

Abstract
Mitochondrial protein quality control requires the action of proteases to remove damaged or unnecessary proteins and perform key regulatory cleavage events. Important components of the quality control network are the mitochondrial AAA proteases, which capture energy from ATP hydrolysis to destabilize and degrade protein substrates on both sides of the inner membrane. Dysfunction of these proteases leads to the breakdown of mitochondrial proteostasis and is linked to the development of severe human diseases. In this review, we will describe recent insights into the structure and motions of the mitochondrial AAA proteases and related enzymes. Together, these studies have revealed the mechanics of ATP-driven protein destruction and significantly advanced our understanding of how these proteases maintain mitochondrial health.
Keywords: mitochondrial AAA proteases, i-AAA, m-AAA, protein quality control, AAA+ proteases
Introduction
Protein degradation is a key arm of the mitochondrial proteostasis network that protects the integrity and operation of the organelle [1, 2]. Current estimates suggest that the mitochondrial proteome contains approximately 1500–2000 proteins, of which greater than 99% are encoded in the nucleus and imported from the cytosol[3]. Upon entry, many of these imported proteins assemble into complexes together with additional subunits encoded by the mitochondrial genome. Thus, key mitochondrial activities such as respiration require the careful coordination of these two protein pools to achieve correct subunit stoichiometry and complex formation [4, 5]. Mitochondria are also a hub of oxidative damage arising from the production of reactive oxygen species (ROS) by the OXPHOS system[6, 7]. Consequently, oxidatively damaged proteins must be quickly removed and replaced to preserve their specific activities and prevent the accumulation of harmful aggregates. Lastly, the mitochondrial proteome undergoes constant resculpting in response to the changing demands of the cell, whether through cell cycle progression, altered energy sources, or in response to cellular stress [8, 9]. All of these processes require the efficient destruction of protein molecules and mitochondria contain a diverse collection of proteases located across all compartments that achieve a constant high rate of protein turnover [10]. Breakdown of mitochondrial proteostasis results in significant negative consequences to the structure and function of the organelle [1, 11]. Unsurprisingly, a number of human diseases are associated with impaired or improper mitochondrial protein degradation, including cancer and numerous neurodegenerative diseases[12–15]. Moreover, decline of mitochondrial proteolytic capacity over time and accumulation of oxidized proteins has been linked to aging[1, 16–18]. Thus, studying the mechanisms of mitochondrial protein degradation, and its role in the wider proteostatic network is crucial for understanding mitochondrial function in both health and disease states.
The AAA+ protease family provides a major route for protein degradation in mitochondria. AAA+ enyzmes (ATPases Associated with diverse cellular Activities) are a broad superfamily of cellular machines that capture energy from the hydrolysis of ATP to power the remodeling of various biological molecules, including the unwinding of DNA helices, the formation of vesicles, and the disassembly of stable protein complexes[19–21]. Within this superfamily, the AAA+ proteases perform ATP-powered extraction, unfolding, and degradation of protein molecules[22]. Mitochondria contain multiple AAA+ proteases that perform degradation across the different compartments of the organelle. Soluble matrix-located family members include Lon and ClpXP, which belong to the HCLR clade within the wider AAA+ superfamily [21, 23–27]. This review will focus on two homologous membrane-anchored proteases that belong to the separate classical AAA clade [21, 27]. These proteases, generically named; intermembrane space-AAA (i-AAA) and matrix-AAA (m-AAA), are permanently anchored to the mitochondrial inner membrane (IM) and provide proteolytic surveillance across both faces of the IM and in the adjoining intermembrane space (IMS) and matrix, respectively [28] (Figure 1). Historically, these membrane-bound proteases have been collectively known as the “mitochondrial AAA proteases”, and we refer to them as such throughout this review. The mitochondrial AAA proteases maintain proteostasis by coordinating levels of respiratory complex subunits, prevent accumulation of potentially harmful damaged proteins, and perform important regulatory proteolytic cleavages[29]. New information gained from high resolution cryoEM structures and solution biochemistry studies has shed light on the architectures of the mitochondrial AAA proteases and the complex motions used by these enzymes to perform ATP-driven protein degradation that preserves mitochondrial function.
Figure 1. Versatile mitochondrial AAA proteases.
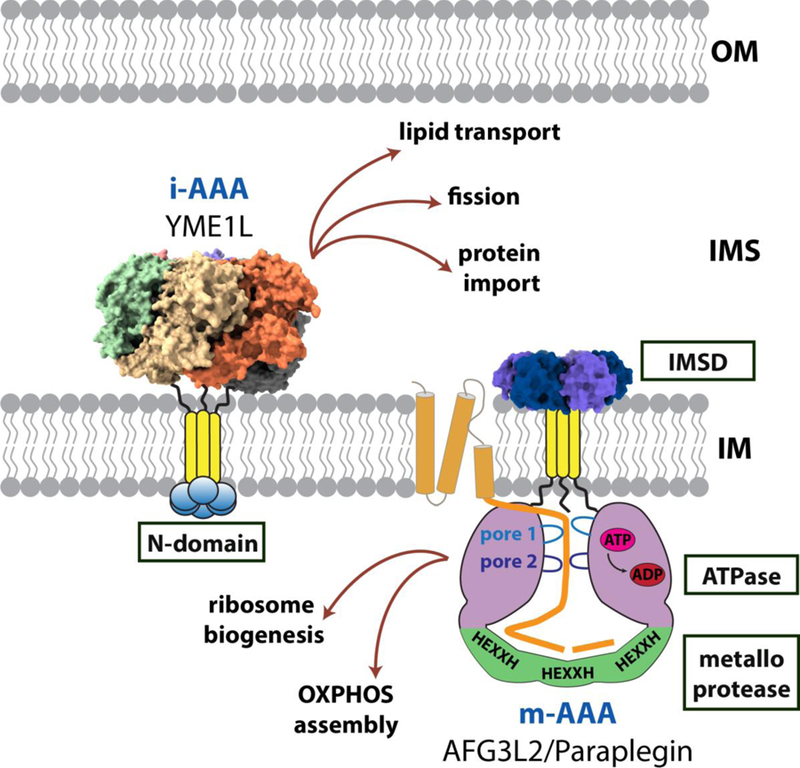
Cartoon showing the diverse mitochondrial activities influenced by proteolysis provided by the mitochondrial AAA proteases. Available structural information is displayed for the catalytic core of Yme1 (PDB ID: 6AZ0) [31] and the analogous periplasmic domain of FtsH (PDB ID: 4V0B) [32].
Domain organization
The mitochondrial AAA proteases belong to a subfamily of membrane-anchored proteases related to the ancestral bacterial FtsH protease[28]. In contrast to some members of the AAA+ protease family, all catalytic domains of the FtsH-like proteases are encoded on a single polypeptide chain[30].
Each subunit contains a transmembrane (TM) domain close to the N-terminus followed by a AAA+ domain and a C-terminal zinc metalloprotease domain [28]. Replacement of this domain by an artificial hexamerizing sequence yielded an active soluble protease, demonstrating that the primary function of the TM domain is to drive oligomerization[33]. Additional small ancillary domains are found flanking the TM domain that locate to the opposite side of the membrane to the catalytic domains (N-domain in i-AAA; IMS domain (IMSD) in m-AAA). Assembly of these multi-domain subunits into active hexameric proteases creates a barrel-shaped catalytic core comprising a ring of six ATPase domains sitting atop a ring of six protease domains. Protein substrates are unfolded and translocated through the center of the ATPase ring into a degradation chamber containing the six sequestered metalloprotease active sites. Upon entry into the chamber, the unfolded substrate polypeptide encounters the active sites and is cleaved into small peptides that are released for further processing and recycling (Figure 1).
All i-AAA proteases are composed of six identical gene products (YME1L in human, Yme1 in yeast). In contrast, the yeast m-AAA protease is an obligate heterohexamer of alternating Yta10 and Yta12 subunits[34]. In humans, the m-AAA protease can assemble into both homooligomers of AFG3L2 subunits or heterooligomers of AFG3L2 and Paraplegin (SPG7)[35]. The distribution of the two mammalian isoforms is tissue specific with a greater proportion of AFG3L2 homohexamers found in neuronal tissues[35]. In mice, a third isoform, AFG3L1, incorporates into m-AAA to add further subunit diversity [35, 36]. However, in humans, AFG3L1 forms an inactive pseudogene [36]. Integration of the transmembrane domains into the IM projects the catalytic cores into the adjoining aqueous compartments. The TM domain of the i-AAA protease contains a single transmembrane span that inserts into the IM placing the catalytic core in the IMS. In contrast, the TM domain of the m-AAA protease spans twice across the membrane and the core is projected into the matrix. These opposing orientations within the IM position the i-AAA and m-AAA proteases to provide protein quality control at both faces of the membrane and within the IMS and matrix, respectively[37].
Diverse functions in mitochondrial quality control
The mitochondrial AAA proteases are notable for their wide diversity of quality control activities[29] (Figure 1). One well-established function is the surveillance of their respective compartments for the appearance of damaged or improperly assembled proteins. Examples of these substrates include multiple components of the respiratory chain and F1-F0 ATPase complexes, and the regulator of the calcium uniporter, EMRE, which can overaccumulate due to imbalance in the expression of the two contributory genomes [38–43]. Oxidatively damaged proteins can emerge as errors in the biogenesis of assembled OXPHOS complexes give rise to unregulated ROS formation [44, 45]. Moreover, the complement of cysteine-rich proteins within the IMS are particularly susceptible to oxidative damage, including the small Tim protein chaperone subunit Tim10, which is degraded by the Yme1 upon disruption of its internal disulfide bonds [46–48].
These house-keeping activities can be considered as generally non-specific quality control to limit the consequences of imbalance or damage and preserve mitochondrial function. In contrast, the mitochondrial AAA proteases can also perform proteolysis of specific substrates as a means of regulating a particular action. An important example of this proteolytic control is seen in human OPA1, the regulator of mitochondrial dynamics, which is cleaved by YME1L to generate a short isoform that acts as a signal for mitochondrial fission [49–51]. Additionally, the human lipid carrier protein PRELID (Ups1/Ups2 in yeast) is degraded by YME1L as a means of controlling the flux of cardiolipin phospholipid precursors to the IM[52]. This specific proteolysis can also be used in the biogenesis of important mitochondrial components, such as the maturation of the mitochondrial ribosomal subunit, MrpL32, by m-AAA through removal of an unstructured N-terminal pre-sequence [53–55]. Lastly, non-proteolytic remodelling of substrates by the mitochondrial AAA proteases has been observed, such as the dislocation of cytochrome c peroxidase from the IM by m-AAA for further proteolytic processing by Pcp1 [56].
The performance of both house-keeping and substrate specific proteolysis requires the mitochondrial AAA proteases to utilize multiple modes of substrate recognition. Most AAA+ proteases recognize specific substrates through degron sequences present at accessible locations on the substrate[57]. Solution studies in vitro have revealed the presence of degrons in the accessible N-termini of the Yme1 substrate, Tim10, and the m-AAA substrate, MrpL32[48, 55]. Interestingly, the mechanism used by Yme1 to recognize the phenylalanine-rich degron present in Tim10 could also provide a means to detect exposed hydrophobic regions of unfolded proteins for non-specific house-keeping proteolysis [29, 58].
Architectures of the mitochondrial AAA proteases
The overall architecture of a mitochondrial AAA protease was revealed by a low resolution (12 Å) cryoEM structure of the detergent solubilized full-length Yta10/Yta12 from S. cerevisiae [59]. This heterohexameric structure showed the expected arrangement of stacked ATPase and protease rings that sequester the proteolytic active sites within the degradation chamber. The TM domains cluster into a tight hexameric bundle that is connected to the ATPase ring by flexible linkers creating approximately 13 Å of space between the membrane face and the ATPase ring[59]. The ancillary IMS domains fan out away from the TM bundle with only limited contacts between them. An NMR structure of the IMS domain from human AFG3L2 revealed strong similiarity with the analogous periplasmic domain of bacterial FtsH[60]. In FtsH, these domains form a close hexameric ring, suggesting the conformation seen in Yta10/Yta12 may not reflect that present in the inner membrane. The resolution of the reconstructions was insufficient to distinguish between the Yta10 and Yta12 subunits and the imposition of C6 symmetry averaging produced a symmetric reconstruction of the catalytic core. Higher resolution information on the catalytic cores of this protease family has been previously provided from multiple crystal structures of truncated FtsH constructs from a variety of bacterial sources [61–65]. The best resolved of these FtsH structures contain ADP molecules bound to all six nucleotide-binding sites and almost all structures display an asymmetry in the ATPase ring that departs from the six-fold symmetry of the domain architecture. However, it is unclear to what degree the fully ADP-bound state or interactions formed during crystal packing induce these arrangements.
A significant recent advance by Puchades and co-workers has been the determination of an atomic resolution (3.4 Å) cryoEM structure of the assembled catalytic core of Yme1 with substrate captured in the process of translocation through the ATPase domains [31] (Figure 2a). A hexamerizing coiled-coil was used to replace the stabilizing interactions usually provided by the TM domains and enable assembly outside of the membrane[66]. In contrast to previous models, the ATPase domains of Yme1 form a right-handed spiral that sits atop a flat planar ring of protease domains. Thus, while both the Yme1 cryoEM structure and FtsH crystal structures display asymmetry in the ATPase ring, the nature of this asymmetry is markedly different. This structure has provided significant new information on how this family of proteases engages and translocates substrates during ATP-dependent protein degradation. The detailed arrangements of the different structural features of the mitochondrial AAA proteases, and the wider field of FtsH-family proteases are discussed below.
Figure 2. Insights from an atomic resolution cryoEM structure of Yme1,
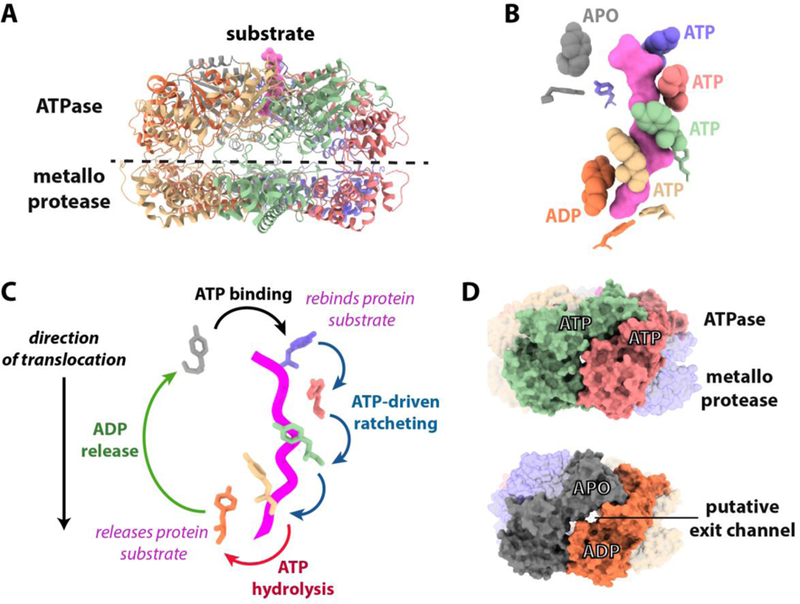
(A) Ribbon model showing the cryoEM structure of the hexameric Yme1 catalytic core[31]. Substrate polypeptide trapped in the ATPase spiral is colored pink. (B) The translocating pore loops surround the substrate in a spiral staircase. The side chains of the substrate-contacting pore loop 1 and pore loop 2 tyrosine residues are shown as balls and sticks, respectively. (C) How cycling of pore loop positions driven by sequential ATP hydrolysis can drive hand-over-hand translocation. Suggested transitions between the positions of pore-loop 1 tyrosines during the ATP hydrolysis cycle are shown based on the hexameric Yme1 structure. The coloring of the tyrosines matches that from 2B and substrate polypeptide is shown as a pink worm. (D) Putative exit channels appear dependent on subunit nucleotide state. Surface representations of Yme1 showing the appearance of lateral channels ~3.5 Å in diameter between apo and ADP bound subunits that are not seen between ATP bound subunits. Channel dimensions were calculated using Caver 3.0[77].
An ATPase ratchet drives protein translocation
The AAA+ ring serves as both the gatekeeper and ATP-driven motor of the protease by selecting substrates for degradation and generating the force required to translocate substrates into the degradation chamber. AAA+ domains can be identified by a series of family specific sequence motifs [19]. The Walker-A and Walker-B motifs are required for ATP binding and hydrolysis, respectively. A glutamate to glutamine substitution in the Walker B motif creates an ‘ATP-trap’ that binds and only very slowly hydrolyzes ATP.
In most AAA+ proteases, two substrate-contacting pore loops (pore loop 1 and pore loop 2) project from the ATPase domains into the central translocating pore to deliver the force for unfolding and translocation[67, 68]. The consensus sequence of the primary force-generating pore loop 1 is [Aromatic]-[hydrophobic]-[Gly], with the aromatic residue believed to directly contact the substrate[69, 70]. Pore loop 2 is less well conserved in both length and sequence but has also been demonstrated to play an important role in substrate translocation in multiple different AAA+ proteins [71–73].
In Yme1, the ATPase domains form a spiral arrangement with a distance of 24 Å between upper and lower positions of the spiral. This spiral arrangement is strikingly similar to recent cryoEM structures of other AAA+ enzymes, including those with non-proteolytic functions[74–76]. This similarity in structure across a diverse protein family suggests conservation of the basic mechanism for ATP-driven substrate remodelling.
The four subunits found at the highest positions of the spiral were bound to ATP, whereas the subunit at the lowest position of the spiral was bound to ADP in a post-hydrolysis state (Figure 2B). A sixth ‘step’ subunit, with an empty nucleotide-binding pocket, adopts an intermediate position between the lowest and highest subunits that is disengaged from the spiral. Subunit conformations similar to the ADP and apo states in Yme1 have also been observed in crystal structures of FtsH[65].
Incorporation of the ‘ATP-trap’ substitution captured co-purified substrate polypeptide traversing through the central axis of the ATPase spiral. The translocating pore loops surround the substrate in a spiral staircase that mirrors the positions of their parent domains (Figure 2A–B). The conserved pore loop 1 aromatic tyrosines of ATP-bound subunits closely contact the backbone atoms of the substrate polypeptide, whereas the identical residue in the ADP-bound subunit is disengaged from the substrate and the pore loop 1 of the ‘step’ subunit is positioned away from the substrate at a significant distance. A second staircase formed from each pore loop 2 is located immediately below the first with an analogous tyrosine in position to accept the incoming polypeptide, albeit without forming substrate contacts in the captured state. Direct contact between pore loop 2 and substrate has been demonstrated biochemically in the AAA+ unfoldase, ClpX, and presumably, translocation of the substrate through the pore loop 1 spiral brings the substrate into contact with the lower staircase [71]. Whether this hand-off occurs in a coordinated fashion through specific loop-loop interactions remains an open question. The relative low sequence conservation of pore loop 2 compared to pore loop 1 suggests this pore loop 2 may function as a specificity determinant in addition to adding grip during substrate translocation.
The nucleotide dependent arrangement of pore loops seen in Yme1 demonstrates important mechanistic principles that can be extended to other AAA+ proteins. Firstly, by contacting the backbone atoms of the substrate polypeptide, the pore loops can perform translocation independent of substrate sequence, a requirement for degrading a diverse pool of substrate proteins. Secondly, using the ATP-bound subunits to hold the substrate provides a mechanism for translocation where the hydrolysis of ATP in a subunit promotes uncoupling of its pore loops from the substrate, allowing the subunit to step up and rejoin the spiral at the top position. Consecutive ATP hydrolysis events create a ratchet that drives unfolding and translocation in a single direction (Figure 2C).
How the sequence of ATP hydrolysis events is controlled in AAA+ enzymes is an open question. The spiral of ATPase domains observed in Yme1 and other AAA+ proteins is consistent with a sequential hydrolysis mechanism whereby subunits ‘fire’ in order around the ring [31, 75, 76, 78]. However, solution biochemistry studies on members of the HCLR clade of AAA+ proteases, ClpXP and HslU, have ruled out a strictly sequential firing order[79, 80]. Any potential mechanism requires the communication of nucleotide state from one subunit to its neighbors. Importantly, the nucleotide-binding pocket of Yme1 is formed largely within a single cis subunit with a smaller number of interactions provided in trans by the clockwise adjacent subunit, thus providing a means of communication[81] (Figure 3). These ‘trans-acting’ elements include a pair of conserved arginine residues known as the ‘arginine fingers’ and an InterSubunit Signaling (ISS) motif, with the consensus sequence [Asp]-[Gly]-[Phe] [82, 83]. Both of these elements are found in the classical AAA clade, which include the mitochondrial AAA proteases, but the ISS motif is absent in the HCLR clade [81, 84, 85]. The function of the arginine fingers was originally proposed based on homology to similar residues in GTPases, whereas the ISS was identified in a genetic screen for residues that communicate the presence of bound ATP between subunits in the Yta10/Yta12 heterohexamer [83]. In Yme1, these trans-acting elements adopt radically different conformations dependent on the nucleotide state of the cis subunit. In ATP-bound subunits, the trans arginine fingers project into the nucleotide-binding pocket and interact with the gamma-phosphate of the ATP. In this position, the arginine fingers can also interact with the aspartate residue of the ISS motif. The ISS phenylalanine then extends across the nucleotide-binding pocket and interacts with a cluster of other phenylalanine residues in the body of the cis subunit. Post ATP hydrolysis, loss of the gamma phosphate alters the positions of the arginine fingers and breaks their interaction with the ISS aspartate. The entire ISS motif retracts back into a helix in the trans subunit, pulling the ISS phenylalanine away from the cis phenylalanine cluster. These rearrangements allow the ‘step’ subunit to move to its position outside of the spiral register (Figure 3). Importantly, similar structural rearrangements of the ISS motif have now been observed in other high-resolution cryoEM structures of classical AAA+ proteins, including the 26S proteasome[74].
Figure 3. Rearrangements of trans-acting elements in the nucleotide-binding pocket.
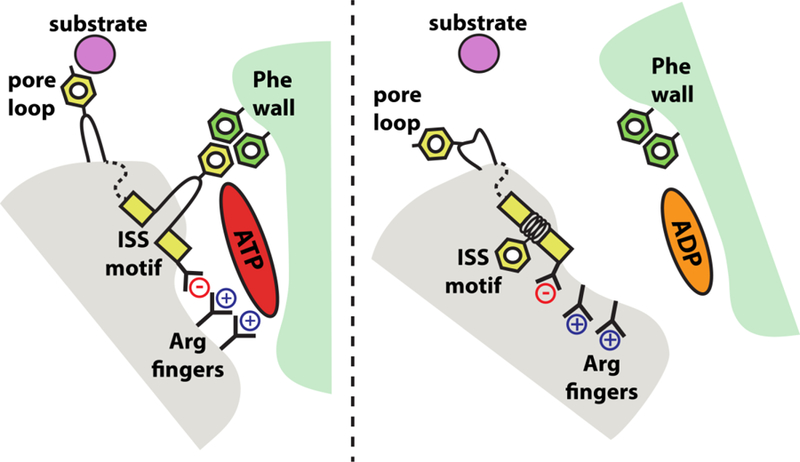
Schematic of the Yme1 nucleotide-binding pocket. When the cis subunit is bound to ATP, the gamma-phosphate is sensed by the trans arginine fingers, and the trans ISS motif extends across the pocket to form tight bridging interactions with cis phenylalanines. After hydrolysis to ADP, interactions between the arginine finger and the now absent gamma phosphate are lost, and the ISS motif winds back into the trans subunit, weakening the cis-trans subunit interface. These rearrangements are allosterically transmitted to the pore loops to break their interaction with the substrate polypeptide. Together, these motions allow the trans AAA+ domain to ‘step’ out from the spiral, unbound to substrate, and reset the ATPase ratchet.
The reorganization of the ISS motif and the breaking of the subunit interface occur simultaneously with a retraction of pore loop 1 belonging to the trans subunit that breaks contact between its tyrosine and the substrate. Alternate pore loop conformations have also been observed in crystal structures of substrate-free FtsH and the related AAA+ disaggregase, Hsp104[65, 85]. Based on these structures, a mechanism for communication can be proposed where loss of the gamma phosphate after ATP hydrolysis is sensed by the arginine fingers and communicated to the ISS motif through the arginine-aspartate interactions. This results in retraction of the ISS motif from the cis subunit and allosterically releases the trans pore loop 1 from the substrate[31, 85].
Proteolysis occurs in a degradation chamber
Assembly of the catalytic core creates a ring of protease domains below the ATPase domains to complete the degradation chamber and places the six proteolytic active sites at the internal periphery of the chamber. These zinc metalloprotease sites are defined by a conserved HEXXH motif that coordinates a catalytic zinc ion and promotes cleavage of peptide bonds[28, 86]. In all structures of assembled FtsH-like proteases, the protease domains form a flat planar ring that exhibits strong six-fold symmetry, indicating that the conformation of these domains does not alter as the ATPase domains above undergo large nucleotide dependent movements. As the substrate is fed into the degradation chamber by the ATPase ratchet, it must find its way to the protease sites at the periphery of the chamber for cleavage. It is unclear if the substrate is directed into a specific site or if cleavage occurs upon stochastic interaction with any unoccupied site.
Many proteases cleave substrates at specific sequences, determined by the precise structure of the active site. The protease site of FtsH-like proteases contain a β-sheet that likely forms backbone interactions with the substrate to position the scissile peptide close to the catalytic zinc ion for cleavage[31]. In Yme1, the active site forms a hydrophobic “island” within the largely hydrophilic degradation chamber that could help recruit hydrophobic substrates to the site for cleavage. Analysis of the product peptides produced from the degradation of a diverse collection of substrates by the human AFG3L2 homohexamer revealed a cleavage preference for sequences containing bulky hydrophobic residues at the P1’ residue, immediately C-terminal of the scissile peptide bond[55]. It is likely that the surrounding structure of the AFG3L2 protease sites contains a pocket suitable for the accommodation of these residues. This combination of non-specific backbone interactions and a cleavage preference for hydrophobic sequences may provide the m-AAA protease with the ability to rapidly degrade a wide variety of proteins that would normally be found within the inner membrane.
Once cleavage has occurred the peptide product must be released from the degradation chamber. Peptides produced from degradation by AFG3L2 ranged between 7 and 30 residues in length with 14-mers found to be the most abundant species[55]. Interestingly, the distance between protease active sites in the Yme1 chamber is approximately ~38 Å, consistent with approximately 12 residues in an extended conformation. This correlation could indicate that the product peptide is quickly released after cleavage, rather than undergoing a second round of proteolysis. Alternatively, this product length may simply reflect the number of residues required to form stable interactions between the substrate and protease sites to promote cleavage. How product peptides are released from the degradation chamber of FtsH-like proteases has not yet been established. Calculation of the Yme1 molecular surface shows that the large nucleotide-dependent conformational changes in the ATPase spiral create lateral openings approximately 3.5 Å wide between the ADP and apo subunits that could potentially serve as exit passages for peptides (Figure 2D). The appearance of these openings in only certain nucleotide states suggests that the exit of peptides could be allosterically coupled to the ATP-hydrolysis cycle.
Flexibility enables protease function
Allowing large-scale conformational changes in the ATPase spiral while simultaneously preserving the planar protease ring requires a hinge. In Yme1, a flexible linker is present between the ATPase and protease domains and adopts strikingly different conformations in different subunits[31]. This linker contains a conserved glycine that has been identified as a putative hinge residue in both Yme1 and FtsH, and mutation of this glycine to a more bulky leucine significantly reduced degradation activity in both enzymes [31, 64]. The exact conformational trajectory that these subunits follow during the ATPase cycle is uncertain. Single molecule FRET experiments on substrate-free FtsH suggested rotations between the ATPase and protease domains create five distinct subunit conformations during the catalytic cycle and that the motions between these states result are driven by thermal fluctuations rather than ATP hydrolysis[87]. However, the signature ATPase spiral now seen in multiple diverse AAA+ enzymes appears only in substrate bound structures, suggesting that the engagement of substrate by the pore loops organizes the ATPase domains into the active spiral conformation not seen when substrate is absent.
Linker regions are also found between the TM domain and ATPase domains that create ~13 Å of space between the membrane face and the ATPase ring in Yta10/Yta12[59]. These linker sequences are poorly conserved but contain large numbers of likely unstructured residues. While inner membrane embedded substrates may project accessible degrons close to the ATPase pore loops, soluble substrates of FtsH-like proteases must travel through the narrow space created by these linkers to gain access to the degradation chamber. These linkers presumably impart a significant degree of flexibility between the membrane face and the body of the ATPase spiral that allows for the accommodation of such substrates.
The mitochondrial AAA proteases have also been suggested to undergo structural transitions in response to stress. YME1L is degraded by the ATP-independent protease OMA1 under conditions that deplete ATP and depolarize the inner membrane[88, 89]. The absence of ATP likely minimizes the interface between ATPase domains and promotes a more open conformation that could be targeted by OMA1. A recent solution study on YME1L has suggested that both low nucleotide concentration and oxidative modification by hydrogen peroxide impart conformational changes that could sensitize the protease for degradation by OMA1[90].
Concluding Remarks
A high-resolution cryoEM structure of an active state mitochondrial AAA protease in combination with in vitro and in vivo biochemical studies has provided fascinating new insights into the mechanism of ATP-driven protein degradation at the faces of the inner membrane. However, many interesting questions remain that could be illuminated by further structural studies. For example, it is possible that these enzymes adopt additional conformations during their catalytic cycle that are yet to be observed in cryoEM studies. These alternative conformations could be explored by integrated structural approaches, such as single-molecule nanometry or hydrogen-deuterium exchange mass spectrometry. Indeed, detailed single molecule analyses of AAA+ enzyme motions have already been performed for other family members[91–93]. The identification of sequence degrons within protease substrates hypothesizes the existence of substrate recognition surfaces that could be revealed by models in complex with physiological substrates. Lastly, available structural information is largely limited to the catalytic cores of these enzymes. Mechanistic studies of the full-length proteases, particularly embedded into a membrane environment, may shed light on the roles of the non-catalytic domains in substrate recognition and processing and cooperation with other mitochondrial components.
Acknowledgements
We thank our collaborators Gabriel Lander and R. Luke Wiseman at The Scripps Research Institute. We apologize to any colleagues whose work we have not been able to include in this review. Work in the author’s lab is funded by NIH award R01GM115898.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interest Statement
The authors declare no competing interests
References
- 1.Moehle EA, Shen K, and Dillin A, Mitochondrial proteostasis in the context of cellular and organismal health and aging. J Biol Chem, 2019. 294(14): p. 5396–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker MJ, Tatsuta T, and Langer T, Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol, 2011. 3(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Calvo SE, Clauser KR, and Mootha VK, MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res, 2016. 44(D1): p. D1251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryan MT and Hoogenraad NJ, Mitochondrial-nuclear communications. Annu Rev Biochem, 2007. 76: p. 701–22. [DOI] [PubMed] [Google Scholar]
- 5.Couvillion MT, et al. , Synchronized mitochondrial and cytosolic translation programs. Nature, 2016. 533(7604): p. 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beckman KB and Ames BN, The free radical theory of aging matures. Physiol Rev, 1998. 78(2): p. 547–81. [DOI] [PubMed] [Google Scholar]
- 7.Ugarte N, Petropoulos I, and Friguet B, Oxidized mitochondrial protein degradation and repair in aging and oxidative stress. Antioxid Redox Signal, 2010. 13(4): p. 539–49. [DOI] [PubMed] [Google Scholar]
- 8.Lebeau J, Rainbolt TK, and Wiseman RL, Coordinating Mitochondrial Biology Through the Stress-Responsive Regulation of Mitochondrial Proteases. Int Rev Cell Mol Biol, 2018. 340: p. 79–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gottlieb RA and Bernstein D, Mitochondrial remodeling: Rearranging, recycling, and reprogramming. Cell Calcium, 2016. 60(2): p. 88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Augustin S, et al. , Characterization of peptides released from mitochondria: evidence for constant proteolysis and peptide efflux. J Biol Chem, 2005. 280(4): p. 2691–9. [DOI] [PubMed] [Google Scholar]
- 11.Brandt T, et al. , Changes of mitochondrial ultrastructure and function during ageing in mice and Drosophila. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rugarli EI and Langer T, Mitochondrial quality control: a matter of life and death for neurons. EMBO J, 2012. 31(6): p. 1336–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulteau AL and Bayot A, Mitochondrial proteases and cancer. Biochim Biophys Acta, 2011. 1807(6): p. 595–601. [DOI] [PubMed] [Google Scholar]
- 14.Chen B, et al. , Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol, 2011. 3(8): p. a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levytskyy RM, Germany EM, and Khalimonchuk O, Mitochondrial Quality Control Proteases in Neuronal Welfare. J Neuroimmune Pharmacol, 2016. 11(4): p. 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruan L, et al. , Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bota DA and Davies KJ, Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol, 2002. 4(9): p. 674–80. [DOI] [PubMed] [Google Scholar]
- 18.Bota DA, Van Remmen H, and Davies KJ, Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett, 2002. 532(1–2): p. 103–6. [DOI] [PubMed] [Google Scholar]
- 19.Hanson PI and Whiteheart SW, AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol, 2005. 6(7): p. 519–29. [DOI] [PubMed] [Google Scholar]
- 20.Sysoeva TA, Assessing heterogeneity in oligomeric AAA+ machines. Cell Mol Life Sci, 2017. 74(6): p. 1001–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neuwald AF, et al. , AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res, 1999. 9(1): p. 27–43. [PubMed] [Google Scholar]
- 22.Sauer RT and Baker TA, AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem, 2011. 80: p. 587–612. [DOI] [PubMed] [Google Scholar]
- 23.Wang N, et al. , A human mitochondrial ATP-dependent protease that is highly homologous to bacterial Lon protease. Proc Natl Acad Sci U S A, 1993. 90(23): p. 11247–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suzuki CK, et al. , Requirement for the yeast gene LON in intramitochondrial proteolysis and maintenance of respiration. Science, 1994. 264(5156): p. 273–6. [DOI] [PubMed] [Google Scholar]
- 25.van Dyck L, et al. , Mcx1p, a ClpX homologue in mitochondria of Saccharomyces cerevisiae. FEBS Lett, 1998. 438(3): p. 250–4. [DOI] [PubMed] [Google Scholar]
- 26.Corydon TJ, et al. , Human and mouse mitochondrial orthologs of bacterial ClpX. Mamm Genome, 2000. 11(10): p. 899–905. [DOI] [PubMed] [Google Scholar]
- 27.Erzberger JP and Berger JM, Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu Rev Biophys Biomol Struct, 2006. 35: p. 93–114. [DOI] [PubMed] [Google Scholar]
- 28.Leonhard K, et al. , AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP-dependent degradation of inner membrane proteins in mitochondria. EMBO J, 1996. 15(16): p. 4218–29. [PMC free article] [PubMed] [Google Scholar]
- 29.Glynn SE, Multifunctional Mitochondrial AAA Proteases. Front Mol Biosci, 2017. 4: p. 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langklotz S, Baumann U, and Narberhaus F, Structure and function of the bacterial AAA protease FtsH. Biochim Biophys Acta, 2012. 1823(1): p. 40–8. [DOI] [PubMed] [Google Scholar]
- 31.Puchades C, et al. , Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science, 2017. 358(6363). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scharfenberg F, et al. , Structure and evolution of N-domains in AAA metalloproteases. J Mol Biol, 2015. 427(4): p. 910–23. [DOI] [PubMed] [Google Scholar]
- 33.Nishimura K, Kato Y, and Sakamoto W, Chloroplast Proteases: Updates on Proteolysis within and across Suborganellar Compartments. Plant Physiology, 2016. 171(4): p. 2280–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arlt H, et al. , The YTA10–12 complex, an AAA protease with chaperone-like activity in the inner membrane of mitochondria. Cell, 1996. 85(6): p. 875–85. [DOI] [PubMed] [Google Scholar]
- 35.Koppen M, et al. , Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol Cell Biol, 2007. 27(2): p. 758–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kremmidiotis G, et al. , Molecular and functional analyses of the human and mouse genes encoding AFG3L1, a mitochondrial metalloprotease homologous to the human spastic paraplegia protein. Genomics, 2001. 76(1–3): p. 58–65. [DOI] [PubMed] [Google Scholar]
- 37.Leonhard K, et al. , Membrane protein degradation by AAA proteases in mitochondria: extraction of substrates from either membrane surface. Mol Cell, 2000. 5(4): p. 629–38. [DOI] [PubMed] [Google Scholar]
- 38.Konig T, et al. , The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol Cell, 2016. 64(1): p. 148–162. [DOI] [PubMed] [Google Scholar]
- 39.Arlt H, et al. , The formation of respiratory chain complexes in mitochondria is under the proteolytic control of the m-AAA protease. Embo Journal, 1998. 17(16): p. 4837–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guzelin E, Rep M, and Grivell LA, Afg3p, a mitochondrial ATP-dependent metalloprotease, is involved in degradation of mitochondrially-encoded Cox1, Cox3, Cob, Su6, Su8 and Su9 subunits of the inner membrane complexes III, IV and V. FEBS Lett, 1996. 381(1–2): p. 42–6. [DOI] [PubMed] [Google Scholar]
- 41.Pearce DA and Sherman F, Degradation of cytochrome oxidase subunits in mutants of yeast lacking cytochrome c and suppression of the degradation by mutation of yme1. J Biol Chem, 1995. 270(36): p. 20879–82. [DOI] [PubMed] [Google Scholar]
- 42.Nakai T, et al. , Multiple genes, including a member of the AAA family, are essential for degradation of unassembled subunit 2 of cytochrome c oxidase in yeast mitochondria. Mol Cell Biol, 1995. 15(8): p. 4441–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stiburek L, et al. , YME1L controls the accumulation of respiratory chain subunits and is required for apoptotic resistance, cristae morphogenesis, and cell proliferation. Mol Biol Cell, 2012. 23(6): p. 1010–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bode M, et al. , Inaccurately assembled cytochrome c oxidase can lead to oxidative stress-induced growth arrest. Antioxid Redox Signal, 2013. 18(13): p. 1597–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khalimonchuk O, Bird A, and Winge DR, Evidence for a pro-oxidant intermediate in the assembly of cytochrome oxidase. J Biol Chem, 2007. 282(24): p. 17442–9. [DOI] [PubMed] [Google Scholar]
- 46.Baker MJ, et al. , Impaired folding of the mitochondrial small TIM chaperones induces clearance by the i-AAA protease. J Mol Biol, 2012. 424(5): p. 227–39. [DOI] [PubMed] [Google Scholar]
- 47.Spiller MP, et al. , Mitochondrial Tim9 protects Tim10 from degradation by the protease Yme1. Biosci Rep, 2015. 35(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rampello AJ and Glynn SE, Identification of a Degradation Signal Sequence within Substrates of the Mitochondrial i-AAA Protease. J Mol Biol, 2017. 429(6): p. 873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anand R, et al. , The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol, 2014. 204(6): p. 919–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song Z, et al. , OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol, 2007. 178(5): p. 749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu SB and Pekkurnaz G, Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J Mol Biol, 2018. 430(21): p. 3922–3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Potting C, et al. , Regulation of mitochondrial phospholipids by Ups1/PRELI-like proteins depends on proteolysis and Mdm35. EMBO J, 2010. 29(17): p. 2888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nolden M, et al. , The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell, 2005. 123(2): p. 277–89. [DOI] [PubMed] [Google Scholar]
- 54.Bonn F, et al. , Presequence-dependent folding ensures MrpL32 processing by the m-AAA protease in mitochondria. EMBO J, 2011. 30(13): p. 2545–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding BJ, et al. , Dissecting Substrate Specificities of the Mitochondrial AFG3L2 Protease. Biochemistry, 2018. 57(28): p. 4225–4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tatsuta T, et al. , m-AAA protease-driven membrane dislocation allows intramembrane cleavage by rhomboid in mitochondria. EMBO J, 2007. 26(2): p. 325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baker TA and Sauer RT, ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci, 2006. 31(12): p. 647–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rampello AJ and Glynn SE, Identification of a Degradation Signal Sequence within Substrates of the Mitochondrial i-AAA Protease. Journal of Molecular Biology, 2017. 429(6): p. 873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee S, et al. , Electron cryomicroscopy structure of a membrane-anchored mitochondrial AAA protease. J Biol Chem, 2011. 286(6): p. 4404–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramelot TA, et al. , NMR structure and MD simulations of the AAA protease intermembrane space domain indicates peripheral membrane localization within the hexaoligomer. FEBS Lett, 2013. 587(21): p. 3522–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suno R, et al. , Structure of the whole cytosolic region of ATP-dependent protease FtsH. Mol Cell, 2006. 22(5): p. 575–85. [DOI] [PubMed] [Google Scholar]
- 62.Bieniossek C, Niederhauser B, and Baumann UM, The crystal structure of apo-FtsH reveals domain movements necessary for substrate unfolding and translocation. Proceedings of the National Academy of Sciences of the United States of America, 2009. 106(51): p. 21579–21584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bieniossek C, et al. , The molecular architecture of the metalloprotease FtsH. Proc Natl Acad Sci U S A, 2006. 103(9): p. 3066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vostrukhina M, et al. , The structure of Aquifex aeolicus FtsH in the ADP-bound state reveals a C-2-symmetric hexamer. Acta Crystallographica Section D-Structural Biology, 2015. 71: p. 1307–1318. [DOI] [PubMed] [Google Scholar]
- 65.Uthoff M and Baumann U, Conformational flexibility of pore loop-1 gives insights into substrate translocation by the AAA(+) protease FtsH. J Struct Biol, 2018. 204(2): p. 199–206. [DOI] [PubMed] [Google Scholar]
- 66.Shi H, Rampello AJ, and Glynn SE, Engineered AAA+ proteases reveal principles of proteolysis at the mitochondrial inner membrane. Nat Commun, 2016. 7: p. 13301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Weibezahn J, et al. , Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell, 2004. 119(5): p. 653–65. [DOI] [PubMed] [Google Scholar]
- 68.Song HK, et al. , Mutational studies on HslU and its docking mode with HslV. Proc Natl Acad Sci U S A, 2000. 97(26): p. 14103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yamada-Inagawa T, et al. , Conserved pore residues in the AAA protease FtsH are important for proteolysis and its coupling to ATP hydrolysis. J Biol Chem, 2003. 278(50): p. 50182–7. [DOI] [PubMed] [Google Scholar]
- 70.Wang J, et al. , Nucleotide-dependent conformational changes in a protease-associated ATPase HsIU. Structure, 2001. 9(11): p. 1107–16. [DOI] [PubMed] [Google Scholar]
- 71.Martin A, Baker TA, and Sauer RT, Diverse pore loops of the AAA+ ClpX machine mediate unassisted and adaptor-dependent recognition of ssrA-tagged substrates. Mol Cell, 2008. 29(4): p. 441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hinnerwisch J, et al. , Loops in the central channel of ClpA chaperone mediate protein binding, unfolding, and translocation. Cell, 2005. 121(7): p. 1029–41. [DOI] [PubMed] [Google Scholar]
- 73.Lee J, et al. , Structural determinants for protein unfolding and translocation by the Hsp104 protein disaggregase. Biosci Rep, 2017. 37(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de la Pena AH, et al. , Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science, 2018. 362(6418): p. 1018-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gates SN, et al. , Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science, 2017. 357(6348): p. 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Monroe N, et al. , Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chovancova E, et al. , CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput Biol, 2012. 8(10): p. e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Enemark EJ and Joshua-Tor L, Mechanism of DNA translocation in a replicative hexameric helicase. Nature, 2006. 442(7100): p. 270–5. [DOI] [PubMed] [Google Scholar]
- 79.Martin A, Baker TA, and Sauer RT, Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines. Nature, 2005. 437(7062): p. 1115–20. [DOI] [PubMed] [Google Scholar]
- 80.Baytshtok V, et al. , Covalently linked HslU hexamers support a probabilistic mechanism that links ATP hydrolysis to protein unfolding and translocation. J Biol Chem, 2017. 292(14): p. 5695–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wendler P, et al. , Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta, 2012. 1823(1): p. 2–14. [DOI] [PubMed] [Google Scholar]
- 82.Chang CW, Lee S, and Tsai FTF, Structural Elements Regulating AAA+ Protein Quality Control Machines. Front Mol Biosci, 2017. 4: p. 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Augustin S, et al. , An intersubunit signaling network coordinates ATP hydrolysis by m-AAA proteases. Mol Cell, 2009. 35(5): p. 574–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Biter AB, et al. , Functional analysis of conserved cis- and trans-elements in the Hsp104 protein disaggregating machine. J Struct Biol, 2012. 179(2): p. 172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Biter AB, et al. , Structural basis for intersubunit signaling in a protein disaggregating machine. Proc Natl Acad Sci U S A, 2012. 109(31): p. 12515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rawlings ND and Barrett AJ, Evolutionary families of metallopeptidases. Methods Enzymol, 1995. 248: p. 183–228. [DOI] [PubMed] [Google Scholar]
- 87.Ruer M, et al. , ATPase and Protease Domain Movements in the Bacterial AAA+ Protease FtsH Are Driven by Thermal Fluctuations. J Mol Biol, 2018. 430(22): p. 4592–4602. [DOI] [PubMed] [Google Scholar]
- 88.Rainbolt TK, et al. , Reciprocal Degradation of YME1L and OMA1 Adapts Mitochondrial Proteolytic Activity during Stress. Cell Rep, 2016. 14(9): p. 2041–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rainbolt TK, Saunders JM, and Wiseman RL, YME1L degradation reduces mitochondrial proteolytic capacity during oxidative stress. EMBO Rep, 2015. 16(1): p. 97–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brambley CA, et al. , Characterization of Mitochondrial YME1L Protease Oxidative Stress-Induced Conformational State. J Mol Biol, 2019. 431(6): p. 1250–1266. [DOI] [PubMed] [Google Scholar]
- 91.Olivares AO, Baker TA, and Sauer RT, Mechanical Protein Unfolding and Degradation. Annu Rev Physiol, 2018. 80: p. 413–429. [DOI] [PubMed] [Google Scholar]
- 92.Ryu JK, Jahn R, and Yoon TY, Review: Progresses in understanding N-ethylmaleimide sensitive factor (NSF) mediated disassembly of SNARE complexes. Biopolymers, 2016. 105(8): p. 518–31. [DOI] [PubMed] [Google Scholar]
- 93.Bhabha G, et al. , How Dynein Moves Along Microtubules. Trends Biochem Sci, 2016. 41(1): p. 94–105. [DOI] [PMC free article] [PubMed] [Google Scholar]