) SVM implementation,23 and we proceeded to experiment with three successive optimizations (Table 1). The first speedup tackles the observation that a significant portion of Percolator execution time is spent on data I/O. In particular, reading in PSMs from disk to system memory required a substantial amount of overall runtime for large datasets. We thus focused on the parallelization of data I/O in Percolator using GNU libstdc++ parallel mode.24 Once enabled through the compiler flag -D GLIBCXX PARALLEL, this mode automatically parallelizes the standard library functions that load features into memory prior to SVM analysis.
Table 1: Percolator optimizations.
Note that in reported results, the optimizations are implemented cumulatively.
| Method | Description |
|---|---|
| Optimization 1 | Parallelize I/O |
| Optimization 2 | Parallelize inner folds of three-fold cross-validation |
| Optimization 3 | Single-threaded optimizations to CGLS as described in Halloran and Rocke11 |
The next speedup deals with the manner in which Percolator’s nested cross-validation is carried out. While the outer folds in Percolator are run in parallel, the inner folds are implemented in a sequential manner. We thus reimplemented this procedure such that the nested inner folds are run in parallel, using a user-specified number of threads. Finally, we optimized the CGLS solver itself using a mixture of low-level linear algebra function calls and software streamlining, as described previously.11
Optimizations are compared against the recently described CGLS multithreaded speedup,11 referred to as CGLS-par. In contrast to the second in our series of optimizations, which uses multiple threads to parallelize runs of CGLS at the cross-validation level, CGLS-par instead uses multiple threads to parallelize computation within the CGLS algorithm.
2.2. Data sets
We analyzed two published drafts of the human proteome and one Plasmodium falciparum data set (Table 2). The mass spectrometry data comprising the Kim et al. draft map of the human proteome16 consists of 25 million high resolution mass spectra contained in raw mass spectrometry data files representing >2,000 acquisitions on 30 histologically normal human cell and tissue types, including 17 adult tissues, 7 fetal tissues, and 6 hematopoietic cell types, fractionated by SDS-PAGE and analyzed on high resolution Fourier transform mass spectrometers (LTQ-Orbitrap Elite and LTQ-Orbitrap Velos) with HCD fragmentation. The data underlying the Wilhelm et al. draft map of the human proteome33 consists of mass spectrometry data files representing 19,433 acquisitions on 31 human tissue types and body fluids from healthy volunteers, fractionated by NuPage gel and analyzed on an LTQ-Orbitrap Elite and LTQ-Orbitrap Velos instruments with HCD fragmentation and LTQ-Orbitrap XL instruments with CID fragmentation. For both human data sets, RAW files were converted to mzML format using msconvert15 with peak-picking and deisotoping filters (--filter “peakPicking vendor msLevel=2” --filter “MS2Deisotope Poisson”).
Table 2:
Data sets.
Both human data sets were searched similarly. We downloaded the Uniprot human reference proteome from http://www.uniprot.org/proteomes/UP000005640 on Feb 17, 2016. We generated a peptide index for database search using the Tide search engine,4 as implemented in Crux,17 allowing for a maximum of two missed tryptic cleavages and two oxidized methionine residues on each peptide. For each peptide in the index, Tide automatically generates a decoy peptide by shuffling residues, leaving N- and C-terminal residues in place. We then used Tide to search each converted data file against the database with the following parameters: 50 ppm precursor tolerance; 0.02 Da fragment bin width; remove precursor peaks from spectra.
The Saraf data set19 data set is generated from a collection of Plasmodium falciparum samples collected at seven time points during the red blood cell stages of the parasite life cycle. The resulting peptide mixtures were analyzed by MudPIT on a Thermo Scientific LTQ Orbitrap. In this study, which focused on detection of histone modifications, a total of 504 MS/MS runs were collected. This data was searched using Tide against a database of 18 Plasmodium histone proteins, digested non-enzymatically and allowing a wide variety of modifications, and up to two modifications per peptide. The corresponding tide-index options are --enzyme no-enzyme --mods-spec 2KR+14.0157,2MLPDHY+15.9949,2LR+27.9949,2KR+28.0314,2K+42.0470,2KST+42.0106,2L+68.0261,2STY+79.9663,2L+114.0429 --nterm-peptide-mods-spec 1MSAV+42.0106,1M+58.0055. This set of modifications results in a database containing 88,143,399 distinct target peptides. Searches were performed against a concatenated target-decoy database (--concat T) with a 10 ppm search window (--precursor-window 10 --precursor-window-type ppm) using exact p-value scoring (--exact-p-value T) and removing precursor peaks (--remove-precursor-peak T).
2.3. Experimental environment
All computational experiments were run using a multicore compute server with one terabyte of RAM, comprised of Intel Xeon E7–4830 v3 CPUs clocked at 2.10 GHz. In order to accurately measure each method’s performance under standard use, Percolator’s outer cross-validation folds were run in parallel for all timing tests.
2.4. Software details
The optimizations described herein are supported for Unix-based platforms and freely available at https://github.com/percolator/percolator/tree/pthread-speedup. To enable these speedups, users can specify the additional command-line option --ncposthreads, which control the number of threads employed within each of the three outermost cross-validation folds. Thus, for a number of threads n specified using --ncposthreads n, the total number of threads utilized is 3n. Our speedups are compared against CGLS-par, the optimized multithreaded solver described in,11 updating the software described therein to the current Percolator release. For the number of threads per cross-validation fold n, CGLS-par was run using k = 3n total threads by setting the flag -nrk.
3. Results
3.1. Large-scale SVM learning timing tests
The methods listed in Table 1 were tested on the Wilhelm, Kim, and Saraf data sets. For the large-scale Kim and Saraf data sets, reported runtimes are the minimum over three separate runs, where original Percolator runtime required 11.85 hours and 7.5 hours, respectively. Due to the long runtime for the massive-scale Wilhelm data set—the original Percolator SVM learning time was 69.86 hours for this data set, and additionally required over eleven hours to load the data—carefully monitored single runtimes are reported. All evaluated methods were run using 2, 4, 6, 8, and 10 threads per each of the three cross-validation folds.
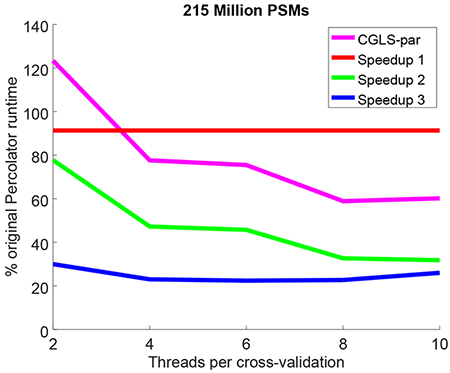
The resulting runtimes (Table 3 and Figure 2) improve after each successive optimization, and the improvement is more pronounced when more cores are used. Furthermore, a comparison to CGLS-par (which only improves SVM learning time) excluding the I/O runtime benefits of optimization 1 shows that our optimizations drastically improve the SVM learning component of Percolator relative to this alternative approach for all thread configurations considered (Figure 3).
Table 3: Comparison of optimizations on three data sets.
The table lists the runtimes (in hours) of five different methods on three data sets. The original Percolator method (“CGLS”) is compared with CGLS optimized for multithreaded use from11 (“CGLS-par”) and the three successive optimizations presented in this work. For the multithreaded methods CGLS-par, Opt. 2, and Opt. 3, the number of threads per cross-validation fold is denoted below the multithreaded methods as “2t,” “4t,” etc. Reported runtimes for the Kim and Saraf data sets are the minimum wall-clock times measured over three runs, and reported runtimes for the much larger Wilhelm data set are from a single run.
| Data Set | CGLS | Opt. 1 | CGLS-par | Opt. 2 | Opt. 3 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2t | 4t | 6t | 8t | 10t | 2t | 4t | 6t | 8t | 10t | 2t | 4t | 6t | 8t | 10t | |||
| Wilhelm | 81.4 | 74.3 | 100.4 | 63.2 | 61.4 | 47.9 | 49.0 | 63.3 | 38.5 | 37.2 | 26.6 | 25.9 | 24.4 | 18.7 | 18.2 | 18.5 | 21.2 |
| Kim | 12.8 | 11.1 | 11.9 | 8.7 | 7.4 | 7.1 | 6.6 | 7.3 | 4.8 | 3.9 | 3.4 | 2.8 | 3.1 | 2.5 | 2.3 | 2.3 | 2.2 |
| Saraf | 22.5 | 20.6 | 18.2 | 14.6 | 14.2 | 14.2 | 14.2 | 12.1 | 8.1 | 7.0 | 7.0 | 6.9 | 7.0 | 6.2 | 6.2 | 6.2 | 6.2 |
Figure 2: Speedups after successive Percolator optimizations.
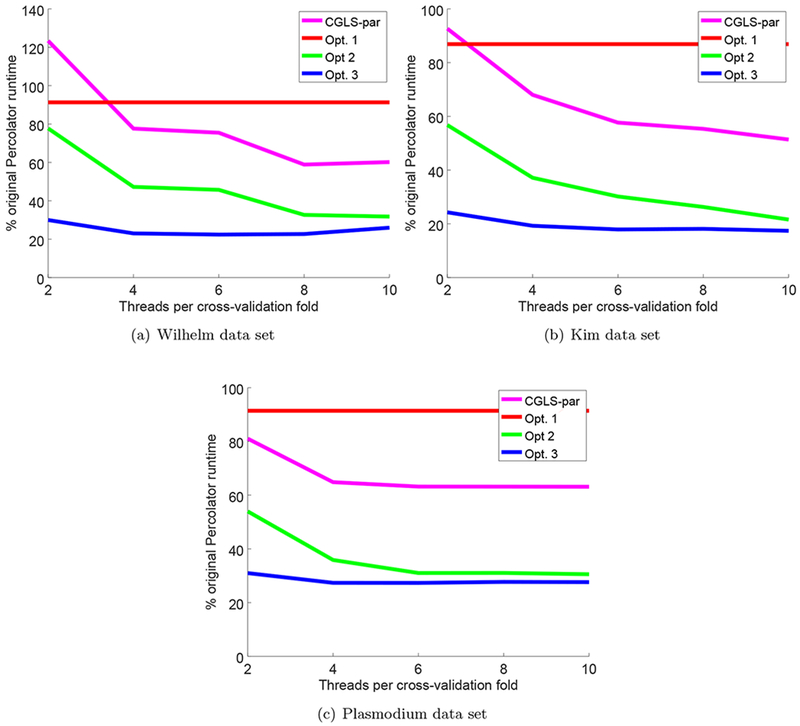
Each panel plots the total runtime divided by the original Percolator runtime, as a function of the number of threads per cross-validation folds. The successive optimizations are compared against the version of CGLS optimized for multithreaded use in,11 referred to as CGLS-par. Reported runtimes for the Kim and Saraf data sets are the minimum wall-clock times measured over three runs and reported runtimes for the much larger Wilhelm data set are the product of a single run.
Figure 3: Speedups only considering SVM solver runtime.
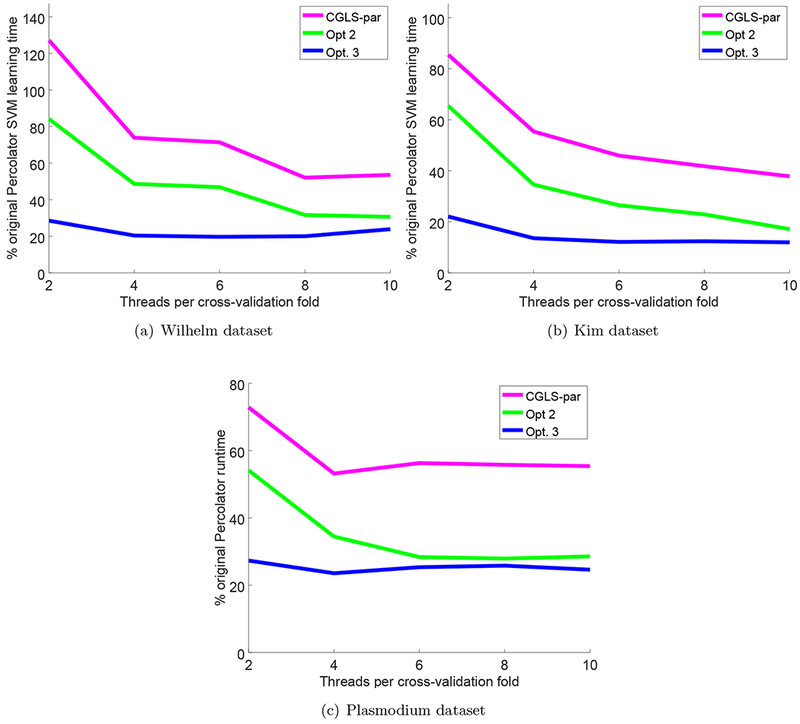
Each panel plots the total SVM learning runtime divided by the original Percolator SVM learning runtime, as a function of the number of threads per cross-validation folds. Optimizations only affecting SVM learning time (i.e., Opt. 2 and Opt. 3) are compared against the SVM learning runtime of the version of CGLS optimized for multithreaded use in,11 referred to as CGLS-par. Reported runtimes for the Kim and Saraf data sets are the minimum wall-clock times measured over three runs and reported runtimes for the much larger Wilhelm data set are the product of a single run.
3.2. Memory consumption
In addition to characterizing the relative speedups of the various optimization methods, we also measured their relative memory footprints. Toward this end, each method was further tested over a random subset of the Kim data set containing approximately eight million PSMs. For each method, memory consumption was measured as the “Maximum resident set size,” as reported by GNU time.
In this experiment, successive optimizations and further speedups required an increase in memory usage, largely owing to the parallelization of solver calls in optimizations 2 and 3. For 2, 4, 6, 8, and 10 threads per each of the Percolator cross-validation folds, the resulting system memory used for successive optimizations 2 and 3 slowly grows with the increased number of threads (Figure 4), where original Percolator memory consumption was 12.2 GB and the maximum memory consumption for optimizations 1, 2, and 3 were 13.7 GB, 23.4 GB, and 41.1 GB, respectively. Thanks to the substantially improved overall runtime, allocated system memory was also quickly released, thus allowing the use of ten times the number of original Percolator threads while only requiring 3.37 times the memory use.
Figure 4: Comparison of memory consumption.
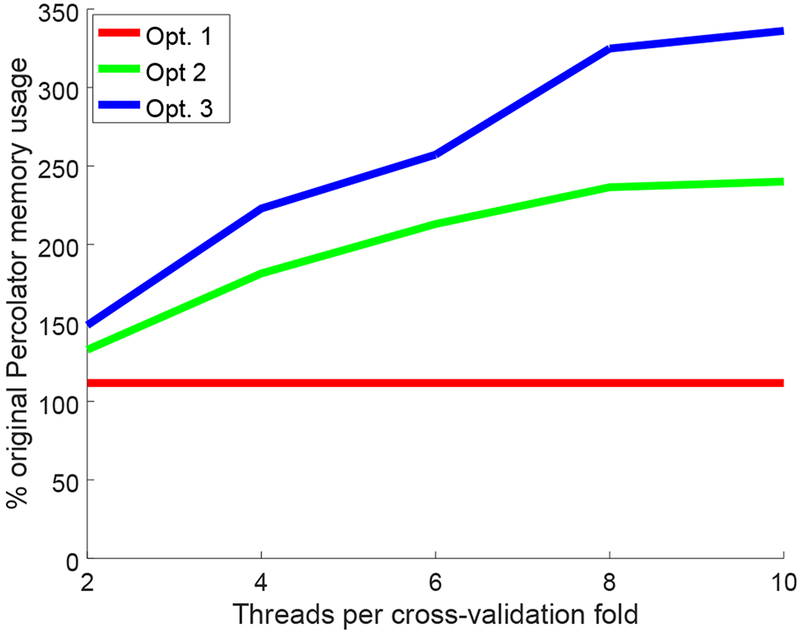
Each panel plots memory consumption of a given method divided by original Percolator memory consumption (y-axis) as a function of the number of threads allocated per cross-validation fold. These results are for a random subset of 7,710,057 PSMs from the Kim data set. Memory consumption was measured as the “Maximum resident set size,” as reported by GNU time.
4. Discussion
Each of our successive optimizations speed up Percolator execution, culminating in a substantial decrease in overall runtime. We compare all optimizations against CGLS-par, the recent multithreaded approach of Halloran and Rocke,11 wherein multiple threads are used to parallelize computation within Percolator’s SVM learning solver (CGLS). In contrast to CGLS-par, our approach to multithreading-wherein multiple threads are used to parallelize calls to CGLS within Percolator’s cross-validation scheme-is much faster irrespective of the number of threads employed. Furthermore, the final optimization rapidly approaches peak performance relative to the number of threads utilized, making these speedups useful for both common compute environments (such as desktops) and higher-end parallel architectures (such as compute clusters).
Owing to the nature of the parallelization exploited herein (i.e., multiple calls to CGLS run in parallel), the presented speedups require higher memory usage, as demonstrated in Figure 4. However, the increased memory usage is far from restrictive when considering the speedups gained, instead offering a flexible tradeoff between speed and memory resources. For instance, peak performance (of optimization 3) for all data sets is nearly obtained using four threads per cross-validation fold, which requires only slightly more than double normal Percolator memory usage to achieve an average 4.6 times faster runtime over the large-scale data sets considered. For even more restrictive memory settings, the minimum multithreaded setting of two threads per cross-validation fold still completes an average 3.6 times faster over the large-scale data sets while only requiring 50% more memory than the original Percolator. In the event multithreading is completely disabled, users will still see an appreciable decrease in runtime owing to optimization 1. For example, I/O for the Wilhelm data set was reduced from 11.5 hours down to just 4.5 hours.
In future work, we plan to explore the use of these optimizations to enable nonlinear classification in Percolator. Percolator currently uses a linear kernel wherein the learned decision boundary between targets and decoys is a hyperplane in the input space. In contrast, analysis of complex data sets—which possibly require more complicated decision boundaries to discriminate targets from decoys—may be improved through the use of non-linear kernels, at the cost of increased runtimes. The optimizations presented herein thus render the exploration of such non-linear kernels feasible for Percolator analysis.
Acknowledgments
We thank Bernhard Kuster and Mathias Wilhelm at Technische Universitaet München for providing us with access to the data from the Wilhelm et al. study. This work was supported, in part, by the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through grant UL1 TR001860 and by National Institutes of Health award R01 GM121818 to W.S.N. L.K. was supported by a grant from the Swedish Research Council (grant 2017-04030).
References
- 1.Anderson DC, Li W, Payan DG, and Noble WS. A new algorithm for the evaluation of shotgun peptide sequencing in proteomics: support vector machine classification of peptide MS/MS spectra and SEQUEST scores. Journal of Proteome Research, 2(2):137–146, 2003. [DOI] [PubMed] [Google Scholar]
- 2.Brosch M, Yu L, Hubbard T, and Choudhary J. Accurate and sensitive peptide identification with Mascot Percolator. Journal of Proteome Research, 8(6):3176–3181, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi H and Nesvizhskii AI. Semisupervised model-based validation of peptide identifications in mass spectrometry-based proteomics. Journal of Proteome Research, 7(1):254–265, 2008. [DOI] [PubMed] [Google Scholar]
- 4.Diament B and Noble WS. Faster SEQUEST searching for peptide identification from tandem mass spectra. Journal of Proteome Research, 10(9):3871–3879, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Du X, Yang F, Manes NP, Stenoien DL, Monroe ME, Adkins JN, States DJ, Purvine SO, Camp DG, and Smith RD. Linear discriminant analysis-based estimation of the false discovery rate for phosphopeptide identifications. Journal of Proteome Research, 7:2195–2203, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eng JK, McCormack AL, and Yates JR III. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry, 5:976–989, 1994. [DOI] [PubMed] [Google Scholar]
- 7.Gonnelli G, Stock M, Verwaeren J, Maddelein D, De Baets B, Martens L, and Degroeve S. A decoy-free approach to the identification of peptides. Journal of Proteome Research, 14:1792–1798, 2015. [DOI] [PubMed] [Google Scholar]
- 8.Granholm V, Kim S, Navarro JCF, Sjölund E, Smith RD, and Käll L. Fast and accurate database searches with MS-GF+Percolator. Journal of Proteome Research, 13:890–897, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Granholm V, Noble WS, and Käll L. A cross-validation scheme for machine learning algorithms in shotgun proteomics. BMC Bioinformatics, 13(Suppl 16):S3, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halloran JT and Rocke DM. Gradients of generative models for improved discriminative analysis of tandem mass spectra. In Advances in Neural Information Processing Systems, pages 5724–5733, 2017. [PMC free article] [PubMed] [Google Scholar]
- 11.Halloran JT and Rocke DM. A matter of time: faster Percolator analysis via efficient SVM learning for large-scale proteomics. Journal of Proteome Research, 17(5):1978–1982, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Käll L, Canterbury J, Weston J, Noble WS, and MacCoss MJ. A semi-supervised machine learning technique for peptide identification from shotgun proteomics datasets. Nature Methods, 4:923–25, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Käll L, Storey J, and Noble WS. Nonparametric estimation of posterior error probabilities associated with peptides identified by tandem mass spectrometry. Bioinformatics, 24(16):i42–i48, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keller A, Nesvizhskii AI, Kolker E, and Aebersold R. Empirical statistical model to estimate the accuracy of peptide identification made by MS/MS and database search. Analytical Chemistry, 74:5383–5392, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Kessner D, Chambers M, Burke R, Agnus D, and Mallick P. Proteowizard: open source software for rapid proteomics tools development. Bioinformatics, 24(21):2534–2536, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim M, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. A draft map of the human proteome. Nature, 509(7502):575–581, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McIlwain S, Tamura K, Kertesz-Farkas A, Grant CE, Diament B, Frewen B, Howbert JJ, Hoopmann MR, Käll L, Eng JK, MacCoss MJ, and Noble WS. Crux: rapid open source protein tandem mass spectrometry analysis. Journal of Proteome Research, 13(10):4488–4491, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore RE, Young MK, and Lee TD. Qscore: An algorithm for evaluating SEQUEST database search results. Journal of the American Society for Mass Spectrometry, 13(4):378–386, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Saraf A, Cervantes S, Bunnik EM, Ponts N, Sariu ME, Chung DD, Prudhomme J, Varberg JM, Wen Z, Washburn MP, Florens L, and Le Roch KG. Dynamic and combinatorial landscape of histone modifications during the intraerythrocytic developmental cycle of the malaria parasite. Journal of Proteome Research, 15(8):2878–2801, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serang O, MacCoss MJ, and Noble WS. Efficient marginalization to compute protein posterior probabilities from shotgun mass spectrometry data. Journal of Proteome Research, 9(10):5346–5357, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Silva AS, Martens L, and Degroeve S. Accurate peptide fragmentation predictions allow data driven approaches to replace and improve upon proteomics search engine scoring functions. bioRxiv, page 428805, 2018. [DOI] [PubMed] [Google Scholar]
- 22.Silva ASC, Palmer A, Kovalev V, Tarasov A, Alexandrov T, Martens L, and Degroeve S. Data-driven rescoring of metabolite annotations significantly improves sensitivity. Analytical Chemistry, 90(19):11636–11642, 2018. [DOI] [PubMed] [Google Scholar]
- 23.Sindhwani V and Keerthi SS. Large scale semi-supervised linear SVMs In SIGIR ‘06: Proceedings of the 29th annual international ACM SIGIR conference on Research and development in information retrieval, pages 477–484, New York, NY, USA, 2006. ACM Press. [Google Scholar]
- 24.Singler J and Konsik B. The gnu libstdc++ parallel mode: software engineering considerations. In Proceedings of the 1st international workshop on Multicore software engineering, pages 15–22. ACM, 2008. [Google Scholar]
- 25.Spivak M, Weston J, Bottou L, Käll L, and Noble WS. Improvements to the Percolator algorithm for peptide identification from shotgun proteomics data sets. Journal of Proteome Research, 8(7):3737–3745, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanner S, Shu H, Frank A, Wang Ling-Chi, Zandi E, Mumby M, Pevzner PA, and Bafna V. InsPecT: Identification of posttranslationally modified peptides from tandem mass spectra. Analytical Chemistry, 77:4626–4639, 2005. [DOI] [PubMed] [Google Scholar]
- 27.The M and Käll L. Focus on the spectra that matter by clustering of quantification data in shotgun proteomics. BioRxiv, page 488015, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.The M, MacCoss MJ, Noble WS, and Käll Lukas. Fast and accurate protein false discovery rates on large-scale proteomics data sets with Percolator 3.0. Journal of the American Society of Mass Spectrometry, 27(11):1719–1727, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tu C, Sheng Q, Li J, Ma D, Shen X, Wang W, Shyr Y, Yi Z, and Qu J. Optimization of search engines and postprocessing approaches to maximize peptide and protein identification for high-resolution mass data. Journal of Proteome Research, 14(11):4662–4673, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Washburn MP, Wolters D, and Yates JR III. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nature Biotechnology, 19:242–247, 2001. [DOI] [PubMed] [Google Scholar]
- 31.Wen B, Du C, Li G, Ghali F, Jones AR, Käll L, Xu S, Zhou R, Ren Z, Feng Q, Xu X, and Wang J. Ipeak: An open source tool to combine results from multiple ms/ms search engines. Proteomics, 15(17):2916–2920, 2015. [DOI] [PubMed] [Google Scholar]
- 32.Wen B, Li G, Wright JC, Du C, Feng Q, Xu X, Choudhary JS, and Wang J. The OMSSAPercolator: an automated tool to validate OMSSA results. Proteomics, 14(9):1011–1014, 2014. [DOI] [PubMed] [Google Scholar]
- 33.Wilhelm Mathias, Schlegl Judith, Hahne Hannes, Gholami Amin Moghaddas, Lieberenz Marcus, Savitski Mikhail M, Ziegler Emanuel, Butzmann Lars, Gessulat Siegfried, Marx Harald, et al. Mass-spectrometry-based draft of the human proteome. Nature, 509(7502):582–587, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Wright JC, Collins MO, Yu L, Käll L, Brosch M, and Choudhary JS. Enhanced peptide identification by electron transfer dissociation using an improved Mascot Percolator. Molecular and Cellular Proteomics, 11(8):478–491, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu M, Li Z, and Li L. Combining Percolator with X!Tandem for accurate and sensitive peptide identification. Journal of Proteome Research, 12(6):3026–3033, 2013. [DOI] [PubMed] [Google Scholar]
- 36.Yang P, Ma J, Wang P, Zhu Y, Zhou BB, and Yang YH. Improving X!Tandem on peptide identification from mass spectrometry by self-boosted Percolator. IEEE/ACM Transactions on Computational Biology and Bioinformatics, 9(5):1273–1280, 2012. [DOI] [PubMed] [Google Scholar]
- 37.Zhang J, Ma J, Dou L, Wu S, Qian X, Xie H, Zhu Y, Zhang FH, Ma J, Dou L, Wu S, Qian X, Xie H, Zhu Y, and He F. Bayesian nonparametric model for the validation of peptide identification in shotgun proteomics. Molecular and Cellular Proteomics, 8(3):547–557, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]