

Abstract
Purpose
We attempted to identify the genes involved in the pathogenesis of uterine leiomyomas, under a hypothesis that the aberrant expression of upstream regulatory genes caused by aberrant DNA methylation is involved in the onset and development of uterine leiomyomas.
Methods
To find such genes, we compared genome-wide mRNA expression and DNA methylation in uterine leiomyomas and adjacent normal myometrium. Analysis of the data by Ingenuity Pathway Analysis software identified SATB2 which is known to be an epigenetic regulator, and NRG1 as candidate upstream regulatory genes. To infer the functions of these genes, human uterine smooth muscle cell lines overexpressing SATB2 or NRG1 genes were established (SATB2 or NRG1 lines), and their transcriptomes and pathways were analyzed.
Results
SATB2 and NRG1 were confirmed to be hypermethylated and upregulated in most uterine leiomyoma specimens (nine to 11 of the 11 cases). Among the established cell lines, morphological changes from spindle-like forms to fibroblast-like forms with elongated protrusions were observed in only the SATB2 line. Pathway analysis revealed that WNT/β-catenin and TGF-β signaling pathways which are related to the pathogenesis of uterine leiomyomas were activated in both SATB2 and NRG1 lines. In addition, signaling of growth factors including VEGF, PDGF, and IGF1, and retinoic acid signaling were activated in the SATB2 and NRG1 lines, respectively.
Conclusions
These results indicate that SATB2 and NRG1 overexpression induced many of the signaling pathways that are considered to be involved in the pathogenesis of uterine leiomyomas, suggesting that these genes have roles as upstream regulatory factors.
Electronic supplementary material
The online version of this article (10.1007/s10815-019-01582-y) contains supplementary material, which is available to authorized users.
Keywords: Uterine leiomyomas, Upstream regulatory genes, SATB2, NGR1
Introduction
Uterine leiomyomas are tumors that originate from uterine smooth muscle cells and are the most common gynecologic neoplasms, occurring in more than 25% of reproductive-age women [1]. Although uterine leiomyomas are benign diseases, they cause severe pelvic pain, menorrhagia, dysmenorrhea, anemia, infertility, and miscarriage, and thus significantly impair the quality of life [1, 2]. Surgery has long been the main mode of therapy for uterine leiomyomas, and hysterectomy is an attractive option to eliminate such problems for women who have completed childbearing. However, in recent years, women with uterine leiomyomas who wish retaining uteri for future pregnancies are increasing in number, since childbearing age is increasing due to changes in women’s lifestyle. Thus, there is a need for a therapy with molecular targeted agents. To identify such agents, it is necessary to elucidate the molecular mechanisms involved in the onset and development of uterine leiomyomas.
A number of factors have been reported to be involved in the pathogenesis of uterine leiomyomas, e.g., estrogen/estrogen receptor 1 (ESR1) [3], progesterone/progesterone receptor (PGR) [3, 4], several growth factors such as transforming growth factor beta (TGFB) [5], insulin-like growth factor (IGF) [6], epidermal growth factor (EGF) [7], platelet-derived growth factor (PDGF) [8], vascular growth factor (VEGF) [9], and fibroblast growth factor (FGF) [10]. In addition, several signaling pathways such as wingless-type (WNT)/beta catenin signaling [5], retinoic acid signaling [11], vitamin D signaling, and peroxisome proliferator-activated receptor gamma (PPARG) signaling [3] are also known to play important roles in the growth of uterine leiomyomas. However, these factors and signaling pathways have not yet fully explained the pathogenesis of uterine leiomyomas.
The risk of uterine leiomyomas has been associated with genetic factors such as African descent [12] and also with acquired factors such as the hormonal environment, nutrition, and lifestyle. Thus, early menarche, high body mass index, a meat diet, and high blood pressure increase the risk, whereas giving birth, a vegetarian diet, and smoking reduce the risk [12–14]. The acquired factors can be involved in the pathogenesis of uterine leiomyomas by causing epigenetic mutations. In fact, to date, DNA methylation and transcriptional analyses by ourselves [15–18] and other research groups [19, 20] have demonstrated that uterine leiomyomas have aberrant DNA methylation and mRNA expression profiles, suggesting that the aberrant gene expression caused by aberrant DNA methylation plays a key role in the pathogenesis of uterine leiomyomas [21]. On the other hand, studies of induced pluripotent stem cells and direct reprogramming have revealed that only a few master genes can determine or alter the cell fate [22, 23]. Only three mutations of driver genes are reported to be responsible for tumor development in lung and colorectal cancers [24]. These findings raise the possibility that several upstream regulatory genes, so-called master genes or driver genes, are preferentially involved in the pathogenesis of uterine leiomyomas. Thus, we hypothesize that the aberrant expression of the upstream regulatory genes caused by aberrant DNA methylation plays a key role in the pathogenesis of uterine leiomyomas.
In this study, to identify potential upstream regulatory genes, we searched for genes with aberrant DNA methylation and mRNA expression based on differences in DNA methylome and transcriptome data from multiple paired samples of uterine leiomyomas and adjacent normal myometrium. Furthermore, we examined by transcriptome and pathway analyses whether overexpression of the upstream regulatory genes actually activates signaling pathways similar to those involved in uterine leiomyomas.
Materials and methods
Tissue samples and cell culture
Paired specimens of uterine leiomyoma and adjacent normal myometrium were obtained from 11 Japanese women. The women underwent hysterectomy, and their ages were from 37 to 49 years old (mean ± SD; 44.5 ± 4.5). None of women had received previous treatment with sex steroid hormones or gonadotropin-releasing hormone analogs. Dissected specimens were immediately immersed in liquid nitrogen and stored at − 80 °C until DNA and RNA isolation.
Human immortalized uterine smooth muscle cell line (hTERT UtSMCs) which was obtained from Drs Konishi and Matsumura [25], were cultured in Dulbecco’s Modified Eagle medium (DMEM), high glucose (Wako, Osaka, Japan) supplemented with 10% FBS and antibiotic-antimycotic (Gibco BRL, Rockville, MD, USA) at 37 °C in CO2 incubator.
The three-dimensional culture was performed with collagen gel-embedded culture method [26, 27]. Cells of each hTERT UtSMC line were made into a single cell by treatment of trypsin, and then mixed with collagen solution [40% rat tail collagen type I (final concentration 1.6 mg/ml; Sigma-Aldrich, Tokyo, Japan), 20% 5 × DMEM, 10% FBS, 10% 0.44 M NaHCO3 and 5% 0.18 N NaOH] at 1 × 107 cells/ml. Two 10 μl-droplets of collagen solution containing cells were made on bottom of 12 well-plate (BD Biosciences, San Jose, Calif., USA) and incubated at 37 °C for 30 min to gel. The resulting cell-collagen clumps were added with the culture medium at 0.5 ml/well, and maintained at 37 °C in CO2 incubator.
Construction of the expression vectors containing SATB2 or NRG1 together with AcGFP
The coding sequence (CDS) of SATB2 (NM_001172509) and NRG1 (NM_013964) was amplified by PCR using cDNA of uterine leiomyoma as a template and primers shown in Supplementary Table 2. PCR was performed using 1.25 units of PrimeSTAR GXL DNA polymerase (Takara, Kyoto, Japan) under the cycling conditions (35 cycles of 98 °C for 10 s, 60 °C for 15 s, and 68 °C for 2.5 min). The IRES-AcGFP fragment was cleaved with restriction enzymes Sal I and Not I from pEF1a-IRES-AcGFP 1 vector (Takara). For the SATB2 expression vector (pLVSIN_SATB2-IRES-AcGFP), the SATB2 CDS treated with Xho I and Sal I and the IRES-AcGFP fragment were co-ligated at the multiple cloning site of pLVSIN-CMV-Hyg vector (Takara). For the NRG1 expression vector (pLVSIN_NRG1-IRES- AcGFP), SATB2 CDS was excised from pLVSIN_SATB2-IRES-AcGFP with Xho I and Sma I, and then NRG1 CDS treated with Xho I and Sma I were ligated into the remaining vector. A mock control (pLVSIN_IRES-AcGFP) in which only IRES-AcGFP fragment was inserted was also constructed.
Establishment of hTERT UtSMC lines overexpressing SATB2 or NRG1
The genes were transferred to hTERT UtSMCs by a lentivirus. The SATB2 expression vector (pLVSIN_SATB2-IRES-AcGFP), NRG1 expression vector (LVSIN-NRG1-IRES-AcGFP), or control vector (pLVSIN_IRES-AcGFP) together with the vectors expressing the lentiviral constitutive proteins (Lenti-X HTX packaging mix; Takara) were co-transfected into HEK293T cells (Takara) using the lipofectamine LTX (Invitrogen, Carlsbad, CA, USA). Two days after the transfection, the culture medium was concentrated to 50 times using a Lenti-X concentrator (Takara) and used as a packaged lentivirus. The packaged lentivirus was added to hTERT UtSMC adjusted to 3 × 104 cells/well (24 well-plate; BD Biosciences). The stable hTERT UtSMC lines overexpressing SATB2 and AcGFP (SATB2 line), NRG1 and AcGFP (NRG1 line), or AcGFP (control) were established by sorting with 10 μg/ml hygromycin for a month.
Genomic DNA and RNA isolation
Genomic DNA and total RNA from tissues were isolated as reported previously [28]. In brief, the genomic DNA was isolated by treatment with proteinase K (Qiagen, Hilden, Germany), followed by phenol/chloroform extraction and ethanol precipitation. Total RNA was isolated by treatment with ISOGEN reagent (Nippon Gene, Tokyo, Japan), followed by chloroform extraction and 2-propanol precipitation.
Total RNA from cell lines was isolated using an RNeasy mini kit (Qiagen,) according to the manufacturers’ instructions.
RT-PCR and real-time RT-PCR
First strand cDNA was synthesized by random hexamers using a QuantiTect Reverse Transcription Kit (Qiagen) as previously reported [28]. For RT-PCR, the synthesized cDNA was amplified by PCR using one unit of Biotaq HS DNA polymerase (Bioline, London, UK) and the primers listed in Supplementary Table 2 under the cycling conditions (28 to 32 cycles of 95 °C for 30 s, 60 °C for 30 s and 72 °C for 30 s, with an initial step of 95 °C for 10 min and a final step of 72 °C for 7 min). GAPDH was used as an internal control for the quantity of the RNA used. For real-time RT-PCR, amplification was performed using TB green premix Ex taq II (Takara) and primers listed in Supplementary Table 2 under the cycling conditions (45 cycles of 95 °C for 10 s, 60 °C for 10 s and 72 °C for 10 s, with an initial step of 95 °C for 30 s). The relative expression levels were determined by the delta-delta Ct method. The average Ct values were calculated from those of triplicate reactions. Delta Ct values were calculated using average Ct values of GAPDH as a reference gene. Delta-delta Ct values were calculated using the delta Ct value of the adjacent normal myometrium for each uterine leiomyoma specimen, and that of the control line for established cell lines, as a reference, respectively.
Combined bisulfite restriction analysis (COBRA)
DNA methylation levels were evaluated by COBRA as previously reported [28, 29]. Sodium bisulfite treatment was performed using an EpiTect Bisulfite kit (Qiagen) according to the conditions as follows: 95 °C for 5 min, 65 °C for 85 min, 95 °C for 5 min, and 65 °C for 175 min. PCR was performed using one unit of Biotaq HS DNA polymerase and the primes shown in Supplementary Table 2 under the cycling conditions (35 to 38 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s, with an initial step of 95 °C for 10 min and a final step of 72 °C for 7 min). A part of the PCR product was digested with the restriction enzyme Taq I (Takara) or HpyCH4IV (New England Biolabs, Ipswich, MA, USA). PCR products from methylated DNA and unmethylated DNA are digested and undigested by the restriction enzyme treatment, respectively. The treated PCR product was electrophoresed. The intensity of the signals was measured by densitometry. Methylation levels (%) were calculated as the ratio of the digested PCR product in the total PCR product. The experiments were done in triplicates. In this study, the aberrant methylation of the loci in uterine leiomyomas was defined as more than 20% higher or lower ratio as compared with that of corresponding myometrium.
Immunofluorescent staining
Cultured cells were fixed with 4% paraformaldehyde in PBS for 20 min. Fixed cells were permeabilize with 0.2% triton-X100 in PBS, and blocked with blocking solution (10% bovine fetal serum and 1% bovine serum albumin in PBST) for 60 min. Then the cells were incubated with rabbit anti-vimentin monoclonal antibody (Abcam, Tokyo, Japan; Cat# ab92547, RRID:AB_10562134) as primary antibody (diluted at 1:500 in the blocking solution) at 4 °C overnight, and incubated with the Alexa Fluor 594 conjugated goat anti-rabbit IgG (Abcam Cat# ab150084, RRID:AB_2734147) as secondary antibody (diluted at 1:1000 in PBS) for 45 min at room temperature. Vimentin is an intermediate filament specific to mesenchymal cells and is one of the markers for smooth muscle cells. Nuclei were stained with DAPI (500 ng/ml) in PBS.
Western blot analyses
Cultured cells were lysed in RIPA buffer (Wako). Samples were boiled for 5 min after SDS sample buffer (New England BioLabs) added. Proteins (10 μg) were electrophoresed on a 10% of SDS-polyacrylamide gel (SDS-PAGE). After SDS-PAGE, the proteins were transferred to a polyvinylidene difluoride membrane (New England BioLabs) by semi-dry blotting. The blotted membrane was incubated with mouse anti-human SATB2 monoclonal antibody (Abcam; Cat# ab51502, RRID:AB_882455), rabbit anti-human NRG1 monoclonal antibody (Abcam; Cat# ab180808, RRID:AB_2732881), or mouse anti-human TUBB monoclonal antibody (Sigma-Aldrich; Cat# T5201, RRID:AB_609915), as primary antibodies (diluted at 1:1000 in the blocking solution), incubated with the peroxidase-conjugated secondary antibody, incubated in ECL-Western blotting detection regents (GE Healthcare, Little Chalfont, Buckinghamshire, UK) for 5 min, and used to expose Hyperfilm-ECL (GE Healthcare).
Microarray and pathway analysis
The transcriptomes of three paired specimens of uterine leiomyoma and adjacent myometrium were reported previously (the data are available at the Gene Expression Omnibus (GEO) Web site; http://www.ncbi.nlm.nih.gov/geo; under accession No. GSE45189) [18]. The genes in which the mean of the expression levels in three leiomyoma specimens was greater than 1.5 or less than 2/3-fold of that in three myometrium specimens, were judged as aberrantly expressed genes. The microarray analysis of the SATB2, NRG1, and control lines was carried out as reported previously [18]. cDNA was prepared from 250 ng of total RNA. Gene expression was analyzed using a GeneChip Human Genome 2.0 ST Array (Affymetrix, Santa Clara, CA, USA) supporting 40,716 genes. Hybridization to the microarrays, washing, staining, and scanning were performed using the GeneChip system (Affymetrix). The scanned data were processed using the Patrek Genomics Suite software program (Partek, Munster, Germany). We selected genes in which the expression in the SATB2 or NRG1 lines was greater than 1.5 or less than 2/3-fold of that in the control line. Then, we analyzed the canonical pathways with Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, Redwood City, CA, USA).
Results
Identification of potential upstream regulatory genes
Using our previous transcriptome data (GEO accession No. GSE45189) [18], 1187 aberrantly expressed genes in uterine leiomyomas (392 upregulated and 795 downregulated genes) were identified. The 1187 genes were then subjected to upstream regulatory factor analysis using IPA software. The software identified 122 candidate genes (27 upregulated and 95 downregulated). According to our hypothesis that aberrant expression of the upstream regulatory genes is caused by aberrant DNA methylation, we screened the 122 genes for ones that were previously identified as aberrantly methylated in our DNA methylome data [18]. Nineteen of the genes (seven upregulated and 12 downregulated) were found (Table 1). Of these 19 genes, nine genes (SATB2, NRG1, EPAS1, NR3C1, GATA2, MEOX2, EBF1, KAT2B, and NR4A2) were known to regulate transcription (transcription regulators or ligand-dependent nuclear receptors) (Table 1). In this study, we focused on two upregulated genes SATB homeobox 2 (SATB2) and neuregulin 1 (NRG1) (Table 1). Their mRNA expressions and DNA methylation status were examined in multiple paired specimens of uterine leiomyoma and adjacent normal myometrium. SATB2 was highly expressed in all of 11 uterine leiomyoma cases and NRG1 was also highly expressed in nine of the 11 cases (except cases #4 and #6) (Fig. 1a). The relative expression levels of SATB2 and NRG1 in uterine leiomyoma specimens were higher than those in adjacent normal myometrium specimens in each case (Fig. 1b). In addition, SATB2 was hypermethylated in all cases but #5 and NRG1 was hypermethylated in all cases (Fig. 1c). Therefore, SATB2 and NRG1 were considered as candidates for the upstream regulatory genes in uterine leiomyomas.
Table 1.
Candidates for the upstream regulatory genes which were identified using
| Gene | Description | Location | Molecule type |
|---|---|---|---|
| Upregulated genes | |||
| SATB2 | SATB homeobox 2 | Nucleus | Transcription regulator |
| NRG1 | Neuregulin 1 | Plasma membrane | Growth factor and transcription regulator |
| CACNA1C | Calcium voltage-gated channel subunit alpha1 C | Plasma membrane | Ion channel |
| COL4A2 | Collagen type IV alpha 2 | Extracellular space | Extracellular matrix |
| EPHB1 | EPH receptor B1 | Plasma membrane | Kinase |
| PRL | Prolactin | Extracellular space | Cytokine |
| VCAN | Versican | Extracellular space | Extracellular matrix |
| Downregulated genes | |||
| EPAS1 | Endothelial PAS domain protein 1 | Nucleus | Transcription regulator |
| NR3C1 | Nuclear receptor subfamily 3 group C member 1 | Nucleus | Ligand-dependent nuclear receptor |
| MET | MET proto-oncogene, receptor tyrosine kinase | Plasma membrane | Kinase |
| GATA2 | GATA binding protein 2 | Nucleus | Transcription regulator |
| SASH1 | SAM and SH3 domain containing 1 | Extracellular Space | Other |
| MEOX2 | Mesenchyme homeobox 2 | Nucleus | Transcription regulator |
| CFB | Complement factor B | Extracellular Space | Peptidase |
| EBF1 | Early B cell factor 1 | Nucleus | Transcription regulator |
| KAT2B | Lysine acetyltransferase 2B | Nucleus | Transcription regulator |
| KDR | Kinase insert domain receptor | Plasma membrane | Kinase |
| NR4A2 | Nuclear receptor subfamily 4 group A member 2 | Nucleus | Ligand-dependent nuclear receptor |
| PLCE1 | Phospholipase C epsilon 1 | Cytoplasm | Enzyme |
Fig. 1.

mRNA expression and DNA methylation status of SATB2 and NRG1 genes in multiple paired samples of uterine leiomyoma and adjacent normal myometrium. aSATB2 and NRG1 mRNA expression which was analyzed by RT-PCR in triplicates in the 11 paired samples of uterine leiomyoma (L) and adjacent normal myometrium (M). GAPDH was used as an internal control. b Relative expression levels of SATB2 and NRG1 mRNA to GAPDH. Values are mean ± SD. c DNA methylation levels of SATB2 and NRG1 genes in the 11 paired samples of uterine leiomyoma and adjacent normal myometrium were evaluated in triplicates by combined bisulfite restriction analysis (COBRA). Values are mean ± SD
Establishment of human immortalized uterine smooth muscle cell lines overexpressing SATB2 or NRG1
In order to infer the functions of SATB2 and NRG1 genes in uterine leiomyomas, uterine smooth muscle cell lines overexpressing each gene were established. We constructed expression vectors harboring GFP (AcGFP) together with SATB2 or NRG1. The vectors were transduced into human telomerase reverse transcriptase immortalized uterine smooth muscle cells (hTERT UtSMC) [25], yielding the SATB2 and NRG1 overexpression lines. Real-time RT-PCR (Fig. 2d) and Western blotting (Fig. 2e) analyses confirmed that the transgenes were strongly expressed in their respective cell lines. No differences in proliferation were observed between the SATB2 and NRG1 lines and their parent lines (data not shown). The cells of SATB2 line had a fibroblast-like morphology with elongated protrusions, whereas the control cells had a spindle-like morphology (Fig. 2a, b). The morphology of NRG1 line did not differ from the control cell line (Fig. 2a, b).
Fig. 2.

Establishment of immortalized uterine smooth muscle cell lines overexpressing SATB2 or NRG1 genes. The expression vectors for the SATB2 and NRG1 lines contained the coding sequences of SATB2 and NRG1, respectively, plus AcGFP. The vectors were transduced into human telomerase reverse transcriptase immortalized uterine smooth muscle cells (hTERT UtSMCs). Cells transduced with only AcGFP were used as a mock control line (control). a Cell morphology of the SATB2 line, NRG1 line, control line, and parental line (parent). Scale bars: 100 μm. b Immunofluorescent staining for vimentin (red) in the SATB2 line, NRG1 line, control line, and parental line. Nuclei were stained with DAPI (violet). Scale bars: 100 μm. c Cell morphology in three-dimensional (3D) culture of the established cell lines. The 3D culture was carried out with a collagen gel-embedded culture [26, 27]. The photographs show the cell morphology of each cell line after 2 days of culture. Dotted lines outline the nodule-like aggregates of cells. Arrows indicate the aggregates of cells. Scale bars: 200 μm. d Relative expression levels of SATB2 and NRG1 mRNA to GAPDH in the established cell lines were analyzed in triplicates by real-time RT-PCR. Values are mean ± SD. e SATB2 and NRG1 protein expression in the established cell line was analyzed by Western blotting. TUBB was used as an internal control
Three-dimensional (3D) culture of the cell lines
When the cells were grown in a single layer, only the SATB2 line cells had the morphological changes (Fig. 2a, b). To see if this was an artifact of 2D culture, we also grew the cells in 3D culture (using collagen gel) [26, 27], which is generally considered to better reflect in vivo environments. After 2 days in culture, the SATB2 line cells extended within the cell-collagen clumps without contact inhibition and formed nodule-like aggregates of cells (outlined by the dotted line) (Fig. 2c), while the cells of the other cell lines formed small aggregates (arrows) (Fig. 2c), suggesting that SATB2 overexpression altered cell adhesion and aggregation.
PGR and ESR1 mRNA expression in the cell lines
The genes for progesterone receptor (PGR) and estrogen receptor 1 (ESR1) were investigated by real-time PCR. ESR1 was not detected in any of the cell lines (data not shown), while PGR was upregulated in the NRG1 line, but not in the SATB2 line (Fig. 3), indicating that NRG1 overexpression induced PGR upregulation.
Fig. 3.
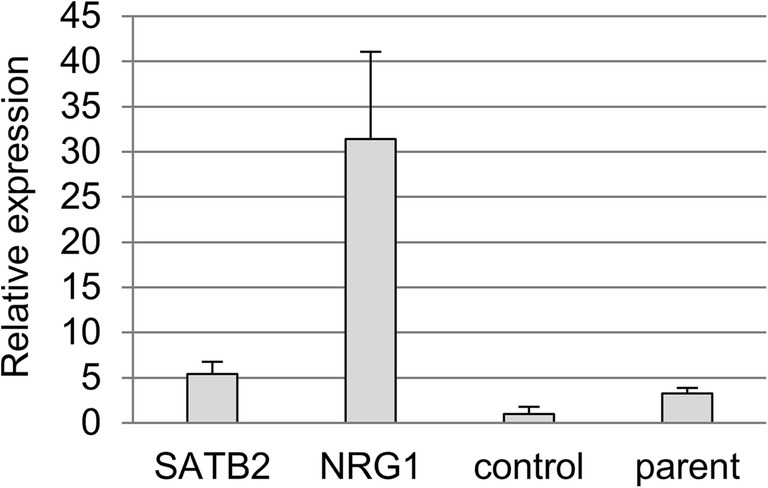
PGR expression in the cell lines. Relative expression levels of PGR mRNA to GAPDH in the established cell lines were analyzed in triplicates by real-time RT-PCR. Values are mean ± SD
Genes with expressions altered by overexpression of SATB2 or NRG1
The transcriptomes were analyzed to identify the genes altered by overexpression of SATB2 or NRG1. Overexpression of SATB2 upregulated and downregulated a total of 566 genes, while overexpression of NRG1 upregulated and downregulated a total of 626 genes (Table 2). In our previous study, 392 genes were upregulated and 795 were downregulated in uterine leiomyomas in comparison with normal myometrium [18] (Table 2). We compared the genes altered by SATB2 or NRG1 overexpression with the aberrantly expressed genes in uterine leiomyomas. In the SATB2 line, 12 upregulated and 51 downregulated genes were consistent with the aberrantly expressed genes in uterine leiomyomas (Table 3). In the NRG1 line, 14 upregulated and 19 downregulated genes were consistent with the aberrantly expressed genes in uterine leiomyomas (Table 3).
Table 2.
Numbers of upregulated and downregulated genes in SATB2 and NRG1 overexpressing cell lines (SATB2 and NRG1 line) compared with the control cell line, and numbers of aberrantly upregulated and downregulated genes in uterine leiomyomas compared with adjacent normal myometrium
| Samples | No. of upregulated genes | No. of downregulated genes | Total no. |
|---|---|---|---|
| SATB2 line | 203 | 363 | 566 |
| NRG1 line | 360 | 266 | 626 |
| Uterine leiomyomasa | 392 | 795 | 1187 |
Table 3.
Upregulated and downregulated genes in the SATB2 or NRG1 line common to aberrantly expressed genes in uterine leiomyomas
| # | Common genes altered in the SATB2 line and uterine leiomyomas | Common genes altered in the NRG1 line and uterine leiomyomas | ||
|---|---|---|---|---|
| Up (12) | Down (51) | Up (14) | Down (19) | |
| 1 | TGFB3 | NEK7 | TGFB3 | NEK7 |
| 2 | TNFRSF19 | TM4SF1 | TNFRSF19 | TM4SF1 |
| 3 | PSD3 | FRMD3 | PSD3 | FRMD3 |
| 4 | KIAA1199 | PTGS1 | KIAA1199 | PTGS1 |
| 5 | SNORD114–3 | FLT1 | TET1 | FLT1 |
| 6 | SATB2 | PTPRB | THSD4 | PTPRB |
| 7 | TYMS | AIM1 | QPRT | AIM1 |
| 8 | MMP16 | PLCB4 | TCEAL7 | PLCB4 |
| 9 | GPSM2 | LGR4 | NBLA00301 | LGR4 |
| 10 | SNORD116–8 | GPC4 | NAV2 | GPC4 |
| 11 | ZMAT3 | GLS | HSPA4L | GCLC |
| 12 | EDA2R | CTGF | SCD | LDB2 |
| 13 | PKD2 | SNORA75 | AHR | |
| 14 | EFHD1 | BEX1 | SERPINE1 | |
| 15 | LYST | PLEKHA6 | ||
| 16 | NDRG4 | PODXL | ||
| 17 | HEG1 | SOCS3 | ||
| 18 | SP110 | S1PR1 | ||
| 19 | RPS6KA3 | LTBP1 | ||
| 20 | ASS1 | |||
| 21 | SEC11C | |||
| 22 | PARD3B | |||
| 23 | GCNT4 | |||
| 24 | APOD | |||
| 25 | BMP4 | |||
| 26 | ITGB3 | |||
| 27 | C1R | |||
| 28 | MYO1D | |||
| 29 | GABARAPL1 | |||
| 30 | LRCH2 | |||
| 31 | SAV1 | |||
| 32 | AKAP12 | |||
| 33 | SNX25 | |||
| 34 | ACSL5 | |||
| 35 | ARHGAP20 | |||
| 36 | C11orf63 | |||
| 37 | MCTP1 | |||
| 38 | ITGBL1 | |||
| 39 | SKAP2 | |||
| 40 | UST | |||
| 41 | ERRFI1 | |||
| 42 | PALMD | |||
| 43 | TNIK | |||
| 44 | CREG1 | |||
| 45 | CCL2 | |||
| 46 | SERPING1 | |||
| 47 | TLR4 | |||
| 48 | LIMCH1 | |||
| 49 | WWC2 | |||
| 50 | ATP9A | |||
| 51 | FABP4 | |||
Up: common genes upregulated in each cell line and uterine leiomyomas
Down: common genes downregulated in each cell line and uterine leiomyomas
The numbers of genes are shown in parentheses
Pathway analysis for the genes altered by SATB2 or NRG1 overexpression
The canonical pathway analysis using IPA software identified 92 pathways associated with the 566 genes altered in the SATB2 line, 58 pathways associated with the 626 genes altered in the NRG1 line and 151 pathways associated with 1187 aberrantly expressed genes in uterine leiomyomas (Fig. 4a and Supplementary Table 1). Of the 92 pathways activated by SATB2 overexpression, 63 were common to the pathways activated by aberrantly expressed genes in uterine leiomyomas (Fig. 4b and Table 4). Thus, 42% (63/151) of the activated pathways in uterine leiomyomas may be caused by SATB2 overexpression (Fig. 4b). Among the 63 pathways, WNT/β-catenin signaling, TGF-β signaling, and signaling of growth factors including VEGF, PDGF, and IGF1 (Table 4) are known to be involved in the pathogenesis of uterine leiomyomas [3, 5, 6, 8, 9]. In addition, the 63 pathways included 12 cancer-related pathways: “molecular mechanisms of cancer,” “regulation of the epithelial-mesenchymal transition pathway,” “UVA-induced MAPK signaling,” “pancreatic adenocarcinoma signaling,” “ovarian cancer signaling,” “role of tissue factor in cancer,” “colorectal cancer metastasis,” “glioma signaling,” “glioma invasiveness,” “glioblastoma multiforme signaling,” “bladder cancer signaling,” and “HER-2 signaling in breast cancer” (Table 4). Of the 58 pathways activated by NRG1 overexpression, 33 were common to the pathways activated by aberrantly expressed genes in uterine leiomyomas (Fig. 4c and Table 4). Thus, 22% (33/151) of activated pathways in uterine leiomyomas may be caused by NRG1 overexpression (Fig. 4c). These 33 pathways included three signaling (four pathways) associated with the pathogenesis of uterine leiomyomas (WNT/β-catenin signaling, TGF-β signaling and retinoic acid signaling: “retinoic acid mediated apoptosis signaling” and “RAR activation”) [3, 5, 11], and five cancer-related pathways: “molecular mechanisms of cancer,” “regulation of the epithelial-mesenchymal transition,” “UVA-induced MAPK signaling,” “pancreatic adenocarcinoma signaling,” and “ovarian cancer signaling” (Table 4).
Fig. 4.

Pathway analysis of genes with altered expressions in the hTERT UtSMC lines overexpressing SATB2 or NRG1 and uterine leiomyomas. a Numbers of signaling pathways of the altered genes in SATB2 line, NRG1 line, and uterine leiomyomas. Canonical pathways were analyzed with Ingenuity Pathway Analysis (IPA) software. Pathways with p values below 0.05 were selected. b Percentage of pathways altered by SATB2 overexpression in the pathways activated in uterine leiomyomas. Gray region indicates the percentage of the pathway common to the SATB2 line and uterine leiomyomas. White region indicates the percentage of the pathway exclusively in uterine leiomyomas. c Percentage of pathways altered by NRG1 overexpression in the pathways activated in uterine leiomyomas. Gray region indicates the percentage of the pathway common to the NRG1 line and uterine leiomyomas. White region indicates the percentage of the pathway exclusively in uterine leiomyomas
Table 4.
Common signaling pathways between uterine leiomyomas and the SATB2 or NRG1 lines
| # | Common pathways in the SATB2 line and uterine leiomyomas (63) | p value for the SATB2 line | Common pathways in the NRG1 line and uterine leiomyomas (33) | p value for the NRG1 line |
|---|---|---|---|---|
| 1 | Axonal guidance signaling | 2.09 × 10−7 | Human embryonic stem cell pluripotency | 1.15 × 10−5 |
| 2 | Hepatic fibrosis/hepatic stellate cell activation | 1.23 × 10−6 | Role of osteoblasts, osteoclasts and chondrocytes in rheumatoid arthritis | 1.66 × 10−5 |
| 3 | Role of Osteoblasts, Osteoclasts and Chondrocytes in Rheumatoid Arthritis | 8.91 × 10−6 | Hepatic fibrosis/hepatic stellate cell activation | 4.07 × 10−5 |
| 4 | Osteoarthritis pathway | 9.33 × 10−6 | G protein coupled receptor signaling | 1.38 × 10−4 |
| 5 | Leukocyte extravasation signaling | 3.55 × 10−5 | Regulation of the Epithelial-mesenchymal transition pathway | 1.70 × 10−4 |
| 6 | Role of Macrophages, Fibroblasts and Endothelial Cells in Rheumatoid Arthritis | 3.80 × 10−5 | Adipogenesis pathway | 1.41 × 10−3 |
| 7 | Pancreatic adenocarcinoma signaling | 8.71 × 10−5 | Role of macrophages, fibroblasts and endothelial cells in rheumatoid arthritis | 1.55 × 10−3 |
| 8 | Human embryonic stem cell pluripotency | 1.38 × 10−4 | Wnt/β-catenin signaling | 2.82 × 10−3 |
| 9 | Granulocyte adhesion and diapedesis | 2.45 × 10−4 | Retinoic acid mediated apoptosis signaling | 4.27 × 10−3 |
| 10 | Molecular Mechanisms of Cancer | 4.90 × 10−4 | TGF-β signaling | 4.57 × 10−3 |
| 11 | Atherosclerosis signaling | 6.46 × 10−4 | Osteoarthritis pathway | 5.62 × 10−3 |
| 12 | Virus Entry via Endocytic Pathways | 7.59 × 10−4 | Axonal guidance signaling | 6.61 × 10−3 |
| 13 | Reelin Signaling in Neurons | 1.12 × 10−3 | HGF Signaling | 6.92 × 10−3 |
| 14 | Complement system | 1.55 × 10−3 | Pancreatic adenocarcinoma signaling | 7.76 × 10−3 |
| 15 | Glioma signaling | 1.58 × 10−3 | Ovarian cancer signaling | 7.94 × 10−3 |
| 16 | Aryl hydrocarbon receptor signaling | 1.78 × 10−3 | NF-κB signaling | 1.12 × 10−2 |
| 17 | Colorectal cancer metastasis signaling | 1.91 × 10−3 | UVA-induced MAPK signaling | 1.35 × 10−2 |
| 18 | Wnt/β-catenin signaling | 1.95 × 10−3 | HMGB1 signaling | 1.38 × 10−2 |
| 19 | PAK signaling | 2.29 × 10−3 | Leukocyte extravasation signaling | 1.45 × 10−2 |
| 20 | Macropinocytosis signaling | 2.34 × 10−3 | p53 signaling | 1.62 × 10−2 |
| 21 | Endothelin-1 signaling | 2.45 × 10−3 | NRF2-mediated oxidative stress response | 1.66 × 10−2 |
| 22 | Regulation of the Epithelial-mesenchymal transition pathway | 3.80 × 10−3 | RAR activation | 1.91 × 10−2 |
| 23 | Glioblastoma multiforme signaling | 4.27 × 10−3 | cAMP-mediated signaling | 1.95 × 10−2 |
| 24 | Agranulocyte adhesion and diapedesis | 4.37 × 10−3 | Xenobiotic metabolism signaling | 2.09 × 10−2 |
| 25 | Neuropathic pain signaling in dorsal horn neurons | 5.62 × 10−3 | Molecular mechanisms of cancer | 2.09 × 10−2 |
| 26 | Ovarian cancer signaling | 5.89 × 10−3 | MSP-RON signaling pathway | 2.95 × 10−2 |
| 27 | HIF1α signaling | 5.89 × 10−3 | Cardiac hypertrophy signaling | 3.16 × 10−2 |
| 28 | Caveolar-mediated Endocytosis Signaling | 6.03 × 10−3 | STAT3 pathway | 3.24 × 10−2 |
| 29 | Phagosome formation | 6.76 × 10−3 | Sphingosine-1-phosphate signaling | 3.39 × 10−2 |
| 30 | Role of tissue factor in cancer | 7.41 × 10−3 | LPS/IL-1 Mediated Inhibition of RXR function | 4.27 × 10−2 |
| 31 | Hmgb1 signaling | 1.07 × 10−2 | Role of pattern recognition Receptors in recognition of bacteria and viruses | 4.47 × 10−2 |
| 32 | Adipogenesis pathway | 1.15 × 10−2 | Prostanoid biosynthesis | 4.79 × 10−2 |
| 33 | Protein kinase a signaling | 1.17 × 10−2 | Coagulation system | 4.90 × 10−2 |
| 34 | NRF2-mediated oxidative stress response | 1.23 × 10−2 | ||
| 35 | Role of pattern recognition Receptors in recognition of bacteria and viruses | 1.26 × 10−2 | ||
| 36 | LPS/IL-1 mediated inhibition of RXR function | 1.38 × 10−2 | ||
| 37 | Growth hormone signaling | 1.38 × 10−2 | ||
| 38 | Paxillin signaling | 1.41 × 10−2 | ||
| 39 | Bladder cancer signaling | 1.45 × 10−2 | ||
| 40 | Xenobiotic metabolism signaling | 1.48 × 10−2 | ||
| 41 | Role of NFAT in cardiac hypertrophy | 1.55 × 10−2 | ||
| 42 | IL-8 signaling | 1.62 × 10−2 | ||
| 43 | Clathrin-mediated endocytosis signaling | 1.66 × 10−2 | ||
| 44 | Tec kinase signaling | 1.70 × 10−2 | ||
| 45 | HGF signaling | 1.74 × 10−2 | ||
| 46 | Agrin interactions at neuromuscular junction | 2.09 × 10−2 | ||
| 47 | PDGF signaling | 2.14 × 10−2 | ||
| 48 | Glioma invasiveness signaling | 2.19 × 10−2 | ||
| 49 | NF-κB signaling | 2.19 × 10−2 | ||
| 50 | eNOS signaling | 2.19 × 10−2 | ||
| 51 | Nitric oxide signaling in the cardiovascular system | 2.40 × 10−2 | ||
| 52 | MSP-RON signaling pathway | 2.45 × 10−2 | ||
| 53 | Sphingosine-1-phosphate signaling | 2.69 × 10−2 | ||
| 54 | UVA-induced MAPK signaling | 3.39 × 10−2 | ||
| 55 | IGF-1 signaling | 3.55 × 10−2 | ||
| 56 | Glucocorticoid receptor signaling | 3.55 × 10−2 | ||
| 57 | VEGF signaling | 3.72 × 10−2 | ||
| 58 | Prostanoid biosynthesis | 4.37 × 10−2 | ||
| 59 | Coagulation system | 4.37 × 10−2 | ||
| 60 | Ethanol degradation IV | 4.68 × 10−2 | ||
| 61 | TGF-β signaling | 4.68 × 10−2 | ||
| 62 | Germ cell-sertoli cell junction signaling | 4.68 × 10−2 | ||
| 63 | HER-2 signaling in breast cancer | 4.90 × 10−2 |
Canonical pathways were analyzed with Ingenuity Pathway Analysis (IPA) software
Pathways with p-values below 0.05 were selected
The numbers of pathways are shown in parentheses
Discussion
In this study, we identified SATB2 and NRG1 as candidates for the upstream regulatory genes in uterine leiomyomas. SATB2 and NRG1 were expressed strongly in approximately 90% and 80% of uterine leiomyoma specimens, respectively. In the pathway analysis, 68% (63/92) of the pathways activated by SATB2 overexpression and 57% (33/57) of the pathways activated by NRG1 overexpression were common to the aberrantly altered pathways in uterine leiomyomas. We previously reported that 120 genes were aberrantly expressed and methylated in uterine leiomyomas and that the pathway most clearly associated with these genes was a cancer-related pathway [18]. Cancer-related pathways were also identified as the ones most closely associated with the genes with altered expressions in the SATB2 and NRG1 lines, suggesting that the signaling pathways activated by overexpression of these genes reflect the aberrantly altered pathways in uterine leiomyomas. The pathways activated by SATB2 and NRG1 overexpression also include WNT/β-catenin and TGF-β signaling pathways, both of which play significant roles in the pathogenesis of uterine leiomyomas [5], while the pathways activated by SATB2 overexpression include VEGF, PDGF, and IGF1 signaling pathways that are involved in the growth of uterine leiomyomas [3, 6, 8, 9], and the pathways activated by NRG1 overexpression included retinoic acid signaling that is involved in the growth of these tumors [3, 11]. Thus, the signaling pathways activated by overexpression of these genes are associated with many of the factors currently known to be involved in the pathogenesis of uterine leiomyomas. The findings that SATB2 and NRG1 are highly expressed specifically in uterine leiomyomas and that their overexpression activates signaling pathways related to the pathogenesis of uterine leiomyomas suggest that these genes are involved in the pathogenesis of these tumors as upstream regulatory factors.
SATB2 is reported to be an epigenetic regulator associated with higher-order chromatin structures, and is a transcription factor involved in the regulation of expression of a broad variety of genes [30, 31]. SATB2 expression is also associated with various types of cancers, colorectal, head and neck, bone, breast, and pancreatic [32–35]. High expression of SATB2 is observed in 85% of colorectal cancers [33] and correlates with poor prognosis in breast cancer [34]. Recently, SATB2 was suggested to drive growth and metastasis of pancreatic cancer [35]. NRG1 encodes at least six types of proteins which function as ligands for signal transduction via receptors of the ERBB family [36, 37]. Although immature NRG1 is a transmembrane-type protein, a soluble intracellular domain (ICD) is produced by its proteolysis [37, 38]. The soluble ICD is transferred to the nucleus from the cytoplasm and interacts with other transcription factors to participate in the regulation of expression of various genes [38]. NRG1 is also suggested to be involved in the development of various cancer types [39].
SATB2 is likely to play roles in the morphological changes that occur in uterine leiomyomas. The phenotypic changes including cell morphology, cell adhesion, and aggregation were caused by SATB2 overexpression but not by NRG1 overexpression. The fibroblast-like morphology caused by SATB2 overexpression resembles that of cultured uterine leiomyomas [26]. The morphological differences between the SATB2 line cells and the cells of other lines in 3D culture may reflect the morphological differences between uterine leiomyoma cells and normal myometrium cells. In the long-term culture of tissue explants, the myometrium-derived cells formed topographically uniform “hills and valleys” reflecting the cell density, as did the cell aggregations in dense places (hills) [40]. In contrast, the leiomyoma-derived cells grew without contact inhibition and as a result formed large nodule-like aggregates of cells [40]. The phenotypic changes can be at least partly explained by differences in the signaling pathways activated by SATB2 and NRG1 overexpression. The common pathways activated in both SATB2 line and uterine leiomyomas included pathways related to morphology (“PAK signaling”), cellular fibrosis (“PDGF signaling,” “IGF1 signaling,” “IL8 signaling,” and “eNOS signaling”) and cell adhesion (“granulocyte adhesion and diapedesis,” “agranulocyte adhesion and diapedesis,” and “Paxillin signaling”) (Table 4). They are all related to cell morphology and adhesion. Since the common pathways activated in both NRG1 line and uterine leiomyomas did not include any of the pathways described above (Table 4), SATB2 must play a central role in regulating the morphology of leiomyoma cells.
It is well known that uterine leiomyomas display significant heterogeneity in terms of histopathology, genetic changes, and gene expressions, suggesting the existence of subtypes of uterine leiomyomas [41, 42]. The variability and inconsistencies frequently seen among samples and studies may be largely explained by different gene expressions [41, 42]. Transcriptional differences in important genes and pathways also can explain the frequently seen differences in clinicopathological features of uterine leiomyomas [42]. The differences in morphology and aggregation between SATB2 and NRG1 cell lines seen in this study may indicate the existence of different types of uterine leiomyomas.
Progesterone signaling has important roles in the growth of uterine leiomyomas [3, 4]. Recently, much attention has been paid to the role of progenitor cells in the onset of uterine leiomyomas. However, expression of the gene for progesterone receptor (PGR) is not detected in the side population cells of uterine leiomyomas, which are considered as the progenitor cells of uterine leiomyomas [43]. Because PGR is highly expressed in uterine leiomyomas, the progenitor cells have to acquire the ability to express PGR during the development of uterine leiomyomas. However, the mechanism of its acquisition is unknown. In this study, the expression level of PGR mRNA was remarkably increased by NRG1 overexpression. This may provide some clues about the acquisition mechanisms of PGR expression in the progenitor cells.
We revealed that SATB2 and NRG1 were highly expressed and hypermethylated in most uterine leiomyoma specimens. The aberrantly methylated regions of the two genes are located in the vicinity of one of their multiple transcription start sites (TSSs). One of the SATB2 variants (NM_001172517) and one of the NRG1 variants (NM_013959) is transcribed from each TSS. In general, DNA hypermethylation in the vicinity of a TSS is thought to suppress expression, although the theory is unlikely to apply to both variants. One possible explanation for the discrepancy is that in uterine leiomyomas, the other variant transcripts except for NM_001172517 and NM_013959 may be transcribed from other TSSs. Indeed, our unpublished data suggest this possibility in SATB2. SATB2 has at least three different TSSs, located approximately 5 kb upstream and 1 kb and 8 kb downstream of the hypermethylated region analyzed in this study. Expression analysis of variants transcribed from each TSS revealed that only one of the SATB2 variants (NM_001172509) is expressed from the TSS located 8 kb downstream of the hypermethylated region in both normal myometrium and uterine leiomyomas.
In this study, we identified SATB2 and NRG1 as candidates of the upstream regulatory genes in uterine leiomyomas. The transcriptome analyses and pathway analyses of the cell lines overexpressing SATB2 and NRG1 indicate that the pathways activated by SATB2 and NRG1 overexpression include the signaling pathways thought to be associated with the onset and development of uterine leiomyomas. These results suggest that SATB2 and NRG1 act as upstream regulatory factors in the onset and development of leiomyomas.
Electronic supplementary material
(PDF 287 kb)
Acknowledgments
We would like to thank Drs. Ikuo Konishi and Noriomi Matsumura (Kyoto University, and Kindai University, respectively) for providing us human immortalized uterine smooth muscle cells (hTERT UtSMCs). This work was supported in part by JSPS KAKENHI Grants 15K10720, 16K11142, 16K20191, 17K11240, 17K11239, and 16K11091 for Scientific Research from the Ministry of Education, Science, and Culture, Japan.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical statement
This study was approved by the Institutional Review Board of Yamaguchi University Graduate School of Medicine. Informed consent was obtained from the patients before the collection of any samples. All of the experiments handling human tissues were performed in accordance with Tenets of the Declaration of Helsinki.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Stewart EA. Uterine fibroids. Lancet. 2001;357:293–298. doi: 10.1016/S0140-6736(00)03622-9. [DOI] [PubMed] [Google Scholar]
- 2.Bajekal N, Li TC. Fibroids, infertility and pregnancy wastage. Hum Reprod Update. 2000;6:614–620. doi: 10.1093/humupd/6.6.614. [DOI] [PubMed] [Google Scholar]
- 3.Borahay MA, Al-Hendy A, Kilic GS, Boehning D. Signaling pathways in leiomyoma: understanding pathobiology and implications for therapy. Mol Med. 2015;21:242–256. doi: 10.2119/molmed.2014.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ishikawa H, Ishi K, Serna VA, Kakazu R, Bulun SE, Kurita T. Progesterone is essential for maintenance and growth of uterine leiomyoma. Endocrinology. 2010;151:2433–2442. doi: 10.1210/en.2009-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ciebiera M, Włodarczyk M, Wrzosek M, Męczekalski B, Nowicka G, Łukaszuk K, Ciebiera M, Słabuszewska-Jóźwiak A, Jakiel G. Role of transforming growth factor b in uterine fibroid biology. Int J Mol Sci. 2017;18:e 2435. doi: 10.3390/ijms18112435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng L, Wen Y, Han Y, Wei A, Shi G, Mizuguchi M, Lee P, Hernando E, Mittal K, Wei JJ. Expression of insulin-like growth factors (IGFs) and IGF signaling: molecular complexity in uterine leiomyomas. Fertil Steril. 2009;91:2664–2675. doi: 10.1016/j.fertnstert.2007.10.083. [DOI] [PubMed] [Google Scholar]
- 7.Ren Y, Yin H, Tian R, Cui L, Zhu Y, Lin W, Tang XD, Gui Y, Zheng XL. Different effects of epidermal growth factor on smooth muscle cells derived from human myometrium and from leiomyoma. Fertil Steril. 2011;96:1015–1020. doi: 10.1016/j.fertnstert.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Suo G, Jiang Y, Cowan B, Wang JY. Platelet-derived growth factor C is upregulated in human uterine fibroids and regulates uterine smooth muscle cell growth. Biol Reprod. 2009;81:749–758. doi: 10.1095/biolreprod.109.076869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang CC, Hsieh YY, Lin WH, Lin CS. Leiomyoma and vascular endothelial growth factor gene polymorphisms: a systematic review. Taiwan J Obstet Gynecol. 2010;49:247–253. doi: 10.1016/S1028-4559(10)60056-3. [DOI] [PubMed] [Google Scholar]
- 10.Helmke BM, Markowski DN, Müller MH, Sommer A, Müller J, Möller C, et al. HMGA proteins regulate the expression of FGF2 in uterine fibroids. Mol Hum Reprod. 2011;17:135–142. doi: 10.1093/molehr/gaq083. [DOI] [PubMed] [Google Scholar]
- 11.Zaitseva M, Vollenhoven BJ, Rogers PA. Retinoids regulate genes involved in retinoic acid synthesis and transport in human myometrial and fibroid smooth muscle cells. Hum Reprod. 2008;23:1076–1086. doi: 10.1093/humrep/den083. [DOI] [PubMed] [Google Scholar]
- 12.Faerstein E, Szklo M, Rosenshein N. Risk factors for uterine leiomyoma: a practice-based case-control study. I. African-American heritage, reproductive history, body size, and smoking. Am J Epidemiol. 2001;153:1–10. doi: 10.1093/aje/153.1.1. [DOI] [PubMed] [Google Scholar]
- 13.Faerstein E, Szklo M, Rosenshein NB. Risk factors for uterine leiomyoma: a practice-based case-control study. II. Atherogenic risk factors and potential sources of uterine irritation. Am J Epidemiol. 2001;153:11–19. doi: 10.1093/aje/153.1.11. [DOI] [PubMed] [Google Scholar]
- 14.Chiaffarino F, Parazzini F, La Vecchia C, Chatenoud L, Di Cintio E, Marsico S. Diet and uterine myomas. Obstet Gynecol. 1999;94:395–398. doi: 10.1016/s0029-7844(99)00305-1. [DOI] [PubMed] [Google Scholar]
- 15.Asada H, Yamagata Y, Taketani T, Matsuoka A, Tamura H, Hattori N, Ohgane J, Hattori N, Shiota K, Sugino N. Potential link between estrogen receptor-alpha gene hypomethylation and uterine fibroid formation. Mol Hum Reprod. 2008;14:539–545. doi: 10.1093/molehr/gan045. [DOI] [PubMed] [Google Scholar]
- 16.Yamagata Y, Maekawa R, Asada H, Taketani T, Tamura I, Tamura H, Ogane J, Hattori N, Shiota K, Sugino N. Aberrant DNA methylation status in human uterine leiomyoma. Mol Hum Reprod. 2009;15:259–267. doi: 10.1093/molehr/gap010. [DOI] [PubMed] [Google Scholar]
- 17.Maekawa R, Yagi S, Ohgane J, Yamagata Y, Asada H, Tamura I, et al. Disease-dependent differently methylated regions (D-DMRs) of DNA are enriched on the X chromosome in uterine leiomyoma. J Reprod Dev. 2011;57:604–612. doi: 10.1262/jrd.11-035A. [DOI] [PubMed] [Google Scholar]
- 18.Maekawa R, Sato S, Yamagata Y, Asada H, Tamura I, Lee L, Okada M, Tamura H, Takaki E, Nakai A, Sugino N. Genome-wide DNA methylation analysis reveals a potential mechanism for the pathogenesis and development of uterine leiomyomas. PLoS One. 2013;8:e66632. doi: 10.1371/journal.pone.0066632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navarro A, Yin P, Monsivais D, Lin SM, Du P, Wei JJ, et al. Genome-wide DNA methylation indicates silencing of tumor suppressor genes in uterine leiomyoma. PLoS One. 2012;7:e33284. doi: 10.1371/journal.pone.0033284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyata T, Sonoda K, Tomikawa J, Tayama C, Okamura K, Maehara K, Kobayashi H, Wake N, Kato K, Hata K, Nakabayashi K. Genomic, Epigenomic, and transcriptomic profiling towards identifying omics features and specific biomarkers that distinguish uterine leiomyosarcoma and leiomyoma at molecular levels. Sarcoma. 2015;2015:412068. doi: 10.1155/2015/412068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Q, Mas A, Diamond MP, Al-Hendy A. The mechanism and function of epigenetics in uterine leiomyoma development. Reprod Sci. 2016;23:163–175. doi: 10.1177/1933719115584449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 23.Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci U S A. 2015;112:118–123. doi: 10.1073/pnas.1421839112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsumura N, Mandai M, Miyanishi M, Fukuhara K, Baba T, Higuchi T, Kariya M, Takakura K, Fujii S. Oncogenic property of acrogranin in human uterine leiomyosarcoma: direct evidence of genetic contribution in in vivo tumorigenesis. Clin Cancer Res. 2006;12:1402–1411. doi: 10.1158/1078-0432.CCR-05-2003. [DOI] [PubMed] [Google Scholar]
- 26.Malik M, Catherino WH. Development and validation of a three-dimensional in vitro model for uterine leiomyoma and patient-matched myometrium. Fertil Steril. 2012;97:1287–1293. doi: 10.1016/j.fertnstert.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 27.Malik M, Britten J, Segars J, Catherino WH. Leiomyoma cells in 3-dimensional cultures demonstrate an attenuated response to fasudil, a rho-kinase inhibitor, when compared to 2-dimensional cultures. Reprod Sci. 2014;21:1126–1138. doi: 10.1177/1933719114545240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato S, Maekawa R, Yamagata Y, Asada H, Tamura I, Lee L, et al. Potential mechanisms of aberrant DNA hypomethylation on the x chromosome in uterine leiomyomas. J Reprod Dev. 2014;60:47–54. doi: 10.1262/jrd.2013-095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato S, Maekawa R, Yamagata Y, Tamura I, Lee L, Okada M, Jozaki K, Asada H, Tamura H, Sugino N. Identification of uterine leiomyoma-specific marker genes based on DNA methylation and their clinical application. Sci Rep. 2016;6:30652. doi: 10.1038/srep30652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobreva G, Chahrour M, Dautzenberg M, Chirivella L, Kanzler B, Fariñas I, Karsenty G, Grosschedl R. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell. 2006;125:971–986. doi: 10.1016/j.cell.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 31.Gyorgy AB, Szemes M, de Juan Romero C, Tarabykin V, Agoston DV. SATB2 interacts with chromatin-remodeling molecules in differentiating cortical neurons. Eur J Neurosci. 2008;27:865–873. doi: 10.1111/j.1460-9568.2008.06061.x. [DOI] [PubMed] [Google Scholar]
- 32.Brocato J, Costa M. SATB1 and 2 in colorectal cancer. Carcinogenesis. 2015;36:186–191. doi: 10.1093/carcin/bgu322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Magnusson K, de Wit M, Brennan DJ, Johnson LB, McGee SF, Lundberg E, et al. SATB2 in combination with cytokeratin 20 identifies over 95% of all colorectal carcinomas. Am J Surg Pathol. 2011;35:937–948. doi: 10.1097/PAS.0b013e31821c3dae. [DOI] [PubMed] [Google Scholar]
- 34.Patani N, Jiang W, Mansel R, Newbold R, Mokbel K. The mRNA expression of SATB1 and SATB2 in human breast cancer. Cancer Cell Int. 2009;9:18. doi: 10.1186/1475-2867-9-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu W, Ma Y, Shankar S, Srivastava RK. Role of SATB2 in human pancreatic cancer: implications in transformation and a promising biomarker. Oncotarget. 2016;7:57783–57797. doi: 10.18632/oncotarget.10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steinthorsdottir V, Stefansson H, Ghosh S, Birgisdottir B, Bjornsdottir S, Fasquel AC, Olafsson O, Stefansson K, Gulcher JR. Multiple novel transcription initiation sites for NRG1. Gene. 2004;342:97–105. doi: 10.1016/j.gene.2004.07.029. [DOI] [PubMed] [Google Scholar]
- 37.Mei L, Nave KA. Neuregulin-ERBB signaling in the nervous system and neuropsychiatric diseases. Neuron. 2014;83:27–49. doi: 10.1016/j.neuron.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Willem M. Proteolytic processing of Neuregulin-1. Brain Res Bull. 2016;126:178–182. doi: 10.1016/j.brainresbull.2016.07.003. [DOI] [PubMed] [Google Scholar]
- 39.Fernandez-Cuesta L, Thomas RK. Molecular pathways: targeting NRG1 fusions in lung cancer. Clin Cancer Res. 2015;21:1989–1994. doi: 10.1158/1078-0432.CCR-14-0854. [DOI] [PubMed] [Google Scholar]
- 40.Kobayashi Y, Nikaido T, Zhai YL, Iinuma M, Shiozawa T, Shirota M, Fujii S. In-vitro model of uterine leiomyomas: formation of ball-like aggregates. Hum Reprod. 1996;11:1724–1730. doi: 10.1093/oxfordjournals.humrep.a019476. [DOI] [PubMed] [Google Scholar]
- 41.Bertsch E, Qiang W, Zhang Q, Espona-Fiedler M, Druschitz S, Liu Y, Mittal K, Kong B, Kurita T, Wei JJ. MED12 and HMGA2 mutations: two independent genetic events in uterine leiomyoma and leiomyosarcoma. Mod Pathol. 2014;27:1144–1153. doi: 10.1038/modpathol.2013.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mehine M, Kaasinen E, Heinonen HR, Mäkinen N, Kämpjärvi K, Sarvilinna N, Aavikko M, Vähärautio A, Pasanen A, Bützow R, Heikinheimo O, Sjöberg J, Pitkänen E, Vahteristo P, Aaltonen LA. Integrated data analysis reveals uterine leiomyoma subtypes with distinct driver pathways and biomarkers. Proc Natl Acad Sci U S A. 2016;113:1315–1320. doi: 10.1073/pnas.1518752113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ono M, Qiang W, Serna VA, Yin P, Coon JS, 5th, Navarro A, Monsivais D, Kakinuma T, Dyson M, Druschitz S, Unno K, Kurita T, Bulun SE. Role of stem cells in human uterine leiomyoma growth. PLoS One. 2012;7:e36935. doi: 10.1371/journal.pone.0036935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 287 kb)