

Abstract
Cognitive functions are dependent upon intercommunications between the cellular components of the neurovascular unit (NVU). Vascular risk factors are associated with a more rapid rate of cognitive decline with aging and cerebrovascular diseases magnify both the incidence and the rate of cognitive decline. The causal relationship between vascular risk factors and injury to the NVU is, however, lacking. We hypothesized that vascular risk factors, such as hypertension and dyslipidemia, promote disruption of the NVU leading to early cognitive impairment. We compared brain structure and cerebrovascular functions of 1-year old (middle-aged) male wild-type (WT) and atherosclerotic hypertensive (LDLr−/−:hApoB+/+, ATX) mice. In addition, mice were subjected, or not, to a transverse aortic constriction (TAC) for 6 weeks to assess the acute impact of an increase in systolic blood pressure on the NVU and cognitive functions. Compared with WT mice, ATX mice prematurely developed cognitive decline associated with cerebral micro-hemorrhages, loss of microvessel density and brain atrophy, cerebral endothelial cell senescence and dysfunction, brain inflammation, and oxidative stress associated with blood-brain barrier leakage and brain hypoperfusion. These data suggest functional disturbances in both vascular and parenchymal components of the NVU. Exposure to TAC-induced systolic hypertension promoted cerebrovascular damage and cognitive decline in WT mice, similar to those observed in sham-operated ATX mice; TAC exacerbated the existing cerebrovascular dysfunctions and cognitive failure in ATX mice. Thus, a hemodynamic stress such as systolic hypertension could initiate the cascade involving cerebrovascular injury and NVU deregulation and lead to cognitive decline, a process accelerated in atherosclerotic mice.
Keywords: Hypertension, 7T-MRI, Senescence, Apoptosis, Transverse aortic constriction, Endothelial function, Blood-brain barrier, Carotid stiffness, VCID
Introduction
Cardiovascular diseases (CVD), such as hypertension, atherosclerosis and stroke, and neurological disorders such as sporadic Alzheimer disease (AD) or consecutive to cerebrovascular diseases such as vascular cognitive impairment and dementia (VCID), commonly occur concomitantly in the elderly (Donnan et al. 2008; Taylor et al. 2017). When combined, CVD and neurological disorders may be the highest determinant of whether individuals develop dementia (DeCarli 2012). Midlife CVD increase the risk of late-life dementia (Fotuhi et al. 2012) and individuals who present a history of vascular risk factors also show a more rapid rate of cognitive decline (O’Donnell et al. 2012). Epidemiological studies in individuals with AD or in the elderly identified vascular complications associated with peripheral or brain diseases as risk factors for AD (Breteler 2000; de la Torre 2002). Altogether, this suggests that cognitive decline may be unveiled by brain vascular abnormalities and in particular brain microvascular abnormalities (de la Torre 2002; de Montgolfier et al. 2019; Zlokovic 2002).
Vascular cells (endothelial, smooth muscle cells (VSMC), pericytes) share intimate interactions with brain tissues (astrocytes, neurons, their axons, and other supporting cells), forming the neurovascular unit (NVU), which is crucial for brain homeostasis (Iadecola 2004). Alterations in vascular integrity evidenced in patients by augmented blood-brain barrier (BBB) permeability (Nation et al. 2019) have downstream effects within the NVU, including changes in blood flow dynamics, cerebrovascular autoregulation, and neuroinflammation (Toth et al. 2013), as well as neuronal and synaptic dysfunctions (Zlokovic 2008). Consequently, a disruption of the NVU, which occurs in aging and in neurodegenerative diseases, is associated with cognitive decline (Kisler et al. 2017). Changes in vascular homeostasis may precede neurodegenerative disorders observed with aging (Nation et al. 2019; Zlokovic 2011), but relatively little is known about the causal relationship between vascular risk factors and disruption of the NVU. It is essential to better understand how vascular risk factors influence brain function to propose preventive interventions and treatments that could protect cognitive skills and abilities in the elderly.
Using our severely dyslipidemic and spontaneously atherosclerotic LDLr−/−:hApoB+/+ (ATX) mice that display mild hypertension and both peripheral and cerebral vascular dysfunction including carotid stiffening (Bolduc et al. 2011; Drouin et al. 2011a), we hypothesized that they were more susceptible to vascular stress, which would lead to premature loss of BBB integrity, NVU uncoupling, and cognitive impairment. We then explored whether an acute exogenous vascular stress induced by a systolic pressure overload, using TAC surgery as a model of isolated systolic hypertension, would result in a cognitive decline in WT mice similar to that observed in naïve ATX mice and lead to aggravated brain damage in ATX mice. Our results suggest that in ATX and in TAC mice, the NVU uncoupling associated with vascular dysfunctions is determinant to the premature decline of brain and cognitive functions. We propose that a hemodynamic vascular stress such as systolic hypertension could initiate the cascade involving cerebrovascular injury and NVU deregulation and lead to cognitive decline, a process accelerated in dyslipidemic and hypertensive ATX mice.
Methods
Animals
We used male dyslipidemic LDLr−/−;ApoB100+/+ (ATX) mice (C57Bl/6J/SJL/129S mixed background) (n = 44) and male C57Bl/6J wild-type (WT) mice (n = 45) (Bolduc et al. 2011). ATX mice develop atherosclerotic lesions in the aorta by the age of 6 months (Bolduc et al. 2011) with elevation in circulating levels of lipids between 3 and 12 months of age (Drouin et al. 2011b); cerebral arteries of 12-month-old ATX mice do not present macroscopic signs of atherosclerosis (Drouin et al. 2011b). ATX mice maximal life expectancy is reduced compared to WT mice (25.2 vs. 32.8 months, p < 0.05; Fig. 1a). All experiments were performed in 1-year-old mice in accordance with the Guide for the Care and Use of Experimental Animals of the Canadian Council on Animal Care and the Guide for the Care and Use of Laboratory Animals of the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and the study was approved by the Montreal Heart Institute Ethics Committee (ET No. 2015-62-01). Mice were kept under standard conditions (24 °C; 12-h:12-h light/dark cycle) and were fed ad libitum with regular chow (2019S; Harlan Laboratories, Madison, WI, US).
Fig. 1.

Survival curve of ATX (n = 40) and WT (n = 20) mice. Median survival is similar in ATX (20.5 month) and in WT mice (21.7 months). ATX mice life expectancy, however, is reduced (25.2 vs. 32.8 months; log-rank (Mantel Cox) test chi-square = 4.092, p = 0.0431). Effect of TAC surgery on b heart/body weight ratio in WT (n = 45) and ATX (n = 44) mice; c operative mortality in WT (n = 45) and ATX (n = 44) mice after sham and TAC surgical procedures. Percentages include the proportion of deaths that occurred during or soon after (24–48 h) the surgical procedure. Data are mean ± SEM. $p < 0.05 vs. sham mice; *p < 0.05 vs. WT mice
Surgical procedure and blood pressure measures
Transverse aortic constriction (TAC) was performed in mice as previously reported (de Montgolfier et al. 2019) using the method initially described by Rockman and collaborators (Rockman et al. 1991). In brief, in 10.5-month-old mice anesthetized with 2% isoflurane via inhalation, TAC-induced hypertension in the right brain hemisphere (ipsilateral side) was performed between truncus anonymous and the left carotid, with a 7.0 nylon suture ligature placed around the aorta. A 27-gauge (0.4 mm diameter) needle was placed against and tied to the aorta with the suture. Then, the needle was removed promptly to yield a constriction with a diameter approximately equal to that of the needle, producing a ~ 60% aortic constriction. Another group of mice underwent the same surgical procedure without realizing aortic stenosis and were used as control (sham). After 6 weeks of constriction (at the age of 1 year), blood pressure (systolic and diastolic pressures) in the right carotid artery was monitored under anesthesia (44 mg/kg ketamine [Bimeda-MTC, Cambridge, ON, Canada] and 2.2 mg/kg xylazine [Bayer, Mississauga, ON, Canada], intra-peritoneal) using a Millar catheter (Millar Instruments, Houston, TX, USA) to confirm an increased in pulse pressure (PP) on the right ipsilateral side (de Montgolfier et al. 2019). Mice were then euthanized and heart weight and total body weight were recorded (Fig. 1b). Perioperative mortality was also noted (Fig.1c).
MRI acquisitions and data analysis
Magnetic resonance imaging (7T-MRI) was performed in anesthetized 1-year-old mice to measure cerebral blood perfusion using a CASL technique as previously described (de Montgolfier et al. 2019). Anatomical scans were also performed using a 3D true free induction with steady-state precession (TFISP) sequence (Pouliot et al. 2017) at 100-μm isotropic resolution. All mice were initially anesthetized with 5% isoflurane in oxygen and then transferred to an animal holder within the magnet where they were maintained under 1.5–2.5% isoflurane in 30% oxygen in air and their body temperature was maintained at 37.0 °C using a warm air fan (SA Instruments, Stony Brook, NY). Respiration (target = 100, allowed range before adjusting isoflurane = 80–120 BPM) and heart rate were monitored, the latter with a pulse oximeter. The computations were performed using Advanced Normalization Tools (ANTs) (Avants et al. 2008; Avants et al. 2011; Tustison et al. 2010) (version 2.1.0), called from Matlab, and run on a server (Briarée, Calcul Québec, Canada). The physical dimensions in the headers of all images were initially multiplied by a factor of 10, thus making a mouse brain of comparable size to a human brain. This helped with running the ANT scripts on mice, suggesting that some dimension parameters in ANTs are hard-coded.
Co-registration
Brains of all mice were manually masked using ITK-SNAP (version 3.6) (Yushkevich et al. 2006). An initial template was then constructed using the antsMultivariate-TemplateConstruction2.sh ANTs script. To help with achieving higher spatial sensitivity, the brains were digitally up-sampled by a factor of 2 in each dimension. To improve on the initial manual masks and on the template construction, a template mask larger than the brain by several voxels in all directions was projected onto all the brains, and the ANTs script was re-run allowing for more iterations at each stage. This resulted in extracted brains, linear and non-linear transformation parameters, and a template with 50-μm isotropic resolution. A 32-μm isotropic resolution ex vivo atlas with 62 structures was co-registered to that template (Dorr et al. 2008) and its labels were projected onto each brain in native space. A thorough visualization of all co-registrations superposed with the atlas labels was made by generating mosaics of cuts in the three standard orthogonal planes.
Segmentation
The antsAtroposN4.sh ANTs script was used to generate a four-tissue segmentation of the template (GM = gray matter, WM = white matter, DGM = deep gray matter). Options used consisted in the HistogramParzenWindow (Avants et al. 2011; Yushkevich et al. 2006) option in ANTs, and setting the mrf parameter to 0.005, while keeping the default 0.25 weight for priors. The output was a segmentation image of the template and tissue probability posteriors. These were then used as priors, after projection to the native space of each brain, for the application of the antsAtroposN4.sh script to each mouse brain. The segmentations were also visualized thoroughly with mosaics as for the co-registrations above.
Volumes of segmented brain regions of interest were normalized to total brain volume within the corresponding genotype. Partial volumes were computed from the intersections of the atlas with the four-tissue segmentation, which was reduced to three tissues by combining deep gray matter with gray matter. To reduce the number of voxels in an atlas region which belonged to a tissue segmentation type different from its main type (e.g., gray matter voxels in the corpus callosum or white matter voxels in the cortex), cerebrospinal fluid voxels from the segmentation were all assigned to ventricles in the atlas, and then two iterations of the following assignment algorithm were applied: each atlas region was grown outward by one voxel and the new atlas region was defined as the intersection of the grown region with the voxels of the segmentation that are of the correct type for that region.
Neurobehavioral tests
Neurobehavioral tests were performed in 1-year-old mice as previously described (de Montgolfier et al. 2019). Animals were assigned to groups randomly. The Morris Water Maze test was performed to assess learning and spatial memory. The water maze apparatus consisted of a white circular pool of 150 cm in diameter and 60 cm in height, filled with water made opaque with non-toxic white paint kept at a temperature of 22 °C. A plastic transparent platform (10 cm in diameter) was placed 1.5 cm below the water surface and 40 cm from the edge of the pool, except on day 1 (habituation phase) where the platform was placed 0.5 cm above the water surface. The entire procedure took 11 days. Mice were individually transferred from the home-cage to the pool. Release points were balanced across four symmetrical positions on the pool perimeter. Each day of the test, mice underwent four trials during which they were allowed to freely swim for 60 s or until they found and climbed onto the platform; each trial was spaced from the other by a 60-min inter-trial interval. Platform finding was defined as staying for at least 3 s on the platform. On day 1, during the habituation phase, mice that did not find the platform were trained in locating it by placing them on the platform for 30 s at the end of the trial. Then, 48 h later, the acquisition phase started on day 3 (and during 5 days in a row); mice that did not find the platform were trained in locating it by placing them on the platform for 30 s at the end of the trial. On the fifth day of the acquisition phase and 48 h after the last acquisition trial, the platform was removed from the pool and each mouse was tested for memory retention in a 60-s probe trial. During the probe trial, the time spent in the target quadrant (TQ) of the maze (where the platform was located) was scored as a reliable measure of memory retention 72 h after the last acquisition trial. The swim path of the mice and the time spent in the target quadrant were recorded by means of a computer-based video-tracking system Smart (version 3.0, Panlab/Harvard Apparatus). For the probe phases, the variables recorded were time spent in each quadrant. All recordings were automatically quantified without human intervention.
The Y-maze test was performed to assess spatial working memory. The Y-maze test is based on spontaneous alternation behavior and is used to measure spatial working memory. The maze consists of three arms (41 cm long, 16 cm high, and 9 cm wide, labeled A, B, or C) diverging at a 120° angle from the central point. The experiments were performed in a dimly illuminated room, and the floor of the maze was cleaned with super hypochlorous water-soaked paper after each mouse to avoid olfactory cues. Each mouse was placed at the end of the starting arm and allowed to move freely through the maze during a 5-min session. The sequence of arm entries was manually recorded with strict criteria (standard operating procedures) to assure independence from the experimenter: (1) a mouse was considered to have entered an arm when all four paws were positioned in the arm runway; (2) an actual alternation was defined as entries into all of the three arms on consecutive occasions. The maximum alternation was subsequently calculated by measuring the total number of arm entries minus 2 and the percentage of alternation was calculated as (actual alternations/maximum alternations)×100. Total number of arms entered during the sessions (reflecting locomotor activity) was also recorded. Mice that entered arms less than eight times during the test were eliminated because the data obtained from those mice were not considered to be representative.
Cerebral vessels and parenchymal isolation
After blood pressure acquisition and euthanasia, brains were rapidly removed and cut sagittally into contralateral and ipsilateral hemispheres as previously described (de Montgolfier et al. 2019). The hemispheres were snap frozen in liquid nitrogen and stored at − 80 °C. Ipsilateral brain hemispheres were homogenized in ice-cold PBS buffer (pH 7.4 supplemented with phosphatase and protease inhibitor cocktail) (Sigma-Aldrich, Oakville, ON, Canada) with a loosely fitting Dounce homogenizer, and centrifuged at 2000g for 5 min at 4 °C. The non-vascular supernatant was removed and stored on ice. The vascular pellet was re-suspended in PBS and centrifuged at 2000g for another 5 min at 4 °C. The supernatant was combined with the first non-vascular supernatant and centrifuged for 10 min at 3000g at 4 °C. The resulting pellet containing the parenchymal fraction was stored at − 80 °C in QUIAZOL buffer (Qiagen, Montreal, QC, Canada) until RNA extraction. The vascular pellets were re-suspended in PBS and centrifuged at 2000g at 4 °C. The supernatant was discarded and the pellet re-suspended in cold PBS (twice). Then, the re-suspended pellet was passed through a 50-μm nylon mesh (CellMicroSieves, Biodesign, Carmel, NY, USA) and microvessels retained on the mesh were washed with cold PBS. Microcerebral vessels were collected in cold PBS and centrifuged for 10 min at 3000g at 4 °C. The resulting pellet containing the cerebral vessels was stored at − 80 °C in RLT buffer (Qiagen) supplemented with β-mercaptoethanol until RNA extraction. Thus, from the same mouse, gene expression was measured in both the microvascular and the parenchymal fractions of the brain. Purity of the two preparations was controlled by Western blot of eNOS and synaptophysin proteins (Fig. 2).
Fig. 2.
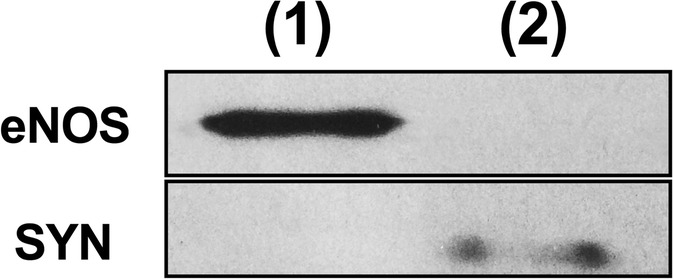
Microvascular versus parenchymal brain fractions. Protein expression of eNOS in the microvascular fraction only, and of synaptophysin (SYN) in the parenchymal fraction only. (1): cerebral microvessels; (2): parenchymal fraction
Endothelial function of cerebral arteries by pressurized arteriography
Freshly isolated brains were placed in ice-cold physiological salt solution (PSS; mmol/L: 130 NaCl; 4.7 KCl; 1.18 KH2 PO4; 1.17 MgSO4; 14.9 NaHCO3; 1.6 CaCl2; 0.023 EDTA; 10 glucose; pH 7.4) aerated with 12% O2, 5% CO2, and 83% N2 at 37 °C. The middle cerebral artery (MCA) of the ipsilateral hemisphere was isolated, transferred to the arteriograph chamber (Living System Instrumentation, St-Alban, VT, USA), cannulated, and pressurized at 60 mmHg for endothelial function assessment as previously described (de Montgolfier et al. 2019; Drouin and Thorin 2009). The artery segment was equilibrated for 45 min, allowing for myogenic tone to develop. Because myogenic tone is very low in cerebral arteries from C57Bl/6 mice (Raignault et al. 2017), arteries were pre-constricted to ~ 25% of the initial diameter with phenylephrine (Sigma-Aldrich; 1 μmol/L) after equilibration and myogenic tone development, as previously reported (Raignault et al. 2017); then, dilatory responses were tested with a single cumulative concentration response curve to acetylcholine (Ach, Sigma-Aldrich; 10−13 mol/L to 10−4 mol/L). Responses to Ach were expressed as percentage of change in vessel diameter from pre-constriction tone.
Compliance of carotid and cerebral arteries
Reactivity and compliance studies were performed on different arterial segments from the ipsilateral hemisphere. Passive pressure-diameter curves were conducted in a Ca2+-free PSS containing 1 mM of EGTA to abolish myogenic tone and to assess the mechanical properties of the arteries (Bolduc et al. 2011; de Montgolfier et al. 2019). Internal and external diameter changes were measured after each increment of intraluminal pressure (from 10 to 120 mmHg with a first 10-mmHg step followed by 20-mmHg steps for cerebral arteries and from 60 to 180 mmHg with 20-mmHg steps for carotid arteries), to calculate mechanical parameters. The circumferential wall strain (strain, %) was calculated according to [(D − Dinitial mmHg) / Dinitial mmHg], where D is the internal diameter at a given pressure and Dinitial mmHg is the initial diameter at the initial pressure (10 mmHg for cerebral arteries and 60 mmHg for carotid arteries) as previously described (Bolduc et al. 2011; de Montgolfier et al. 2019).
Arterial remodeling
Arterial remodeling was measured in isolated pressurized carotid and MCA segments used for the reactivity studies, and it was assessed by measuring the change in wall thickness (μm), as previously described (de Montgolfier et al. 2019).
Perfusion with Evans blue dye for brain barrier permeability
Evans blue perfusion was performed as previously described (de Montgolfier et al. 2019). Briefly, mice were anesthetized with isoflurane, the thoracic cavity was opened, and the cardiac perfusion was performed first using PBS, followed by Evans blue cocktail (1% of Evans Blue in 4% PFA). Then, mice were decapitated and the brains were rapidly removed, post-fixed 4 h in 4% PFA, cryoprotected in 30% sucrose overnight at 4 °C, snap frozen in liquid nitrogen, and stored at − 80 °C. Cryotome 20-μm-thick coronal brain sections were fixed in 4% PFA for 1 h at RT (room temperature). After rinsing twice with PBS, slides were incubated for 20 min at RT in DAPI solution for nuclei staining (1:600, Thermo Fisher Scientific, Saint-Laurent, QC, Canada). After washing, slides were covered by mounting medium and coverslips for imaging. Blood-brain barrier permeability of the ipsilateral hemisphere was visualized and analyzed as Evans Blue fluorescence (excitation at 543 nm and emission at 680 nm) extravasation from microvessels in the parenchyma (de Montgolfier et al. 2019).
Histology and immunostaining
Coronal 30-μm-thick cryosections were mounted on glass slides (superfrost Plus, Fisherbrand, USA) and dried overnight. Four sections per mouse, located at + 1.32, − 0.82 (cortex), − 1.64 and − 2.92 (hippocampus and cortex) from Bregma were analyzed. Fluorescence was visualized using confocal microscopy (Zeiss LSM 510; Carl Zeiss, Dorval, QC, Canada). All images were analyzed with the ImageJ software (version 2.0, NIH).
Microvessel density
Fixed brain sections were washed with PBS and incubated at 4 °C for 24 h in PBS containing the polyclonal rabbit anti-collagen IV antibody, (1:100, Abcam ab19808, Abcam Inc., Toronto, ON, Canada) supplemented with 1% BSA). Collagen IV is an established marker of the vessel basal lamina (Schmidt and von Hochstetter 1995). After washing, slides were incubated at RT for 2 h in PBS containing Alexa Fluor 647 donkey anti-rabbit IgG (1:500, Abcam) and DAPI solution (1:600, Thermo Fisher Scientific), supplemented with 1% BSA. Finally, slides were washed with PBS and covered by mounting medium and coverslips for imaging using confocal microscopy. Microvessel density was quantified as a percentage of collagen IV positive area per region of interest (ROI).
Astrogliosis
Slides were incubated at 4 °C for 24 h in PBS containing the polyclonal rabbit anti-GFAP antibody, (1:1200, Abcam ab7260) supplemented with 1% BSA. After rinsing with PBS, slides were incubated at RT for 2 h in PBS containing Alexa Fluor 488 donkey anti-rabbit IgG (1:1600, Abcam) and DAPI solution (1:600, Thermo Fisher Scientific), supplemented with 1% BSA. After rinsing twice with PBS for 5 min, slides were covered by mounting medium and coverslips for imaging using confocal microscopy. Astrocyte reactivity was quantified as a percentage of GFAP-positive area per ROI.
Dihydroethidium staining
After fixation in 4% PFA, brain slides were incubated with 5 μM dihydroethidium (DHE) solution (Sigma-Aldrich) and DAPI solution (1:600, Thermo Fisher Scientific) in a light-protected humidified chamber at 37 °C for 30 min, covered with mounting medium and coverslips for imaging using confocal microscopy. Reactive oxygen species production and damage was quantified as a percentage of DHE relative fluorescence.
Prussian blue staining for micro-hemorrhage
Prussian blue staining was performed as previously described (de Montgolfier et al. 2019). Brain sections were fixed in 4% PFA. After rinsing twice with PBS for 5 min, slides were incubated for 30 min at RT in the fume hood with freshly mixed 4% potassium ferrocyanide with 4% hydrochloric acid. Slides were rinsed under running tap water for 30 s. Next, slides were counter-stained using nuclear fast red at RT for 5 min, rinsed under running tap water for 1 min, dehydrated quickly in 95% and 100% ethanol, cleared in xylene, and covered by mounting medium and coverslips for imaging (Olympus BX45, Montreal, QC, Canada). The number of events per section was counted.
Total RNA extraction and real-time quantitative polymerase chain reaction
Total RNA was extracted from ipsilateral hemispheres of both the microvessels and the parenchymal fraction using the RNeasy mini-kit (Qiagen) and RNeasy Lipid Tissue Mini Kit (Qiagen), respectively, according to the manufacturer’s protocol. Total RNA was quantified using a NanoDrop ND-100 spectrophotometer (Thermo Fisher Scientific). Total RNA was reverse transcribed into first-strand complementary DNA with M-MLV reverse transcriptase (Thermo Fisher Scientific, Invitrogen) using random hexamer primers. The qPCR reactions were carried out on diluted reverse transcribed products by using the DNA-binding dye SYBR Green PCR Master Mix (Thermo Fisher Scientific) to detect PCR products with StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). The primers of target genes (Tnfα, Il1β, Il6, Icam-1, Gfap, p21, p16, Sod2, Nox2, Nox4, Bax, Bcl2, Zo-1, Ocln, Cldn5, Pdgfrβ, Syp, Psd-95, Bdnf, NeuN, CycloA) were designed using Clone Manager software (Table 1). All samples were run in duplicate and the fold changes in gene expression were calculated by the ΔΔCT method using cyclophilin A as the housekeeping gene.
Table 1.
List of primers used for real-time quantitative PCR
| Target gene | Primers | Sequences |
|---|---|---|
| Tnfα | Forward | AAGCCTGTAGCCCACGTCGTA |
| Reverse | GGCACCACTAGTTGGTTGTCTTTG | |
| Il1β | Forward | TGCCACCTTTTGACAGTGATG |
| Reverse | TGATGTGCTGCTGCGAGATT | |
| Il6 | Forward | GGATACCACTCCCAACAGACC |
| Reverse | TTCTGCAAGTGCATCATCGT | |
| Icam-1 | Forward | CAATTCACACTGAATGCCAGCTC |
| Reverse | CAAGCAGTCCGTCTCGTCCA | |
| Gfap | Forward | CGAGATCGCCACCTACAGGA |
| Reverse | CGGATCTGGAGGTTGGAGAA | |
| p21 | Forward | TGTCGCTGTCTTGCACTCT |
| Reverse | AGACCAATCTGCGCTTGGA | |
| p16 | Forward | GTTCTTGGTCACTGTGAGGATTCAG |
| Reverse | CCATCATCATCACCTGGTCCAG | |
| Sod2 | Forward | ACCGAGGAGAAGTACCACGA |
| Reverse | TGGGTTCTCCACCACCCT TA | |
| Nox2 | Forward | AGTGCCCAGTACCAAAGT TC |
| Reverse | GTCCCACCTCCATCTTGAATC | |
| Nox4 | Forward | ATCCTTTTACCTATGTGCCGG |
| Reverse | CTTTCTGGGATCCTCATTCTGG | |
| Bax | Forward | AGCAAACTGGTGCTCAAGGC |
| Reverse | CCACAAAGATGGTCACTGTC | |
| Bcl2 | Forward | GTGGTGGAGGAACTCTTCAG |
| Reverse | GTTCCACAAAGGCATCCCAG | |
| Zo-1 | Forward | TGTCCCTGTGAGTCCTTCAG |
| Reverse | ACTCAACACACCACCATTGC | |
| Ocln | Forward | GCGGAAAGAGTTGACAGTCC |
| Reverse | CTTCAGGCACCAGAGGTGTT | |
| Cldn5 | Forward | TCGGGTGAGCATTCAGTCTT |
| Reverse | GTCACGATGTTGTGGTCCAG | |
| Pdgfrβ | Forward | GGGAGATCTTCACACTGGGT |
| Reverse | CATAGATCTCGTCGGAGGCA | |
| Syp | Forward | AGGGTGGTTATCAACCCGAT |
| Reverse | TGGGCTTCACTGACCAGATT | |
| Psd-95 | Forward | CCTGTGTCCCTCATACGACA |
| Reverse | TACTGGCCAGCCTCAATGAA | |
| Bdnf | Forward | AAGCCGCAAAGAAGTTCCAC |
| Reverse | CTTGTCCGTGGACGTTTACT | |
| NeuN | Forward | CACAGCCCATTGCTGGGA |
| Reverse | TGAACCGGAAGGGGATGTTG | |
| CycloA | Forward | CCGATGACGAGCCCTTGG |
| Reverse | GCCGCCAGTGCCATTATG |
Statistical analysis
Statistical analyses were performed with the software Prism (Prism 7.0, GraphPad, San Diego, CA, USA). Statistical analysis of behavioral tests was performed with the program Smart (Smart 3.0, Panlab). Data are expressed as means ± standard error of the mean (SEM). All group comparisons were analyzed using two-way ANOVA; Sidak’s multiple comparisons tests were performed when the interaction between “genotype” (WT versus ATX) and “surgery” (sham versus TAC) was significant (p < 0.05). Experimenters were not blinded to groups during data analysis except for confocal image analyses. N numbers reflects the number of mice included in each experiment. Group sizes were determined according to previous studies (Bolduc et al. 2011; de Montgolfier et al. 2019; Drouin et al. 2011a; Drouin et al. 2011b).
Results
Mild hypertension, increased pulse pressure, and carotid stiffness in ATX mice are associated with cognitive impairment
When compared to sham-WT mice, 1-year-old sham-ATX mice, exhibiting 5-fold higher cholesterol levels and ≈ 70% aortic atherosclerotic plaque (Drouin et al. 2011b), are characterized by mild systolic and diastolic hypertension, a high pulse pressure (Fig. 3a), and carotid stiffening illustrated by a thickening of the carotid wall and a decrease in compliance (Fig. 3b). As expected, TAC surgery increased heart/body weight ratio after 6 weeks of aortic constriction in both WT and ATX mice (Fig. 1b). Of note, ATX mice exhibited a higher (p < 0.05) mortality rate after surgery than WT mice (22% operative mortality rate in sham- and TAC-ATX mice vs. 9% in sham- and TAC-WT mice, p < 0.05; the surviving mice were all healthy) (Fig. 1c). In addition, TAC increased ipsilateral (right side, i.e., the ipsi side to TAC-induced pressure overload) carotid systolic (but not diastolic) and pulse pressures in both WT and ATX mice (Fig. 3a). TAC increased wall thickness and reduced compliance of carotids in WT mice but did not further alter these parameters in ATX mice (Fig. 3b). In ipsilateral cerebral arteries, TAC surgery tended (p = 0.078) to increase MCA wall thickness in WT mice and to a lesser extent in ATX mice (Fig. 4a), suggesting that TAC-induced pressure overload propagated to the brain vasculature. Arterial compliance measured in MCA was not different between WT and ATX mice and was not affected by TAC (Fig. 4b). In addition, wall thickness in MCA of sham-ATX mice was similar to that of sham-WT mice (Fig. 4a), confirming the lack of intimal thickening and atherosclerosis in these intracranial arteries. Indeed, an apparent “protection” of intracranial arteries from atherosclerosis has been reported based on the fact that atherosclerotic lesions are rarely observed in cerebral arteries from animal models (Didion et al. 2001; Drouin et al. 2011b; Stewart-Lee and Burnstock 1991).
Fig. 3.

ATX mice display mild hypertension, carotid wall thickening, and stiffening. a Systolic (left), diastolic (middle), and pulse (right) pressures measured by Millar catheter in the ipsilateral carotid of sham- (n = 4) and TAC-WT (n = 7) and sham- (n = 4) and TAC-ATX (n = 5) mice. b Wall thickness (left) and compliance (right) of ipsilateral carotid arteries isolated from WT (n = 6–8) and ATX (n = 6) mice. Data are mean ± SEM *p < 0.05 vs. WT mice; $p < 0.05 vs. sham mice (within the corresponding genotype)
Fig. 4.

a Wall thickness and b compliance of ipsilateral middle cerebral arteries isolated from WT (n = 4–6) and ATX mice (n = 4). Data are mean ± SEM
Regarding cognitive abilities, the percentage of alternation behavior in the Y maze was significantly decreased in 1-year-old sham-ATX mice (Fig. 5a) compared to age-matched sham-WT mice, suggestive of a premature dysfunctional working memory. TAC-induced pressure overload promoted a reduction in the percentage of alternation behavior in WT mice and worsened dysfunctional working memory in ATX mice that failed the Y maze test (Fig. 5a) (the percent of alternation behavior of TAC-ATX mice was not statistically different from 50%). In addition, compared to sham-WT mice, sham-ATX mice showed longer latency to reach the hidden platform during the learning phase of the MWM test (Fig. 5b) and spent significantly less time in the target quadrant during the probe test (Fig. 5c), revealing learning and memory deficiencies. TAC reduced learning and memory capacities in WT mice as evidenced by a significantly increased latency (Fig. 5b) and a decrease in the time spent in the target quadrant (Fig. 5c). In ATX mice, TAC did not further exacerbate the already low cognitive capacities (Fig. 5b, c).
Fig. 5.

Mild hypertension and vascular stiffening in ATX mice are associated with cognitive impairment in learning and memory abilities. a Evaluation of the working memory ability by the spontaneous exploratory behavior using the Y-maze test in WT (n = 12–13) and ATX (n = 9–12) mice. b Evaluation of the learning and spatial memory capacities of WT (n = 12–13) and ATX (n = 12–14) mice using the Morris Water Maze test and c the probe test. Data are mean ± SEM; *p < 0.05 vs. WT mice; $p < 0.05 vs. sham mice (within the corresponding genotype); †p < 0.05 vs. 50% alternation in the Y maze; †p < 0.05 vs. day 1 in the MWM test
Of note, in the sham-WT control group, the alternation behavior percentage was statistically different from 50% in the Y maze behavioral test (Fig. 5a) and the latency to reach the platform decreased significantly between day 1 and day 5 in the MWM test (Fig. 5b), validating both tests. No change in the number of arm entries in the Y maze (right panel in Fig. 5a) nor in swim speed in the MWM test (right panel in Fig. 5c) were noticed between groups of mice, suggesting that neither the surgery nor the genotype had an impact on motor functions and anxiety.
Altogether, these data show that cardiovascular risk factors are associated with memory and learning impairment in mice, and that pressure overload induces (in WT mice) or exacerbates (in ATX mice) these cognitive dysfunctions.
Brains from ATX mice display a decrease in volume of specific areas involved in learning and memory
Anatomical MRI scans revealed that sham-ATX mice exhibited differences in brain volumes compared to sham-WT mice: although no change in total brain volume was recorded between groups, ATX mice displayed smaller volumes in the hippocampus, cortex (entorhinal, occipital, parietotemporal), and septum, while the volume of the corpus callosum increased significantly in ATX mice (Table 2). No change in gray or white matter volumes was recorded in WT and ATX mice 6 weeks post-TAC (data not shown).
Table 2.
Comparison of anatomical brain volumes in sham WT and ATX mice measured by 7-T MRI
| Normalized brain area volumes | WT (n = 11) | ATX (n = 12) | % of change | p value |
|---|---|---|---|---|
| Total brain | 1.000 ± 0.000 | 1.000 ± 0.000 | 0.00 | 0.600 |
| Amygdala | 0.110 ± 0.003 | 0.112 ± 0.003 | 1.82 | 0.026 |
| Cerebellum | 0.410 ± 0.016 | 0.414 ± 0.009 | 0.98 | 0.559 |
| Corpus callosum | 0.097 ± 0.007 | 0.113 ± 0.004 | 16.49 | < 0.0001 |
| Cortex | 0.982 ± 0.005 | 0.968 ± 0.004 | − 1.43 | < 0.0001 |
| Deep gray matter | 1.333 ± 0.054 | 1.332 ± 0.021 | − 0.08 | 0.934 |
| Entorhinal cortex | 0.077 ± 0.002 | 0.072 ± 0.002 | − 6.49 | < 0.0001 |
| Frontal cortex | 0.301 ± 0.004 | 0.300 ± 0.005 | − 0.33 | 0.621 |
| Hippocampus | 0.195 ± 0.004 | 0.190 ± 0.007 | − 2.56 | 0.018 |
| Occipital cortex | 0.049 ± 0.002 | 0.046 ± 0.002 | − 6.12 | 0.002 |
| Olfactory bulb | 0.245 ± 0.010 | 0.244 ± 0.004 | − 0.41 | 0.621 |
| Parietotemporal cortex | 0.555 ± 0.007 | 0.549 ± 0.005 | − 1.08 | 0.020 |
| Septum | 0.030 ± 0.001 | 0.029 ± 0.001 | − 3.33 | 0.033 |
| White matter | 0.239 ± 0.011 | 0.246 ± 0.016 | 2.93 | 0.231 |
ATX mice were smaller than WT mice but brain weight-to-body weight ratio was identical in WT (0.012 ± 0.000 g/g) and ATX (0.012 ± 0.000 g/g) mice. Each volume from the brain region of interest was normalized to the total brain volume within the corresponding genotype to avoid bias in change in cerebral volume due to smaller body size. Data are mean ± SEM. p < 0.05 vs. WT mice
ATX mice display cerebral hypoperfusion, cerebral endothelial dysfunction, loss of microvessel density, and brain micro-hemorrhages
Sham-ATX mice exhibited a significant decrease in cerebral perfusion in the whole brain and in cortical and hippocampal areas, compared to sham-WT mice (Fig. 6). The TAC surgery significantly reduced cerebral perfusion in the ipsilateral side of the brain in WT mice, but had no additive effect in ATX mice (Fig. 6).
Fig. 6.

ATX mice display brain hypoperfusion. Quantification of ipsilateral cerebral perfusion (mL/g/min), assessed in vivo by 7-T MRI in sham-(n = 8) or TAC-WT (n = 10) and sham- (n = 10) and TAC-ATX (n = 8) mice. Typical 7-T MRI images are shown
Endothelium-dependent dilations to acetylcholine measured in isolated MCA were decreased in sham-ATX mice compared to sham-WT mice (Fig. 7a). TAC impaired endothelium-dependent dilation in WT mice and further reduced dilation in MCA isolated from the ipsilateral hemisphere of ATX mice (Fig. 7a). On the other hand, smooth muscle contractility to phenylephrine was similar among groups but tended to increase after TAC (Fig. 7b).
Fig. 7.

ATX mice display cerebral endothelial dysfunction, loss of microvascular density, and micro-hemorrhages. a Endothelium-dependent dilatory responses (%) to acetylcholine (ACh) were measured in ipsilateral middle cerebral arteries isolated from both WT (n = 5–8) and ATX (n = 5) sham and TAC mice. b Levels of pre-contraction induced by phenylephrine in ipsilateral middle cerebral arteries isolated from WT (n = 4–6) and ATX mice (n = 4). c Cerebral microvessel density (% of positive staining per region of interest, ROI) was assessed by immunohistochemistry staining of collagen IV in brain coronal cryosections in the ipsilateral side of the cortex and hippocampal areas from WT (n = 6) and ATX (n = 6) sham and TAC mice. Typical confocal microscope fluorescent images are shown; scale bar = 100 μm, for all images. d The presence of micro-bleeds (capillary hemorrhages) was visualized by Prussian blue staining of coronal slices in the ipsilateral side of the brain from both WT (n = 3) and ATX (n = 3) sham and TAC mice. Typical images are shown; scale bar = 0.5 mm, for all images. Data are mean ± SEM. *p < 0.05 vs. WT mice; $p < 0.05 vs. sham mice (within the corresponding genotype)
The density of cortical and hippocampal microvessels, assessed by collagen IV immunostaining, was reduced in sham-ATX compared to sham-WT mice (Fig. 7c). TAC reduced microvessels density in the ipsilateral cortex and hippocampus of WT mice and accentuated the loss of microvessels in ATX mice (Fig. 7c). No Prussian blue-positive staining, a marker of micro-hemorrhages, was observed in sham-WT mice (Fig. 7d); in contrast, the presence of some micro-bleeds was detected in the cortex, but not the hippocampus, of sham-ATX mice (Fig. 7d). TAC surgery induced micro-bleeds in the ipsilateral cortex of WT mice and accentuated their number in ATX mice (Fig. 7d).
The cerebral vasculature of ATX mice is characterized by senescent markers and an increase in blood-brain barrier permeability
Gene expression of the inflammatory markers Tnfα and Il1ß and of the oxidative stress marker Nox4 tended to increase in microvessels from the right ipsilateral hemisphere of sham-ATX mice compared to sham-WT mice (Table 3). Expression of the senescent marker p16 was significantly increased in sham-ATX compared to sham-WT mice (Table 3). There was no change in mRNA expression of the anti-apoptotic Bcl2 or pro-apoptotic Bax between WT and ATX mice (Table 3).
Table 3.
Expression of inflammatory, oxidative, senescent, apoptotic, and synaptic gene markers in the ipsilateral hemisphere microvasculature of WT and ATX mice
| WT mice (n = 12) | ATX mice (n = 12) | |||
|---|---|---|---|---|
| Sham (n = 6) | TAC (n = 6) | Sham (n = 6) | TAC (n = 6) | |
| Tnfα | 1.00 ± 0.04 | 1.20 ± 0.21 | 1.72 ± 0.15 | 2.37 ± 0.45 *,$ |
| Il1ß | 1.00 ± 0.07 | 1.90 ± 0.43 | 1.74 ± 0.30 | 1.58 ± 0.15 |
| Il6 | 1.00 ± 0.03 | 1.31 ± 0.11 | 1.37 ± 0.14 | 2.00 ± 0.42 $ |
| Icam-1 | 1.00 ± 0.04 | 1.35 ± 0.14 $ | 1.15 ± 0.06 | 1.45 ± 0.13 |
| Sod2 | 1.00 ± 0.05 | 0.71 ± 0.09 | 0.93 ± 0.02 | 0.73 ± 0.10 |
| Nox2 | 1.00 ± 0.15 | 1.65 ± 0.19 $ | 1.44 ± 0.18 | 1.92 ± 0.12 |
| Nox4 | 1.00 ± 0.22 | 1.70 ± 0.40 | 1.70 ± 0.22 | 3.59 ± 0.62 *,$ |
| p21 | 1.00 ± 0.07 | 0.35 ± 0.04 $ | 0.71 ± 0.08 | 0.77 ± 0.14 |
| p16 | 1.00 ± 0.03 | 0.42 ± 0.10 $ | 1.39 ± 0.07 * | 0.48 ± 0.09 $ |
| Bcl2 | 1.00 ± 0.08 | 0.66 ± 0.09 | 1.03 ± 0.08 | 0.63 ± 0.05 $ |
| Bax | 1.00 ± 0.01 | 1.47 ± 0.20 $ | 1.06 ± 0.05 | 1.38 ± 0.07 |
| Bax/Bcl2 | 1.00 ± 0.06 | 2.44 ± 0.43 $ | 1.00 ± 0.06 | 2.19 ± 0.28 $ |
| Zo-1 | 1.00 ± 0.08 | 0.52 ± 0.04 $ | 1.05 ± 0.10 | 0.83 ± 0.05 |
| Occludin | 1.00 ± 0.11 | 0.37 ± 0.09 $ | 0.77 ± 0.08 | 0.42 ± 0.07 $ |
| Claudin-5 | 1.00 ± 0.04 | 0.55 ± 0.07 | 1.66 ± 0.24 * | 0.63 ± 0.08 $ |
| Pdgfrß | 1.00 ± 0.03 | 0.55 ± 0.13 $ | 0.86 ± 0.07 | 0.58 ± 0.07 $ |
Data are mean ± SEM. *p < 0.05 vs. WT; $p < 0.05 vs. sham (within the corresponding genotype)
In the ipsilateral microcirculation of WT mice, TAC surgery induced an inflammatory (higher Icam1 expression) and oxidative stress (higher Nox2 expression) response, while the markers of senescence were reduced (lower p16 and p21 expression) and paralleled by an increase in Bax/Bcl2 ratio suggestive of apoptosis (Table 3). In ATX mice, TAC further increased inflammation (higher Tnfα and Il6 expression), oxidative stress (higher Nox4 expression), while reducing senescence and activating apoptosis (Table 3).
To further characterize the impact of vascular risk factors on the NVU, we next looked at the integrity of the blood-brain barrier (BBB). Transcript levels of brain endothelial tight junction proteins Zo-1 and Occludin were similar between sham-ATX and sham-WT mice, whereas the expression of Claudin-5 was significantly greater in ATX mice; mRNA expression of the pericyte marker Pdgfrß was similar in the two groups (Table 3). In addition, fluorescence microscopy brain images from sham-WT mice showed the presence of Evans blue dye only in blood vessels (Fig. 8), whereas Evans blue staining in sham-ATX mice was also detected in the brain parenchyma (in both the cortex and hippocampus) (Fig. 8), demonstrating leakage of the dye across the BBB suggestive of an increased BBB permeability. TAC surgery induced disruption of the BBB in WT mice as shown by Evans blue extravasation in the cortex (but not in the hippocampus) (Fig. 8), and by the decrease in transcript levels of the tight junction proteins Zo-1, Occludin, and Claudin-5, and of Pdgfrß mRNA expression (Table 3). Similarly, TAC in ATX mice reduced gene expression of the tight junction proteins (Table 3) and accentuated Evans blue diffusion into the parenchyma (Fig. 8), demonstrating that TAC-induced pressure overload worsened BBB dysfunction and leakage in ATX mice.
Fig. 8.

ATX mice exhibit an increase in BBB permeability. Representative images of Evans blue extravasation (% of Evans blue fluorescence/ROI) in the ipsilateral brain parenchyma of sham- and TAC-WT and -ATX mice are shown; scale bar = 50 μm, for all images. Data of sham-WT (n = 5) and sham-ATX (n = 5) mice and TAC-WT (n = 3) and TAC-ATX (n = 3) are summarized as mean ± SEM. *p < 0.05 vs. WT mice; $p < 0.05 vs. sham mice (within the corresponding genotype)
Brain tissues of ATX mice are characterized by an increase in inflammatory and oxidative markers and by the loss of synaptic and cognitive markers
Immunostaining of the astrocytic protein GFAP in the cortex and the hippocampus was similar in sham-ATX compared to sham-WT mice (Fig. 9a), but Gfap and Il6 transcript levels were increased in the parenchyma of sham-ATX compared to sham-WT mice (Table 4). Staining of the DHE superoxide marker revealed an increase in brain superoxide anion levels in the cortex and hippocampus of sham-ATX compared to sham-WT mice (Fig. 9b). In addition, Nox2 mRNA expression was greater in the brain parenchyma of sham-ATX mice compared to sham-WT mice (Table 4). There was no change in the expression of the senescence or apoptotic markers in sham-ATX compared to sham-WT mice (Table 4). Expressions of synaptophysin (Syp) and brain-derived neurotrophic factor (Bdnf) transcripts were decreased in sham-ATX compared to sham-WT mice (Table 4).
Fig. 9.

ATX mice exhibit an increase in inflammation and oxidative stress in the brain. a Immunostaining of GFAP-positive cells (% of positive staining per region of interest, ROI) in the ipsilateral cortex and hippocampus of sham- and TAC-WT (n = 4) and sham- and TAC-ATX (n = 4) mice. Typical images are shown; scale bar = 100 μm, for all images. b Immunostaining of DHE fluorescence (relative fluorescence units) in the ipsilateral cortex and hippocampus of WT (n = 4–5) and ATX (n = 5–6) sham and TAC mice. Typical images are shown; scale bar = 50 μm, for all images. Data are mean ± SEM. *p < 0.05 vs. WT mice; $p < 0.05 vs. sham mice (within the corresponding genotype)
Table 4.
Expression of inflammatory, oxidative, senescence, apoptotic, and synaptic gene markers in the ipsilateral hemisphere parenchyma of WT and ATX mice
| WT (n = 12) | ATX (n = 12) | |||
|---|---|---|---|---|
| Sham (n = 6) | TAC (n = 6) | Sham (n = 6) | TAC (n = 6) | |
| Tnfα | 1.00 ± 0.02 | 1.54 ± 0.20 $ | 1.14 ± 0.02 | 1.62 ± 0.19 |
| Il1ß | 1.00 ± 0.09 | 1.72 ± 0.22 $ | 1.23 ± 0.11 | 1.50 ± 0.11 |
| Il6 | 1.00 ± 0.03 | 1.19 ± 0.14 | 1.43 ± 0.14 * | 0.78 ± 0.13 *,$ |
| Gfap | 1.00 ± 0.11 | 1.31 ± 0.10 | 1.54 ± 0.07 * | 1.65 ± 0.06 |
| Sod2 | 1.00 ± 0.12 | 0.89 ± 0.08 | 0.81 ± 0.12 | 0.58 ± 0.07 * |
| Nox2 | 1.00 ± 0.07 | 1.59 ± 0.11 $ | 1.30 ± 0.06 * | 1.68 ± 0.09 $ |
| Nox4 | 1.00 ± 0.08 | 1.81 ± 0.32 $ | 1.52 ± 0.21 | 1.43 ± 0.06 |
| p21 | 1.00 ± 0.01 | 1.35 ± 0.12 $ | 1.19 ± 0.06 | 1.37 ± 0.10 |
| p16 | 1.00 ± 0.07 | 1.47 ± 0.17 $ | 1.01 ± 0.15 | 0.55 ± 0.13 *,$ |
| Bcl2 | 1.00 ± 0.20 | 0.80 ± 0.18 | 0.71 ± 0.14 | 0.93 ± 0.21 |
| Bax | 1.00 ± 0.10 | 1.04 ± 0.14 | 0.93 ± 0.17 | 0.94 ± 0.10 |
| Bax/Bcl2 | 1.00 ± 0.21 | 1.23 ± 0.19 | 1.09 ± 0.17 | 0.92 ± 0.19 |
| Syp | 1.00 ± 0.05 | 0.69 ± 0.07 $ | 0.64 ± 0.11 * | 0.50 ± 0.03 |
| Psd-95 | 1.00 ± 0.12 | 1.15 ± 0.21 | 1.22 ± 0.03 | 0.63 ± 0.14 *,$ |
| NeuN | 1.00 ± 0.04 | 0.79 ± 0.06 | 0.78 ± 0.11 | 0.81 ± 0.12 |
| Bdnf | 1.00 ± 0.07 | 0.83 ± 0.07 | 0.67 ± 0.04 * | 0.62 ± 0.10 |
Data are mean ± SEM. *p < 0.05 vs. WT; $p < 0.05 vs. sham (within the corresponding genotype)
In WT mice, TAC induced ipsilateral brain inflammation (increased GFAP staining (Fig. 9a), higher Tnfα and Il1β expression (Table 4)), senescence (higher p21 and p16 expression (Table 4)), and oxidative stress (increased DHE staining (Fig. 9b) and higher Nox2 and Nox4 expression (Table 4)) and reduced Syp expression (Table 4) compared to sham-WT. In ATX mice, TAC increased ipsilateral brain inflammation (increased GFAP staining (Fig. 9a)), further increased oxidative stress and damage (higher Nox2 expression, lower Sod2 expression (Table 4) and increased DHE staining (Fig. 9b)) and lowered senescence (lower p16 expression (Table 4)) compared to sham-ATX. TAC also induced a decreased in the expression of the Psd-95 synaptic marker (Table 4).
Discussion
This study aimed to demonstrate the causal contribution of vascular risk factors on the incidence and the progression of cognitive decline in middle-aged mice. We found that 1-year-old ATX mice that cumulate dyslipidemia, aortic plaque, mild hypertension, central vascular stiffness and elevated pulse pressure exhibit early cognitive impairment. We also found that ATX mice are characterized by multiple cerebrovascular dysfunctions: compared to WT mice, whole brain hypoperfusion, in particular in the regions of the cortex and the hippocampus, micro-hemorrhages and loss of microvessels were observed in association with cerebrovascular endothelial dysfunction and increased BBB permeability. Such cerebrovascular dysfunctions combined with vascular and parenchymal inflammation and oxidative stress could, altogether, reflect a disruption in the NVU and promote brain atrophy. In parallel, induction of isolated systolic hypertension by a TAC surgery in 1-year-old WT mice promoted cerebrovascular and brain damages reminiscent of the neurovascular phenotype of naïve ATX mice and induced cognitive dysfunction; these damages were magnified in TAC-ATX mice. Altogether, our data suggest that the critical step in NVU deregulation that lead to cognitive decline is the cerebrovascular injury generated by hemodynamic abnormalities induced by the isolated systolic hypertension (TAC model) and magnified in the presence of vascular risk factors (ATX mice).
Vascular aging in humans is characterized by endothelial dysfunction and by stiffening of large arteries, systolic hypertension, and increased pulse pressure (Fleg and Strait 2012). Stiffness of conductance arteries such as the carotid artery is strongly associated with cognitive decline in humans (Iadecola et al. 2016; O’Rourke and Safar 2005). Accordingly, stiffening of carotid arteries associated with intimal thickening was observed in our model of atherosclerosis in ATX mice; interestingly, this remodeling did not affect the pial cerebral arteries of ATX mice at this age as suggested by the normal MCA wall thickness compared to WT mice. Endothelial dysfunction in the MCA, however, was present in ATX mice and was accentuated by TAC surgery, suggesting that an impaired endothelium is present before intimal thickening. Endothelial dysfunction is prematurely found in subjects with risk factors for CVD (Cai and Harrison 2000) and in patients with neurodegenerative diseases such as AD (Katusic and Austin 2014). Accordingly, ATX mice develop prematurely the two hallmarks of vascular aging, endothelial dysfunction and carotid stiffening that may predispose them to early cognitive decline.
Microvessel rarefaction occurs in aging (Farkas and Luiten 2001) and is premature with CVD (Moore et al. 2015). A recent study demonstrates that BBB impairment is prodromal of cognitive dysfunction in humans (Nation et al. 2019). The disturbance in BBB function combined with microvessel rarefaction may contribute to cerebral hypoperfusion and cognitive decline observed in the brain of ATX mice and of WT-TAC mice. In ATX mice, TAC surgery worsened cerebral endothelial dysfunction and BBB disruption and augmented the incidence of micro-hemorrhages and loss of microvessels, accelerating memory and learning deficiencies. These damaging effects of TAC on NVU demonstrate that a vascular hemodynamic component directly contributes to cognitive decline, as we recently proposed (de Montgolfier et al. 2019); similar deleterious effects on NVU are observed in hypertensive atherosclerotic ATX mice, suggesting that the process could be promoted by hypertension, a strong vascular risk factor for brain injury (Csiszar et al. 2017; Tucsek et al. 2017).
The deterioration of the cerebral endothelial function leads not only to lower dilatory and barrier functions but also to a reduced anti-inflammatory function and antioxidant capacities (Toth et al. 2017; Toth et al. 2013). These changes were evident in the brain of ATX mice: expression of inflammatory markers increased while oxidative stress-related damages accumulated in the brain. The latter were associated with a tendency of increased expression of free radical-generating enzymes in the microcirculation, and a clear increased expression in the parenchymal fraction of the brain in ATX mice. Hence, brains of middle-age ATX mice are suffering from a pro-atherogenic environment, a pro-inflammatory and pro-oxidant phenotype that can be mimicked in TAC-WT mice and accentuated in TAC-ATX mice. The aging brain exhibit a pro-inflammatory state mostly through microglial and astrocytic activation revealed by the production of pro-inflammatory cytokines (Godbout and Johnson 2009). For example, marker of activated astrocytes GFAP is higher in the brain of old humans and rodents (Godbout and Johnson 2009). In many neurological diseases, a common feature of NVU injury is glial activation (Iadecola and Nedergaard 2007). Inflammation is associated with ROS production and neurons are particularly sensitive to excessive oxidative stress, which lead to neuronal damage (Uttara et al. 2009). We previously reported that an early intervention with an antioxidant treatment prevented endothelial dysfunction and the rise in inflammation and oxidative stress in ATX mice (Drouin et al. 2011b; Gendron et al. 2010), which was associated with a normalization of NVU and cognitive functions (Drouin et al. 2011a).
The defects in neurovascular functions detected in sham-ATX mice were associated with a decrease in the volume of specific regions of the brain, mostly in areas implicated in learning and memory processes. It is known, indeed, that the size of the brain decreases with age (Harada et al. 2013) and affects both white and gray matter regions (Salat et al. 1999). A drop in volume is most prominent in the prefrontal cortex and moderate in the hippocampus (Fotuhi et al. 2012). The loss in white matter strongly correlates with hypertension and stroke (Kennedy and Raz 2009), and CVD have been linked to hippocampus atrophy, which is prodromal to mild cognitive impairment, AD, and dementia (Fotuhi et al. 2012). Nonetheless, this structural change cannot fully explain the decline in cognitive functions since 6 weeks of TAC surgery induced a significant loss of cognitive functions without affecting brain volume neither in ATX nor in WT mice; hence, volume restriction is likely the consequence of chronic stress on the brain vessels and parenchyma. Brain atrophy is linked to structural changes and loss of synapses in the nervous system is a key structural marker of aging (Masliah et al. 1993), of hypertension (Tucsek et al. 2017), and of neurodegenerative diseases (Bishop et al. 2010). We observed a decrease in synaptophysin mRNA expression in the parenchymal brain fraction of ATX mice, suggestive of a reduced synaptic density. Moreover, Bdnf expression, a neurotrophic and survival factor required in learning mechanisms (Kaplan and Miller 2000) that is reduced with aging and in AD (Nagahara et al. 2009), was also decreased in ATX mice. In addition, TAC worsened inflammation and oxidative stress of the parenchyma of ATX mice. Collectively, these changes would disrupt cell-to-cell signaling in the NVU and induce hypoperfusion, leading to chronic neuronal stress; the chronic damage likely translates into a loss of glial cells and neurons that lead with time to cognitive decline and to smaller volume of highly “used” areas of the brain.
The mechanism underlying chronic NVU dysfunction in aging ATX mice could be the progressive accumulation of vascular senescent cells. Senescent cells accumulate with aging and contribute to the general decline of the physiological functions (Baker et al. 2016) and to the development of neurological (Bussian et al. 2018) and vascular (Childs et al. 2016) diseases. In microvessels of ATX mice, p16 gene expression was higher, suggestive of the presence of senescent cells (Kuilman et al. 2010). In contrast, after TAC surgery in both ATX and WT mice, p16 gene expression in cerebral microvessels decreased, a process associated with a concomitant apoptotic response (increased Bax/Bcl2 ratio). An increase in oxidative stress has been reported to promote senescence in the cerebral vasculature (Fulop et al. 2018); oxidative stress-dependent damage could therefore stimulate the senescence damage response (increase in p16 expression) observed in microvessels of ATX mice, while TAC-induced severe damage would lead to apoptosis of microvascular cells (increased Bax/Bcl2 ratio). Nonetheless, TAC was associated with a reduced expression of the antioxidant defense enzyme Sod2 and a further increase in that of the pro-oxidative enzyme Nox2 in the parenchyma. Altogether, these data suggest that vascular senescence and apoptosis are part of a graded damage response, senescence in response to chronic low-grade injury (aging, dyslipidemia, mild hypertension), and apoptosis in response to a severe acute injury (TAC-induced hypertensive micro-hemorrhage) (de Montgolfier et al. 2019). Most importantly, these series of events could contribute to the progressive decline in the volume of memory-associated brain areas with age, a phenomenon accelerated by vascular risk factors.
There are limitations to our study. One is the absence of female mice. Sexual dimorphism in vascular cognitive impairment and dementia is not well understood (Mielke et al. 2014) and this should be addressed in specific follow-up studies. In addition, our ATX mice are not only atherosclerotic but also hypertensive: are the cerebrovascular damage observed in 12-month-old ATX mice more related to severe dyslipidemia or to hypertension? We pose that the NVU disruption observed in ATX mice, largely mimicked in WT-TAC mice and aggravated in ATX-TAC mice, is dependent on the interaction between chronic hemodynamic stress and severe dyslipidemia (Bolduc et al. 2011).
In conclusion, we presented evidence that aging ATX mice recapitulate the phenotype of VCID; we demonstrated that vascular risk factors cause cumulative damages to the NVU in mice, leading to cognitive failure. In addition, we demonstrate that a cerebrovascular hemodynamic stress alone affects brain functions: indeed, in WT mice, TAC surgery generates the functional and molecular phenotype measured in sham-ATX mice, including cognitive decline (Fig. 10). Similarly to what we observed in WT mice following TAC, which damage solely depends on high systolic blood pressure, middle-age ATX mice spontaneously develop micro-bleeds, capillary rarefaction and senescence, hypoperfusion, and cognitive decline. In ATX mice, TAC aggravated both NVU injury and cognition without affecting brain area volumes, suggesting that it is the chronicity of the damage that determines brain remodeling. The present study shows that it is the age-dependent accumulation of small changes in cerebral functions at the cellular and molecular levels that interact and synergize to affect the outcome of cognitive dysfunction. Finally, our findings using ATX mice have clinical relevance: middle-age ATX mice could be used to test novel therapeutics and preventive approaches to alleviate the burden of VCID.
Fig. 10.

Schematization of the putative cellular and molecular events linking risk factors for cardiovascular diseases, cerebral microcirculation, and the development of cognitive decline in middle-age mice. Cerebrovascular cells, astrocytes, and perivascular neurons constitute the interconnected neurovascular unit (NVU) that maintains proper brain functions. Vascular risk factors accelerate the rate of cognitive decline, but their impact on the NVU is not clear. We show that hypertensive, atherosclerotic (LDLr−/−;hApoB100+/+) mice prematurely develop cognitive decline associated with cerebral micro-bleeds, reduced microvessel density, and endothelial dysfunction. These alterations were associated with blood-brain barrier leakage, brain hypoperfusion, senescence and inflammation, and brain atrophy. Induction of severe systolic hypertension in wild-type mice altered both vascular and neuronal functions. Thus, vascular risk factors disrupt the NVU and contribute to premature cognitive failure
Grants
This research was supported by the Canadian Institutes of Health Research (MOP 133649, E.T.) and by the Foundation of the Montreal Heart Institute (E.T.).
Compliance with Ethical Standards
The study was approved by the Montreal Heart Institute ethics committee (ET No 2015-62-01).
Conflict of Interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Avants BB, Epstein CL, Grossman M, Gee JC. Symmetric diffeomorphic image registration with cross-correlation: evaluating automated labeling of elderly and neurodegenerative brain. Med Image Anal. 2008;12:26–41. doi: 10.1016/j.media.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Song G, Cook PA, Klein A, Gee JC. A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage. 2011;54:2033–2044. doi: 10.1016/j.neuroimage.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, A. Saltness R, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–535. doi: 10.1038/nature08983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc V, Drouin A, Gillis MA, Duquette N, Thorin-Trescases N, Frayne-Robillard I, Des Rosiers C, Tardif JC, Thorin E. Heart rate-associated mechanical stress impairs carotid but not cerebral artery compliance in dyslipidemic atherosclerotic mice. Am J Physiol Heart Circ Physiol. 2011;301:H2081–H2092. doi: 10.1152/ajpheart.00706.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breteler MM. Vascular risk factors for Alzheimer’s disease: an epidemiologic perspective. Neurobiol Aging. 2000;21:153–160. doi: 10.1016/S0197-4580(99)00110-4. [DOI] [PubMed] [Google Scholar]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018;562:578–582. doi: 10.1038/s41586-018-0543-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.RES.87.10.840. [DOI] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csiszar A, Tarantini S, Fülöp GA, Kiss T, Valcarcel-Ares MN, Galvan V, Ungvari Z, Yabluchanskiy A. Hypertension impairs neurovascular coupling and promotes microvascular injury: role in exacerbation of Alzheimer’s disease. Geroscience. 2017;39:359–372. doi: 10.1007/s11357-017-9991-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Torre JC. Alzheimer disease as a vascular disorder: nosological evidence. Stroke. 2002;33:1152–1162. doi: 10.1161/01.STR.0000014421.15948.67. [DOI] [PubMed] [Google Scholar]
- de Montgolfier O, Pinçon A, Pouliot P, Gillis MA, Bishop J, Sled JG, Villeneuve L, Ferland G, Lévy BI, Lesage F, Thorin-Trescases N, Thorin É. High systolic blood pressure induces cerebral microvascular endothelial dysfunction, neurovascular unit damage, and cognitive decline in mice. Hypertension. 2019;73:217–228. doi: 10.1161/HYPERTENSIONAHA.118.12048. [DOI] [PubMed] [Google Scholar]
- DeCarli C. Cerebrovascular disease: assessing the brain as an end-organ of vascular disease. Nat Rev Cardiol. 2012;9:435–436. doi: 10.1038/nrcardio.2012.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didion SP, Heistad DD, Faraci FM. Mechanisms that produce nitric oxide-mediated relaxation of cerebral arteries during atherosclerosis. Stroke. 2001;32:761–766. doi: 10.1161/01.STR.32.3.761. [DOI] [PubMed] [Google Scholar]
- Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371:1612–1623. doi: 10.1016/S0140-6736(08)60694-7. [DOI] [PubMed] [Google Scholar]
- Dorr AE, Lerch JP, Spring S, Kabani N, Henkelman RM. High resolution three-dimensional brain atlas using an average magnetic resonance image of 40 adult C57Bl/6J mice. Neuroimage. 2008;42:60–69. doi: 10.1016/j.neuroimage.2008.03.037. [DOI] [PubMed] [Google Scholar]
- Drouin A, Bolduc V, Thorin-Trescases N, Bélanger É, Fernandes P, Baraghis E, Lesage F, Gillis MA, Villeneuve L, Hamel E, Ferland G, Thorin E. Catechin treatment improves cerebrovascular flow-mediated dilation and learning abilities in atherosclerotic mice. Am J Physiol Heart Circ Physiol. 2011;300:H1032–H1043. doi: 10.1152/ajpheart.00410.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin A, Farhat N, Bolduc V, Thorin-Trescases N, Gillis MA, Villeneuve L, Nguyen A, Thorin E. Up-regulation of thromboxane A(2) impairs cerebrovascular eNOS function in aging atherosclerotic mice. Pflugers Arch. 2011;462:371–383. doi: 10.1007/s00424-011-0973-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin A, Thorin E. Flow-induced dilation is mediated by Akt-dependent activation of endothelial nitric oxide synthase-derived hydrogen peroxide in mouse cerebral arteries. Stroke. 2009;40:1827–1833. doi: 10.1161/STROKEAHA.108.536805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64:575–611. doi: 10.1016/S0301-0082(00)00068-X. [DOI] [PubMed] [Google Scholar]
- Fleg JL, Strait J. Age-associated changes in cardiovascular structure and function: a fertile milieu for future disease. Heart Fail Rev. 2012;17:545–554. doi: 10.1007/s10741-011-9270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotuhi M, Do D, Jack C. Modifiable factors that alter the size of the hippocampus with ageing. Nat Rev Neurol. 2012;8:189–202. doi: 10.1038/nrneurol.2012.27. [DOI] [PubMed] [Google Scholar]
- Fulop GA, Kiss T, Tarantini S, Balasubramanian P, Yabluchanskiy A, Farkas E, Bari F, Ungvari Z, Csiszar A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience. 2018;40:513–521. doi: 10.1007/s11357-018-0047-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron ME, Théorêt JF, Mamarbachi AM, Drouin A, Nguyen A, Bolduc V, Thorin-Trescases N, Merhi Y, Thorin E. Late chronic catechin antioxidant treatment is deleterious to the endothelial function in aging mice with established atherosclerosis. Am J Physiol Heart Circ Physiol. 2010;298:H2062–H2070. doi: 10.1152/ajpheart.00532.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godbout JP, Johnson RW. Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Immunol Allergy Clin N Am. 2009;29:321–337. doi: 10.1016/j.iac.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Harada CN, Natelson Love MC, Triebel KL. Normal cognitive aging. Clin Geriatr Med. 2013;29:737–752. doi: 10.1016/j.cger.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature. Nat Neurosci. 2007;10:1369–1376. doi: 10.1038/nn2003. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Yaffe K, Biller J, Bratzke LC, Faraci FM, Gorelick PB, Gulati M, Kamel H, Knopman DS, Launer LJ, Saczynski JS, Seshadri S, Zeki al Hazzouri A. Impact of hypertension on cognitive function: a scientific statement from the American Heart Association. Hypertension. 2016;68:e67–e94. doi: 10.1161/HYP.0000000000000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/S0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Katusic ZS, Austin SA. Endothelial nitric oxide: protector of a healthy mind. Eur Heart J. 2014;35:888–894. doi: 10.1093/eurheartj/eht544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy KM, Raz N. Pattern of normal age-related regional differences in white matter microstructure is modified by vascular risk. Brain Res. 2009;1297:41–56. doi: 10.1016/j.brainres.2009.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, Lazic D, Tsai PS, Zhao Z, Zhou Y, Boas DA, Sakadžić S, Zlokovic BV. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci. 2017;20:406–416. doi: 10.1038/nn.4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, DeTeresa R, Terry RD. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology. 1993;43:192–197. doi: 10.1212/WNL.43.1_Part_1.192. [DOI] [PubMed] [Google Scholar]
- Mielke MM, Vemuri P, Rocca WA. Clinical epidemiology of Alzheimer’s disease: assessing sex and gender differences. Clin Epidemiol. 2014;6:37–48. doi: 10.2147/CLEP.S37929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SM, Zhang H, Maeda N, Doerschuk CM, Faber JE. Cardiovascular risk factors cause premature rarefaction of the collateral circulation and greater ischemic tissue injury. Angiogenesis. 2015;18:265–281. doi: 10.1007/s10456-015-9465-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, Benzinger TLS, Fagan AM, Ringman JM, Schneider LS, Morris JC, Chui HC, Law M, Toga AW, Zlokovic BV. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270–276. doi: 10.1038/s41591-018-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell M, Teo K, Gao P, Anderson C, Sleight P, Dans A, Marzona I, Bosch J, Probstfield J, Yusuf S. Cognitive impairment and risk of cardiovascular events and mortality. Eur Heart J. 2012;33:1777–1786. doi: 10.1093/eurheartj/ehs053. [DOI] [PubMed] [Google Scholar]
- O’Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney: cause and logic of therapy. Hypertension. 2005;46:200–204. doi: 10.1161/01.HYP.0000168052.00426.65. [DOI] [PubMed] [Google Scholar]
- Pouliot P, Gagnon L, Lam T, Avti PK, Bowen C, Desjardins M, Kakkar AK, Thorin E, Sakadzic S, Boas DA, Lesage F. Magnetic resonance fingerprinting based on realistic vasculature in mice. Neuroimage. 2017;149:436–445. doi: 10.1016/j.neuroimage.2016.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raignault A, Bolduc V, Lesage F, Thorin E. Pulse pressure-dependent cerebrovascular eNOS regulation in mice. J Cereb Blood Flow Metab. 2017;37:413–424. doi: 10.1177/0271678X16629155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J, Chien KR. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci U S A. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salat DH, Kaye JA, Janowsky JS. Prefrontal gray and white matter volumes in healthy aging and Alzheimer disease. Arch Neurol. 1999;56:338–344. doi: 10.1001/archneur.56.3.338. [DOI] [PubMed] [Google Scholar]
- Schmidt D, von Hochstetter AR. The use of CD31 and collagen IV as vascular markers. A study of 56 vascular lesions. Pathol Res Pract. 1995;191:410–414. doi: 10.1016/S0344-0338(11)80727-2. [DOI] [PubMed] [Google Scholar]
- Stewart-Lee AL, Burnstock G. Changes in vasoconstrictor and vasodilator responses of the basilar artery during maturation in the Watanabe heritable hyperlipidemic rabbit differ from those in the New Zealand White rabbit. Arterioscler Thromb. 1991;11:1147–1155. doi: 10.1161/01.ATV.11.5.1147. [DOI] [PubMed] [Google Scholar]
- Taylor CA, Greenlund SF, McGuire LC, Lu H, Croft JB. Deaths from Alzheimer’s disease - United States, 1999-2014. MMWR Morb Mortal Wkly Rep. 2017;66:521–526. doi: 10.15585/mmwr.mm6620a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth P, Tarantini S, Csiszar A, Ungvari Z. Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol. 2017;312:H1–H20. doi: 10.1152/ajpheart.00581.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth P, Tucsek Z, Sosnowska D, Gautam T, Mitschelen M, Tarantini S, Deak F, Koller A, Sonntag WE, Csiszar A, Ungvari Z. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab. 2013;33:1732–1742. doi: 10.1038/jcbfm.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucsek Z, Noa Valcarcel-Ares M, Tarantini S, Yabluchanskiy A, Fülöp G, Gautam T, Orock A, Csiszar A, Deak F, Ungvari Z. Hypertension-induced synapse loss and impairment in synaptic plasticity in the mouse hippocampus mimics the aging phenotype: implications for the pathogenesis of vascular cognitive impairment. Geroscience. 2017;39:385–406. doi: 10.1007/s11357-017-9981-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tustison NJ, Avants BB, Cook PA, Zheng Y, Egan A, Yushkevich PA, Gee JC. N4ITK: improved N3 bias correction. IEEE Trans Med Imaging. 2010;29:1310–1320. doi: 10.1109/TMI.2010.2046908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 2006;31:1116–1128. doi: 10.1016/j.neuroimage.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Vascular disorder in Alzheimer’s disease: role in pathogenesis of dementia and therapeutic targets. Adv Drug Deliv Rev. 2002;54:1553–1559. doi: 10.1016/S0169-409X(02)00150-3. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]