

Abstract
The clinical relevance of the Human Genome Project and next generation sequencing technology was demonstrated for the first time in 2009, when whole-exome sequencing (WES) provided the definitive diagnosis of congenital chloride diarrhea in an infant with presumed renal salt-wasting disease. Over the past decade, numerous studies have shown the utility of WES for clinical diagnosis as well as for discovery of novel genetic disorders through analysis of a single or a handful of informative pedigrees. Hence, advances in improving the speed, accuracy and computational analysis combined with exponential decrease in the cost of sequencing the human genome is transforming the practice of medicine. The impact of WES has been most noticeable in pediatric disorders and oncology, but its utility in the liver clinic is recently emerging. Here, we assess the current status of WES for clinical diagnosis and acceleration of translation research to enhance care of patients with liver disease.
Over the past decade, advances in human genetics and genomics through next generation sequencing (NGS) technology is transforming the practice of medicine. The first draft of the human genome sequence was released in 2001(1, 2) and completed in 2003. It revealed approximately 20,000 protein-coding genes that comprise the human genome. Simultaneously, convergence of advances on several fronts have improved the speed, cost and accuracy of sequencing of all 20,000 genes through the development and optimization of NGS technology. NGS(3) is also referred to as massively parallel sequencing or deep sequencing, and whole-exome-sequencing (WES) is one of its applications. Here, we focus on the diagnostic utility of WES in the hepatology clinic and its potential to refine our contemporary taxonomy of liver disease with far-reaching implications.
A decade of whole-exome sequencing for clinical diagnosis and human disease gene discovery
A landmark in history of human genomics occurred in 2009 when WES was first applied for clinical diagnosis(4, 5), including an unanticipated diagnosis of congenital chloride diarrhea in an infant with suspected renal salt-wasting disease(4), and in another patient, to discover a novel genetic defect underlying a multisystemic syndrome of unknown cause(6). Since then, multiple studies have demonstrated the value of WES in clinical diagnosis and human disease gene discovery(7). Moreover, these discoveries have led to novel insights into disease mechanisms, provided new diagnostic tests, as well as identified new therapeutic targets. Thus far, the phenotypic consequence of genetic variants in only 20% of genes comprising the human genome have been delineated(7). Considering that up to 30% of human genes when mutated are likely to be embryonically lethal; there are as many as 10,000 genes which when mutated could impact human health in ways we have yet to understand(8). Moreover, the full phenotypic spectrum of most gene alterations remains to be defined as the majority of genotype/phenotype correlations are confined to the classical phenotypes only. Hence, it is expected that there are many unrecognized Mendelian traits embedded within the contemporary taxonomy of liver diseases.
What should hepatologists understand about whole-exome sequencing?
The exome corresponds to the coding regions (called exons) of the ~20,000 genes that comprise the human genome. The exome represents only ~1% of the entire genome, yet it is estimated to harbor the majority (~85%) of DNA alterations that cause human disease(4). Genomic DNA can be isolated from a diverse array of materials, such as blood, buccal swabs, saliva, paraffin embedded tissue blocks and dry blood spots. The latter is particularly useful if samples require shipping long distance from resource-limited areas of the world. WES, which consists in sequencing the coding part of most of our 20,000 genes, comprises two key components: (i) the library preparation from source DNA, including exome capture, followed by next generation sequencing, and (ii) the bioinformatics processing and analysis of WES data. In brief, the computational pipeline encompasses (i) the mapping and alignment of the individual WES data to the reference human genome, (ii) the variant calling, that permits identification of the nucleotide bases in the proband that are different from the reference human genome sequence, and (iii) the annotation of the variants for minor allele frequency (MAF), amino acid changes, prediction of deleteriousness using in silico prediction methods [e.g. SIFT(9, 10), PolyPhen-2(11), CADD score(12, 13), MetaSVM(14)], amino acid conservation across species, tissue expression, among others. Variants are then prioritized based on this information. The general prerequisites for a gene variant to be disease causing (pathogenic) are two-fold. First, the genetic variant must be rare (MAF<1%) in the general (unaffected) population; in other words, the higher the frequency, the lower the probability for it to be causal of a rare disease. Second, the genetic variant must affect protein function; thus, damaging mutations, such as premature termination, frameshift and splice-site mutations are prioritized, followed by missense mutations predicted to be deleterious by aforementioned in silico algorithms. Genome aggregation database (gnomAD), the largest publicly available genome database, contains the information of 125,748 exome sequences and 15,708 whole-genome sequences(15). The goal of WES is to identify rare (MAF<1%) disease-causing protein-coding gene variant(s), establishing genotype-phenotype causality. It is important to highlight the difference between WES and genome wide association studies (GWAS), which aims to identify an association between common variants and complex diseases. In contrast to rare pathogenic variants that predominantly occur in exons, common genetic variants typically occur in non-coding regions of the genome (introns) - but may occur in coding regions - have a small effect size, and therefore are insufficient to independently cause disease. A classic example is the PNPLA3 polymorphism (p.I148M, rs738409) that confers risk for non-alcoholic fatty liver disease (NAFLD) and alcoholic liver disease(16, 17).
Where could hepatologists be missing liver-related genetic traits?
Children with chronic liver disease represent an enriched population with single gene disorders, and therefore many of known genetic causes of liver diseases present during childhood(18). Thus, genetic liver disorders are traditionally associated with pediatric hepatology practice, where NGS for timely diagnosis has been progressively embraced over the past years(19). There is extensive experience of application of WES in non-hepatic pediatric disorders. In 2013, WES of 250 consecutive participants referred for evaluation of a possible genetic condition yielded a genetic diagnosis in a quarter of these patients(20); and in 2016, deep phenotyping and WES in 41 probands with intellectual developmental disorder and unexplained metabolic abnormalities led to a diagnosis in as many as 68% of the cases(21), one of the highest diagnostic rates reported. Other independent studies have consistently reported WES diagnostic rates of 20% or higher (22–25). Furthermore, we and others have successfully combined WES with deep phenotyping (i.e. detailed characterization of each patient’s phenotypic features) to identify the underlying genetic defects in infants and children with idiopathic liver diseases, including children with liver failure of indeterminate etiology(18, 19, 26–32). Astute clinical annotation (i.e., comprehensive phenotype description of the patient) is central to harnessing the maximal potential of genomic data(29, 33). Notably, the vast majority of these studies have been performed in pediatric populations and in cancer patients. Hence, the value of WES in the diagnosis and management of adult patients with non-oncological diseases is only recently emerging(25, 33–36). In fact, in adult hepatology clinic, Wilson disease, hemochromatosis and alpha-1 antitrypsin deficiency, are routinely investigated as part of a comprehensive work-up for liver test abnormalities and/or advanced liver disease by using single gene and/or gene panel test(s). When this work-up is unrevealing it usually stops there. As an example, hemochromatosis could be due to loss-of-function mutations in five distinct genes, namely HFE, HJV, HAMP, TRF2, SLC40A1, but only HFE gene is typically sequenced. In a recent study of 19 patients with idiopathic liver disease despite a comprehensive work-up performed by a hepatologist, WES provided a genetic diagnosis in five of these patients (~25%), uncovering four distinct monogenic disorders. This study identified five patients with up to two decades of misdiagnosis who in fact harbored Mendelian disorders previously unrecognized and highlight the utility of WES in patients with undiagnosed liver disease. Therefore, appropriate use of WES has the potential to reveal the contribution of monogenic disorders in adult hepatology practice with immediate implications for timely diagnosis and adequate clinical management targeted to the underlying molecular pathogenesis of disease. An obvious subset of patients that may harbor unrecognized genetic disorders affecting the liver are those suffering from liver disease of unknown cause. Additionally, patients who are labeled as suffering from alcoholic liver disease, but their alcohol drinking habits do not support the severity of liver disease; or from NAFLD, but were never overweight, should be recognized by hepatologist(s) as undiagnosed and avoid the temptation to assign them into a classical category of liver disease. Incorrect diagnosis can hinder access to optimal care and in case of alcohol wrongly stigmatize the patient. Moreover, in adult medicine if cursory family history is unrevealing, genetic disorders are often not further considered. Our experience leads us to predict that we will identify a significant number of sporadic cases of liver disease due to de novo mutation(s), which are DNA alteration(s) that occur de novo in the affected individual and therefore are not inherited from biological parents(33); the recognition and identification of such de novo mutations have been facilitated by the advent of NGS.
When should WES be used in adult hepatology clinical practice?
Based on experience from our group(33), the Mayo Clinic Center for Individualized Medicine(37) and data from investigators in other medical subspecialties(25, 34–36), WES should be incorporated in the evaluation of adult patients with (i) a chronic and/or severe phenotype of unclear cause with presentation in early adulthood; (ii) atypical clinical presentation [e.g. lean NAFLD/ non-alcoholic steatohepatitis (NASH)]; (iii) presence of congenital and/or syndromic features and (iv) multi-systemic involvement. Also, patients with liver disease who are offspring of a consanguineous union or who have a positive family history for liver dysfunction and/or hepatocellular carcinoma should be evaluated for a genetic disorder (Figure 1). However, the absence of positive family history should not deter any physician from pursuing genomic analysis if any of the other features are present. In fact, in our series none of the five adult patients with idiopathic liver disease who attained a genetic diagnosis(33) reported family history of liver disease. Lastly, as WES is getting more widely used in clinical practice, it is already replacing the conventional single-gene and gene-panel tests in large academic centers. WES creates the possibility of screening most relevant liver disease genes at once, providing genetic diagnoses across diverse medical subspecialties (Table 1), as liver disease can be a presentation of a liver-focused disorder or the main clinical manifestation of a systemic, multi-organ, disease as illustrated in(33). Moreover, there are cases in which WES identified mutations in genes known to cause a disease distinct from the initial clinical diagnosis(22). For example, a pathogenic homozygous mutation in MPV17 that causes a hepatocerebral mitochondrial depletion syndrome was identified in a patient with a (mis)diagnosis of Wilson disease based on scoring systems described in AASLD and EASL guidelines(29). This example underscores the importance of performing molecular studies for the diagnosis of Wilson disease and in the absence of bi-allelic mutations in ATP7B, an alternative diagnosis should be considered. It is important to recognize the known limitations of WES, which are mainly three-fold: (i) False negatives. Some segments of the genome are not amenable to capture(38); (ii) Failure to detect large genomic deletions and/or insertions; (iii) Failure to detect structural or chromosomal abnormalities. Thus, for specific cases where these limitations are known barriers to diagnosis, whole-genome sequencing is being evaluated as an alternative to fill in these gaps. However, for most clinical scenarios, WES is a valuable tool for clinical diagnosis and acceleration of translation research that will benefit the patient. Importantly, it is enabling the recognition of an expanded range of liver-related phenotypes not previously appreciated by physicians focusing on strict correlation of classical phenotypes with genotypes(39). Hence, new genotype-phenotype knowledge is constantly being generated. For this reason, reanalysis of “negative” WES should be performed in the clinical management of patients with undiagnosed liver disease, where this new information is incorporated and updated analytical pipelines might be used. In fact, WES reanalysis has been shown to improve diagnostic rates in patients that remain without a molecular diagnosis(40). An important prerequisite for use of genomic diagnosis in the clinic is the need for genetic counseling pre- and post-whole exome sequencing test in order to address potential personal and familial implications of incidental findings unrelated to the disease under investigation and to discuss the meaning of a “negative” or “positive” WES report, respectively. This aspect of patient management is especially important when the need arises to expand the analysis from the ‘clinical exome’ (i.e., interrogating only the list of known genes relevant to patient’s clinical signs and symptoms) to the rest of the exome. The cost of WES is comparable to an abdomen MRI of the liver, and its turn-around time varies from a few days (for urgent/emergent cases) to a few weeks (for routine cases). Together with multiple examples where unbiased WES provided a timely diagnosis and ended a long and costly medical odyssey, most of the medical insurances are moving towards providing coverage for clinical WES, for which a pre-authorization form filled by the physician is typically required(24). Importantly, the genetic information nondiscrimination act (GINA) signed in 2008 protects individuals against discrimination from health insurance companies and employers on the basis of genetic information. However, this federal law has the caveat of not being applicable to life, disability and long-term care insurance companies.
Figure 1.
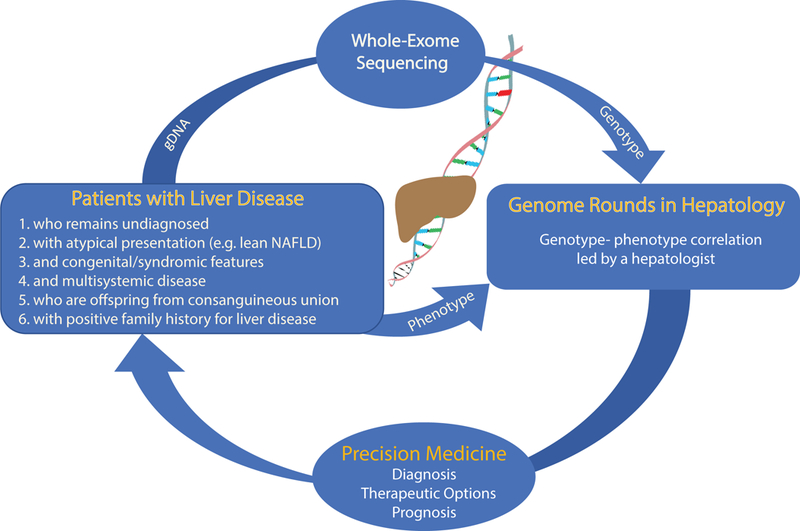
Schematic representation of Genome Rounds and Precision Medicine in Hepatology.
Table 1.
Examples of a diverse array of pediatric and adult liver-related disorders in which WES provides a definitive diagnosis.
| Clinical Applicability of WES in diagnosis and management of liver disease in… | |
|---|---|
| Adults | Children |
| MDR3 deficiency | |
| Mitochondrial disorders | |
| Lipid and lipoprotein metabolism disorders | |
| Hereditary hemochromatosis | |
| Wilson disease | |
| Alpha-1-antitrypsin deficiency | |
| Lysosomal storage disorders | |
| Familial lipodystrophy disorders | |
| Porphyria | |
| Neonatal/Infantile cholestasis due to loss-of-function mutations in ATP8B, ABCB11, TJP2, NR1H4, MYO5B, JAG1, NOTCH2, DCDC2, KIF12… | |
| Bile acid synthesis defects | |
| Metabolic disorders | |
| Acute liver failure due to loss-of-function mutations in NBAS, LARS… | |
Genome Rounds in Hepatology
Traditionally, hepatologists have excelled in detailed annotation of the pattern and natural history of liver disease in their patients. Now with the advent of genomic analysis in the clinic, it is possible to interrogate genomic data with the unique perspective of the clinician (Figure 1). Moreover, increasingly sophisticated ability to annotate liver diseases at the phenotypic and genotypic levels underscore the unprecedented opportunity to discover novel human mutations to advance our understanding of liver diseases. Generally, we recognize the importance of multidisciplinary Radiology and Pathology Rounds to discuss patient data in individual context to frequently yield new insights not previously appreciated. We posit that inclusion of WES results in such multidisciplinary forums, the Genome Rounds, will advance patient-centered care for individuals with liver disease. Like most pathological or imaging reports, clinical WES reports, which contain information on genetic variants found in genes that are related to the phenotype, require clinical correlation. The classification and interpretation of these variants (as pathogenic, likely pathogenic or variant of uncertain significance) reflects the current state of scientific understanding at the time the report is issued. Hence, it is essential that the genetic findings are integrated with clinical features in such forums (Figure 1). In our experience, consideration of the most likely candidate gene variant can often lead to recognition of clinical signs and symptoms not previously recognized, even by seasoned physicians(29, 33). It should be kept in mind that a variant of uncertain significance (VUS) correspond to a genetic alteration which current information is insufficient to determine pathogenicity and therefore a VUS should not be used in clinical decision making. However, whenever possible, efforts to resolve the classification of such a variant as pathogenic or benign should be undertaken. This might include to pursuing familial segregation studies or to conducting functional studies if a variant occurs in a candidate gene which a priori may underlie the patient’s phenotype. However, for many VUS, the increased availability of variant frequency data from large population(s) paired with phenotypic information will lead to the re-classification of VUS as benign(41). If no differences from standard human genome reference sequence were found or only genetic variants known to be benign polymorphisms or synonymous variants not predicted to affect splicing were identified, the clinical WES report may state “no clinically significant genetic variant related to this patient’s phenotype was identified” as data on benign variants are not routinely included. On the other hand, certain common genetic variants in PNPLA3(16, 42) and TM6SF2(43) are associated with an increased risk for hepatic steatosis and advanced liver disease whereas two common variants in HSD17B13(44, 45) have been identified as protective alleles to the development of steatohepatitis and hepatic fibrosis. Future studies are required to evaluate the interplay between these common variants and rare liver-related disease genetic variants in clinical presentation and outcomes. Therefore, time is ripe for developing an integrated forum where patient’s phenotype (including laboratory, imaging and pathological findings) and genotype information are discussed. Such, Genome Rounds, will advance the delivery of individualized medical care in hepatology, the development of best clinical practices for application of genetic analysis in the evaluation of adult liver disease as well as the design of clinical research and clinical trials. Genomic Rounds merge basic, translational and clinical research, generate new genotype-phenotype knowledge, foster scientific inquiry in trainees and colleagues, together, promoting the delivery of excellent clinical care in the field of Hepatology(46).
Precision medicine delivered by a hepatologist
The Precision Medicine Initiative launched 4 years ago is defined as “an approach for disease treatment and prevention that takes into account individual variability in genes, environment, and lifestyle for each person”. We now have the technology and knowledge to integrate detailed genetic and molecular evaluation in clinical hepatology practice. By studying the individual DNA alterations in adults with idiopathic liver disease, WES analysis yielded a genetic diagnosis in approximately 25% of the cases(33). In each case, the molecular pathogenesis of their underlying liver disease was decoded, which led to discussion of available targeted therapeutic options, preventive medicine interventions and adequate family counseling. To establish a genetic diagnosis, additional knowledge in human genetics and genomics might be required if no similar pathogenic variants has been reported. Forums such as Genomic Rounds in hepatology would be the appropriate setting to integrate genotype with phenotype information (Figure 1). Further studies are ongoing to assess the applicability of these findings to an ample liver disease population.
On the other hand, another illustrative example of application of precision medicine in hepatology beyond rare liver diseases, is through the recognition that the genetic risk for hepatic steatosis conferred by common variants encoding PNPLA3 p.I148M, TM6SF2 p.E167K, and GCKR p.P446L is amplified by adiposity(42). Thus, patients who are carriers of these variants should be informed and counseled about the known synergy between their (high) BMI and genotype in the onset and progression of advanced liver disease and its complications, such as hepatocellular carcinoma. In addition, since severity of disease varies with genotype, this information should be integrated at the time of design and report of clinical trials in NAFLD.
Concluding Remarks
By combining unbiased genomic analysis with exquisite deep phenotyping across liver disease patients, we will advance our understanding of genetic contributions to adult liver diseases. The increasing application of this approach will delineate liver disease at the molecular level, enabling its stratification into distinct groups informed by genotype and will refine the taxonomy of liver diseases. Ultimately, all stakeholders, patients, care providers and payors, will benefit from such individualized, genome-based approach to diagnosis and management of liver disease.
ACKNOWLEDGMENTS
We thank João Pedro Pereira for critically reading the manuscript. S.V. is supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number K08DK113109. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest: The authors declare no conflict of interests related to this study.
REFERENCES
- 1.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, et al. Initial sequencing and analysis of the human genome. Nature 2001;409:860–921. [DOI] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, et al. The sequence of the human genome. Science 2001;291:1304–1351. [DOI] [PubMed] [Google Scholar]
- 3.Morey M, Fernandez-Marmiesse A, Castineiras D, Fraga JM, Couce ML, Cocho JA. A glimpse into past, present, and future DNA sequencing. Mol Genet Metab 2013;110:3–24. [DOI] [PubMed] [Google Scholar]
- 4.Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci U S A 2009;106:19096–19101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009;461:272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet 2010;42:30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Posey JE, O’Donnell-Luria AH, Chong JX, Harel T, Jhangiani SN, Coban Akdemir ZH, Buyske S, et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet Med 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chong JX, Buckingham KJ, Jhangiani SN, Boehm C, Sobreira N, Smith JD, Harrell TM, et al. The Genetic Basis of Mendelian Phenotypes: Discoveries, Challenges, and Opportunities. Am J Hum Genet 2015;97:199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 10.Vaser R, Adusumalli S, Leng SN, Sikic M, Ng PC. SIFT missense predictions for genomes. Nat Protoc 2016;11:1–9. [DOI] [PubMed] [Google Scholar]
- 11.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47:D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong C, Wei P, Jian X, Gibbs R, Boerwinkle E, Wang K, Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 2015;24:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet 2010;42:21–23. [DOI] [PubMed] [Google Scholar]
- 18.Karlsen TH, Lammert F, Thompson RJ. Genetics of liver disease: From pathophysiology to clinical practice. J Hepatol 2015;62:S6–S14. [DOI] [PubMed] [Google Scholar]
- 19.Nicastro E, D’Antiga L. Next generation sequencing in pediatric hepatology and liver transplantation. Liver Transpl 2018;24:282–293. [DOI] [PubMed] [Google Scholar]
- 20.Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013;369:1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tarailo-Graovac M, Shyr C, Ross CJ, Horvath GA, Salvarinova R, Ye XC, Zhang LH, et al. Exome Sequencing and the Management of Neurometabolic Disorders. N Engl J Med 2016;374:2246–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dixon-Salazar TJ, Silhavy JL, Udpa N, Schroth J, Bielas S, Schaffer AE, Olvera J, et al. Exome sequencing can improve diagnosis and alter patient management. Sci Transl Med 2012;4:138ra–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, Das K, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA 2014;312:1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Splinter K, Adams DR, Bacino CA, Bellen HJ, Bernstein JA, Cheatle-Jarvela AM, Eng CM, et al. Effect of Genetic Diagnosis on Patients with Previously Undiagnosed Disease. N Engl J Med 2018;379:2131–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lata S, Marasa M, Li Y, Fasel DA, Groopman E, Jobanputra V, Rasouly H, et al. Whole-Exome Sequencing in Adults With Chronic Kidney Disease: A Pilot Study. Ann Intern Med 2018;168:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vilarinho S, Sari S, Yilmaz G, Stiegler AL, Boggon TJ, Jain D, Akyol G, et al. Recurrent recessive mutation in deoxyguanosine kinase causes idiopathic noncirrhotic portal hypertension. Hepatology 2016;63:1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vilarinho S, Sari S, Mazzacuva F, Bilguvar K, Esendagli-Yilmaz G, Jain D, Akyol G, et al. ACOX2 deficiency: A disorder of bile acid synthesis with transaminase elevation, liver fibrosis, ataxia, and cognitive impairment. Proc Natl Acad Sci U S A 2016;113:11289–11293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vilarinho S, Erson-Omay EZ, Harmanci AS, Morotti R, Carrion-Grant G, Baranoski J, Knisely AS, et al. Paediatric hepatocellular carcinoma due to somatic CTNNB1 and NFE2L2 mutations in the setting of inherited bi-allelic ABCB11 mutations. J Hepatol 2014;61:1178–1183. [DOI] [PubMed] [Google Scholar]
- 29.Vilarinho S, Choi M, Jain D, Malhotra A, Kulkarni S, Pashankar D, Phatak U, et al. Individual exome analysis in diagnosis and management of paediatric liver failure of indeterminate aetiology. J Hepatol 2014;61:1056–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Unlusoy Aksu A, Das SK, Nelson-Williams C, Jain D, Ozbay Hosnut F, Evirgen Sahin G, Lifton RP, et al. Recessive Mutations in KIF12 Cause High Gamma-Glutamyltransferase Cholestasis. Hepatol Commun 2019;3:471–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sambrotta M, Strautnieks S, Papouli E, Rushton P, Clark BE, Parry DA, Logan CV, et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet 2014;46:326–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maddirevula S, Alhebbi H, Alqahtani A, Algoufi T, Alsaif HS, Ibrahim N, Abdulwahab F, et al. Identification of novel loci for pediatric cholestatic liver disease defined by KIF12, PPM1F, USP53, LSR, and WDR83OS pathogenic variants. Genet Med 2019;21:1164–1172. [DOI] [PubMed] [Google Scholar]
- 33.Hakim A, Zhang X, DeLisle A, Oral EA, Dykas D, Drzewiecki K, Assis DN, et al. Clinical utility of genomic analysis in adults with idiopathic liver disease. J Hepatol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seidelmann SB, Smith E, Subrahmanyan L, Dykas D, Abou Ziki MD, Azari B, Hannah-Shmouni F, et al. Application of Whole Exome Sequencing in the Clinical Diagnosis and Management of Inherited Cardiovascular Diseases in Adults. Circ Cardiovasc Genet 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, Li Y, et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N Engl J Med 2019;380:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee EJ, Dykas DJ, Leavitt AD, Camire RM, Ebberink E, Garcia de Frutos P, Gnanasambandan K, et al. Whole-exome sequencing in evaluation of patients with venous thromboembolism. Blood Adv 2017;1:1224–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinto EVF, Lazaridis KN. Individualized medicine comes to the liver clinic. J Hepatol 2019. [DOI] [PubMed] [Google Scholar]
- 38.Katsanis SH, Katsanis N. Molecular genetic testing and the future of clinical genomics. Nat Rev Genet 2013;14:415–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu JT, Campeau PM, Lee BH. Genotype-phenotype correlation--promiscuity in the era of next-generation sequencing. N Engl J Med 2014;371:593–596. [DOI] [PubMed] [Google Scholar]
- 40.Ewans LJ, Schofield D, Shrestha R, Zhu Y, Gayevskiy V, Ying K, Walsh C, et al. Whole-exome sequencing reanalysis at 12 months boosts diagnosis and is cost-effective when applied early in Mendelian disorders. Genet Med 2018. [DOI] [PubMed] [Google Scholar]
- 41.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet 2017;49:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, Vogt TF, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abul-Husn NS, Cheng X, Li AH, Xin Y, Schurmann C, Stevis P, Liu Y, et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N Engl J Med 2018;378:1096–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kozlitina J, Stender S, Hobbs HH, Cohen JC. HSD17B13 and Chronic Liver Disease in Blacks and Hispanics. N Engl J Med 2018;379:1876–1877. [DOI] [PubMed] [Google Scholar]
- 46.Armstrong K, Ranganathan R, Fishman M. Toward a Culture of Scientific Inquiry - The Role of Medical Teaching Services. N Engl J Med 2018;378:1–3. [DOI] [PubMed] [Google Scholar]