

Abstract
Objective
To identify the rate of change of clinical outcome measures in children with 2 types of congenital muscular dystrophy (CMD), COL6-related dystrophies (COL6-RDs) and LAMA2-related dystrophies (LAMA2-RDs).
Methods
Over the course of 4 years, 47 individuals (23 with COL6-RD and 24 with LAMA2-RD) 4 to 22 years of age were evaluated. Assessments included the Motor Function Measure 32 (MFM32), myometry (knee flexors and extensors, elbow flexors and extensors), goniometry (knee and elbow extension), pulmonary function tests, and quality-of-life measures. Separate linear mixed-effects models were fitted for each outcome measurement, with subject-specific random intercepts.
Results
Total MFM32 scores for COL6-RDs and LAMA2-RDs decreased at a rate of 4.01 and 2.60 points, respectively, each year (p < 0.01). All muscle groups except elbow flexors for individuals with COL6-RDs decreased in strength between 1.70% (p < 0.05) and 2.55% (p < 0.01). Range-of-motion measurements decreased by 3.21° (p < 0.05) at the left elbow each year in individuals with LAMA2-RDs and 2.35° (p < 0.01) in right knee extension each year in individuals with COL6-RDs. Pulmonary function demonstrated a yearly decline in sitting forced vital capacity percent predicted of 3.03% (p < 0.01) in individuals with COL6-RDs. There was no significant change in quality-of-life measures analyzed.
Conclusion
Results of this study describe the rate of change of motor function as measured by the MFM32, muscle strength, range of motion, and pulmonary function in individuals with COL6-RDs and LAMA2-RDs.
Congenital muscular dystrophies (CMDs) comprise a clinically and genetically heterogeneous group of congenital muscle disorders, with characteristic dystrophic features on muscle biopsy.1 COL6-related dystrophies (COL6-RDs) and LAMA2-related dystrophies (LAMA2-RDs) are disorders of the muscle extracellular matrix involving collagen type VI and laminin α2, respectively, which together represent the most common CMD subtypes with an estimated prevalence of 0.1 to 0.9 per 100,000.1–4 The COL6-RDs and LAMA2-RDs are CMD subtypes, each with a wide phenotypic spectrum; however, both subtypes are characterized by progressive muscle weakness, respiratory insufficiency, and joint contractures.1,5
The 173rd European Neuromuscular Centre International Workshop on CMD Outcome Measures made the recommendation that motor, respiratory, and overall performance of individuals with CMD be studied with a battery of outcome measures.6 Following these recommendations, a 2-year pilot study of COL6-RDs and LAMA2-RDs patient populations identified forced vital capacity percent predicted (FVCpp), muscle strength, functional performance, and quality-of-life assessments as feasible, reliable, and likely valid outcome measures.7
This CMD outcome measures pilot study served as the basis for this longitudinal, prospective, natural history study in COL6-RDs and LAMA2-RDs.7 The focus of this extension study was on the core outcome measures of FVCpp, the Motor Function Measure 32 (MFM32), myometry, goniometry, and Pediatric Quality of Life Inventory (PedsQL) with the objectives of describing the annual rate of change and variability for the outcome measures tested and using data obtained from the MFM32 to perform a power calculation for future clinical trials in the COL6-RDs and LAMA2-RDs.
Methods
Participants
A convenience (i.e., recruited on the basis of accessibility to the study and targeted age range) sample of 51 individuals between the ages of 4 and 22 years were prospectively evaluated from 2010 to 2014. Forty-seven individuals were included in the final data analysis, 23 with COL6-RD and 24 with LAMA2-RD. Diagnosis was established on the basis of clinical phenotype and confirmed by muscle biopsy findings of decreased collagen VI or laminin α2 expression and/or genetic testing results revealing causative mutations in the COL6 genes (COL6A1, COL6A2, and COL6A3) or the LAMA2 gene. Twenty-two individuals participated in year 1, 32 in year 2, 38 in years 3 and 4, and 36 in year 5. In year 2, it was determined that 1 participant presented with unconvincing mutation, 1 participant did not have COL6-RD or LAMA2-RD, and 2 others were lost to attrition and were not included in the analysis. All participants were seen for data collection at the same time during the summers at baseline plus 4 follow-up visits from 2010 through 2014. The average follow-up time per participant was 3 years with a total of 148 person-years of follow-up.
Standard protocol approvals, registration, and patient consents
Participants were recruited at the NIH through 2 ClinicalTrials.gov protocols (NCT01568658 and NCT00004568). Both studies were approved by the NIH Combined Neurosciences Institutional Review Board. Written informed consent and assent were obtained for each participant at the time of enrollment.
All assessments were performed by neuromuscular experts (physicians, physical therapists, nurse practitioners, and psychologists). Each year, before assessing all participants, all clinical evaluators reviewed standardized methods for test administration, and interrater reliability between the clinicians was established. The assessments administered included the following.
Motor Function Measure 32
The MFM32 is a 32-item standardized assessment validated in children and adults 6 to 60 years of age in congenital myopathies, CMDs, Duchenne muscular dystrophy, and various other neuromuscular disorders.8–10 The MFM32 was administered by pediatric physical therapists.
Forced vital capacity
Pulmonary function tests (PFTs) for years 2 through 5 of the study were performed in the Pulmonary Function Laboratory of the National Heart, Lung, and Blood Institute of the NIH as part of an annual evaluation of participants in the above protocols and using the VMAXTM Encore PFT system, software version 21.1A (Care-Fusion Corp, San Diego, CA). American Thoracic Society/European Respiratory Society guidelines were followed. Testing included FVC in the sitting and supine positions, with results reported in both absolute values (in liters) and FVCpp using established reference equations.11–13 If a younger child was unable to complete the PFT, the technician would indicate that the test was incomplete or of poor quality. Only PFT data of adequate quality (according to American Thoracic Society guidelines) were included in the analyses. PFT data from the first year of the study (2010) were not included in the analyses given concerns that results from the use of an ambulatory spirometry unit outside of the pulmonary function laboratory may be less comparable to the results obtained in the laboratory.
Quantitative strength testing
A handheld dynamometer (Microfet, Hoggan Health, Inc, West Jordan, UT) was used as a measure of muscle strength in Newtons of force.14 Raw measures were normalized on the basis of age, sex, and weight. Strength measurements were performed by pediatric physical therapists.
Goniometry measurements
A standard 2-arm goniometer was used to measure passive joint range of motion in a standardized position.15 Joint angles were reported relative to full motion at 0°, with a positive number referring to a hyperextended joint and a negative number referring to a joint contracture. Goniometry measurements of bilateral hips, knees, elbows, and ankles were completed by pediatric physical therapists.
The PedsQL 3.0 neuromuscular module
This 25-item instrument contains 3 subscales: About My/My Child's Neuromuscular Disease, related to the disease process (17 items); Communication, related to communications with health care providers and others about his/her illness (3 items); and About Our Family Resources, related to family financial and social support (5 items). Items for each scale were added and then divided by the number of items answered to create a total score for each scale and an overall total score.16–19
Statistical methods
Separate linear mixed-effects models were fitted for each outcome measurement, with subject-specific random intercepts.20 Models estimated yearly change in the outcome over continuous time in each diagnosis subtype (COL6-RDs and LAMA2-RDs). Time was defined as time from baseline for an individual on a specific outcome measure, and the model permitted missed annual visits. Models were adjusted for sex and time-varying covariates of weight, height, and age. Myometry and PFT models remained unadjusted because the measurements already adjust for demographics in a standardized way. Further analyses expanded diagnosis subtype into 4 clinically distinct groups (ambulant COL6-RDs, nonambulant COL6-RDs, ambulant LAMA2-RDs, and nonambulant LAMA2-RDs), where ambulant was defined as the ability to walk 10 m without orthotics or assistive devices. Changes over time were re-estimated using the same method as above. Plots showing subject-specific trajectories were used to aid in the interpretation of the model results. A Pearson correlation matrix plot was also created using data from the participants' enrollment years to describe the correlations among clinical tests. This plot is an easy way to show many pairwise correlations to gain an understanding of the relationships between variables. Color denotes the strength of the relationship, and the presence of an X denotes nonsignificance. Statistical tests were conducted with a significance level of p < 0.05. All analyses were performed with SAS version 9.4 (SAS Institute Inc, Cary, NC) and R version 3.3.1 (R Foundation for Statistical Computing, Vienna, Austria).
Data availability
Deidentified data will be shared on request with any qualified investigator.
Results
The participants are classified by year, diagnosis, ambulation status, and sex in table 1.
Table 1.
Annual demographics: disease subtype, ambulant status, respiratory support status, and feeding tube status

To further describe the clinical presentation of our cohort, we included the number of individuals who required a gastrostomy tube for nutrition and used nocturnal bilevel positive airway pressure (BiPAP) devices. Joint contractures were present in many of the participants, particularly of the hips, knees, elbows, and ankles. Table e-1 (available from Dryad, doi.org/10.5061/dryad.5p684d0) provides causative genetic variants and muscle biopsy results of each participant. Siblings of the same family are indicated as A/B with A being the older sibling.
The correlation plot (figure 1) demonstrates that all of the outcome measures evaluated correlate with each other with the exception of elbow extensor and flexor strength and the PedsQL measures. Similar groupings of measurements (MFM32, myometry, goniometry, FVC) show moderate to high correlations within themselves either on the subscores or across laterality (right vs left). Of note, the PedsQL Parent scales correlated only moderately (≈0.5–0.6) with some of the strength and functional measures, while the children's PedsQL scales did not correlate with any measures except the PedsQL Parent.
Figure 1. Correlation plot of clinical measures.
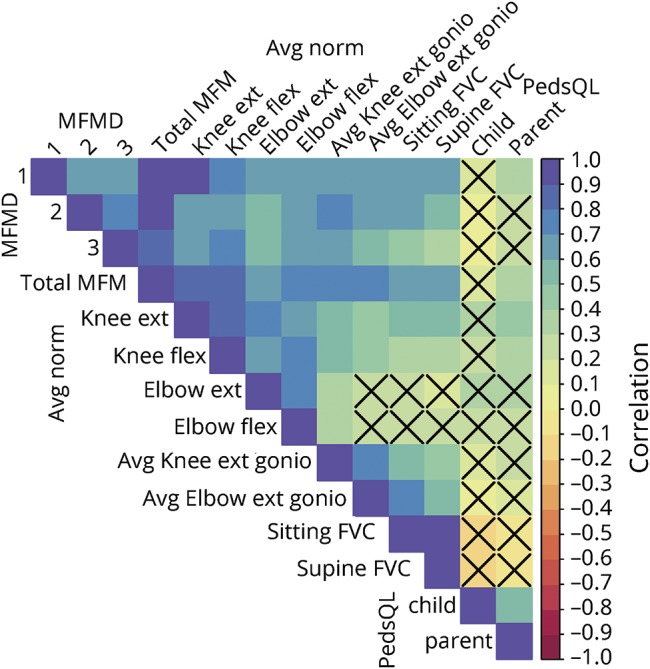
Blue indicates positive correlation; red indicates negative correlation. FVC = forced vital capacity; MFM = Motor Function Measure; PedsQL = Pediatric Quality of Life Inventory.
Rate of change of each outcome measure in participants with COL6-RD and LAMA2-RD
Table 2 shows the rate of change per year of the various outcome measures, stratified by diagnosis type of COL6-RD or LAMA2-RD. In the individuals with COL6-RD, a consistent, annual decrease in MFM32 scores was identified for all 3 MFM32 domain scores and the total MFM32 score, which showed a decline of −4.01 points per year (p < 0.01). On myometry testing, individuals with COL6-RD also demonstrated a loss of strength in all muscle groups measured except elbow flexors. For COL6-RDs, a significant yearly decline was observed in the FVCpp sitting of −3.03 percentage points (p < 0.01). Individuals with LAMA2-RD presented with a statistically significant yearly decrease in the MFM32 scores for the D2 domain and MFM32 total score, which decreased by −2.60 points per year (p < 0.01). In individuals with LAMA2-RD, strength remained stable except for knee flexor strength, which decreased by −2.30%/y (p < 0.01). Joint contractures, as measured by goniometry, reached statistical significance only for loss of range of motion for elbow extension on the left in individuals with LAMA2-RD and right knee extension in individuals with COL6-RD. No change in scores over time were noted on the PedsQL scale in either disease subtype.
Table 2.
Two-group rates of change for key outcome measures

Rate of change of outcome measures in COL6-RD and LAMA2-RD by ambulatory status
To describe the effects of ambulation on an individual's functional capabilities and on rates of decline, we further stratified the 2 CMD subtype groups into 4 groups based on ambulatory status at the time of entry into the study (table 3). Ambulatory status was defined as being able to walk 10 m without assistance.
Table 3.
Four-group rates of change for key outcome measures

Motor Function Measure 32
For the total MFM32 score, both ambulatory and nonambulatory individuals with COL6-RD showed statistically significant declines, whereas for individuals with LAMA2-RD, only those who were nonambulatory showed significant yearly declines (−2.16 points, p < 0.01). Given that the number of ambulant participants with LAMA2-RD was limited, this lack of statistical significance may be due to small sample size. Our cohort of nonambulatory individuals with COL6-RD had the greatest rate of decline of −4.54 points per year (p < 0.01), compared to −2.89 points per year (p < 0.01) for ambulant individuals with COL6-RD. Graphic representation of the MFM32 further displays the annual change in MFM32 total scores for each of the 2 subtypes of CMD, stratified by ambulatory status (figure 2).
Figure 2. MFM domain and total scores of each subject by subcategory.

(A) Motor Function Measure (MFM) D1 scores, (B), MFM D2 domain scores, (C) MFM D3 domain scores, and (D) total MFM scores.
Myometry
The pattern of decline of strength measurement varied by disease subtype and ambulatory status. Individuals with COL6-RD (ambulatory and nonambulatory) showed a significant annual rate of decline in average knee extensor strength (−2.48 percentage points, p < 0.01; −1.96, p < 0.05). A loss of knee flexor strength was demonstrated in ambulatory individuals with COL6-RD (−2.36%, p < 0.01) and nonambulatory individuals with LAMA2-RD (−2.47%, p < 0.01). Nonambulatory individuals with COL6-RD lost elbow flexor strength (−1.65%, p < 0.05) and elbow extensor strength (−2.67%, p < 0.05). One noted exception was in children with LAMA2-RD who were ambulatory, in whom the average elbow extensor strength improved. Of note, because there were very few ambulatory individuals with LAMA2-RD in our cohort, this increase in elbow extensor strength was driven by 1 individual's strength measures, while the others remained relatively stable.
Goniometry
Results differed between the 2 CMD subtypes. Nonambulatory individuals with COL6-RD demonstrated a significant annual rate of decline in both bilateral knee extension (−4.78°, p < 0.01) and bilateral elbow extension (−4.25°, p < 0.01). Nonambulatory individuals with LAMA2-RD showed an annual rate of contracture development in left elbow extension (−4.11°, p < 0.01) but no change in knee range of motion. Neither individuals in the ambulatory COL6-RD or ambulatory LAMA2-RD subgroups showed significant changes in elbow or knee range of motion.
Forced vital capacity
Likewise, FVC sitting differed between the 2 CMD subtypes. A significant annual rate of decline in FVCpp sitting was observed only in individuals with COL6-RD who were ambulatory (−3.44 percentage points per year; p < 0.01). The decrease in FVCpp sitting was not statistically significant in nonambulatory individuals with COL6-RD or in ambulatory or nonambulatory individuals with LAMA2-RD. The annual rate of change in FVCpp supine and the annual rate of relative change from sitting to supine (expressed as a percentage change) were not statistically significant in either COL6-RD or LAMA2-RD. Ambulatory status did not demonstrate a significant effect on annual rates of change of FVC.
Pediatric Quality of Life Inventory
PedsQL total scores did not show statistically significant changes over time for child and parent.
Ambulatory status and loss of ambulation
Over the course of the study, of the 20 participants who were ambulant at the time of entry, 16 (80%) were diagnosed with COL6-RD, and 12 (60%) were female. Five participants lost ambulation during the study (table e-1 available from Dryad, doi.org/10.5061/dryad.5p684d0), all of whom had COL6-RD and 4 of whom were female. In the entire cohort, 2 participants lost ambulation at the age of 8 years, 1 participant at the age of 10 years, and 2 participants at the age of 12 years. Of the participants with LAMA2-RD who were ambulatory in the first year of participation in the study, none lost ambulation.
Discussion
This prospective natural history and comparative outcome measure study of 2 common forms of CMD was designed with 2 main distinguishing features: (1) the incorporation of a 2-year pilot phase to identify feasible, reliable, and valid outcome measures that were carried forward into a study for an additional 3 years, resulting in a 5-year study, and (2) the collection of outcome measures by a team of trained evaluators at a yearly meeting of all study participants at a single site, the NIH Clinical Center. The outcome measures evaluated in detail here are the MFM32, myometry, goniometry, FVC, and PedsQL. Our primary goal was to evaluate the annual rate of change in each of these capacity measures in these 2 subtypes of CMD to establish the rate of disease progression and to evaluate their utility for future clinical trial planning and execution. Overall, across the multiple outcome measures assessed for sensitivity to change in this particular cohort, nonambulatory individuals with COL6-RD had a greater rate of decline than ambulatory individuals with COL6-RD or ambulatory and nonambulatory individuals with LAMA2-RD.
Table 4 summarizes the outcome measures that detected a significant change over 1 year in ambulatory and nonambulatory individuals for both CMD subtypes.
Table 4.
Summary of significant results by outcome measure, disease subtype, and ambulant status

For the 2-group analysis, that is, when only the subtype of CMD was considered, without further stratification for the functional status at trial entry, the MFM32 showed a significant rate of decline per year in the total score and in all domains (D1, D2, D3) for participants with COL6-RD but a decrease only in the D2 domain and total scores in individuals with LAMA2-RD. The MFM32 was sensitive to change in ambulatory as well as nonambulatory individuals in each disease subtype in total score and in at least 1 subdomain. We thus found the MFM32 to be the outcome measure that is most suitable to detect change over a period of 1 year across the spectrum of functional severity in LAMA2-RDs and COL6-RDs. Because the MFM32 covers a wide age range, spans children and adults of both ambulatory and nonambulatory ability, and reflects 3 different motor domains separately, our results support the MFM32 as a sensitive and versatile functional motor outcome measure for both COL6-RD and LAMA2-RD forms of CMD. Thus, we would recommend the use of the MFM32 as an outcome measure in clinical trials in these populations.
Myometry, goniometry, and FVC also showed selective sensitivity to capture change across both ambulatory and nonambulatory individuals with COL6-RD. While the changes in myometry were statistically significant, the changes in each muscle group in isolation may have been too small to be considered functionally meaningful. While individuals with COL6-RD showed decline in almost all myometry categories, nonambulatory individuals with LAMA2-RD demonstrated statistically significant decreases in knee flexion strength only.
All goniometry measures decreased in nonambulatory participants with COL6-RD, but only the decrease in elbow extension was statistically significant in nonambulatory participants with LAMA2-RD. In contrast, goniometry did not decrease significantly in ambulatory individuals with either CMD subtype. As is the case in myometry measurements, the change in range of motion is statistically significant but may not be functionally meaningful. A loss of 3° to 4° may not significantly affect a person's motor function; however, because incremental changes are cumulative over time, progressive contractures can significantly affect independence with function. For instance, the need for functional elbow range of motion was demonstrated to be from 75° to 120° to perform traditional activities of daily living.21 A minimum arc of elbow range of motion of 23° has been reported for contemporary tasks such as using a cell phone.22 The combination of decreased strength with decreased range of motion due to joint contractures contributes to further loss of function compared to decreased strength alone. For example, with muscle weakness, it may be possible to ambulate with gait aids (e.g., ankle-foot orthoses, gait trainers); however, with worsening joint contractures, the biomechanical alignment changes, often resulting in loss of function such as loss of ambulation.
Over the 5 annual time points evaluated in this study, 5 individuals lost ambulation, which occurred between the ages of 8 and 12 years in individuals with COL6-RD, 4 of whom were female. While only individuals with COL6-RD lost ambulation in this study, it is notable that there were 13 more ambulatory individuals with COL6-RD than LAMA2-RD. The 5 children who lost the ability to ambulate showed a decline in all 3 domains of their MFM32 scores, while myometry and PFT results either remained stable or may have improved for one of the time points over the course of the study. The MFM32 scale was the only assessment that exhibited a consistent decline over the 4 years. Similar to previous findings, the mean age at loss of ambulation in COL6-RD was ≈10 years.23,24 The loss of ambulation is less well understood to date in individuals with the partial merosin deficiency form of LAMA2-RD; however, a previous report and our findings suggest that individuals with LAMA2-RD lose ambulation at a later mean age than those with COL6-RD.25 In our ambulant cohort, D1 of the MFM32 continued to worsen in contrast to stable scores on D2 and D3. The opposite was noted in nonambulant individuals (figure 2, A–D).
In this study, the annual rate of decline of FVCpp in the upright (sitting) position was statistically significant only in ambulatory individuals with COL6-RD, while the annual rate of decline of FVCpp was not significant in nonambulatory individuals with COL6-RD. This is in contrast to previous natural history data reported in an international series of 145 individuals with COL6-RD. In that retrospective study, nonambulatory individuals with Ullrich CMD (UCMD) who did not achieve independent ambulation demonstrated a decline in sitting FVC of 4.2% predicted per year (p < 0.0001). Those with UCMD who achieved walking with assistance declined only in sitting FVC by 2.1%/y (p = 0.0003)23,26 This difference may, in part, be due to the much smaller sample size in this 5-year prospective natural history study. A natural history study of pulmonary function in 65 individuals with LAMA2-RD has revealed an annual rate of decline of sitting FVC of ≈1.5% predicted per year in nonambulatory individuals (unpublished data).
It is also important to note that the individuals with COL6-RD or LAMA2-RD evaluated as part of this study have restrictive lung disease secondary to extrinsic causes (causes extrinsic to the lung parenchyma), namely diaphragmatic weakness, intercostal muscle weakness, decreased compliance of the chest wall, and thoracic deformities related to scoliosis, resulting in a progressive decline in pulmonary function and the need for noninvasive ventilation in the form of BiPAP.27,28 In fact, respiratory insufficiency is the leading cause of morbidity and mortality in COL6-RD and LAMA2-RD, particularly if unrecognized or underrecognized. For this reason, pulmonary function should be carefully assessed and monitored in any natural history study or clinical trial of individuals with COL6-RDs or LAMA2-RDs, with consideration given to the fact that individual trajectories of decline in pulmonary function may not be linear but variable at different time points. Furthermore, there may be different rates of decline in FVC during various age ranges; previously reported rates of decline in sitting FVC in individuals with UCMD were 2.6% predicted per year overall (p < 0.0001) between the ages of 4 and 30 years but 3.5%/y between the ages of 5 and 15 years (p < 0.0001).23
One contributing factor that may explain the lack of decline in sitting or supine FVC in individuals with COL6-RD or LAMA2-RD evaluated during this study could be that nonambulatory participants who participated in this study had already reached a nadir of their restrictive lung disease, in which case low FVC values may not have demonstrated further decline during the course of participation in this natural history study. In addition, the use of noninvasive ventilation may play a significant role in potentially stabilizing pulmonary function. Therefore, established BiPAP use in nonambulant individuals with COL6-RD and LAMA2-RD should be documented, given that it may have a potential stabilizing effect on PFT results in individuals with COL6-RD or LAMA2-RD.
Quality of life, as measured by PedsQL, did not demonstrate statistically significant changes. This could be a result of a phenomenon described as the disability paradox, in which an individual with a disability rates his or her quality of life as good while others such as family member or parents rate it as poor.29 Multiple studies report that children with Duchenne muscular dystrophy30,31 or spinal muscular atrophy32,33 rate themselves with higher quality-of-life scores than their parents/guardians. These studies are cross-sectional in nature, with a paucity of longitudinal studies assessing quality-of-life changes over time. The question remains as to whether, in a treatment trial, quality-of-life measures would improve if anchored to another outcome measure such as the MFM32. This is an important consideration given the emphasis by the Food and Drug Administration on patient-reported outcomes.
As observed in some of the plots (figure 2, B–D), several individual trajectories appear at the edges of the scale, and thus an absence of a statistically significant change may be due to a floor or ceiling effect of the measurement. We argue that this is not a flaw in the measurement or scale itself, as is the case for standard floor or ceiling effects, but rather points toward a natural clinical bound for the measurement in question. For example, participants who are nonambulatory and thus perform poorly on MFM32-D1 at baseline are not expected to show any further decline in function in that domain, and thus, the domain measurement accurately reflects their function without blunting the sensitivity of the measure. This is best illustrated in the comparative analysis of the 4-group stratification (ambulant/nonambulant by subtype). While the MFM32-D1 alone would not be sufficient to show a decline in a nonambulant person, the other domains of the MFM32 provide the so-called functional bandwidth to compensate for this. For instance, when the D1 domain may not decline further, the D2 domain may show a decline (i.e., the individual still has functionality in D2 that can worsen to a degree that could be measured). Thus, an ambulant individual who is still “too functional” in domains D2 and D3 would potentially show a decline that would be adequately captured in domain D1 in which the functional loss is most sensitively recorded. As stated, the patterns of bounded longitudinal changes in some scales may be an artifact due to changes in patient function being bounded naturally and not necessarily restricted by the scales themselves.
The design of this 4-year study, which took place at a single site (NIH Clinical Center), allowed the standardized collection of data on a set of measures of motor function, pulmonary function, and quality of life. The study results enabled the identification of outcome measures best suited for capturing annual changes in individuals with COL6-RDs or LAMA2-RDs, the most common subtypes of CMD. Given the recent completion of the first clinical trial for these populations (Congenital Muscular Dystrophy Ascending Multiple Dose Cohort Study Analyzing Pharmacokinetics at Three Dose Levels in Children and Adolescents With Assessment of Safety and Tolerability of Omigapil, NCT01805024) and the prospect of further clinical trials in these subtypes, further delineation of their natural history and identification of viable outcome measures are essential for planning clinical trials for these rare diseases, which remain without therapeutic options.
Our study results may further contribute to clinical trial readiness for the COL6-RDs and the LAMA2-RDs by providing information for guiding sample size calculations and clinical trial design. Power calculations presented here are based on the observations made during this longitudinal study. For example, from our data, we estimate the average yearly decline in the total MFM32 score for COL6-RDs to be −4.01 points with an SD of 4.36. Using an α of 0.05 and power of 0.80, we would need 12 participants to detect a 4.01-point increase in total MFM32, to give a mean slope of 0 (i.e., stabilization of disease). In our study, 23 of 24 individuals with COL6-RD had longitudinal data available on this measure (5% dropout); thus, we would inflate the number of participants needed to 13. For rare diseases such as the COL6-RDs and LAMA2-RDs, these are feasible numbers for clinical trial recruitment. It will, of course, be important to consider whether this required effect size for an intervention under investigation can be reasonably expected. An intervention expected to be minimally efficacious may increase trends by only 2 points, for example. This would result in a slowing of disease progression by −2.01 per year, which would then require more individuals (43, accounting for dropouts). Conversely, in a study with an intervention that is expected to be highly efficacious, yearly trends may increase the MFM32 by 6 points to result in a disease improvement of +1.99 per year. This type of clinical trial scenario would require fewer participants (8, accounting for dropouts). An estimation of the minimal clinically important difference for the MFM32 for both types of CMD, including sensitivity to change analyses, is in preparation. This may help arrive at a more functionally anchored sample size calculation directed at the detection of the smallest clinically meaningful change.
This example helps demonstrate the utility of our COL6-RD and LAMA2-RD natural history dataset and its application to the estimation of sample size (for a single disease subtype, i.e., COL6-RD or LAMA2-RD). It uses a simplified model in that it involves only 1-sample t test on the estimated improvement in slopes against our known population trajectory. In a clinical trial, a mixed statistical model would likely be incorporated rather than a t test.
Taken together, the results of this study demonstrate the value and importance of natural history and outcome measure studies in rare disease populations such as COL6-RDs and LAMA2-RDs and specifically provide the tools for making clinical trials design feasible in these patient populations.
Acknowledgment
The authors thank the affected individuals and their families for their devoted participation to this study, without whom this study would not have been possible. They are grateful to the nurses who staffed the outpatient pediatric clinic and the radiology and phlebotomy technicians who performed blood draws, PFTs, and MRIs. They acknowledge Dr. Andrew Arai, Dr. Jessica Witherspoon, and Clara Jolley for their assistance with this study.
Glossary
- BiPAP
bilevel positive airway pressure
- CMD
congenital muscular dystrophy
- COL6-RD
COL6-related dystrophy
- FVCpp
forced vital capacity percent predicted
- LAMA2-RD
LAMA2-related dystrophy
- MFM32
Motor Function Measure 32
- PedsQL
Pediatric Quality of Life Inventory
- PFT
pulmonary function test
- UCMD
Ullrich congenital muscular dystrophy
Author contributions
Minal S. Jain: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Katherine Meilleur: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, study supervision. Eunhee Kim: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, statistical analysis. Gina Norato: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, statistical analysis. Melissa Waite and Leslie Nelson: data acquisition, accepts responsibility for conduct of research and will give final approval. Michelle McGuire: drafting/revising the manuscript, data acquisition, accepts responsibility for conduct of research and will give final approval. Tina Duong: drafting/revising the manuscript, data acquisition, study concept or design, accepts responsibility for conduct of research and will give final approval. Katherine Keller: data acquisition, accepts responsibility for conduct of research and will give final approval. Donovan J. Lott, Allan Glanzman, and Kristy Rose: drafting/revising the manuscript, data acquisition, accepts responsibility for conduct of research and will give final approval. Marion Main: study concept or design, accepts responsibility for conduct of research and will give final approval, expert opinion. Courtney Fiorini and Irene Chrismer: data acquisition, accepts responsibility for conduct of research and will give final approval. Melody Linton and Monal Punjabi: data acquisition, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Jeffrey Elliott: data acquisition, accepts responsibility for conduct of research and will give final approval. Fatoumata Tounkara: data acquisition, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Ruhi Vasavada: drafting/revising the manuscript, data acquisition, accepts responsibility for conduct of research and will give final approval, statistical analysis. Ranjani Logaraj: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Jocelyn Winkert: drafting/revising the manuscript, study concept or design, accepts responsibility for conduct of research and will give final approval, acquisition of data, statistical analysis. Sandra Donkervoort: data acquisition, study concept or design, accepts responsibility for conduct of research and will give final approval. Meganne Leach: data acquisition, accepts responsibility for conduct of research and will give final approval, contribution of vital reagents/tools/patients. Jahannaz Dastgir: data acquisition, study concept or design, accepts responsibility for conduct of research and will give final approval, study supervision. Linda Hynan: drafting/revising the manuscript, accepts responsibility for conduct of research and will give final approval, acquisition of data, statistical analysis. Carmel Nichols: data acquisition, study concept or design, accepts responsibility for conduct of research and will give final approval, study supervision. Elizabeth Hartnett and Gilberto M. Averion: data acquisition, accepts responsibility for conduct of research and will give final approval. James C. Collins: drafting/revising the manuscript, data acquisition, accepts responsibility for conduct of research and will give final approval. Eunice S. Kim: data acquisition, accepts responsibility for conduct of research and will give final approval. Angela Kokkinis: data acquisition, accepts responsibility for conduct of research and will give final approval, study supervision. Alice Schindler: data acquisition, study concept or design, accepts responsibility for conduct of research and will give final approval, study supervision. Kristen Zukosky: drafting/revising the manuscript, data acquisition, accepts responsibility for conduct of research and will give final approval. Robert Fee: data acquisition, accepts responsibility for conduct of research and will give final approval. Veronica Hinton: data acquisition, study concept or design, accepts responsibility for conduct of research and will give final approval, study supervision. Payam Mohassel: data acquisition, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Diana Bharucha-Goebel: data acquisition, accepts responsibility for conduct of research and will give final approval. Carole Vuillerot: drafting/revising the manuscript, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, contribution of vital reagents/tools/patients. Peter McGraw and Mark Barton: data acquisition, accepts responsibility for conduct of research and will give final approval. Joseph Fontana: analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Anne Rutkowski: study concept or design, accepts responsibility for conduct of research and will give final approval, contribution of vital reagents/tools/patients, study supervision. A. Reghan Foley: drafting/revising the manuscript, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval. Carsten G. Bönnemann: drafting/revising the manuscript, data acquisition, study concept or design, analysis or interpretation of data, accepts responsibility for conduct of research and will give final approval, study supervision, obtaining funding.
Study funding
This study was supported by the Divisions of Intramural Research of National Institute of Neurological Disorders and Stroke and National Institute of Nursing Research, the NIH Clinical Center, and CureCMD.
Disclosure
M. Jain, K. Meilleur, E. Kim, G. Norato, M. Waite, L. Nelson, M. McGuire, T. Duong, K. Keller, and D. Lott report no disclosures relevant to the manuscript. A. Glanzman serves as a board member for Biogen Pharmaceuticals. K. Rose, M. Main, C. Fiorini, I. Chrismer, M. Linton, M. Punjabi, J. Elliott, F. Tounkara, R. Vasavada, R. Logaraj, J. Winkert, S. Donkervoort, and M. Leach report no disclosures relevant to the manuscript. J. Dastgir is a consultant for Avexis, Sarepta, and Biogen Pharmaceuticals. L. Hynan, C. Nichols, E. Hartnett, G. Averion, J. Collins, E. Kim, A. Kokkinis, A. Schindler, K. Zukosky, R. Fee, V. Hinton, P. Mohassel, D. Bharucha-Goebel, C. Vuillerot, P. McGraw, M. Barton, J. Fontana, A. Rutkowski, A. Foley, and C. Bönnemann report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Wang CH, Bönnemann CG, Rutkowski A, et al. Consensus statement on standard of care for congenital muscular dystrophies. J Child Neurol 2010;25:1559–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graziano A, Bianco F, D'Amico A, et al. Prevalence of congenital muscular dystrophy in Italy: a population study. Neurology 2015;84:904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Norwood FLM, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain 2009;132:3175–3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lisi M, Cohn R. Congenital muscular dystrophies: new aspects of an expanding group of disorders. Biochim Biophys Acta 2007;1772:159–172. [DOI] [PubMed] [Google Scholar]
- 5.Bönnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol 2011;7:379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bönnemann CG, Rutkowski A, Mercuri E, Muntoni F; CMD Outcomes Consortium. 173rd ENMC International Workshop: congenital muscular dystrophy outcome measures 5-7 March 2010, Naarden, the Netherlands. Neuromusc Disord 2011;21:513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meilleur KG, Jain MS, Hynan LS, et al. Results of a two-year pilot study of clinical outcome measures in collagen VI-related myopathy and LAMA2-related muscular dystrophy. Neuromuscul Disord 2015;25:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berard C, Payan C, Hodgkinson I, Fermanian J; MFM Collaborative Study Group. A motor function measure scale for neuromuscular diseases: construction and validation study. Neuromuscul Disord 2005;15:463–470. [DOI] [PubMed] [Google Scholar]
- 9.Vuillerot C, Payan C, Girardot F, et al. Responsiveness of the motor function measure in neuromuscular diseases. Arch Phys Med Rehabil 2012;93:2251–2256. [DOI] [PubMed] [Google Scholar]
- 10.Vuillerot C, Rippert P, Kinet V, et al. Rasch analysis of the motor function measure in patients with congenital muscle dystrophy and congenital myopathy. Arch Phys Med Rehabil 2014;95:2086–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller MR, Handkinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J 2005;26:319–338. [DOI] [PubMed] [Google Scholar]
- 12.Hankinson JL, Odencrantz JR, Fedan KB. Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med 1999;159:179–187. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Dockery DW, Wypij D, Fay ME, Ferris BG Jr. Pulmonary function between 6 and 18 years of age. Pediatr Pulmonol 1993;15:75–88. [DOI] [PubMed] [Google Scholar]
- 14.Beenakker EAC, van der Hoeven JH, Fock JM, Maurits NM. Reference values of maximum isometric muscle force obtained in 270 children aged 4–16 years by hand-held dynamometry. Neuromusc Disord 2001;11:441–446. [DOI] [PubMed] [Google Scholar]
- 15.Pandya S, Florence JM, King WM, Robison JD, Oxman M, Province MA. Reliability of goniometric measurements in patients with Duchenne muscular dystrophy. Phys Ther 1985;65:1339–1342. [DOI] [PubMed] [Google Scholar]
- 16.Iannaccone ST, Hynan LS, Morton A, Buchanan R, Limbers CA, Varni JW; AmSMART Group. The Peds QL in pediatric patients with spinal muscular atrophy; feasibility, reliability, and validity of the pediatric Quality of Life Inventory Generic Core Scales and Neuromuscular Module. Neuromuscul Disord 2009;19:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis SE, Hynan LS, Limbers CA, et al. The PedsQL in pediatric patients with Duchenne muscular dystrophy: feasibility, reliability, and validity of the pediatric quality of life Inventory neuromuscular module and generic core scales. J Clin Neuromuscul Dis 2010;11:97–109. [DOI] [PubMed] [Google Scholar]
- 18.Hu J, Jiang L, Hong S, Cheng L, Kong M, Ye Y. Reliability and validity of the Chinese version of the Pediatric Quality of Life Inventory (PedsQL) 3.0 Neuromuscular Module in children with Duchenne muscular dystrophy. Health Qual Life Outcomes 2013;11:47, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dunaway S, Montes J, Montgomery M, et al. Reliability of telephone administration of the PedsQL Generic Quality of Life Inventory and neuromuscular Module in spinal muscular atrophy (SMA). Neuromuscul Disord 2010;20:162–165. [DOI] [PubMed] [Google Scholar]
- 20.Laird NM, Ware JH. Random-effects models for longitudinal data. Biometrics 1982;38:963–974. [PubMed] [Google Scholar]
- 21.Vasen AP, Lacey SH, Keith MW, Shaffer JW. Functional Range of motion of the elbow. J Hand Surg 1995;20:288–292. [DOI] [PubMed] [Google Scholar]
- 22.Sardelli M, Tashjian RZ, MacWilliams BA. Functional elbow range of motion for contemporary tasks. J Bone Joint Surg Am 2011;93:471–477. [DOI] [PubMed] [Google Scholar]
- 23.Foley AR, Quijano-Roy S, Collins J, et al. Natural history of pulmonary function in collagen-VI related myopathies. Brain 2013;136:3625–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nadeau A, Kinali M, Main M, et al. Natural history of Ullrich congenital muscular dystrophy. Neurology 2009;73:25–31. [DOI] [PubMed] [Google Scholar]
- 25.Geranmayeh F, Clement E, Feng L, et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromusc Disord 2010;20:241–250. [DOI] [PubMed] [Google Scholar]
- 26.Meilleur KG, Linton M, Fontana J, et al. Comparison of sitting and supine forced vital capacity in collagen VI-related dystrophy and laminin alpha2 related dystrophy. Pediatr Pulmonol 2017;52:524–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laghi F, Tobin MJ. Disorders of the respiratory muscles. Am J Respir Crit Care Med 2003;168:10–48. [DOI] [PubMed] [Google Scholar]
- 28.Smith PE, Calverley PM, Edwards RH, Evans GA, Campbell EJ. Practical problems in the respiratory care of patients with muscular dystrophy. N Engl J Med 1987;316:1197–1205. [DOI] [PubMed] [Google Scholar]
- 29.Albrecht GL, Devlieger PJ. The disability paradox: high quality of life against all odds. Soc Sci Med 1999; 38: 977–988. [DOI] [PubMed] [Google Scholar]
- 30.Landfeldt E, Lindgren P, Bell CF, et al. Health-related quality of life in patients with Duchenne muscular dystrophy: a multinational, cross-sectional study. Dev Med Child Neurol 2015; 58: 508–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uzark K, King E, Cripe L, et al. Health-related quality of life in children and adolescents with Duchenne muscular dystrophy. Pediatrics 2012;130:e1559–e1566. [DOI] [PubMed] [Google Scholar]
- 32.de Oliveira CM, Araujo AP. Self-reported quality of life has no correlation with functional status in children and adolescents with spinal muscular atrophy. Eur J Paediatr Neurol 2011;15:36–39. [DOI] [PubMed] [Google Scholar]
- 33.Kocova H, Dvorackova O, Vondracek P, Haberlova J. Health-related quality of life in children and adolescents with spinal muscular atrophy in the Czech Republic. Pediatr Neurol 2014;50:591–594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Deidentified data will be shared on request with any qualified investigator.